Tunicamycin-Induced ER Stress is Accompanied with Oxidative Stress via Abrogation of Sulfur Amino Acids Metabolism in the Liver

Abstract
:1. Introduction
2. Results
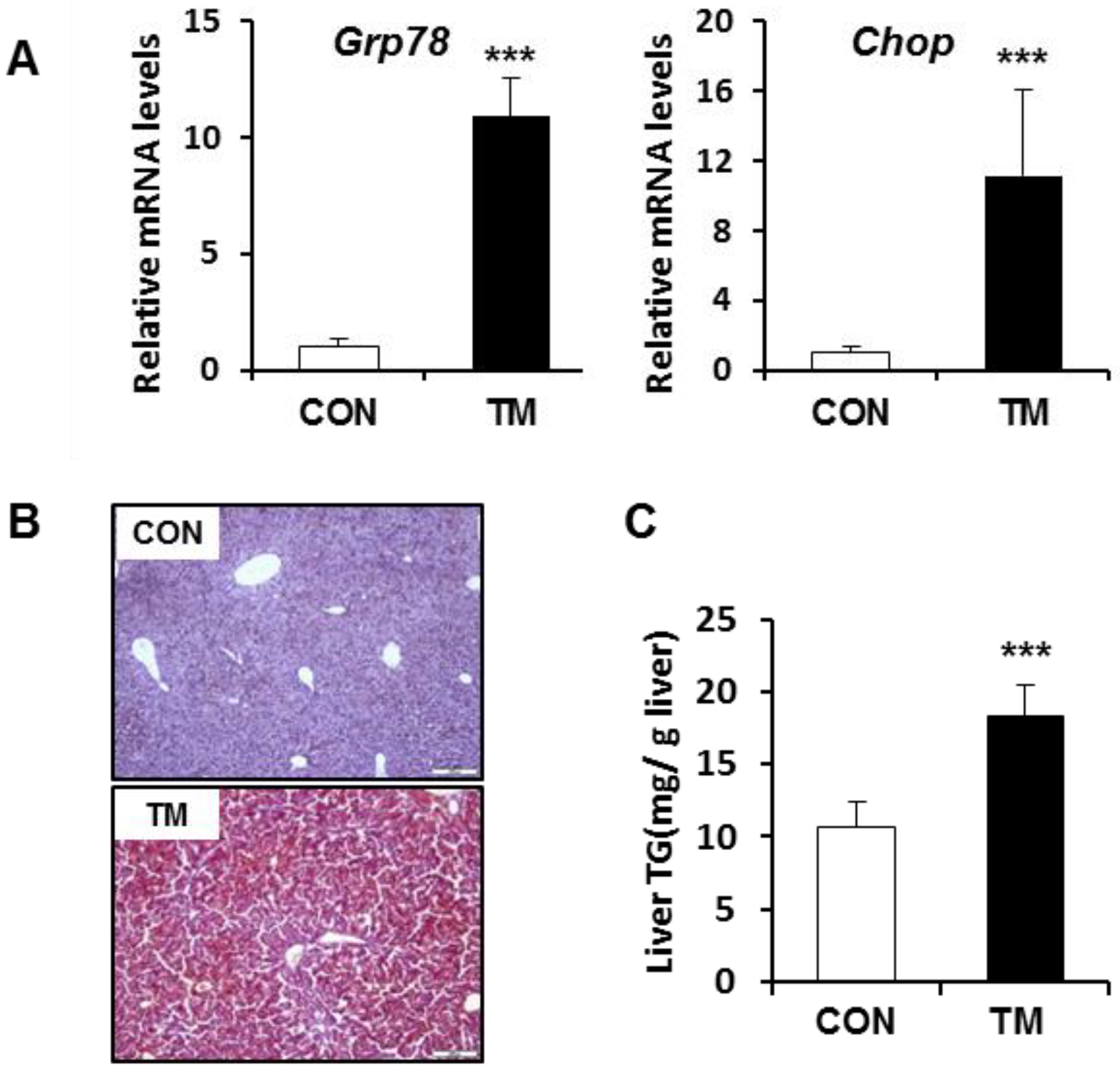
2.1. ER Stress-Mediated Lipid Accumulation in the Liver of Tunicamycin (TM)-Treated Mice
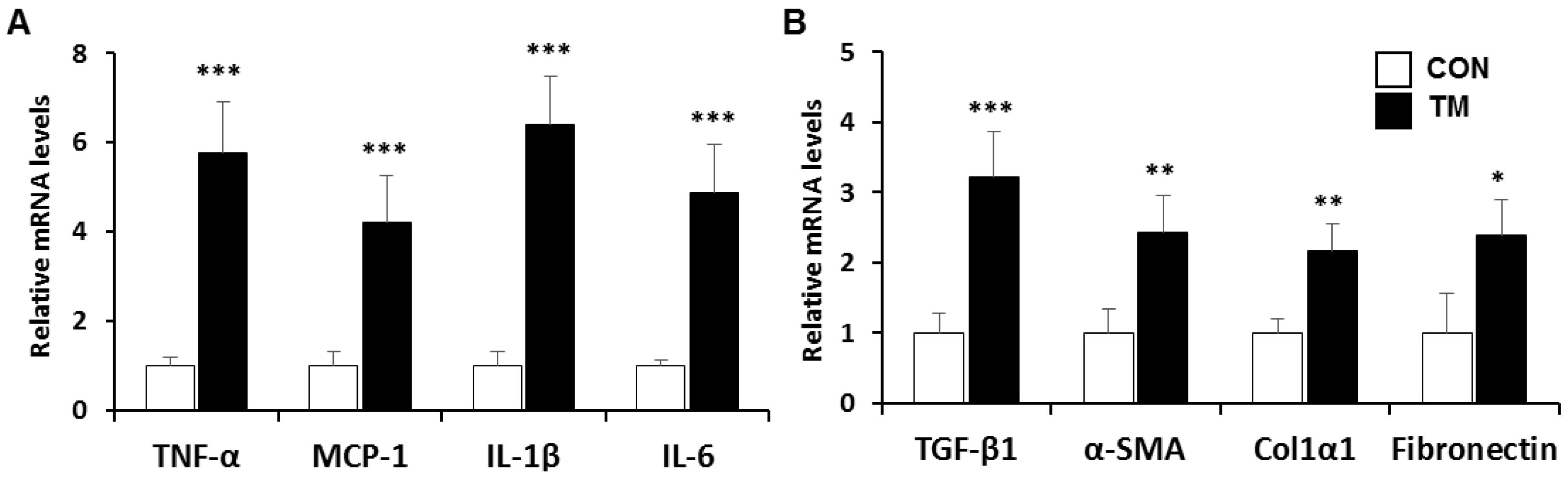
2.2. Changes in mRNA Level Related with Inflammatory Response and Fibrogenesis in the Liver of TM-Treated Mice
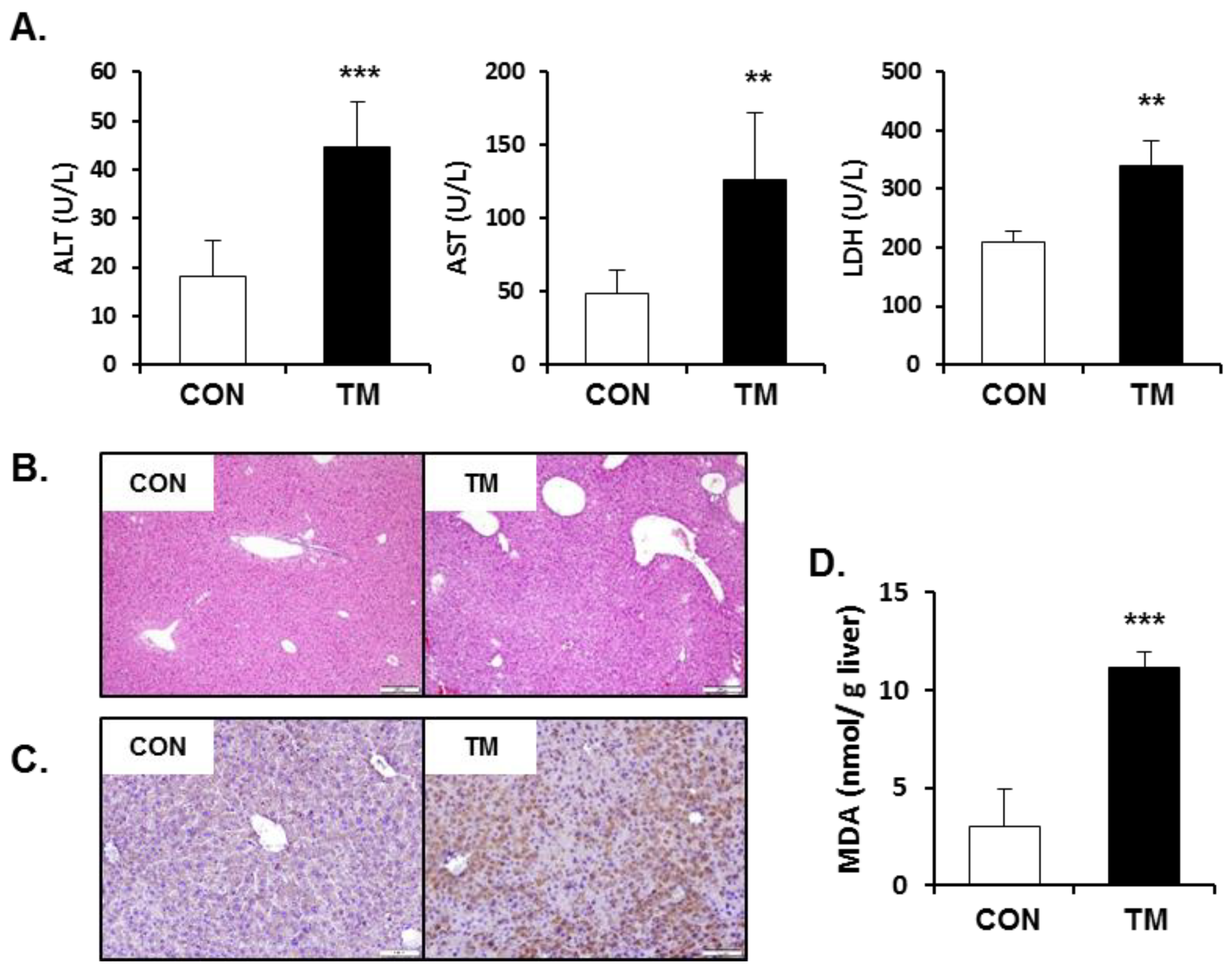
2.3. Increased Hepatotoxicity Accompanied with Oxidative Stress due to TM
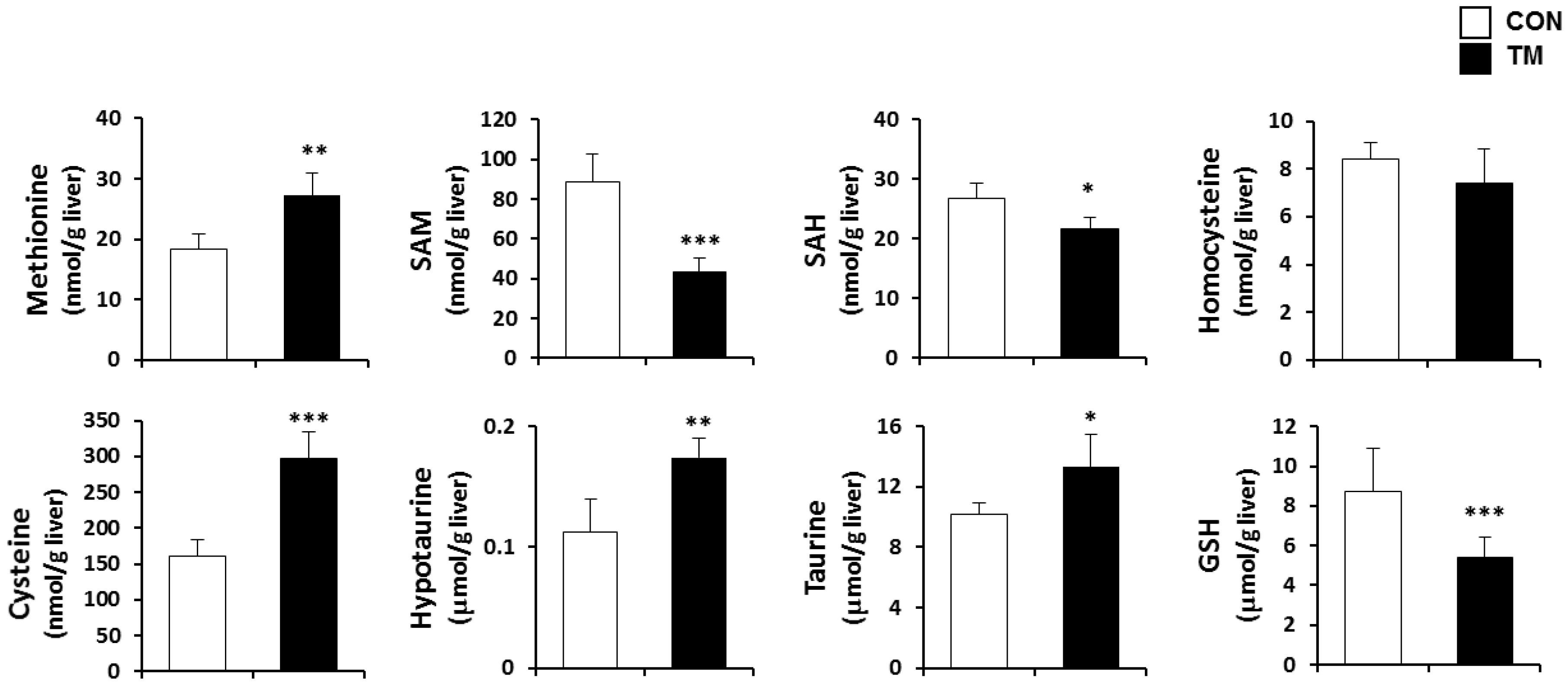
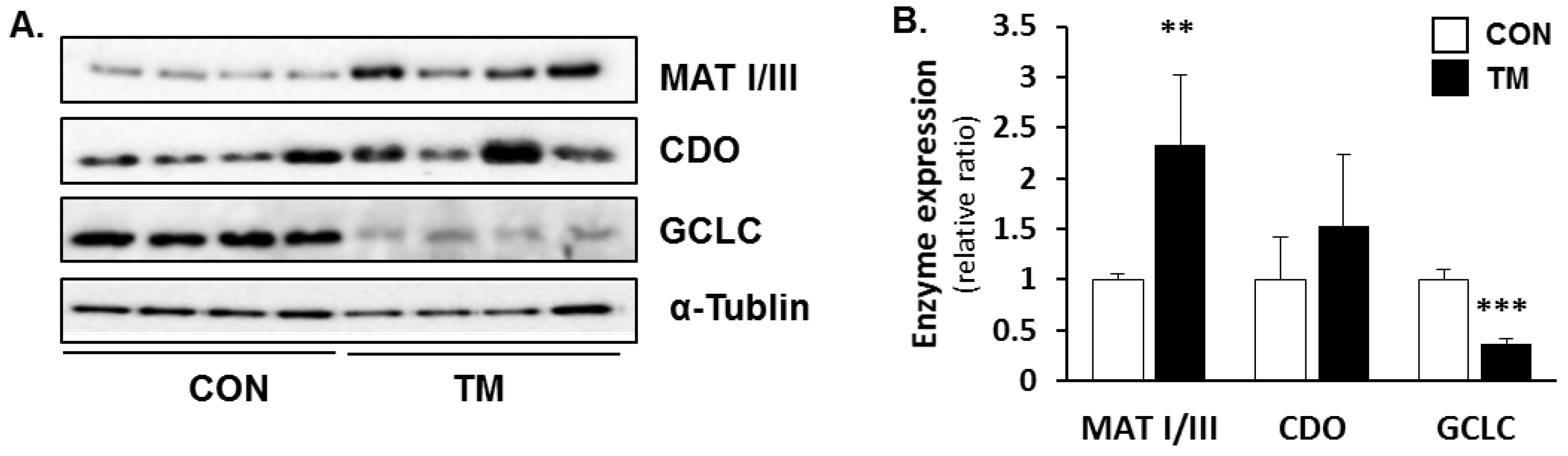
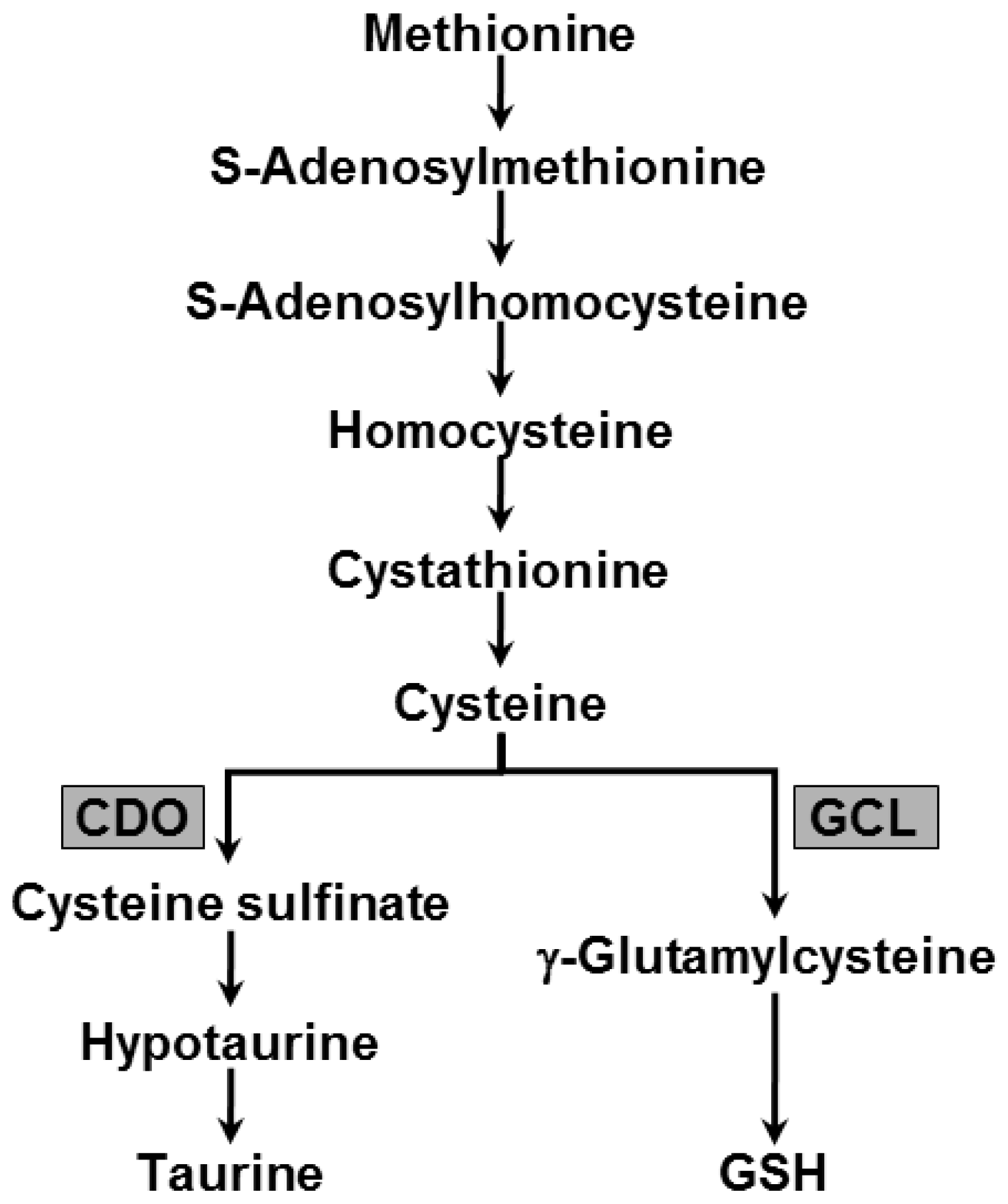
2.4. Changes in Hepatic Metabolism of Sulfur Containing Substances due to TM Treatment
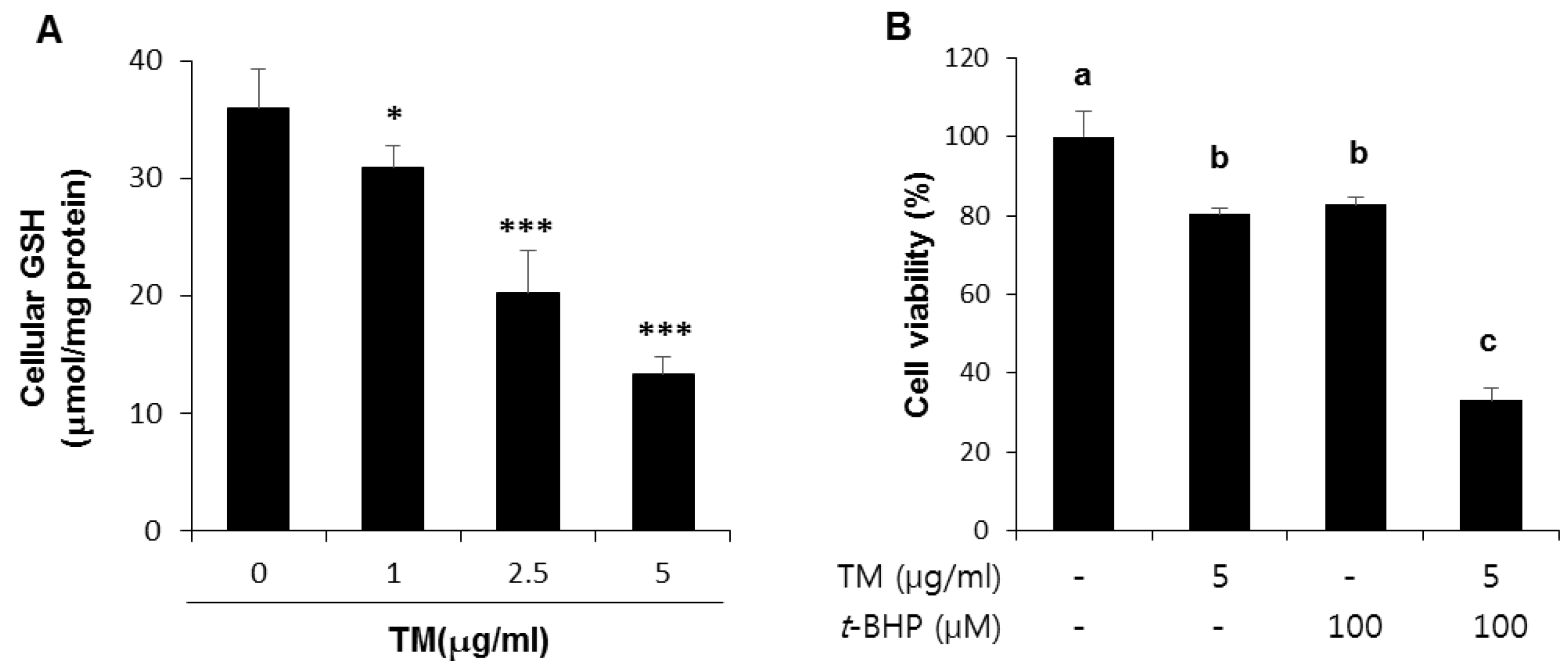
2.5. Decreased Levels of Cellular GSH by TM Treatment in a Dose-Dependent Manner and Potentiation of t-BHP Induced Cell Death by Pre-Exposure of TM in HepG2 Liver Cells
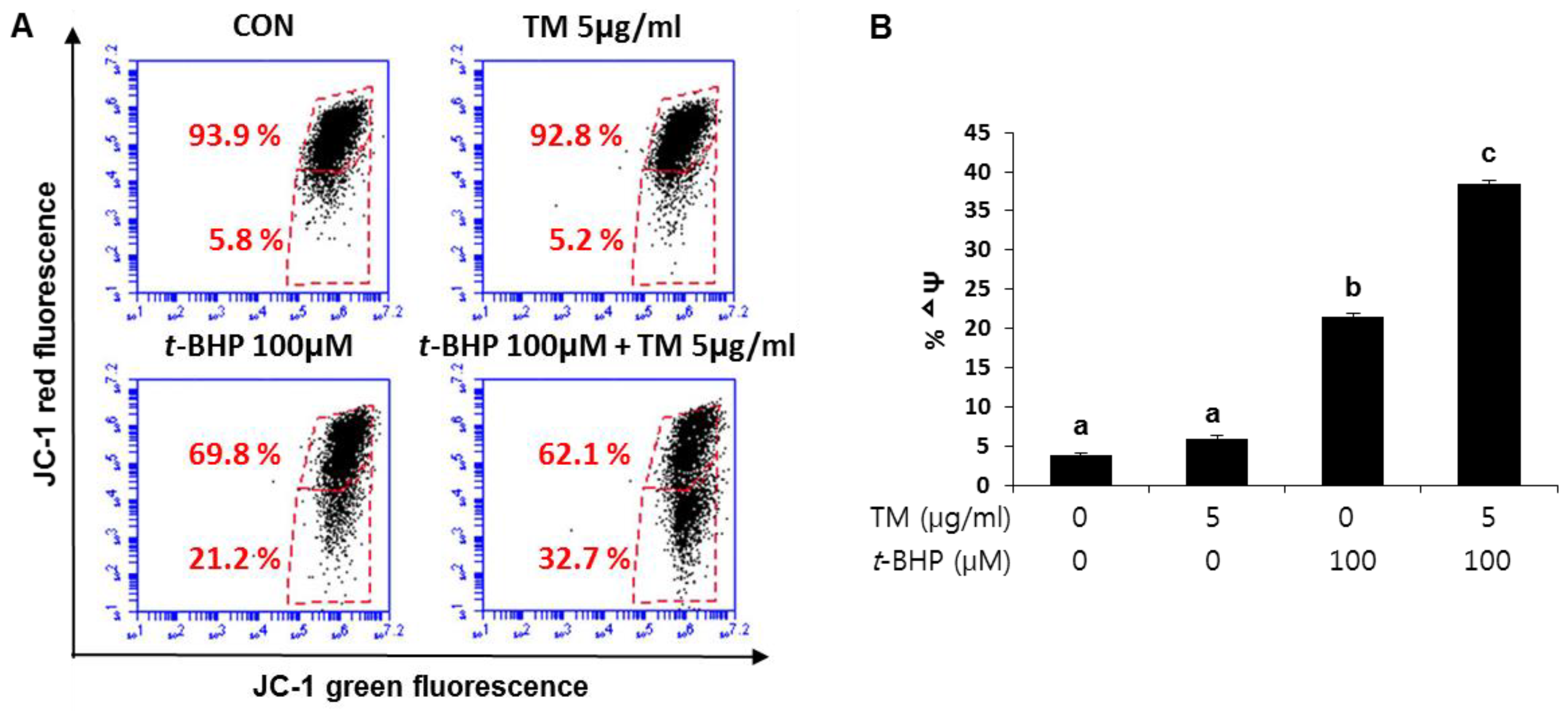
2.6. Involvement of Mitochondrial Dysfunction in the Enhancement of t-BHP-Induced Mitochondrial Dysfunction in the TM Pre-Exposed HepG2 Liver Cells
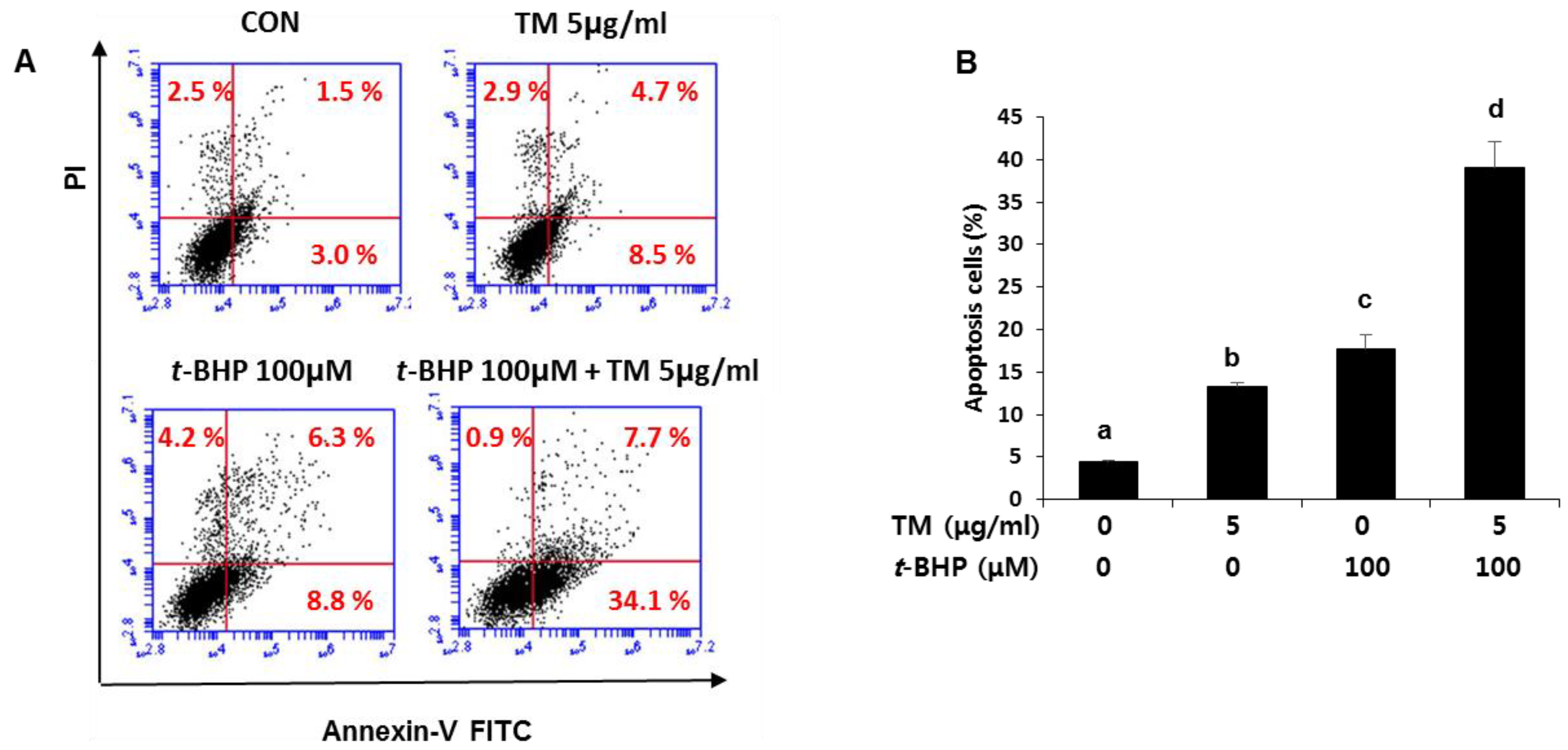
2.7. Enhancement of t-BHP-Induced Apoptotic Cell Death by Pre-Exposure of TM in HepG2 Liver Cells
3. Discussion
4. Materials and Methods
4.1. Animals Experiment
4.2. Hematological Evaluation Indicating Hepatotoxicity
4.3. Histopathological Analysis
4.4. Determination of Triglyceride (TG) in the Liver
4.5. Examination of Lipid Peroxidation in the Liver
4.6. Real-Time Reverse Transcription-Polymerase Chain Reaction (RT-PCR)
4.7. Western Blotting
4.8. Determination of Sulfur-Containing Substances
4.9. Cell Culture
4.10. Determination of Cell Viability
4.11. Analysis of Mitochondrial Membrane Potential (∆ψ)
4.12. Determination of Apoptotic Cells Using FACS Analysis
4.13. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| NAFLD | Non-alcoholic fatty liver disease |
| NASH | Non-alcoholic steatohepatitis |
| TM | tunicamycin |
| ER | endoplasmic reticulum |
| ROS | reactive oxygen species |
| GSH | glutathione |
| t-BHP | tert-butyl hydroperoxide |
References
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Diehl, A.M.; Brunt, E.M.; Cusi, K.; Charlton, M.; Sanyal, A.J. The diagnosis and management of non-alcoholic fatty liver disease: Practice guideline by the American Gastroenterological Association, American Association for the Study of Liver Diseases, and American College of Gastroenterology. Gastroenterology 2012, 142, 1592–1609. [Google Scholar] [CrossRef] [PubMed]
- Loomba, R.; Sanyal, A.J. The global NAFLD epidemic. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 686–690. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Allen, A.M.; Wang, Z.; Prokop, L.J.; Murad, M.H.; Loomba, R. Fibrosis progression in nonalcoholic fatty liver vs nonalcoholic steatohepatitis: A systematic review and meta-analysis of paired-biopsy studies. Clin. Gastroenterol. Hepatol. 2015, 13, 643–654. [Google Scholar] [CrossRef] [PubMed]
- Browning, J.D.; Horton, J.D. Molecular mediators of hepatic steatosis and liver injury. J. Clin. Investig. 2004, 114, 147–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, J.C.; Horton, J.D.; Hobbs, H.H. Human fatty liver disease: Old questions and new insights. Science 2011, 332, 1519–1523. [Google Scholar] [CrossRef] [PubMed]
- Day, C.P.; James, O.F. Steatohepatitis: A tale of two "hits"? Gastroenterology 1998, 114, 842–845. [Google Scholar] [CrossRef]
- Lebeaupin, C.; Vallee, D.; Hazari, Y.; Hetz, C.; Chevet, E.; Bailly-Maitre, B. Endoplasmic reticulum stress signalling and the pathogenesis of non-alcoholic fatty liver disease. J. Hepatol. 2018, 69, 927–947. [Google Scholar] [CrossRef] [PubMed]
- Ghemrawi, R.; Battaglia-Hsu, S.F.; Arnold, C. Endoplasmic Reticulum Stress in Metabolic Disorders. Cells 2018, 7, 63. [Google Scholar] [CrossRef] [PubMed]
- Tariq, Z.; Green, C.J.; Hodson, L. Are oxidative stress mechanisms the common denominator in the progression from hepatic steatosis towards non-alcoholic steatohepatitis (NASH)? Liver Int. 2014, 34, e180–e190. [Google Scholar] [CrossRef] [PubMed]
- Koek, G.H.; Liedorp, P.R.; Bast, A. The role of oxidative stress in non-alcoholic steatohepatitis. Clin. Chim. Acta 2011, 412, 1297–1305. [Google Scholar] [CrossRef] [PubMed]
- Del Ben, M.; Polimeni, L.; Carnevale, R.; Bartimoccia, S.; Nocella, C.; Baratta, F.; Loffredo, L.; Pignatelli, P.; Violi, F.; Angelico, F. NOX2-generated oxidative stress is associated with severity of ultrasound liver steatosis in patients with non-alcoholic fatty liver disease. BMC Gastroenterol. 2014, 14, 81. [Google Scholar] [CrossRef] [PubMed]
- Pessayre, D. Role of mitochondria in non-alcoholic fatty liver disease. J. Gastroenterol. Hepatol. 2007, 22 (Suppl. S1), S20–S27. [Google Scholar] [CrossRef] [PubMed]
- Novo, E.; Busletta, C.; Bonzo, L.V.; Povero, D.; Paternostro, C.; Mareschi, K.; Ferrero, I.; David, E.; Bertolani, C.; Caligiuri, A.; et al. Intracellular reactive oxygen species are required for directional migration of resident and bone marrow-derived hepatic pro-fibrogenic cells. J. Hepatol. 2011, 54, 964–974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singal, A.K.; Jampana, S.C.; Weinman, S.A. Antioxidants as therapeutic agents for liver disease. Liver Int. 2011, 31, 1432–1448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardy, T.; Oakley, F.; Anstee, Q.M.; Day, C.P. Nonalcoholic Fatty Liver Disease: Pathogenesis and Disease Spectrum. Annu. Rev. Pathol. 2016, 11, 451–496. [Google Scholar] [CrossRef] [PubMed]
- Spahis, S.; Delvin, E.; Borys, J.M.; Levy, E. Oxidative Stress as a Critical Factor in Nonalcoholic Fatty Liver Disease Pathogenesis. Antioxid. Redox Signal. 2017, 26, 519–541. [Google Scholar] [CrossRef] [PubMed]
- Marciniak, S.J.; Yun, C.Y.; Oyadomari, S.; Novoa, I.; Zhang, Y.; Jungreis, R.; Nagata, K.; Harding, H.P.; Ron, D. CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev. 2004, 18, 3066–3077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, K.; Shen, X.; Wu, J.; Sakaki, K.; Saunders, T.; Rutkowski, D.T.; Back, S.H.; Kaufman, R.J. Endoplasmic reticulum stress activates cleavage of CREBH to induce a systemic inflammatory response. Cell 2006, 124, 587–599. [Google Scholar] [CrossRef] [PubMed]
- Rutkowski, D.T.; Wu, J.; Back, S.H.; Callaghan, M.U.; Ferris, S.P.; Iqbal, J.; Clark, R.; Miao, H.; Hassler, J.R.; Fornek, J.; et al. UPR pathways combine to prevent hepatic steatosis caused by ER stress-mediated suppression of transcriptional master regulators. Dev. Cell 2008, 15, 829–840. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.; Takahara, K.; Oyadomari, S.; Okada, T.; Sato, T.; Harada, A.; Mori, K. Induction of liver steatosis and lipid droplet formation in ATF6alpha-knockout mice burdened with pharmacological endoplasmic reticulum stress. Mol. Biol. Cell 2010, 21, 2975–2986. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Wang, S.; Malhotra, J.; Hassler, J.R.; Back, S.H.; Wang, G.; Chang, L.; Xu, W.; Miao, H.; Leonardi, R.; et al. The unfolded protein response transducer IRE1alpha prevents ER stress-induced hepatic steatosis. EMBO J. 2011, 30, 1357–1375. [Google Scholar] [CrossRef] [PubMed]
- Jung, T.W.; Hwang, H.J.; Hong, H.C.; Choi, H.Y.; Yoo, H.J.; Baik, S.H.; Choi, K.M. Resolvin D1 reduces ER stress-induced apoptosis and triglyceride accumulation through JNK pathway in HepG2 cells. Mol. Cell. Endocrinol. 2014, 391, 30–40. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.S.; Tsai, C.T.; Huangfu, C.A.; Huang, W.Y.; Lei, H.Y.; Lin, C.F.; Su, I.J.; Chang, W.T.; Wu, P.H.; Chen, Y.T.; et al. ACSL3 and GSK-3beta are essential for lipid upregulation induced by endoplasmic reticulum stress in liver cells. J. Cell. Biochem. 2011, 112, 881–893. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Vera, L.; Fischer, W.H.; Montminy, M. The CREB coactivator CRTC2 links hepatic ER stress and fasting gluconeogenesis. Nature 2009, 460, 534–537. [Google Scholar] [CrossRef] [PubMed]
- Feng, B.; Huang, X.; Jiang, D.; Hua, L.; Zhuo, Y.; Wu, D. Endoplasmic Reticulum Stress Inducer Tunicamycin Alters Hepatic Energy Homeostasis in Mice. Int. J. Mol. Sci. 2017, 18, 1710. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.; Zhang, C.; Zhang, K. Measurement of ER stress response and inflammation in the mouse model of nonalcoholic fatty liver disease. Methods Enzymol. 2011, 489, 329–348. [Google Scholar] [PubMed]
- Lee, J.S.; Zheng, Z.; Mendez, R.; Ha, S.W.; Xie, Y.; Zhang, K. Pharmacologic ER stress induces non-alcoholic steatohepatitis in an animal model. Toxicol. Lett. 2012, 211, 29–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koo, J.H.; Lee, H.J.; Kim, W.; Kim, S.G. Endoplasmic Reticulum Stress in Hepatic Stellate Cells Promotes Liver Fibrosis via PERK-Mediated Degradation of HNRNPA1 and Up-regulation of SMAD2. Gastroenterology 2016, 150, 181–193. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.S. Metabolism of Sulfur-Containing Amino Acids in the Liver: A Link between Hepatic Injury and Recovery. Biol. Pharm. Bull. 2015, 38, 971–974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.H.; Kim, S.H.; Lee, S.; Kim, K.M.; Jung, J.C.; Son, T.G.; Ki, S.H.; Seo, W.D.; Kwak, J.H.; Hong, J.T.; et al. Antioxidant Effect of Barley Sprout Extract via Enhancement of Nuclear Factor-Erythroid 2 Related Factor 2 Activity and Glutathione Synthesis. Nutrients 2017, 9, 1252. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Yoon, D.; Lee, Y.H.; Lee, J.; Kim, N.D.; Kim, S.; Jung, Y.S. Transformation of liver cells by 3-methylcholanthrene potentiates oxidative stress via the downregulation of glutathione synthesis. Int. J. Mol. Med. 2017, 40, 2011–2017. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.S.; Kaufman, R.J. Endoplasmic reticulum stress and oxidative stress in cell fate decision and human disease. Antioxid. Redox Signal. 2014, 21, 396–413. [Google Scholar] [CrossRef] [PubMed]
- Day, C.P.; James, O.F. Hepatic steatosis: Innocent bystander or guilty party? Hepatology 1998, 27, 1463–1466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosso, N.; Chavez-Tapia, N.C.; Tiribelli, C.; Bellentani, S. Translational approaches: From fatty liver to non-alcoholic steatohepatitis. World J. Gastroenterol. 2014, 20, 9038–9049. [Google Scholar] [PubMed]
- Berlanga, A.; Guiu-Jurado, E.; Porras, J.A.; Auguet, T. Molecular pathways in non-alcoholic fatty liver disease. Clin. Exp. Gastroenterol. 2014, 7, 221–239. [Google Scholar] [PubMed]
- Hansen, H.H.; Feigh, M.; Veidal, S.S.; Rigbolt, K.T.; Vrang, N.; Fosgerau, K. Mouse models of nonalcoholic steatohepatitis in preclinical drug development. Drug Discov. Today 2017, 22, 1707–1718. [Google Scholar] [CrossRef] [PubMed]
- Buzzetti, E.; Pinzani, M.; Tsochatzis, E.A. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metabolism 2016, 65, 1038–1048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sumida, Y.; Niki, E.; Naito, Y.; Yoshikawa, T. Involvement of free radicals and oxidative stress in NAFLD/NASH. Free Radic. Res. 2013, 47, 869–880. [Google Scholar] [CrossRef] [PubMed]
- Irie, M.; Sohda, T.; Iwata, K.; Kunimoto, H.; Fukunaga, A.; Kuno, S.; Yotsumoto, K.; Sakurai, K.; Iwashita, H.; Hirano, G.; et al. Levels of the oxidative stress marker gamma-glutamyltranspeptidase at different stages of nonalcoholic fatty liver disease. J. Int. Med. Res. 2012, 40, 924–933. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Shi, W.; Li, G.; Jin, B.; Chen, Y.; Hu, H.; Liu, L.; Xie, F.; Chen, K.; Yin, D. Plasma reactive carbonyl species levels and risk of non-alcoholic fatty liver disease. J. Gastroenterol. Hepatol. 2011, 26, 1010–1015. [Google Scholar] [CrossRef] [PubMed]
- Nobili, V.; Parola, M.; Alisi, A.; Marra, F.; Piemonte, F.; Mombello, C.; Sutti, S.; Povero, D.; Maina, V.; Novo, E.; et al. Oxidative stress parameters in paediatric non-alcoholic fatty liver disease. Int. J. Mol. Med. 2010, 26, 471–476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pirgon, O.; Bilgin, H.; Cekmez, F.; Kurku, H.; Dundar, B.N. Association between insulin resistance and oxidative stress parameters in obese adolescents with non-alcoholic fatty liver disease. J. Clin. Res. Pediatr. Endocrinol. 2013, 5, 33–39. [Google Scholar] [PubMed]
- Jung, Y.S.; Kim, S.J.; Kwon, D.Y.; Kim, Y.C. Comparison of the effects of buthioninesulfoximine and phorone on the metabolism of sulfur-containing amino acids in rat liver. Biochem. Biophys. Res. Commun. 2008, 368, 913–918. [Google Scholar] [CrossRef] [PubMed]
- Volpi, N.; Tarugi, P. Improvement in the high-performance liquid chromatography malondialdehyde level determination in normal human plasma. J. Chromatogr. B Biomed. Sci. Appl. 1998, 713, 433–437. [Google Scholar] [CrossRef]
- Ide, T. Simple high-performance liquid chromatographic method for assaying cysteinesulfinic acid decarboxylase activity in rat tissue. J. Chromatogr. B Biomed. Sci. Appl. 1997, 694, 325–332. [Google Scholar] [CrossRef]
- Nolin, T.D.; McMenamin, M.E.; Himmelfarb, J. Simultaneous determination of total homocysteine, cysteine, cysteinylglycine, and glutathione in human plasma by high-performance liquid chromatography: Application to studies of oxidative stress. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2007, 852, 554–561. [Google Scholar] [CrossRef] [PubMed] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genes | Primer Sequences | |
|---|---|---|
| Grp78 | F: TGGTATTCTCCGAGTGACAGC | R: AGTCTTCAATGTCCGCATCC |
| Chop | F: CACGCACATCCCAAAGCC | R: GGGCACTGACCACTCTGTT |
| TNFα | F: GGCCTCTCTACCTTGTTGCC | R: CAGCCTGGTCACCAAATCAG |
| CCL2 | F: CCAGCAAGATGATCCCAATG | R: CTTCTTGGGGTCAGCACAGA |
| IL1β | F: TTCACCATGGAATCCGTGTC | R: GTCTTGGCCGAGGACTAAGG |
| IL6 | F: TTGCCTTCTTGGGACTGATG | R: CCACGATTTCCCAGAGAACA |
| TGFβ1 | F: GCCCTGGATACCAACTATTGC | R: TGTTGGACAGCTGCTCCACCT |
| αSMA | F: GGCTCTGGGCTCTGTAAGG | R: CTCTTGCTCTGGGCTTCATC |
| Col1a1 | F: ACCTGTGTGTTCCCTACTCA | R: GACTGTTGCCTTCGCCTCTG |
| Fibronectin | F: ATGACGATGGGAAGACCTAC | R: GGCTGGAAAGATTACTCTCG |
| GAPDH | F: GTTGTCTCCTGCGACTTCA | R: GGTGGTCCAGGGTTTCTTA |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, S.H.; Kwon, D.-y.; Kwak, J.-H.; Lee, S.; Lee, Y.-H.; Yun, J.; Son, T.G.; Jung, Y.-S. Tunicamycin-Induced ER Stress is Accompanied with Oxidative Stress via Abrogation of Sulfur Amino Acids Metabolism in the Liver. Int. J. Mol. Sci. 2018, 19, 4114. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19124114
Kim SH, Kwon D-y, Kwak J-H, Lee S, Lee Y-H, Yun J, Son TG, Jung Y-S. Tunicamycin-Induced ER Stress is Accompanied with Oxidative Stress via Abrogation of Sulfur Amino Acids Metabolism in the Liver. International Journal of Molecular Sciences. 2018; 19(12):4114. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19124114
Chicago/Turabian StyleKim, Sou Hyun, Do-young Kwon, Jae-Hwan Kwak, Seunghyun Lee, Yun-Hee Lee, Jieun Yun, Tae Gen Son, and Young-Suk Jung. 2018. "Tunicamycin-Induced ER Stress is Accompanied with Oxidative Stress via Abrogation of Sulfur Amino Acids Metabolism in the Liver" International Journal of Molecular Sciences 19, no. 12: 4114. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19124114