Localization Microscopy of Actin Cytoskeleton in Human Platelets

 ,
, {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Platelet Attachment Protocol
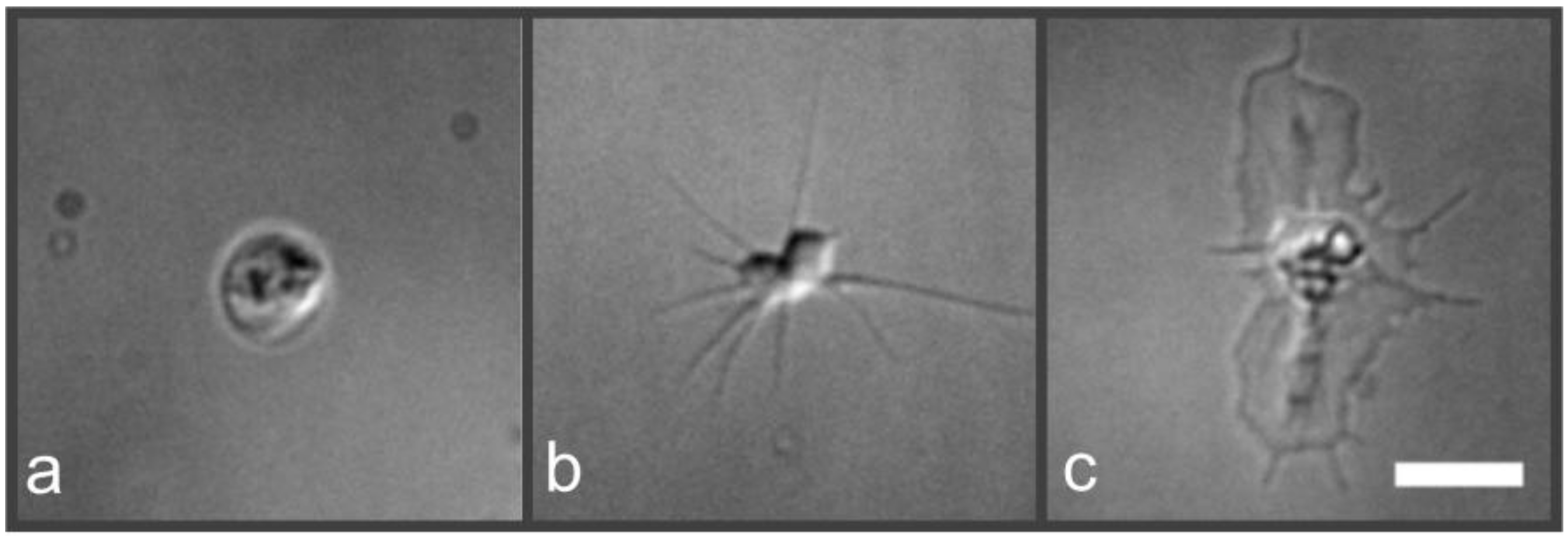
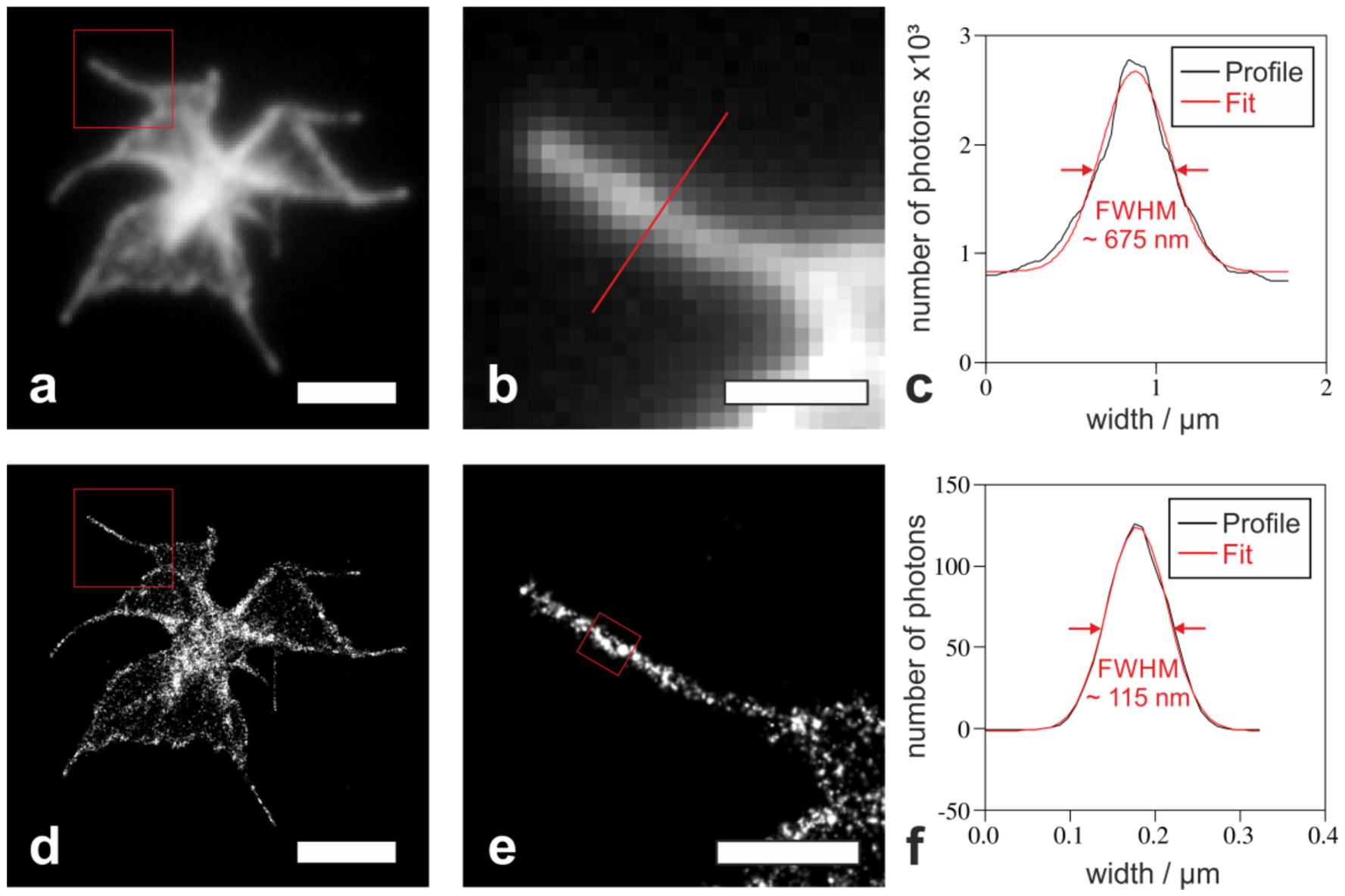
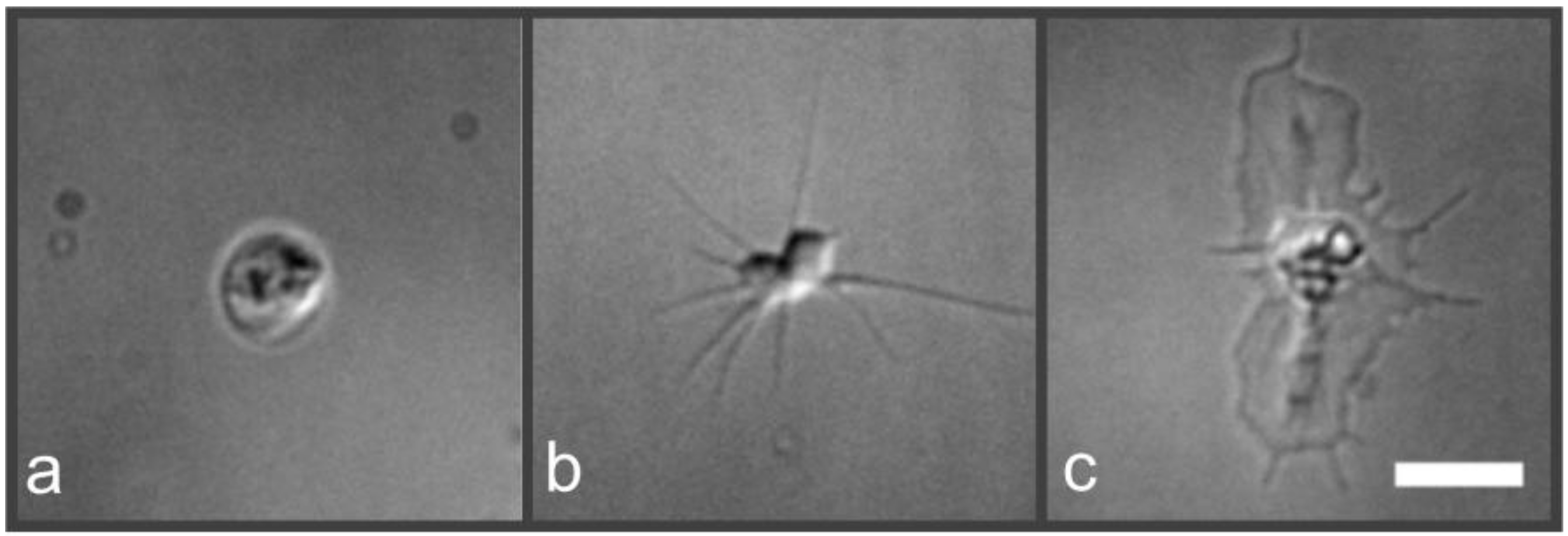
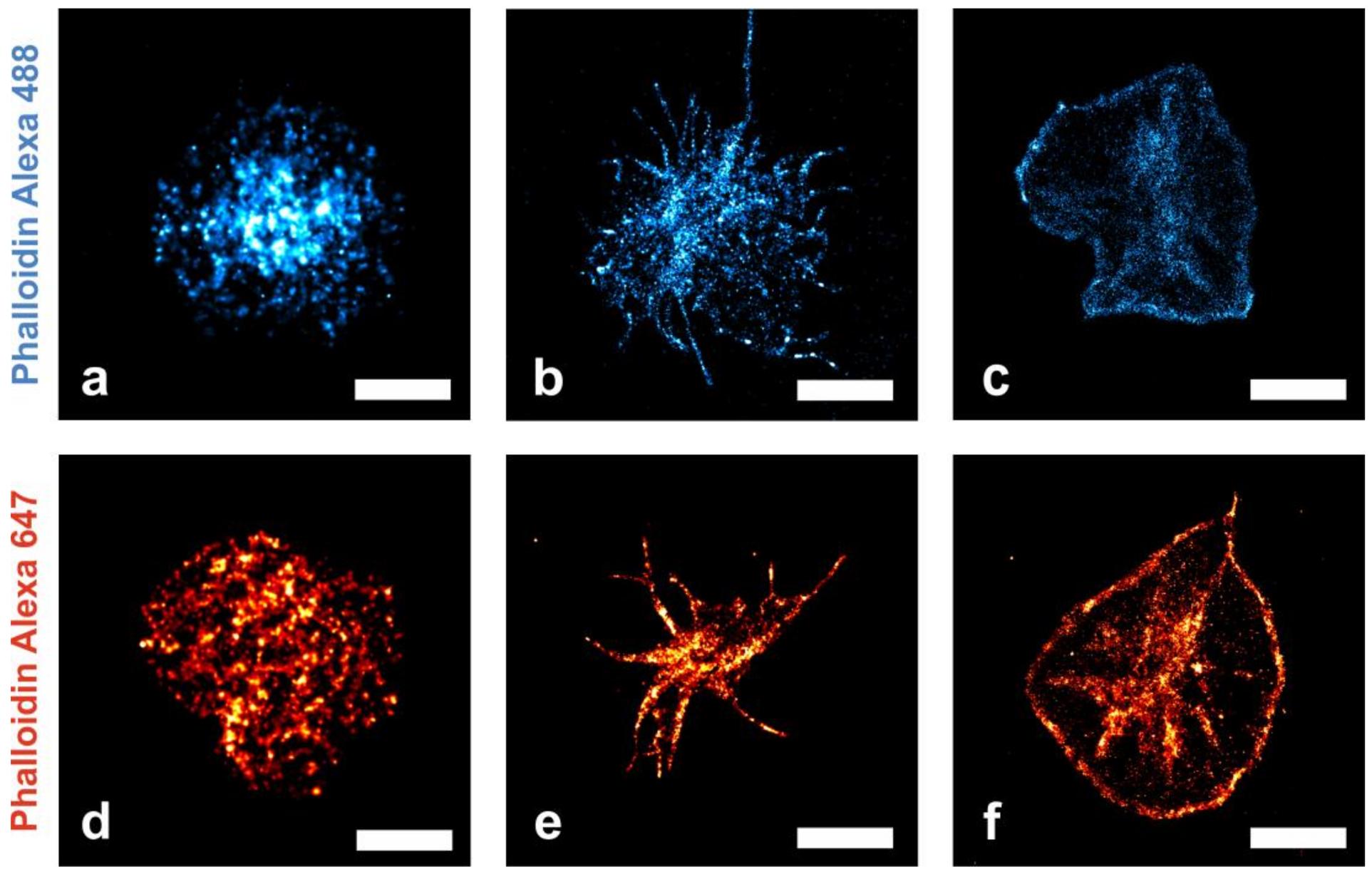
2.2. Nanoscopic Localization of the Actin Cytoskeleton in Platelets
3. Discussion and Conclusions
4. Materials and Methods
4.1. Human Platelet Concentrates
4.2. Statement on the Use of Human Platelet Concentrate Samples
4.3. Platelet Spreading, CD62p and Actin Staining
4.4. Fluorescence Microscope
4.5. Imaging Protocols
4.6. Image Analysis
4.7. Drift Correction
4.7.1. Drift Calculation Method
4.7.2. Drift Correction Algorithm
4.8. Grouping Process
4.9. Smoothing
- Fitting ‘Smoothing-Spline’ with parameter 10−4 only in the discrete time window (sliding along the time-axis).
- Fitting method ‘Smoothing-Spline’ with parameter 10−6 for the entire time line.
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| dSTORM | Direct stochastic optical reconstruction microscopy |
| FWHM | Full width at half maximum |
| DIC | Differential interference contrast |
| TCEP | Tris(2-carboxyethyl)phosphine |
| MEA | β-mercaptoethylamine |
| MV | Methylviologen |
| CBS | Cytoskeleton buffer with sucrose |
| ACD-A | Acid citrate dextrose and adenosine |
| CF | Cost function |
| PSF | Point spread function |
References
- Michelson, A.D. Platelets, 2nd ed.; Elsevier: San Diego, CA, USA, 2006. [Google Scholar]
- Italiano, J.E.; Bergmeier, W.; Tiwari, S.; Falet, H.; Hartwig, J.H.; Hoffmeister, K.M.; André, P.; Wagner, D.D.; Shivdasani, R.A. Mechanisms and implications of platelet discoid shape. Blood 2003, 101, 4789–4796. [Google Scholar] [CrossRef] [PubMed]
- Bender, M.; Eckly, A.; Hartwig, J.H.; Elvers, M.; Pleines, I.; Gupta, S.; Krohne, G.; Jeanclos, E.; Gohla, A.; Gurniak, C.; et al. ADF/n-cofilin-dependent actin turnover determines platelet formation and sizing. Blood 2010, 116, 1767–1775. [Google Scholar] [CrossRef] [PubMed]
- Bach, M.; Savini, C.; Krufczik, M.; Cremer, C.; Röesl, F.; Hausmann, M. Super-resolution localization microscopy of γ-H2AX and heterochromatin after folate deficiency. Int. J. Mol. Sci. 2017, 18, 1726. [Google Scholar] [CrossRef] [PubMed]
- Borgmann, D.M.; Mayr, S.; Polin, H.; Schaller, S.; Dorfer, V.; Obritzberger, L.; Endmayr, T.; Gabriel, C.; Winkler, S.M.; Jacak, J. Single Molecule Fluorescence Microscopy and Machine Learning for Rhesus D Antigen Classification. Sci. Rep. 2016, 6, 32317. [Google Scholar] [CrossRef] [PubMed]
- Poulter, N.S.; Pollitt, A.Y.; Davies, A.; Malinova, D.; Nash, G.B.; Hannon, M.J.; Pikramenou, Z.; Rappoport, J.Z.; Hartwig, J.H.; Owen, D.M.; et al. Platelet actin nodules are podosome-like structures dependent on Wiskott—Aldrich syndrome protein and ARP2/3 complex. Nat. Commun. 2015, 6, 7254. [Google Scholar] [CrossRef] [PubMed]
- Sams, M.; Silye, R.; Göhring, J.; Muresan, L.; Schilcher, K.; Jacak, J. Spatial cluster analysis of nanoscopically mapped serotonin receptors for classification of fixed brain tissue. J. Biomed. Opt. 2013, 19, 11021. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Jones, S.A.; Brandenburg, B.; Zhuang, X. Whole-cell 3D STORM reveals interactions between cellular structures with nanometer-scale resolution. Nat. Methods 2008, 5, 1047–1052. [Google Scholar] [CrossRef] [PubMed]
- Dani, A.; Huang, B.; Bergan, J.; Dulac, C.; Zhuang, X. Superresolution Imaging of Chemical Synapses in the Brain. Neuron 2010, 68, 843–856. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.; Babcock, H.P.; Zhuang, X. Dual-objective STORM reveals three-dimensional filament organization in the actin cytoskeleton. Nat. Methods 2012, 9, 185–188. [Google Scholar] [CrossRef] [PubMed]
- Van De Linde, S.; Löschberger, A.; Klein, T.; Heidbreder, M.; Wolter, S.; Heilemann, M.; Sauer, M. Direct stochastic optical reconstruction microscopy with standard fluorescent probes. Nat. Protoc. 2011, 6, 991–1009. [Google Scholar] [CrossRef] [PubMed]
- Dempsey, G.T.; Vaughan, J.C.; Chen, K.H.; Bates, M.; Zhuang, X. Evaluation of fluorophores for optimal performance in localization-based super-resolution imaging. Nat. Methods 2011, 8, 1027–1036. [Google Scholar] [CrossRef] [PubMed]
- Van de Linde, S.; Krstić, I.; Prisner, T.; Doose, S.; Heilemann, M.; Sauer, M. Photoinduced formation of reversible dye radicals and their impact on super-resolution imaging. Photochem. Photobiol. Sci. 2011, 10, 499–506. [Google Scholar] [CrossRef] [PubMed]
- Gunsolus, I.L.; Hu, D.; Mihai, C.; Lohse, S.E.; Lee, C.S.; Torelli, M.D.; Hamers, R.J.; Murhpy, C.J.; Orr, G.; Haynes, C.L. Facile method to stain the bacterial cell surface for super-resolution fluorescence microscopy. Analyst 2014, 139, 3174–3178. [Google Scholar] [CrossRef] [PubMed]
- Nahidiazar, L.; Agronskaia, A.V.; Broertjes, J.; den van Broek, B.; Jalink, K. Optimizing imaging conditions for demanding multi-color super resolution localization microscopy. PLoS ONE 2016, 11, e0158884. [Google Scholar] [CrossRef] [PubMed]
- Bearer, E.L.; Prakash, J.M.; Li, Z. Actin dynamics in platelets. Int. Rev. Cytol. 2002, 217, 137–182. [Google Scholar] [PubMed]
- Hartwig, J.H.; DeSisto, M. The cytoskeleton of the resting human blood platelet: Structure of the membrane skeleton and its attachment to actin filaments. J. Cell Biol. 1991, 112, 407–425. [Google Scholar] [CrossRef] [PubMed]
- Italiano, J.E.; Lecine, P.; Shivdasani, R.A.; Hartwig, J.H. Blood Platelets Are Assembled Principally at the Ends of Proplatelet Processes Produced by Differentiated Megakaryocytes 7. J. Cell Biol. 1999, 147, 1299–1312. [Google Scholar] [CrossRef] [PubMed]
- Falet, H.; Barkalow, K.L.; Pivniouk, V.I.; Barnes, M.J.; Geha, R.S.; Hartwig, J.H. Roles of SLP-76, phosphoinositide 3-kinase, and gelsolin in the platelet shape changes initiated by the collagen receptor GPVI/FcR gamma-chain complex. Blood 2000, 96, 3786–3792. [Google Scholar] [PubMed]
- Watson, S.P. Platelet activation by extracellular matrix proteins in haemostasis and thrombosis. Curr. Pharm. Des. 2009, 15, 1358–1372. [Google Scholar] [CrossRef] [PubMed]
- Mellor, H. The role of formins in filopodia formation. Biochim. Biophys. Acta Mol. Cell Res. 2010, 1803, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Grimm, J.B.; Klein, T.; Kopek, B.G.; Shtengel, G.; Hess, H.F.; Sauer, M.; Lavis, L.D. Synthesis of a Far-Red Photoactivatable Silicon-Containing Rhodamine for Super-Resolution Microscopy. Angew. Chem. Int. Ed. 2016, 55, 1723–1727. [Google Scholar] [CrossRef] [PubMed]
- Heilemann, M.; Margeat, E.; Kasper, R.; Sauer, M.; Tinnefeld, P. Carbocyanine dyes as efficient reversible single-molecule optical switch. J. Am. Chem. Soc. 2005, 127, 3801–3806. [Google Scholar] [CrossRef] [PubMed]
- Symons, M.H.; Mitchison, T.J. Control of actin polymerization in live and permeabilized fibroblasts. J. Cell Biol. 1991, 114, 503–513. [Google Scholar] [CrossRef] [PubMed]
- Wolter, S.; Löschberger, A.; Holm, T.; Aufmkolk, S.; Dabauvalle, M.C.; Van De Linde, S.; Sauer, M. rapidSTORM: Accurate, fast open-source software for localization microscopy. Nat. Methods 2012, 9, 1040–1041. [Google Scholar] [CrossRef] [PubMed]
- Jacak, J.; Schaller, S.; Borgmann, D.; Winkler, S.M. Characterization of the Distance Relationship Between Localized Serotonin Receptors and Glia Cells on Fluorescence Microscopy Images of Brain Tissue. Microsc. Microanal. 2015, 21, 826–836. [Google Scholar] [CrossRef] [PubMed]
- Göhring, J.; Fulcher, N.; Schilcher, K.; Barta, A.; Jacak, J. Suitable transfection methods for single particle tracing in plant suspension cells. Plant Methods 2014, 10, 15. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.; Schwartz, S.L.; Byars, J.M.; Lidke, K.A. Simultaneous multiple-emitter fitting for single molecule super-resolution imaging. Biomed. Opt. Express 2011, 2, 1377. [Google Scholar] [CrossRef] [PubMed]
- Dennis, J.E.; Mei, H.W. Two new unconstrained optimization algorithms which use function and gradient values. J. Optim. Theory Appl. 1979, 28, 453–482. [Google Scholar] [CrossRef]
- Mortensen, K.I.; Churchman, H.L.S.; Spudich, J.A.; Flyvbjerg, H. Optimized localization-analysis for single-molecule tracking and super-resolution microscopy. Nat. Methods 2010, 7, 377–381. [Google Scholar] [CrossRef] [PubMed]
- Han, R.; Wang, L.; Xu, F.; Zhang, Y.; Zhang, M.; Liu, Z.; Ren, F.; Zhang, F. Drift correction for single-molecule imaging by molecular constraint field, a distance minimum metric. BMC Biophys. 2015, 8, 1. [Google Scholar] [CrossRef] [PubMed]
- Rosenblatt, M. Remarks on Some Nonparametric Estimates of a Density Function. Ann. Math. Stat. 1956, 27, 832–837. [Google Scholar] [CrossRef]
- Parzen, E. On estimation of a probability density function and mode. Ann. Math. Stat. 1962, 33, 1065–1076. [Google Scholar] [CrossRef]

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mayr, S.; Hauser, F.; Peterbauer, A.; Tauscher, A.; Naderer, C.; Axmann, M.; Plochberger, B.; Jacak, J. Localization Microscopy of Actin Cytoskeleton in Human Platelets. Int. J. Mol. Sci. 2018, 19, 1150. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19041150
Mayr S, Hauser F, Peterbauer A, Tauscher A, Naderer C, Axmann M, Plochberger B, Jacak J. Localization Microscopy of Actin Cytoskeleton in Human Platelets. International Journal of Molecular Sciences. 2018; 19(4):1150. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19041150
Chicago/Turabian StyleMayr, Sandra, Fabian Hauser, Anja Peterbauer, Andreas Tauscher, Christoph Naderer, Markus Axmann, Birgit Plochberger, and Jaroslaw Jacak. 2018. "Localization Microscopy of Actin Cytoskeleton in Human Platelets" International Journal of Molecular Sciences 19, no. 4: 1150. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19041150