Inhibition of GLO1 in Glioblastoma Multiforme Increases DNA-AGEs, Stimulates RAGE Expression, and Inhibits Brain Tumor Growth in Orthotopic Mouse Models

 , ,
, ,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
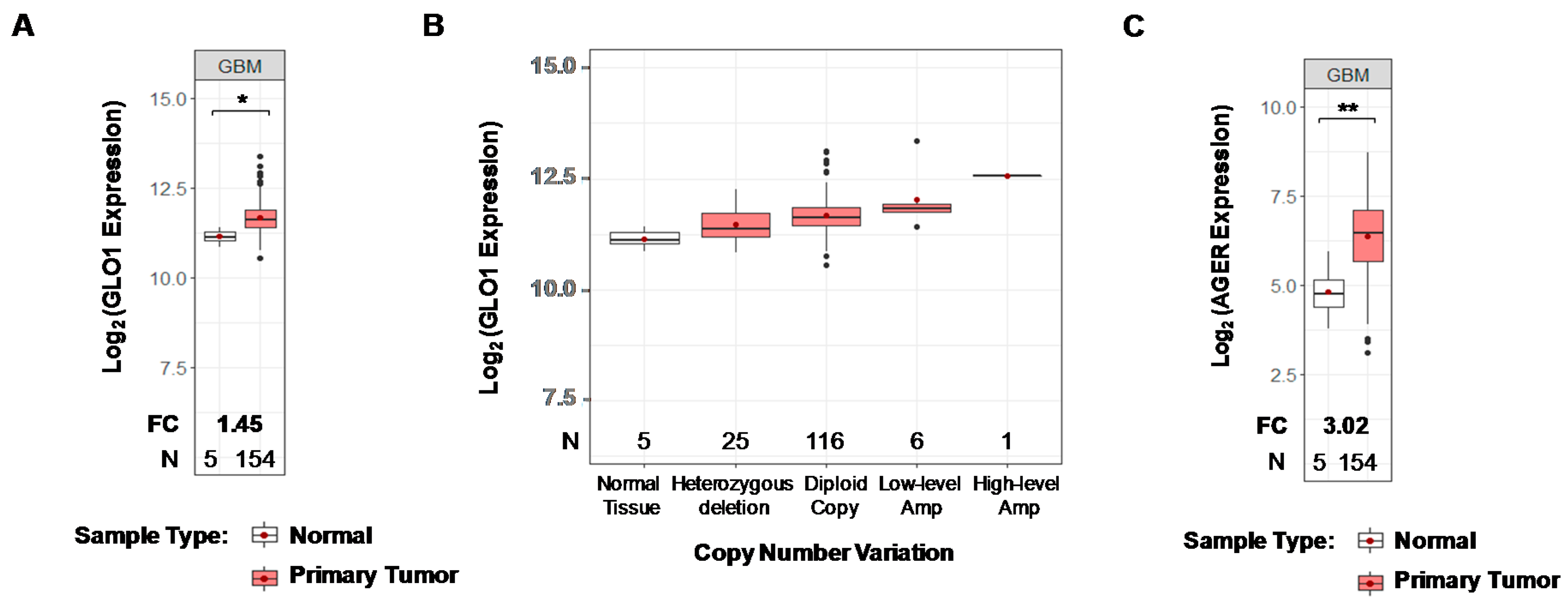
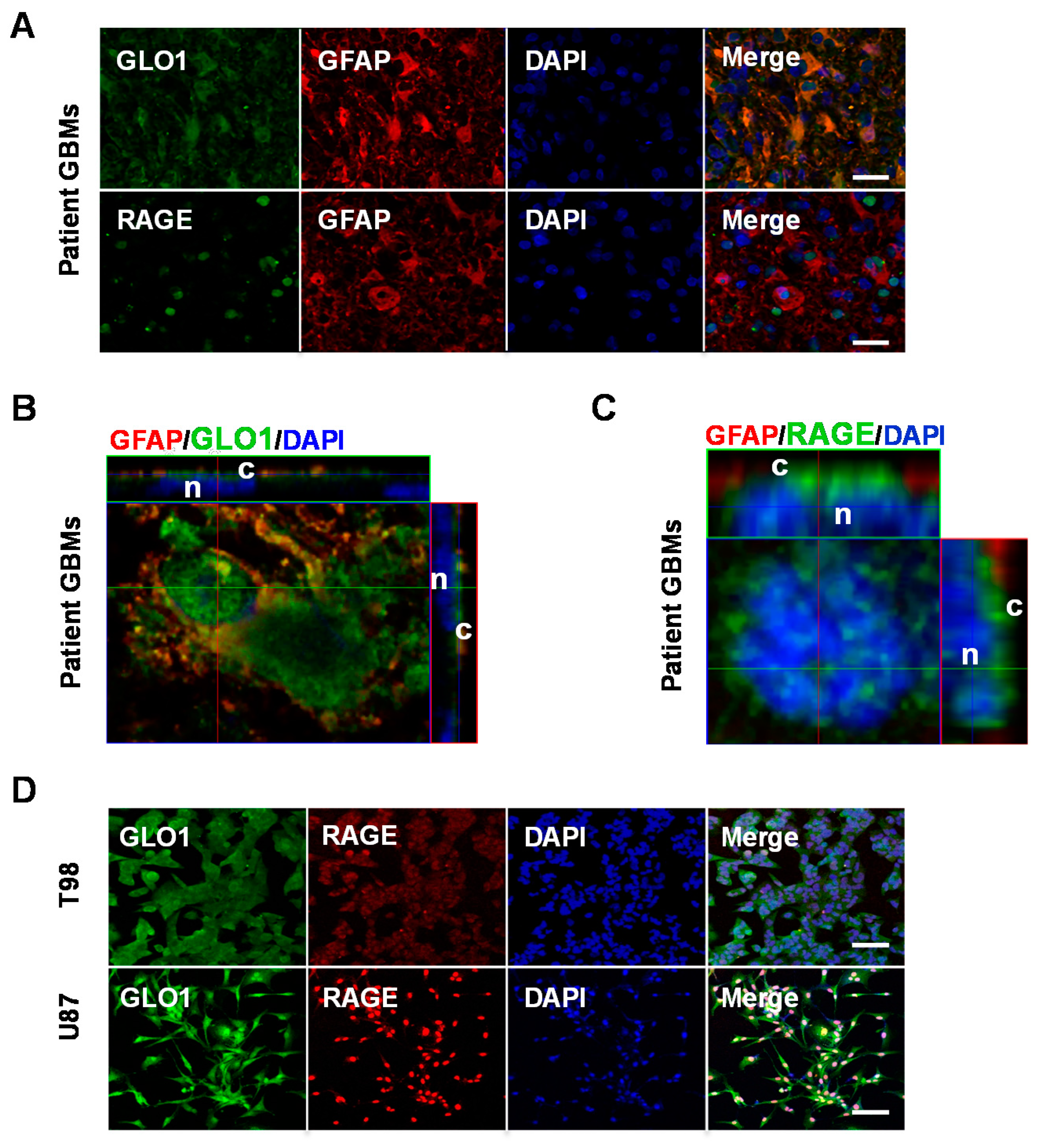
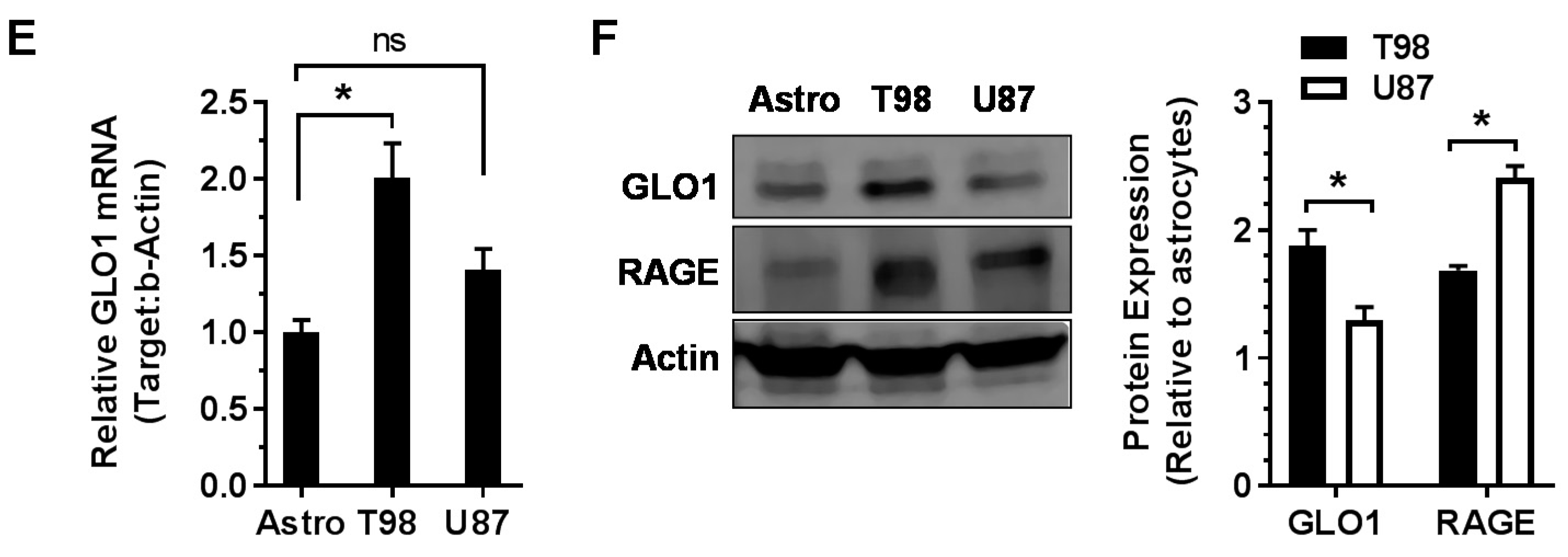
2.1. GLO1 and RAGE Are Overexpressed in GBM
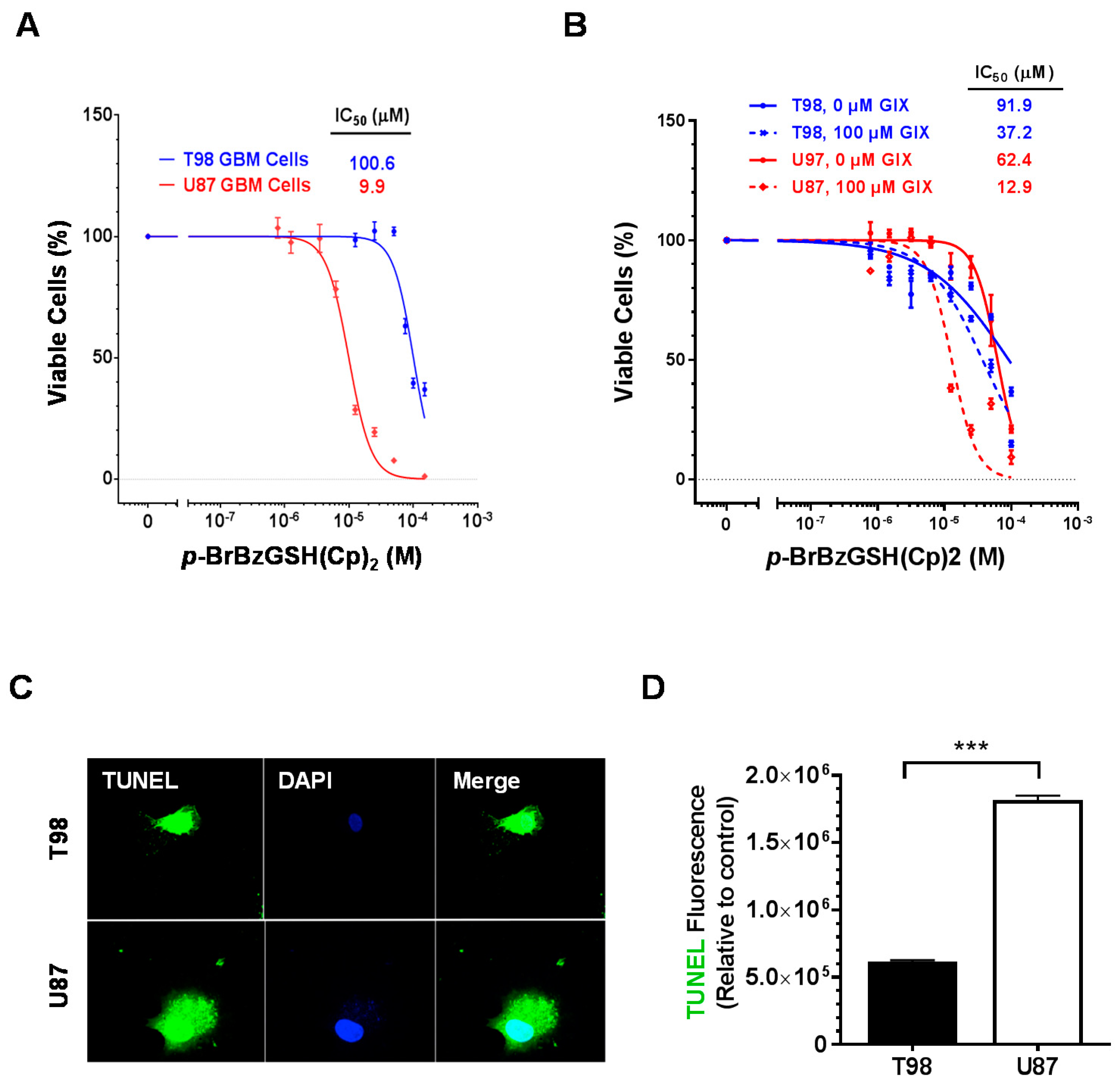
2.2. Small Molecule Inhibition of GLO1 Is Cytotoxic and Induces Apoptosis
2.3. Inhibition of ADAM10 Increases Sensitivity to p-BrBzGSH(Cp)2 and Implicates Partial Protection by sRAGE
2.4. GLO1 Inhibition Increases Nuclear MG and DNA-AGE Levels
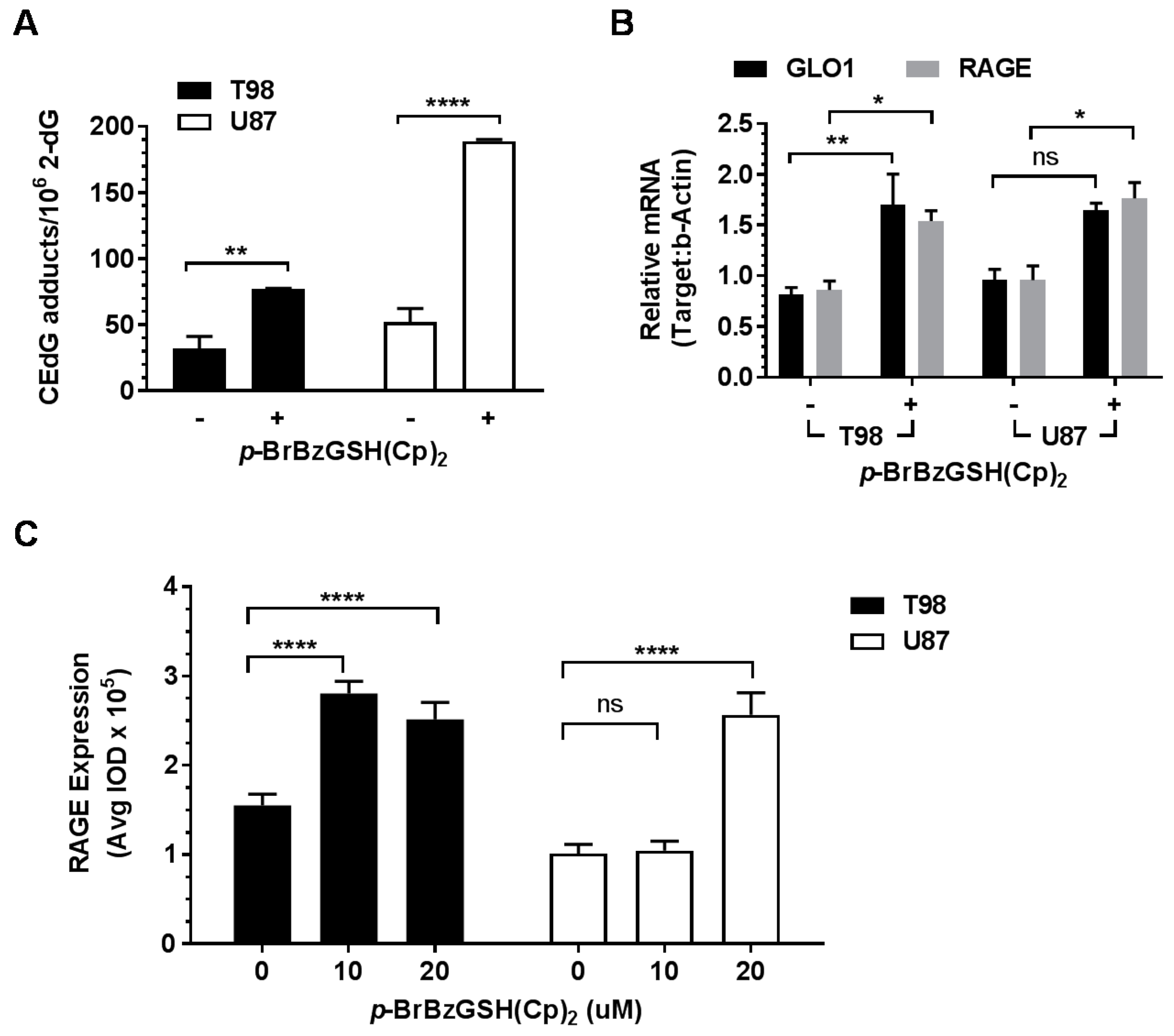
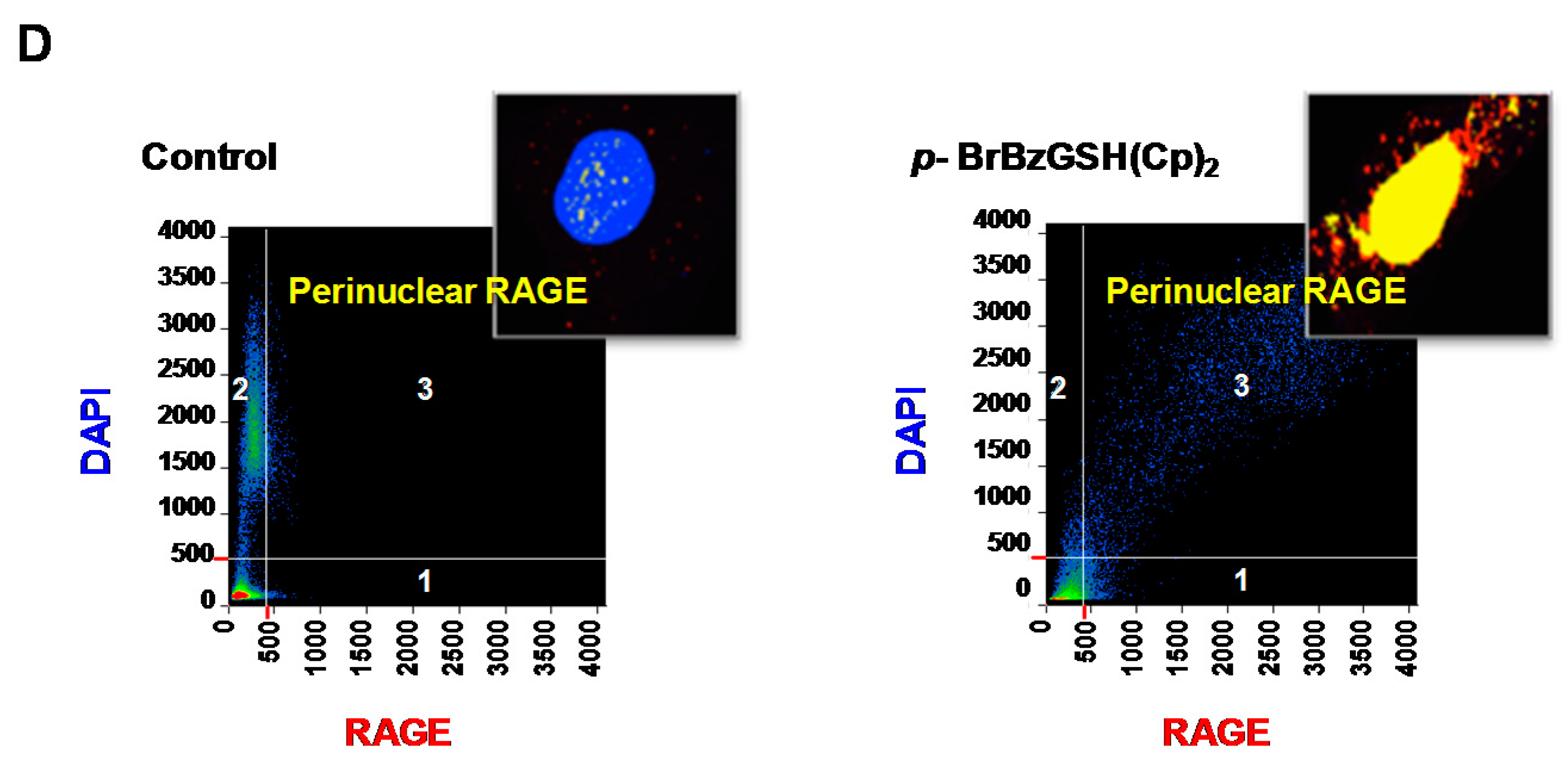
2.5. p-BrBzGSH(Cp)2 Treatment Induces Expression of GLO1 and RAGE
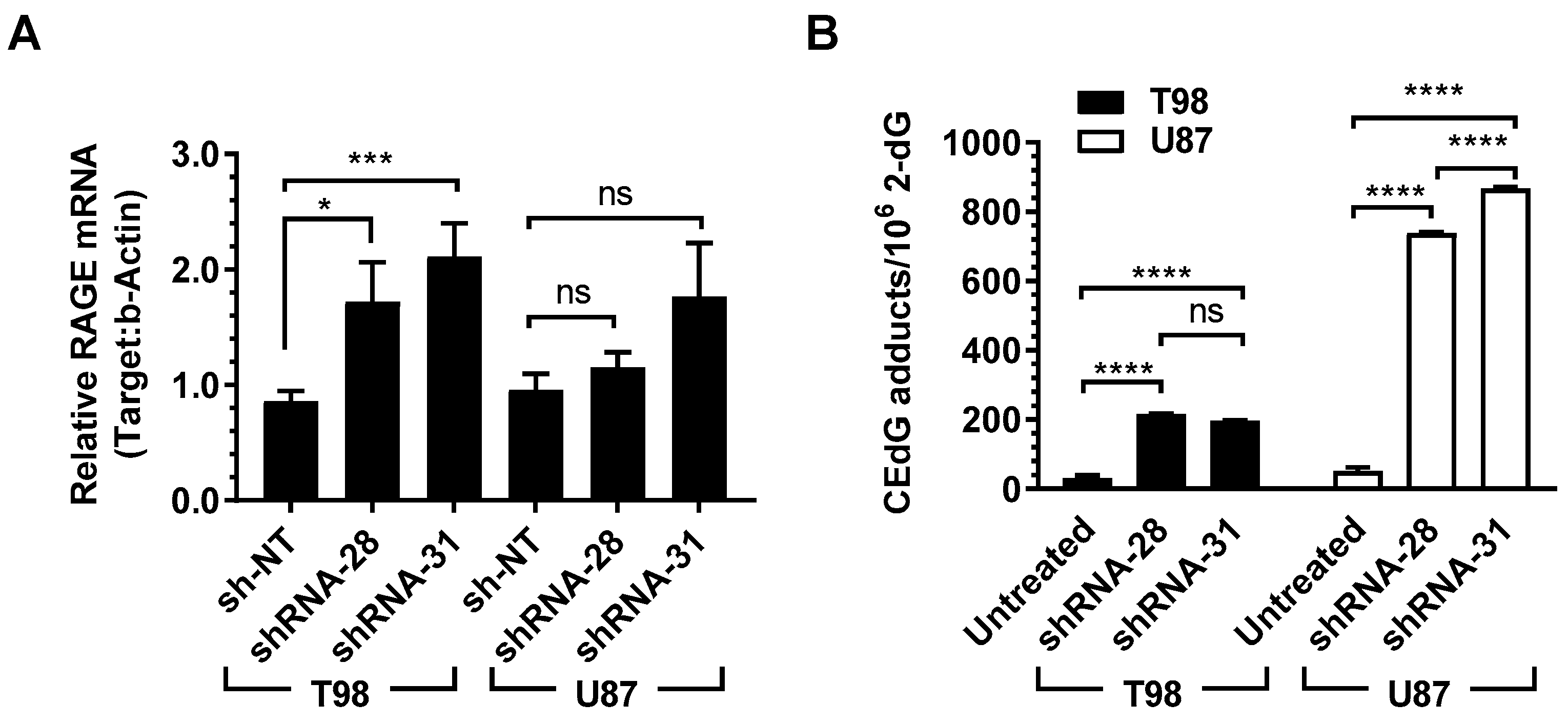
2.6. shRNA Inhibition of GLO1 in GBM Induces RAGE Expression and Increases DNA-AGEs
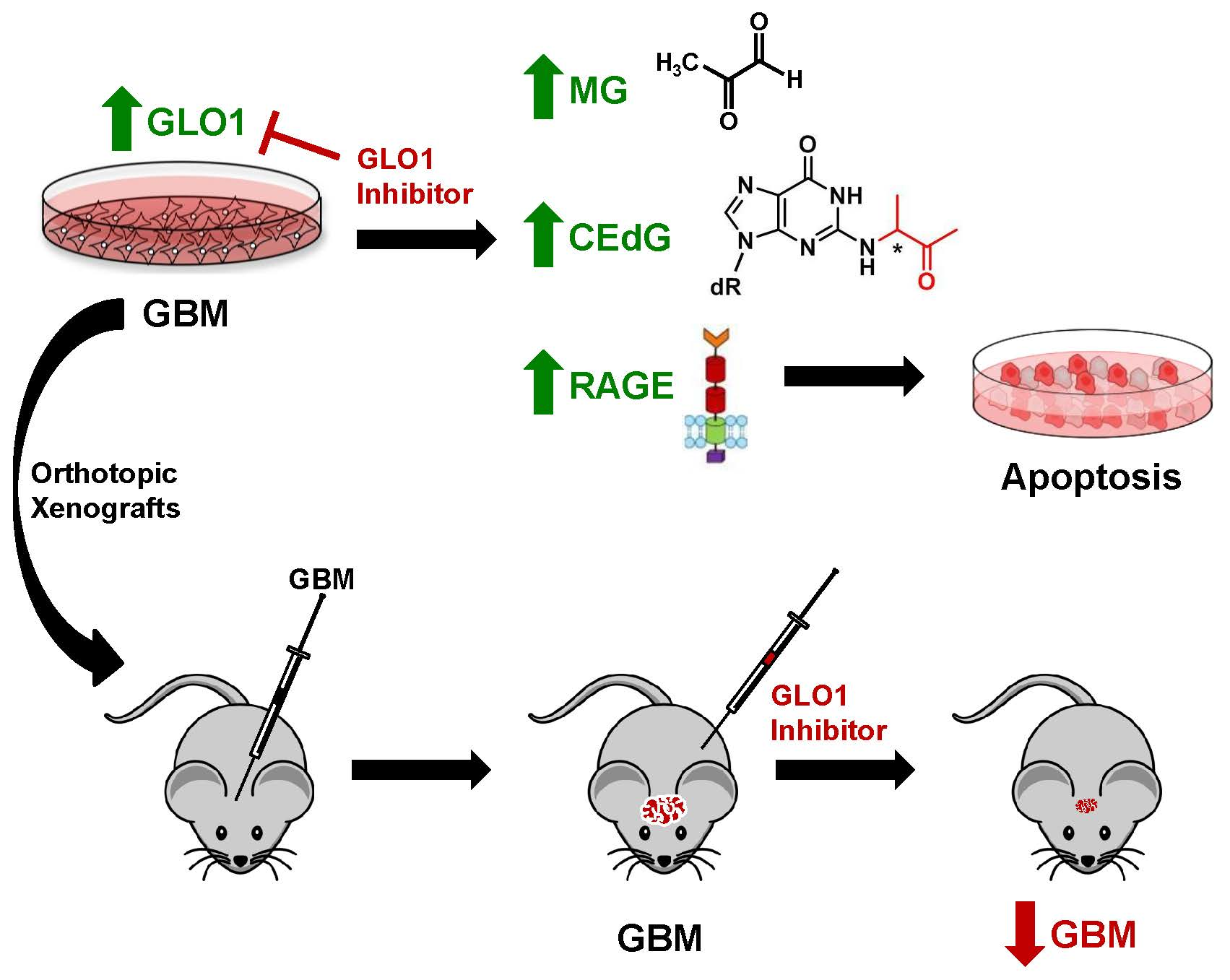
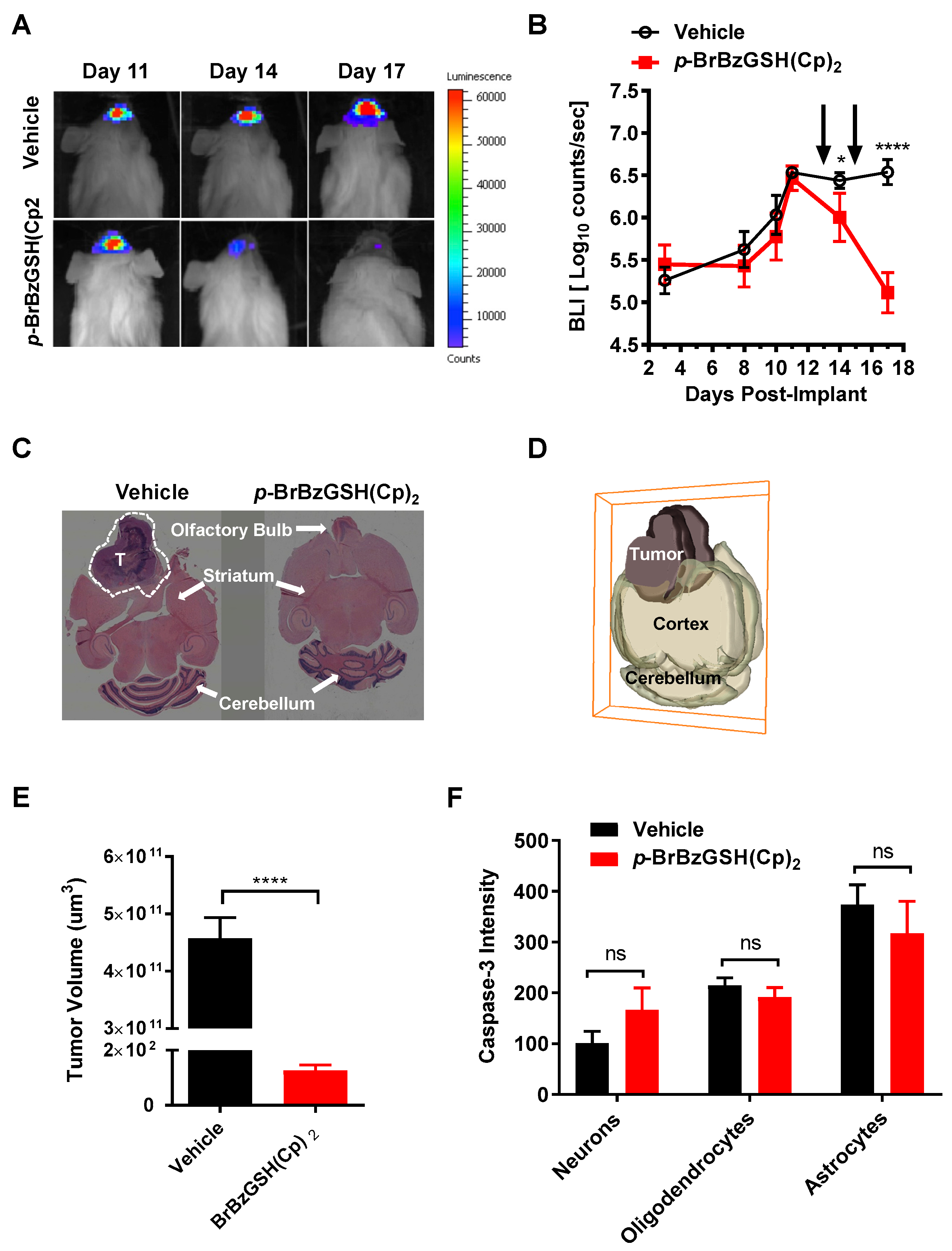
2.7. Inhibition of GLO1 Abrogates GBM Tumor Growth In Vivo
3. Discussion
4. Materials and Methods
4.1. GLO1 and AGER Gene Expression and the Overall Survival Analyses in Public Datasets
4.2. GBM Cell Culture
4.3. Real-Time PCR (RT-qPCR)
4.4. Western Blot Analysis
4.5. Patient GBM Tissues and Immunohistochemical (IHC) Staining
4.6. Immunocytochemistry (ICC)
4.7. Immunofluorescence Intensity Quantification
4.8. 3-D Brain Reconstruction
4.9. Cytotoxicity Assay
4.10. Terminal Deoxynucleotidyl Transferase dUTP Nick End Labeling (TUNEL)
4.11. shRNA Knockdown of GLO1 Expression
4.12. Isolation of DNA for CEdG Analysis
4.13. Preparation of DNA and Quantification of (R, S) CEdG by Mass Spectrometry
4.14. GBM Xenograft Assay
4.15. Statistical Analyses
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| AGEs | advanced glycation endproducts |
| CEdG | N2-1-(carboxyethyl)-2′-deoxyguanosine |
| 3-DG | 3-deoxyglucosone |
| 18FDG | 2-18fluoro-2′-deoxy-d-glucose |
| GBM | glioblastoma multiforme |
| GFAP | glial fibrillary acidic protein |
| GLO1 | glyoxalase 1 |
| GLUT1 | glucose transporter 1 |
| GSH | glutathione |
| 1H-MRS | proton magnetic resonance spectroscopy |
| LC-ESI-MS/MS | liquid chromatography electrospray ionization tandem mass spectrometry |
| MAP2 | microtubule associated processes |
| MBP | myelin basic protein |
| MG | methylglyoxal |
| mRNA | messenger RNA |
| NeuN | neuronal nuclear antigen |
| NOD/SCID | Non-Obese Diabetic/Severe Combined Immunodeficiency |
| p-BrBzGSH(Cp)2 | S-(p-bromobenzyl) glutathione dicyclopentyl ester |
| PET | positron emission tomography |
| RAGE | Receptor for advanced glycation endproducts |
| esRAGE | endogenous secretory form of RAGE |
| fl-RAGE | full-length form of RAGE |
| sRAGE | soluble form of RAGE |
| RT-qPCR | real-time reverse transcription polymerase chain reaction |
| shRNA | short hairpin RNA |
| TCGA | The Cancer Genome Atlas |
| TUNEL | terminal deoxynucleotidyl transferase dUTP nick end-labeling |
References
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 world health organization classification of tumors of the central nervous system: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed]
- Omuro, A.; DeAngelis, L.M. Glioblastoma and other malignant gliomas: A clinical review. JAMA 2013, 310, 1842–1850. [Google Scholar] [CrossRef] [PubMed]
- Cairns, R.A.; Harris, I.S.; Mak, T.W. Regulation of cancer cell metabolism. Nat. Rev. 2011, 11, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Ciminera, A.K.; Jandial, R.; Termini, J. Metabolic advantages and vulnerabilities in brain metastases. Clin. Exp. Metastasis 2017, 34, 401–410. [Google Scholar] [CrossRef] [PubMed]
- Younes, M.; Lechago, L.V.; Somoano, J.R.; Mosharaf, M.; Lechago, J. Wide expression of the human erythrocyte glucose transporter glut1 in human cancers. Cancer Res. 1996, 56, 1164–1167. [Google Scholar] [PubMed]
- La Fougère, C.; Suchorska, B.; Bartenstein, P.; Kreth, F.-W.; Tonn, J.-C. Molecular imaging of gliomas with pet: Opportunities and limitations. Neuro Oncol. 2011, 13, 806–819. [Google Scholar] [CrossRef] [PubMed]
- Workman, P.; Aboagye, E.O.; Chung, Y.-L.; Griffiths, J.R.; Hart, R.; Leach, M.O.; Maxwell, R.J.; McSheehy, P.M.J.; Price, P.M.; Zweit, J.; et al. Minimally invasive pharmacokinetic and pharmacodynamic technologies in hypothesis-testing clinical trials of innovative therapies. J. Nat. Cancer Inst. 2006, 98, 580–598. [Google Scholar] [CrossRef] [PubMed]
- Gatenby, R.A.; Gillies, R.J. Why do cancers have high aerobic glycolysis? Nat. Rev. 2004, 4, 891–899. [Google Scholar] [CrossRef] [PubMed]
- Odani, H.; Shinzato, T.; Matsumoto, Y.; Usami, J.; Maeda, K. Increase in three α,β-dicarbonyl compound levels in human uremic plasma: Specific in vivo determination of intermediates in advanced maillard reaction. Biochem. Biophys. Res. Commun. 1999, 256, 89–93. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Méndez, J.D.; Méndez-Valenzuela, V.; Aguilar-Hernández, M.M. Cellular signalling of the receptor for advanced glycation end products (rage). Cell Signal. 2013, 25, 2185–2197. [Google Scholar] [CrossRef] [PubMed]
- Talesa, V.N.; Ferri, I.; Bellezza, G.; Love, H.D.; Sidoni, A.; Antognelli, C. Glyoxalase 2 is involved in human prostate cancer progression as part of a mechanism driven by pten/pi3k/akt/mtor signaling with involvement of pkm2 and eralpha. Prostate 2017, 77, 196–210. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Bukulin, M.; Kojro, E.; Roth, A.; Metz, V.V.; Fahrenholz, F.; Nawroth, P.P.; Bierhaus, A.; Postina, R. Receptor for advanced glycation end products is subjected to protein ectodomain shedding by metalloproteinases. J. Biol. Chem. 2008, 283, 35507–35516. [Google Scholar] [CrossRef] [PubMed]
- Raucci, A.; Cugusi, S.; Antonelli, A.; Barabino, S.M.; Monti, L.; Bierhaus, A.; Reiss, K.; Saftig, P.; Bianchi, M.E. A soluble form of the receptor for advanced glycation endproducts (rage) is produced by proteolytic cleavage of the membrane-bound form by the sheddase a disintegrin and metalloprotease 10 (adam10). FASEB J. 2008, 22, 3716–3727. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Reverdatto, S.; Frolov, A.; Hoffmann, R.; Burz, D.S.; Shekhtman, A. Structural basis for pattern recognition by the receptor for advanced glycation end products (rage). J. Biol. Chem. 2008, 283, 27255–27269. [Google Scholar] [CrossRef] [PubMed]
- Kostova, N.; Zlateva, S.; Ugrinova, I.; Pasheva, E. The expression of hmgb1 protein and its receptor rage in human malignant tumors. Mol. Cell. Biochem. 2010, 337, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Fritz, G. Rage: A single receptor fits multiple ligands. Trends Biochem. Sci. 2011, 36, 625–632. [Google Scholar] [CrossRef] [PubMed]
- Creighton, D.J.; Hamilton, D.S.; Kavarana, M.J.; Sharkey, E.M.; Eiseman, J.L. Glyoxalase enzyme system as a potential target for antitumor drug development. Drugs Future 2000, 25, 385–392. [Google Scholar] [CrossRef]
- Sakamoto, H.; Mashima, T.; Kizaki, A.; Dan, S.; Hashimoto, Y.; Naito, M.; Tsuruo, T. Glyoxalase I is involved in resistance of human leukemia cells to antitumor agent-induced apoptosis. Blood 2000, 95, 3214–3218. [Google Scholar] [PubMed]
- Santarius, T.; Bignell, G.R.; Greenman, C.D.; Widaa, S.; Chen, L.; Mahoney, C.L.; Butler, A.; Edkins, S.; Waris, S.; Thornalley, P.J.; et al. Glo1—A novel amplified gene in human cancer. Genes Chromosom. Cancer 2010, 49, 711–725. [Google Scholar] [CrossRef] [PubMed]
- Mearini, E.; Romani, R.; Mearini, L.; Antognelli, C.; Zucchi, A.; Baroni, T.; Porena, M.; Talesa, V.N. Differing expression of enzymes of the glyoxalase system in superficial and invasive bladder carcinomas. Eur. J. Cancer 2002, 38, 1946–1950. [Google Scholar] [CrossRef]
- Helgager, J.; Li, J.; Lubensky, I.A.; Lonser, R.; Zhuang, Z. Troglitazone reduces glyoxalase i protein expression in glioma and potentiates the effects of chemotherapeutic agents. J. Oncol. 2010, 2010, 373491. [Google Scholar] [CrossRef] [PubMed]
- Antognelli, C.; Baldracchini, F.; Talesa, V.N.; Costantini, E.; Zucchi, A.; Mearini, E. Overexpression of glyoxalase system enzymes in human kidney tumor. Cancer J. 2006, 12, 222–228. [Google Scholar] [CrossRef] [PubMed]
- Thornalley, P.J.; Rabbani, N. Glyoxalase in tumourigenesis and multidrug resistance. Semin. Cell Dev. Biol. 2011, 22, 318–325. [Google Scholar] [CrossRef] [PubMed]
- Ronen, S.M.; DiStefano, F.; McCoy, C.L.; Robertson, D.; Smith, T.A.D.; Al-Saffar, N.M.; Titley, J.; Cunningham, D.C.; Griffiths, J.R.; Leach, M.O.; et al. Magnetic resonance detects metabolic changes associated with chemotherapy-induced apoptosis. Br. J. Cancer 1999, 80, 1035–1041. [Google Scholar] [CrossRef] [PubMed]
- Synold, T.; Xi, B.; Wuenschell, G.E.; Tamae, D.; Figarola, J.L.; Rahbar, S.; Termini, J. Advanced glycation end products of DNA: Quantification of n2-(1-carboxyethyl)-2′-deoxyguanosine in biological samples by liquid chromatography electrospray ionization tandem mass spectrometry. Chem. Res. Toxicol. 2008, 21, 2148–2155. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Research Network; Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.M.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M. The cancer genome atlas pan-cancer analysis project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar]
- Goswami, C.P.; Nakshatri, H. Proggenev2: Enhancements on the existing database. BMC Cancer 2014, 14, 970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.; Scheck, A.C.; Cloughesy, T.F.; Lai, A.; Dong, J.; Farooqi, H.K.; Liau, L.M.; Horvath, S.; Mischel, P.S.; Nelson, S.F. Gene expression analysis of glioblastomas identifies the major molecular basis for the prognostic benefit of younger age. BMC Med. Gen. 2008, 1, 52. [Google Scholar] [CrossRef] [PubMed]
- Phillips, H.S.; Kharbanda, S.; Chen, R.; Forrest, W.F.; Soriano, R.H.; Wu, T.D.; Misra, A.; Nigro, J.M.; Colman, H.; Soroceanu, L.; et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell 2006, 9, 157–173. [Google Scholar] [CrossRef] [PubMed]
- Costa, B.M.; Smith, J.S.; Chen, Y.; Chen, J.; Phillips, H.S.; Aldape, K.D.; Zardo, G.; Nigro, J.; James, C.D.; Fridlyand, J.; et al. Reversing hoxa9 oncogene activation by pi3k inhibition: Epigenetic mechanism and prognostic significance in human glioblastoma. Cancer Res. 2010, 70, 453–462. [Google Scholar] [CrossRef] [PubMed]
- Buckley, S.T.; Ehrhardt, C. The receptor for advanced glycation end products (rage) and the lung. J. Biomed. Biotechnol. 2010, 2010, 917108. [Google Scholar] [CrossRef] [PubMed]
- Kierdorf, K.; Fritz, G. Rage regulation and signaling in inflammation and beyond. J. Leukoc. Biol. 2013, 94, 55–68. [Google Scholar] [CrossRef] [PubMed]
- Gebhardt, C.; Riehl, A.; Durchdewald, M.; Németh, J.; Fürstenberger, G.; Müller-Decker, K.; Enk, A.; Arnold, B.; Bierhaus, A.; Nawroth, P.P.; et al. Rage signaling sustains inflammation and promotes tumor development. J. Exp. Med. 2008, 205, 275–285. [Google Scholar] [CrossRef] [PubMed]
- Herold, K.; Moser, B.; Chen, Y.; Zeng, S.; Yan, S.F.; Ramasamy, R.; Emond, J.; Clynes, R.; Schmidt, A.M. Receptor for advanced glycation end products (rage) in a dash to the rescue: Inflammatory signals gone awry in the primal response to stress. J. Leukoc. Biol. 2007, 82, 204–212. [Google Scholar] [CrossRef] [PubMed]
- Vince, R.; Daluge, S.; Wadd, W.B. Studies on the inhibition of glyoxalase i by s-substituted glutathiones. J. Med. Chem. 1971, 14, 402–404. [Google Scholar] [CrossRef] [PubMed]
- Thornalley, P.J.; Edwards, L.G.; Kang, Y.; Wyatt, C.; Davies, N.; Ladan, M.J.; Double, J. Antitumour activity of s-p-bromobenzylglutathione cyclopentyl diester in vitro and in vivo: Inhibition of glyoxalase I and induction of apoptosis. Biochem. Pharmacol. 1996, 51, 1365–1372. [Google Scholar] [CrossRef]
- Phillips, S.A.; Thornalley, P.J. The formation of methylglyoxal from triose phosphates. Eur. J. Biochem. 1993, 212, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Lapolla, A.; Flamini, R.; Tonus, T.; Fedele, D.; Senesi, A.; Reitano, R.; Marotta, E.; Pace, G.; Seraglia, R.; Traldi, P. An effective derivatization method for quantitative determination of glyoxal and methylglyoxal in plasma samples by gas chromatography/mass spectrometry. Rapid Commun. Mass Spectrom. 2003, 17, 876–878. [Google Scholar] [CrossRef] [PubMed]
- Thornalley, P.J.; Battah, S.; Ahmed, N.; Karachalias, N.; Agalou, S.; Babaei-Jadidi, R.; Dawnay, A. Quantitative screening of advanced glycation endproducts in cellular and extracellular proteins by tandem mass spectrometry. Biochem. J. 2003, 375, 581–592. [Google Scholar] [CrossRef] [PubMed]
- Papoulis, A.; Al-Abed, Y.; Bucala, R. Identification of N2-(1-carboxyethyl)guanine (ceg) as a guanine advanced glycosylation end product. Biochemistry 1995, 34, 648–655. [Google Scholar] [CrossRef] [PubMed]
- Schneider, M.; Thoss, G.; Hübner-Parajsz, C.; Kientsch-Engel, R.; Stahl, P.; Pischetsrieder, M. Determination of glycated nucleobases in human urine by a new monoclonal antibody specific for n2-carboxyethyl-2′-deoxyguanosine. Chem. Res. Toxicol. 2004, 17, 1385–1390. [Google Scholar] [CrossRef] [PubMed]
- Vaca, C.E.; Fang, J.-L.; Conradi, M.; Hou, S.-M. Development of a 32p-postlabelling method for the analysis of 2’-deoxyguanosine-3’-monophosphate and DNA adducts of methylglyoxal. Carcinogenesis 1994, 15, 1887–1894. [Google Scholar] [CrossRef] [PubMed]
- Bidmon, C.; Frischmann, M.; Pischetsrieder, M. Analysis of DNA-bound advanced glycation end-products by lc and mass spectrometry. J. Chromatogr. B 2007, 855, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Tamae, D.; Lim, P.; Wuenschell, G.E.; Termini, J. Mutagenesis and repair induced by the DNA advanced glycation end product n2-1-(carboxyethyl)-2′-deoxyguanosine in human cells. Biochemistry 2011, 50, 2321–2329. [Google Scholar] [CrossRef] [PubMed]
- Hirahara, I.; Ishibashi, Y.; Kaname, S.; Kusano, E.; Fujita, T. Methylglyoxal induces peritoneal thickening by mesenchymal-like mesothelial cells in rats. Nephrol. Dial. Transplant. 2009, 24, 437–447. [Google Scholar] [CrossRef] [PubMed]
- Amicarelli, F.; Bucciarelli, T.; Poma, A.; Aimola, P.; Di Ilio, C.; Ragnelli, A.M.; Miranda, M. Adaptive response of human melanoma cells to methylglyoxal injury. Carcinogenesis 1998, 19, 519–523. [Google Scholar] [CrossRef] [PubMed]
- Hosoda, F.; Arai, Y.; Okada, N.; Shimizu, H.; Miyamoto, M.; Kitagawa, N.; Katai, H.; Taniguchi, H.; Yanagihara, K.; Imoto, I.; et al. Integrated genomic and functional analyses reveal glyoxalase I as a novel metabolic oncogene in human gastric cancer. Oncogene 2015, 34, 1196–1206. [Google Scholar] [CrossRef] [PubMed]
- Neman, J.; Choy, C.; Kowolik, C.M.; Anderson, A.; Duenas, V.J.; Waliany, S.; Chen, B.T.; Chen, M.Y.; Jandial, R. Co-evolution of breast-to-brain metastasis and neural progenitor cells. Clin. Exp. Metastasis 2013, 30. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Butler, E.B.; Tan, M. Targeting cellular metabolism to improve cancer therapeutics. Cell Death Dis. 2013, 4, e532. [Google Scholar] [CrossRef] [PubMed]
- Szent-Györgyi, A.; Együd, L.G.; McLaughlin, J.A. Keto-aldehydes and cell division. Science 1967, 155, 539–541. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.-F.; Miyata, S.; Hirota, Y.; Higo, S.; Miyazaki, H.; Fukunaga, M.; Hamada, Y.; Ueyama, S.; Muramoto, O.; Uriuhara, A.; et al. Methylglyoxal induces apoptosis through activation of p38 mitogen-activated protein kinase in rat mesangial cells. Kidney Int. 2003, 63, 947–957. [Google Scholar] [CrossRef] [PubMed]
- Fukunaga, M.; Miyata, S.; Liu, B.F.; Miyazaki, H.; Hirota, Y.; Higo, S.; Hamada, Y.; Ueyama, S.; Kasuga, M. Methylglyoxal induces apoptosis through activation of p38 mapk in rat schwann cells. Biochem. Biophys. Res. Commun. 2004, 320, 689–695. [Google Scholar] [CrossRef] [PubMed]
- Xue, M.; Rabbani, N.; Momiji, H.; Imbasi, P.; Anwar, M.M.; Kitteringham, N.; Park, B.K.; Souma, T.; Moriguchi, T.; Yamamoto, M.; et al. Transcriptional control of glyoxalase 1 by Nrf2 provides a stress-responsive defence against dicarbonyl glycation. Biochem. J. 2012, 443, 213–222. [Google Scholar] [CrossRef] [PubMed]
- Nomura, W.; Inoue, Y. Methylglyoxal activates the target of rapamycin complex 2-protein kinase c signaling pathway in saccharomyces cerevisiae. Mol. Cell Biol. 2015, 35, 1269–1280. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Schmidt, A.M. Characterization and functional analysis of the promoter of rage, the receptor for advanced glycation end products. J. Biol. Chem. 1997, 272, 16498–16506. [Google Scholar] [CrossRef] [PubMed]
- Pardridge, W.M. Drug transport across the blood–brain barrier. J. Cereb. Blood Flow Metab. 2012, 32, 1959–1972. [Google Scholar] [CrossRef] [PubMed]
- Kannan, R.; Kuhlenkamp, J.F.; Jeandidier, E.; Trinh, H.; Ookhtens, M.; Kaplowitz, N. Evidence for carrier-mediated transport of glutathione across the blood-brain barrier in the rat. J. Clin. Investig. 1990, 85, 2009–2013. [Google Scholar] [CrossRef] [PubMed]
- Mermel, C.H.; Schumacher, S.E.; Hill, B.; Meyerson, M.L.; Beroukhim, R.; Getz, G. Gistic2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 2011, 12, R41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neman, J.; de Vellis, J. A method for deriving homogenous population of oligodendrocytes from mouse embryonic stem cells. Dev. Neurobiol. 2012, 72, 777–788. [Google Scholar] [CrossRef] [PubMed]








© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jandial, R.; Neman, J.; Lim, P.P.; Tamae, D.; Kowolik, C.M.; Wuenschell, G.E.; Shuck, S.C.; Ciminera, A.K.; De Jesus, L.R.; Ouyang, C.; et al. Inhibition of GLO1 in Glioblastoma Multiforme Increases DNA-AGEs, Stimulates RAGE Expression, and Inhibits Brain Tumor Growth in Orthotopic Mouse Models. Int. J. Mol. Sci. 2018, 19, 406. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19020406
Jandial R, Neman J, Lim PP, Tamae D, Kowolik CM, Wuenschell GE, Shuck SC, Ciminera AK, De Jesus LR, Ouyang C, et al. Inhibition of GLO1 in Glioblastoma Multiforme Increases DNA-AGEs, Stimulates RAGE Expression, and Inhibits Brain Tumor Growth in Orthotopic Mouse Models. International Journal of Molecular Sciences. 2018; 19(2):406. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19020406
Chicago/Turabian StyleJandial, Rahul, Josh Neman, Punnajit P. Lim, Daniel Tamae, Claudia M. Kowolik, Gerald E. Wuenschell, Sarah C. Shuck, Alexandra K. Ciminera, Luis R. De Jesus, Ching Ouyang, and et al. 2018. "Inhibition of GLO1 in Glioblastoma Multiforme Increases DNA-AGEs, Stimulates RAGE Expression, and Inhibits Brain Tumor Growth in Orthotopic Mouse Models" International Journal of Molecular Sciences 19, no. 2: 406. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19020406