1. Introduction
Recessive dystrophic epidermolysis bullosa (RDEB) is a prototypical recessive condition and is characterized by mutations to the
COL7A1 gene on chromosome 3.
COL7A1 is likewise a prototypical large gene and spans ~31 kb and contains 118 exons with an open reading frame of ~9 kb [
1,
2]. RDEB causative mutations occur over the span of the gene and the resultant phenotype is characterized by diminished/absent type VII collagen (C7) protein causing mucocutaneous disease manifestations. Severe, chronic skin blistering occurs along with esophageal strictures, mitten deformities, dental anomalies, corneal scarring, and increased incidence for aggressive squamous cell carcinomas [
3]. Therapeutic benefit can be achieved by the delivery of functional C7 protein. Sources of C7 include transplant of allogeneic or gene corrected autologous cells and/or recombinant C7 protein injection.
Woodley and colleagues delivered recombinant C7 protein by intravenous injection showing that C7 produced locally or from a distance can mediate a functional benefit [
4]. However, repetitive injections of recombinant peptide over the course of a patient’s lifetime are fiscally burdensome, making cellular sources an attractive option. Allogeneic cellular injections have resulted in improved skin integrity; however, the low expression levels of
COL7A1 from the endogenous promoter results in poor delivery beyond the site of injection [
5]. Further, allogeneic cells may not persist long term due to host immune-mediated clearance [
6]. Autologous cellular engineering is highly promising due to the lowered risk of immune rejection, and
COL7A1 gene expression has been restored in patient derived cells using gene therapy and gene editing [
7,
8].
To encode, deliver, and express
COL7A1, gamma retroviral and lentiviral expression vectors have been developed and deployed that result in supraphysiological
COL7A1 gene expression. However, the large size of the
COL7A1 cDNA can result in lowered titers that can make effective delivery a challenge [
5,
9,
10,
11,
12]. Efforts have been undertaken to use less size-restricted platforms such as the phiC31 integrase, or Sleeping Beauty, transposon; however, the effective delivery of these vectors can similarly be challenging [
5,
13,
14]. Additionally, the semi-random genomic integration profiles of these systems in the premalignant RDEB phenotype represents a significant safety concern due to insertional mutagenesis [
15,
16,
17].
To capitalize on the precise targeting capabilities afforded by gene editing, we have targeted the
COL7A1 gene with transcription activator like effector nucleases (TALEN) and the clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9 system derived from
Streptococcus pyogenes [
8,
18]. Along with zinc finger nucleases and meganucleases, TALENs and CRISPR/Cas9 represent programmable reagents capable of generating single or double stranded DNA breaks at user-defined loci [
19,
20]. This stimulates homology directed repair (HDR) from an exogenous template allowing for precision genome modification. In situ gene correction maximizes safety but gene control is regulated by the comparatively weak
COL7A1 promoter. As such, the systemic therapeutic impact may be incomplete due to the limited distribution of C7 protein.
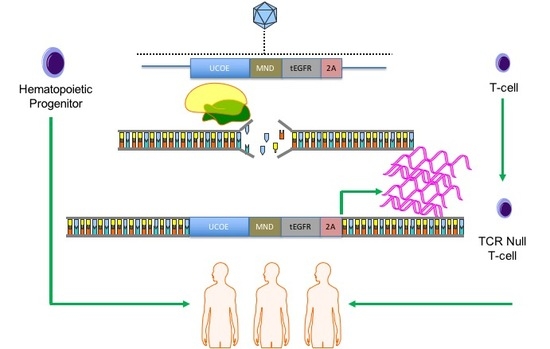
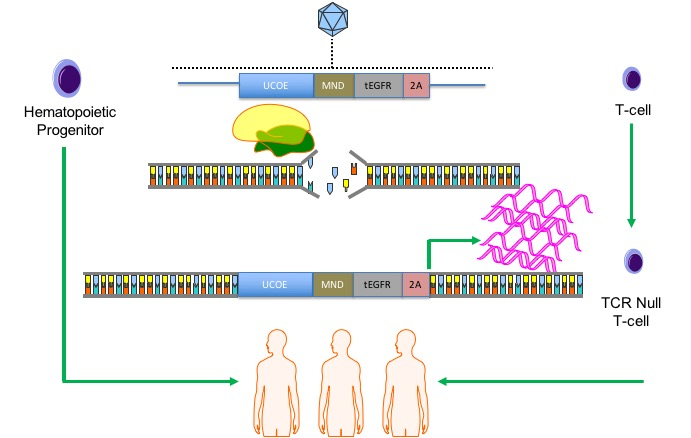
We hypothesized that we could synergize the attributes of gene therapy and gene editing: supraphysiological gene expression and a high degree of specificity. Previous efforts to accomplish this have centered on “safe harbor” site incorporation of a candidate gene driven by exogenous regulatory elements [
21]. Delivering a cargo as large as the ~9 kb
COL7A1 cDNA can be challenging making this approach sub-optimal. To address this, we devised a strategy whereby we could incorporate a powerful transcriptional activator into the native
COL7A1 locus. This resulted in profound upregulation of the endogenous
COL7A1 gene. Because our approach relies on a functional gene embedded in the genome, we pursued our strategy in cells with a favorable immunological profile in that they either innately, or can be engineered to, have a low frequency and incidence of immune-based side effects. Umbilical cord blood (UCB) derived hematopoietic stem cells (HSC) are effective for allogeneic therapy and display reduced rates of graft versus host disease (GVHD) [
22,
23]. Here we show robust
COL7A1 gene activation in UCB HSCs with maintenance of their multi-lineage differentiation potential in colony forming assays. In parallel, we pursued T-cell engineering and observed
COL7A1 expression levels that surpassed those of wild type keratinocytes. By subsequently ablating the T-cell receptor complex we generated a stable population of T-cells with a low risk of triggering GVHD. Collectively, our engineering approach allows for targeted gene upregulation with HDR dependent co-expression of a clinically relevant cell surface marker that serves as a selection agent and suicide gene. This strategy represents a streamlined translational engineering platform readily and rapidly transferrable to multiple model systems for user-defined transcriptional modification.
3. Discussion
Here we report a strategy for targeted gene upregulation using the CRISPR/Cas9 system for homology directed repair mediated insertion of a transcriptional promoting element we termed UMET. This powerful platform employs a ubiquitous chromatin opening element, the MND promoter, and translationally relevant EGFR reporter to drive endogenous gene overexpression. As proof of principle, we targeted the COL7A1 gene that serves as a surrogate for genetic conditions involving large open reading frames that are difficult to deliver as full cDNA expression constructs. This novel engineering methodology allowed for high level COL7A1 expression in allogeneic umbilical cord blood hematopoietic stem cells and peripheral blood T-cells. This robust and user-friendly platform is readily deployable for targeted gene overexpression in multiple applications.
Gene therapy using exogenous gene encoding regulatory elements can result in supraphysiological gene expression in autologous or allogeneic cells. This approach can be highly effective particularly in disease phenotypes where therapeutic peptide(s) produced from a circulating depot population of cells can facilitate systemic therapeutic benefit. A crucial determinant for efficient gene therapy-based cellular engineering is the choice of delivery vehicle which can be broadly categorized into viral and non-viral systems that either integrate into the genome or are maintained as extrachromosomal species. The size of the gene is a key consideration for the choice of vector as many systems are restricted by the limits of cargo they can effectively accommodate and deliver. Larger cargo (5+ kb open reading frames) are generally more difficult to deliver and can be associated with higher rates of toxicity.
Non-integrating expression vectors can be maintained as a non-integrating episome that in cells and tissues with a low proliferative capacity can be maintained long term. However, ex vivo modification of cells with limited growth potential is challenging due to scalability and therapeutic dose of cells able to be delivered. Integrating vectors result in permanent insertion into the genome and long-term expression in parental and progeny cells making them well suited for sustained supranormal gene expression. There is an offsetting risk; however, of genomic sequence disruption and potential for insertional mutagenesis. A further important consideration for gene therapy applications is the potential for adverse immunological side effects. The use of autologous cells that are educated in the host environment represents a desirable approach for minimizing the occurrence of transplant associated complications. A major hurdle to the realization of autologous therapies is efficient and uniform ex vivo engineering of patient cells. The numbers of cells and frequency of gene transfer can be highly variable such that complete therapeutic benefit may not be achieved. Certain disorders such as RDEB that have large open reading frames make delivery of the therapeutic cargo a compounding challenge. Thus, there is a gap in the field of gene and cellular therapy for generating a standardized population of cells with a low propensity for immune-based side effects that express supraphysiological levels of a candidate gene in a manner that maintains the integrity of the genome. To address this deficiency, we instituted a line of study to determine the optimal conditions for candidate gene expression using the native gene with targeted introduction of powerful gene activation components upstream of the start codon. We hypothesized that lymphohematopoietic cells, due to their broad circulatory potential and relative ease of obtainment, would be an ideal population for engineering. We tested our hypothesis in umbilical cord blood hematopoietic stem cells and peripheral blood T-cells. These cells are of high translational impact and have a favorable innate or acquired immunological profile for broad transplant application. UCB hematopoietic progenitors have a lowered risk of causing graft versus host disease allowing for greater donor:recipient mismatches in the allogeneic transplant setting. T-cells, through a secondary engineering step to ablate the T-cell receptor complex, can be rendered inert in regard to antigen recognition capability. In these cells we optimized the engineering parameters for gene expression upregulation for the generation of a uniform pool of cells as an off the shelf product with the potential to be given to multiple recipients with a low risk of allogeneic transplant associated side effects.
The emergence of easily produced programmable nucleases such as the CRISPR/Cas9 system have allowed for precision gene targeting with a specificity that greatly mitigates collateral “off target” genomic damage. CRISPR/Cas9 is comprised of two components: the Cas9 nuclease and a small guide RNA transcript. The functional complex of Cas9 and the gRNA bind the target DNA sequence and generates a single or double stranded DNA break that can be repaired from an exogenous repair template by the error free homology directed repair DNA pathway. While gene editing represents a transformative strategy for cellular engineering, it has been limited to date in its ability to uniformly produce therapeutic levels of gene expression. Editing-based gene correction has centered on two strategies: site specific repair or incorporation of a full open reading frame coding sequence and regulatory elements at safe harbor loci. The correction of a disease-causing base in situ results in maintenance of gene expression from the endogenous regulatory elements that may not drive gene expression levels to the therapeutic threshold. Alternatively, safe harbor locus incorporation of an exogenous promoter, candidate cDNA, and polyadenylation signal can drive high level gene expression. The most common safe harbor is the AAVS1 locus on chromosome 19 that has served as what could be termed a landing pad for user defined open reading frame/regulatory element sequence incorporation. However, the ability to incorporate genes in excess of five kilobases into this locus in therapeutically relevant cells can be challenging. Therefore, we hypothesized that targeted knock in of transcriptional elements and a reporter gene upstream and in frame with an endogenous gene would facilitate gene upregulation and selection of modified cells.
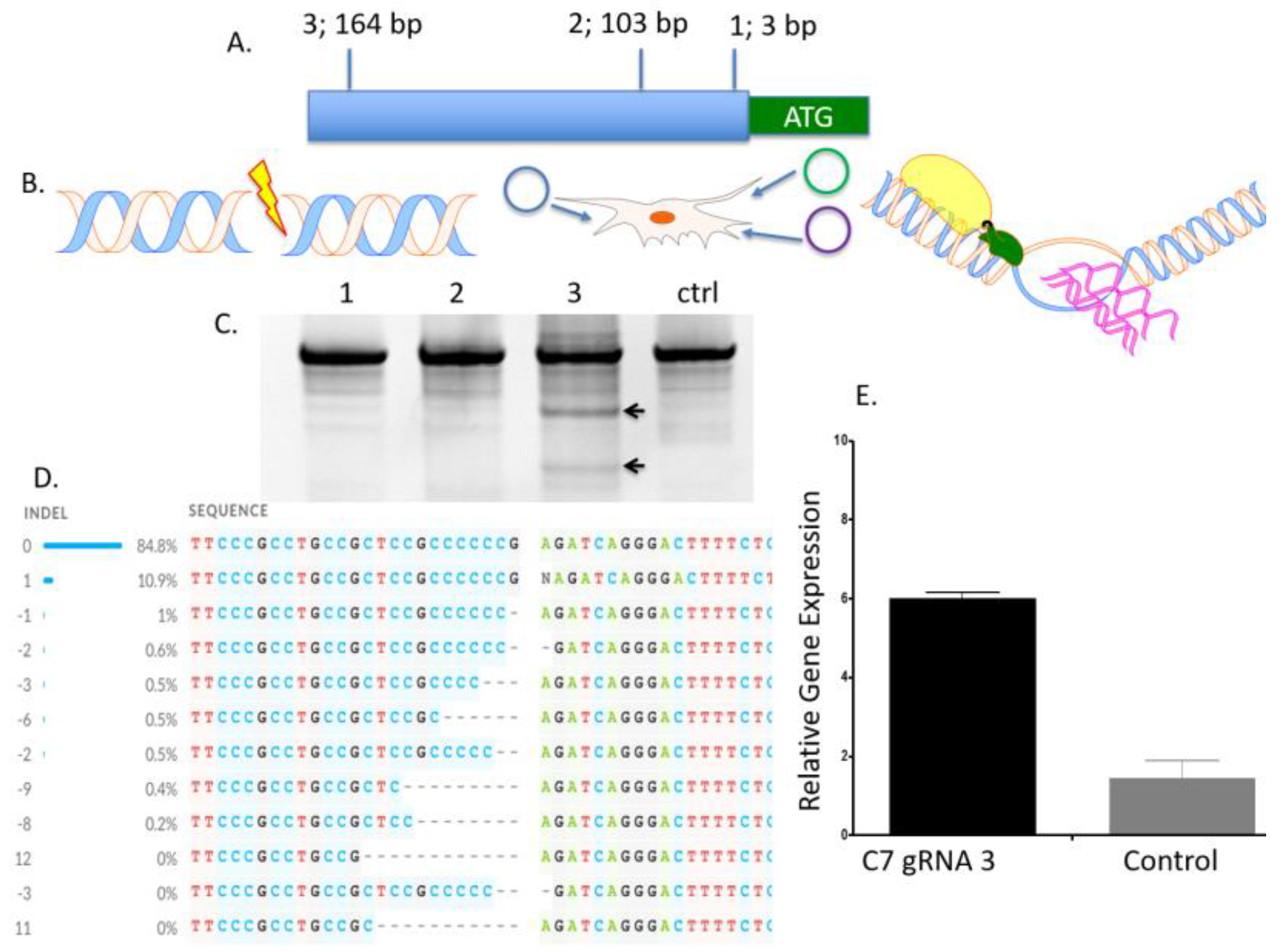
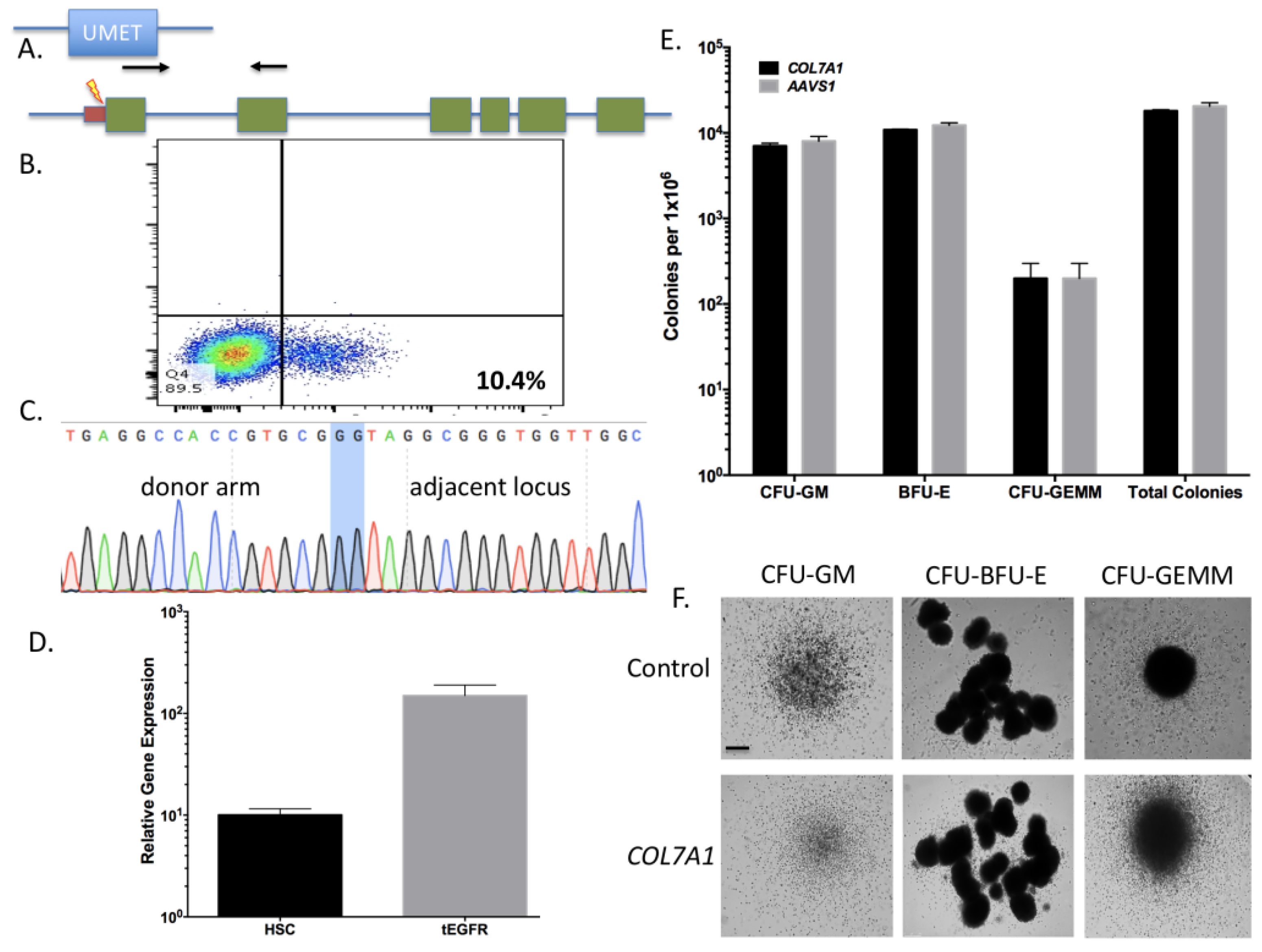
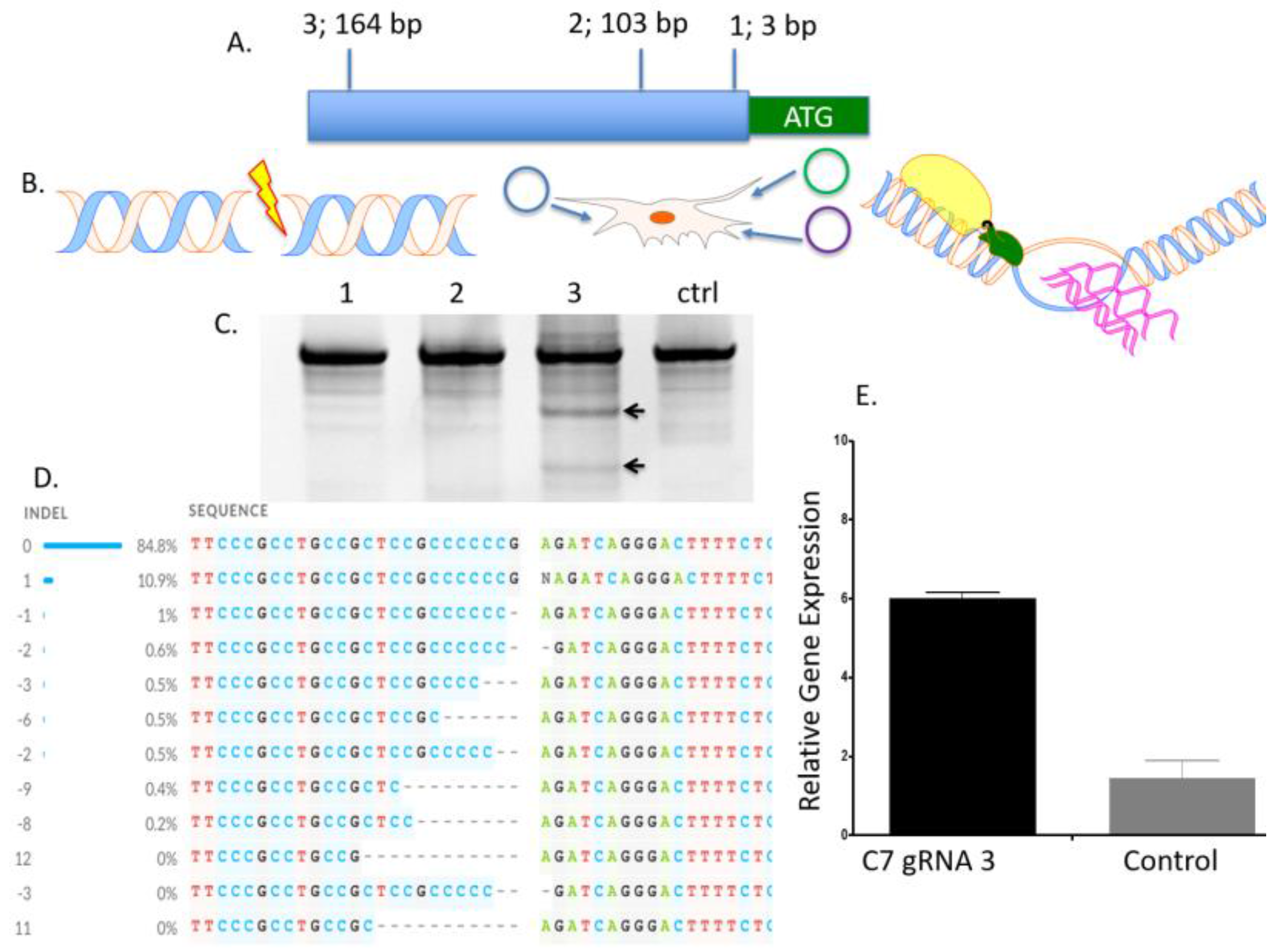
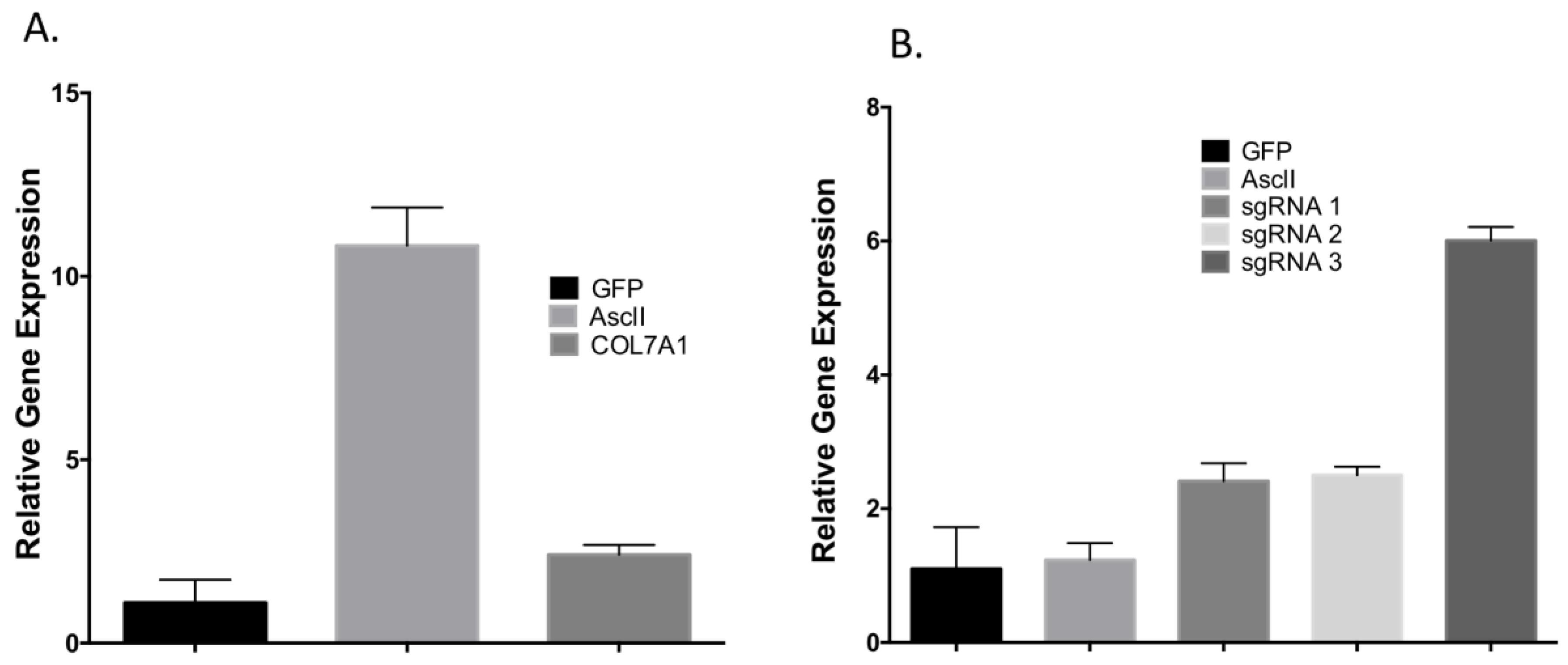
To determine the optimal position to insert the UMET element we screened three guide RNA candidates in a ~200 bp window immediately upstream of the native
COL7A1 transcriptional start site (
Figure 1A). One candidate showed demonstrable activity as assessed by Surveyor assay and sequencing (
Figure 1C,D). To confirm that this candidate would be suitable for promoting gene activity, we introduced a nuclease inactive Cas9 that retains DNA binding ability and is fused to a VP64 transcriptional activator [
25]. Using this system, we observed a ~6-fold increase in gene expression in HEK 293T cells, demonstrating that this portion of the
COL7A1 locus was favorable for promoting transcriptional upregulation (
Figure 1E). Importantly, this version of Cas9 is >5 kb making it difficult to deliver. Our design strategy with the UMET targeting construct represents a novel and streamlined approach for candidate gene upregulation.
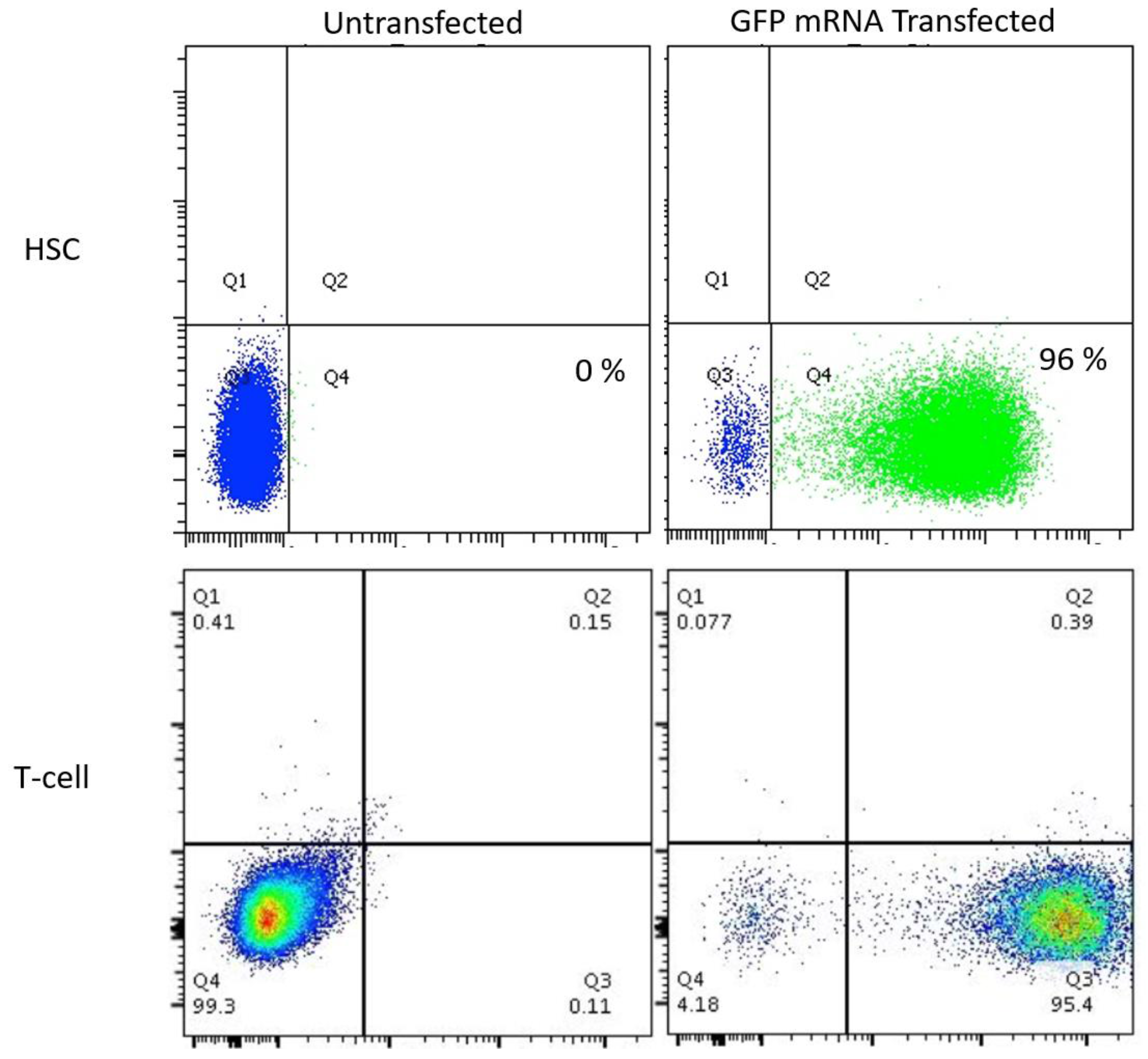
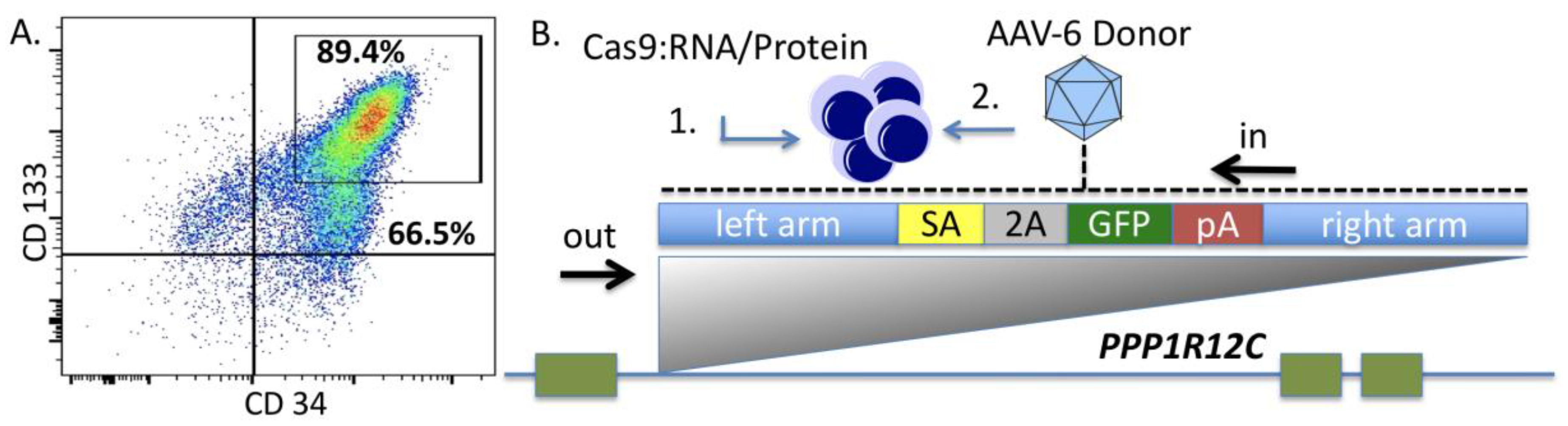
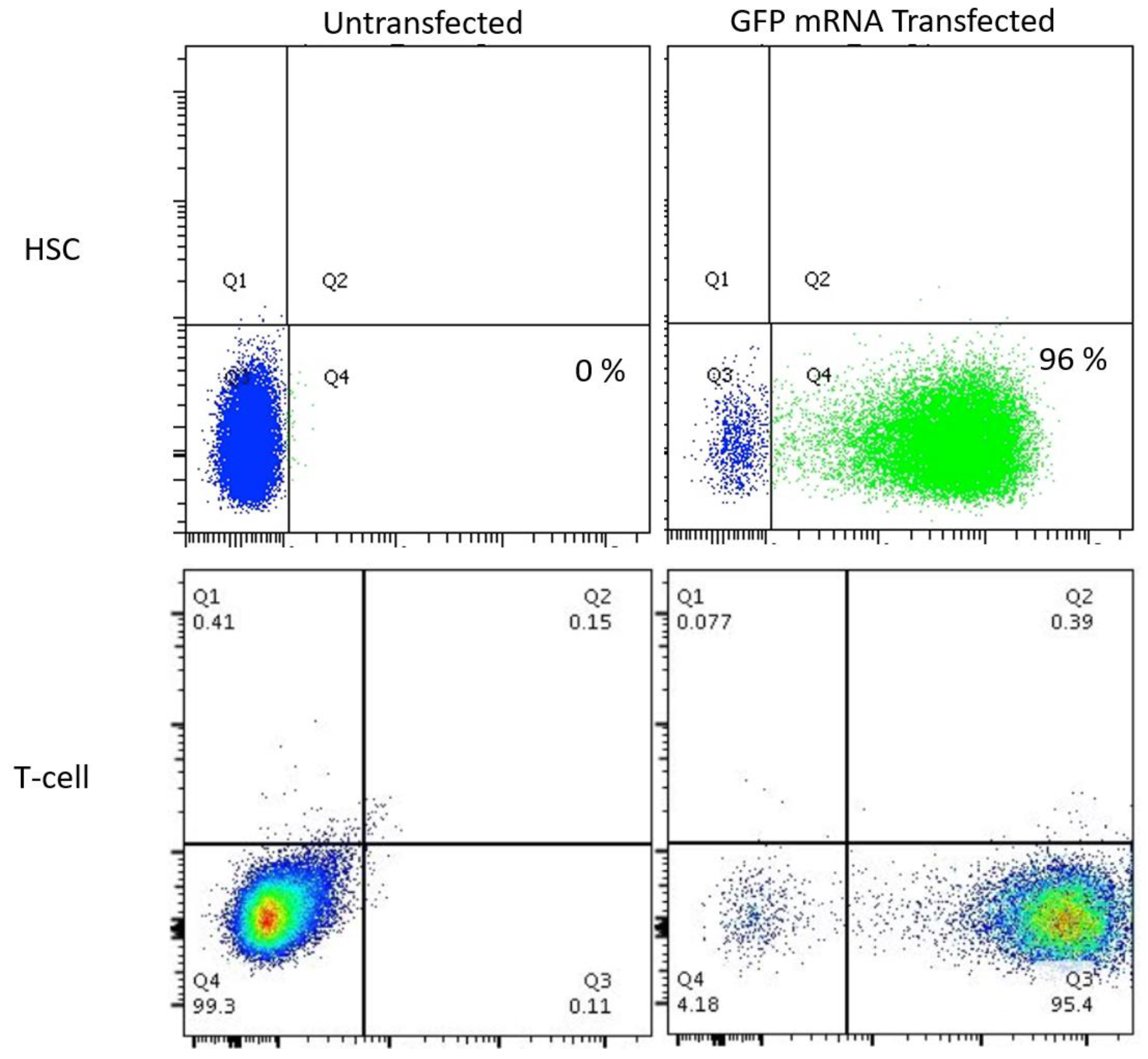
Because few reports show UCB HSC modification with CRISPR/Cas9, we rigorously optimized the conditions for HDR in this cell population that possesses a greater ability to be delivered to disparate patients in allogeneic transplant. The donor units of HSCs routinely showed a high degree of CD34
+CD133
+ cells that are characteristic of HSCs (
Figure 2A). The Cas9 delivery format was then assessed for HDR ability, efficiency, and kinetics. Comparisons between Cas9 mRNA and Cas9 recombinant peptide were performed using a homologous recombination reporter system that is well characterized for the
AAVS1 safe harbor locus (
Figure 2B) [
26]. It is designed to express GFP only upon HDR-based insertion into the first intron of the
PPP1R12C locus. Delivery of the donor was accomplished by AAV-6 transduction, a serotype shown to be highly effective at transducing CD34
+ HSCs [
32]. We observed that a Cas9:gRNA ribonucleoprotein complex was more efficient at facilitating HDR when the AAV-6 particles were added in a narrow time window after electroporation (
Figure 2C). The analyses in
Figure 2C were performed three days after gene transfer and to rigorously assess the timing of HDR post-electroporation and immediate AAV-6 donor addition, we screened cells at defined time points. Based on the data in
Figure 2D we show that the cells undergo HDR modification in a ~24-h time period. This narrow window in which editing occurs is of tremendous importance as it allows for the ex vivo/in vitro culture time to be streamlined in order to maintain the phenotype of the stem cell population. The optimal dose of donor was next assessed and we observed that a dose of 5 × 10
5 was capable of mediating robust HDR (
Figure 2E). The CRISPR/Cas9 modified cells were then placed into semi-solid methycellulose for colony forming unit potential. There were no differences between control, unmodified cells and those that underwent Cas9 RNP electroporation, AAV-6 transduction, and
AAVS1 locus HDR (
Figure 2F). These data demonstrated that the optimized gene editing conditions did not perturb HSC function and lineage commitment in vitro.
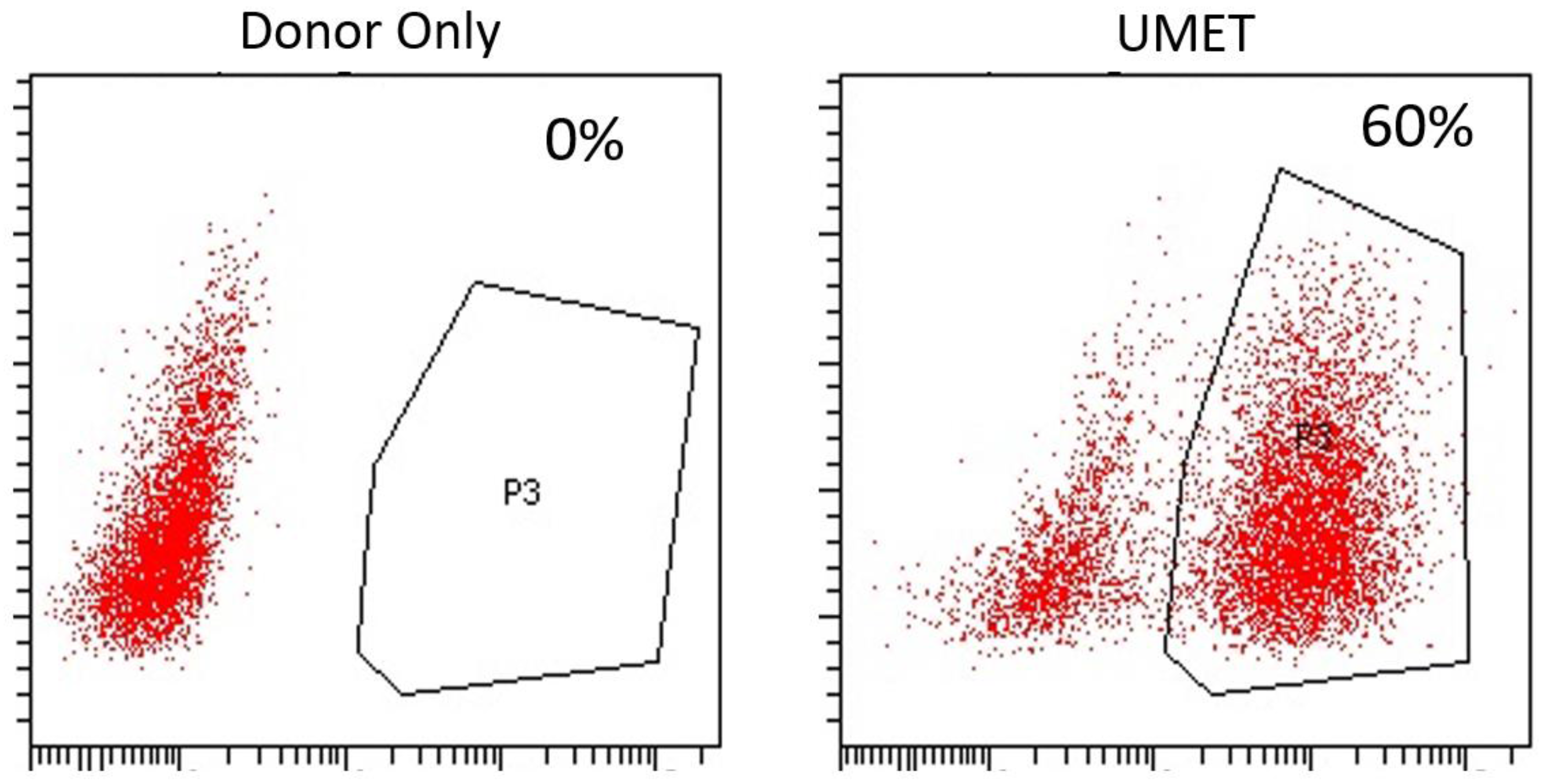
Using the parameters defined in
Figure 2 we then designed, built, and tested a novel donor (UMET) for targeted
COL7A1 gene upregulation. It consisted of a UCOE sequence that has been shown to maintain an open chromatin profile [
28], the MND promoter that is well defined for strong activity in T-cells and HSCs [
29], and the tEGFR that serves for both selection and downstream considerations for safety by allowing for targeted depletion as a suicide gene. The tEGFR was placed in frame with the endogenous
COL7A1 gene by virtue of a T2A sequence (
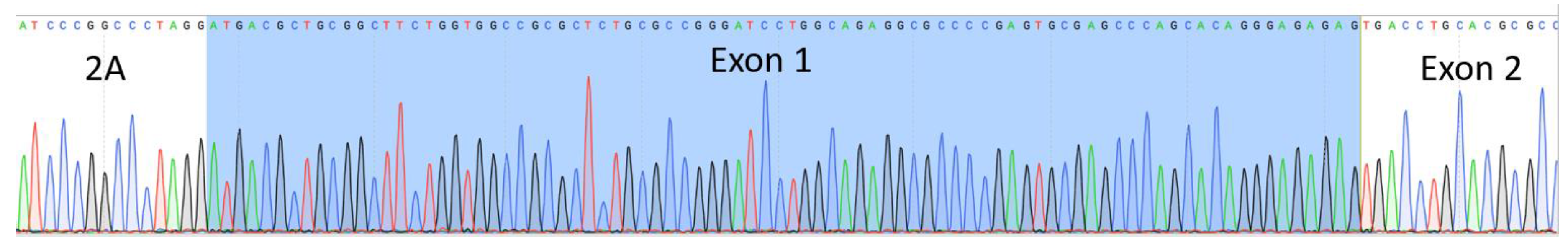
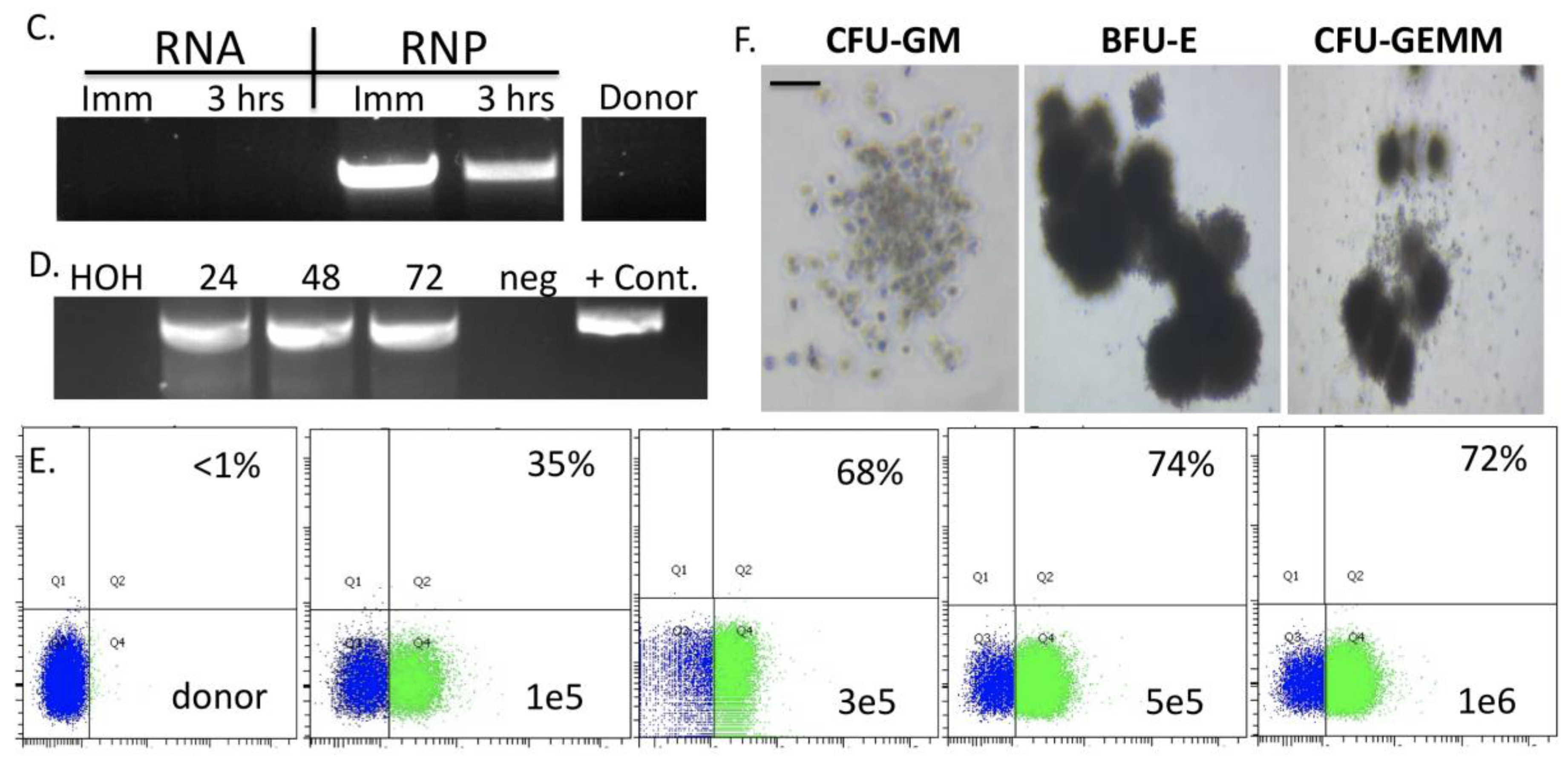
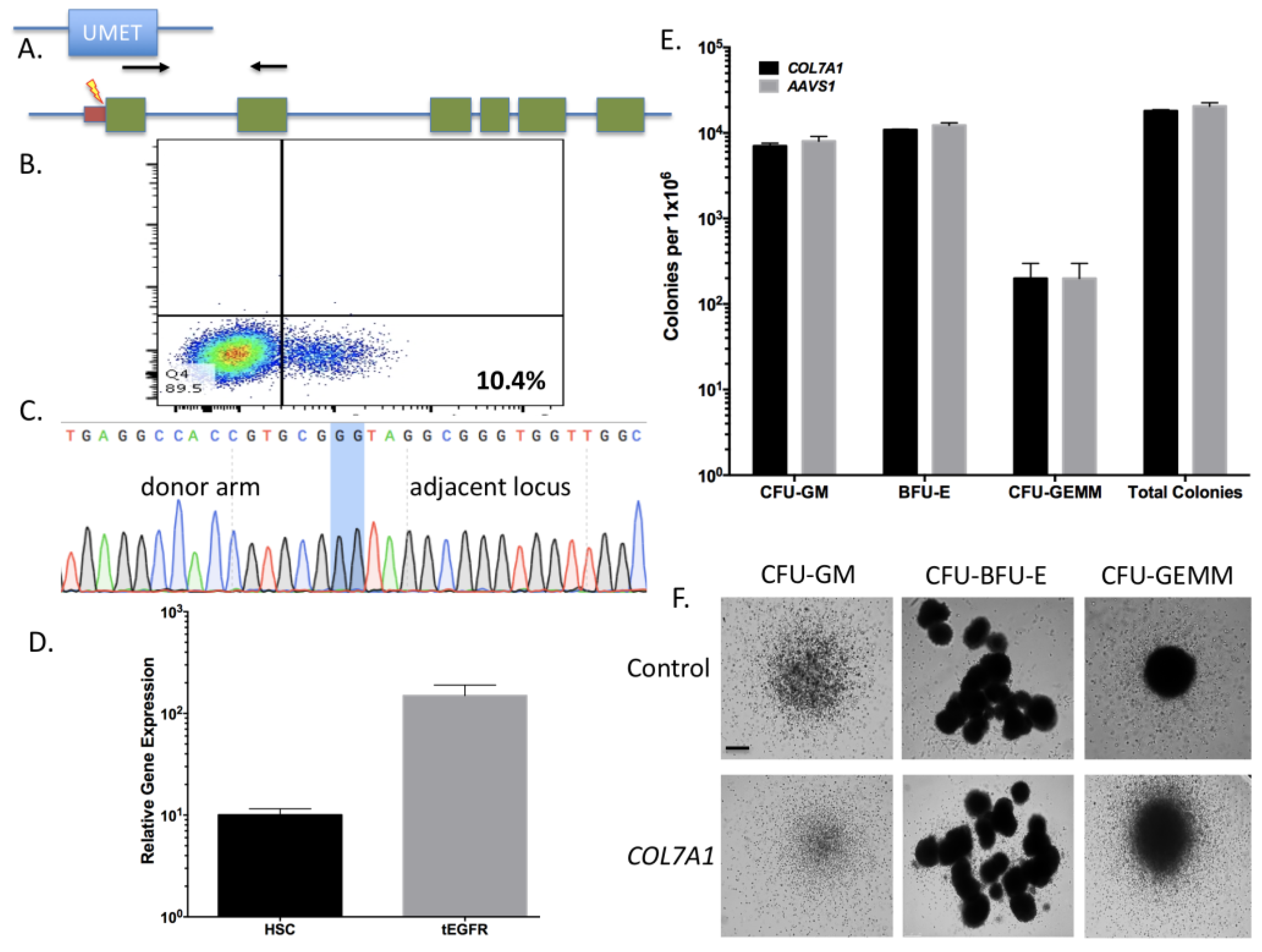
Figure 3A). The entire targeting cassette was ~4 kb to allow for efficient AAV packaging. Cas9 RNP electroporation followed by donor addition resulted in ~10% HSC gene modification rates that allowed for recovery of a pure population of tEGFR
+ CD34
+ cells for molecular and phenotypic characterization (
Figure 3B). Inside out PCR showed
COL7A1 locus HDR (
Figure 3C) and qRT-PCR showed that donor incorporation drove a 15-fold increase in
COL7A1 expression in HSCs (
Figure 3D). The modified cells showed both a normal distribution and morphology of hematopoietic lineage derivatives in vitro demonstrating that supraphysiological
COL7A1 expression did not impact CFU potential (
Figure 3E,F). These data show that
COL7A1 locus targeting and gene expression did not appreciably affect the phenotype and properties of the HSCs. While a diminished rate of
COL7A1 gene targeting compared to
AAVS1 was observed, we propose that this is related to the differential locus accessibility to the CRISPR/Cas9 reagents. The
COL7A1 locus is not predicted to be highly active in HSCs and thus may be condensed with chromatin making it less amenable to targeting than
AAVS1. It is for these reasons we included a UCOE element in our design in order to promote greater gene expression in areas of the genome with diminished transcriptional levels. Importantly, our targeting efficiencies are in line with previous HSC gene editing experiments and the unique inclusion of the tEGFR allows for cell isolation and, paired with scaling, the engineering process is viable for downstream (e.g., clinical) application(s). Collectively, our defined engineering conditions observations are crucial for clinical scale up and application and our optimized protocol promotes a rapid turnaround time for HSC engineering and delivery.
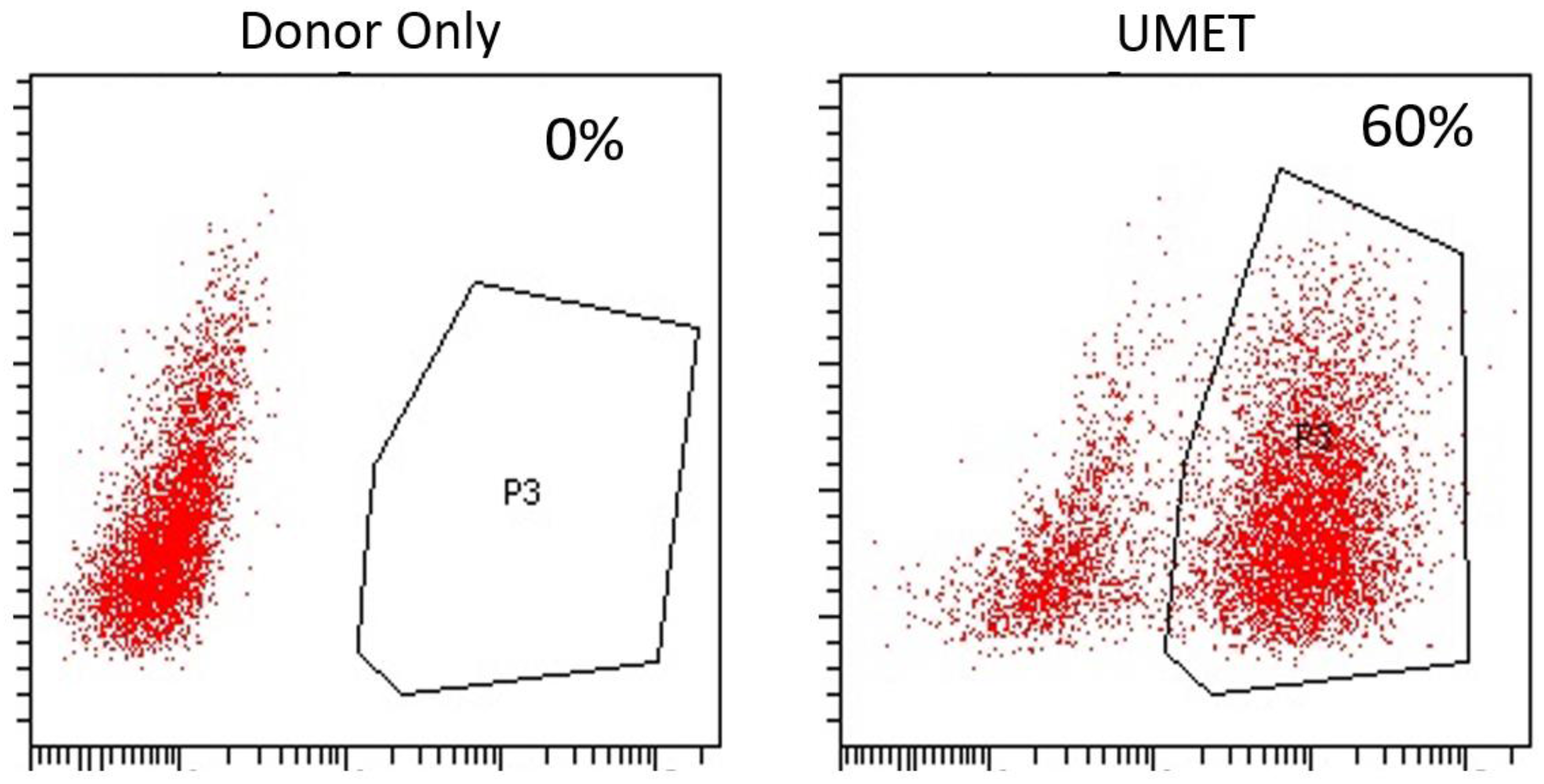
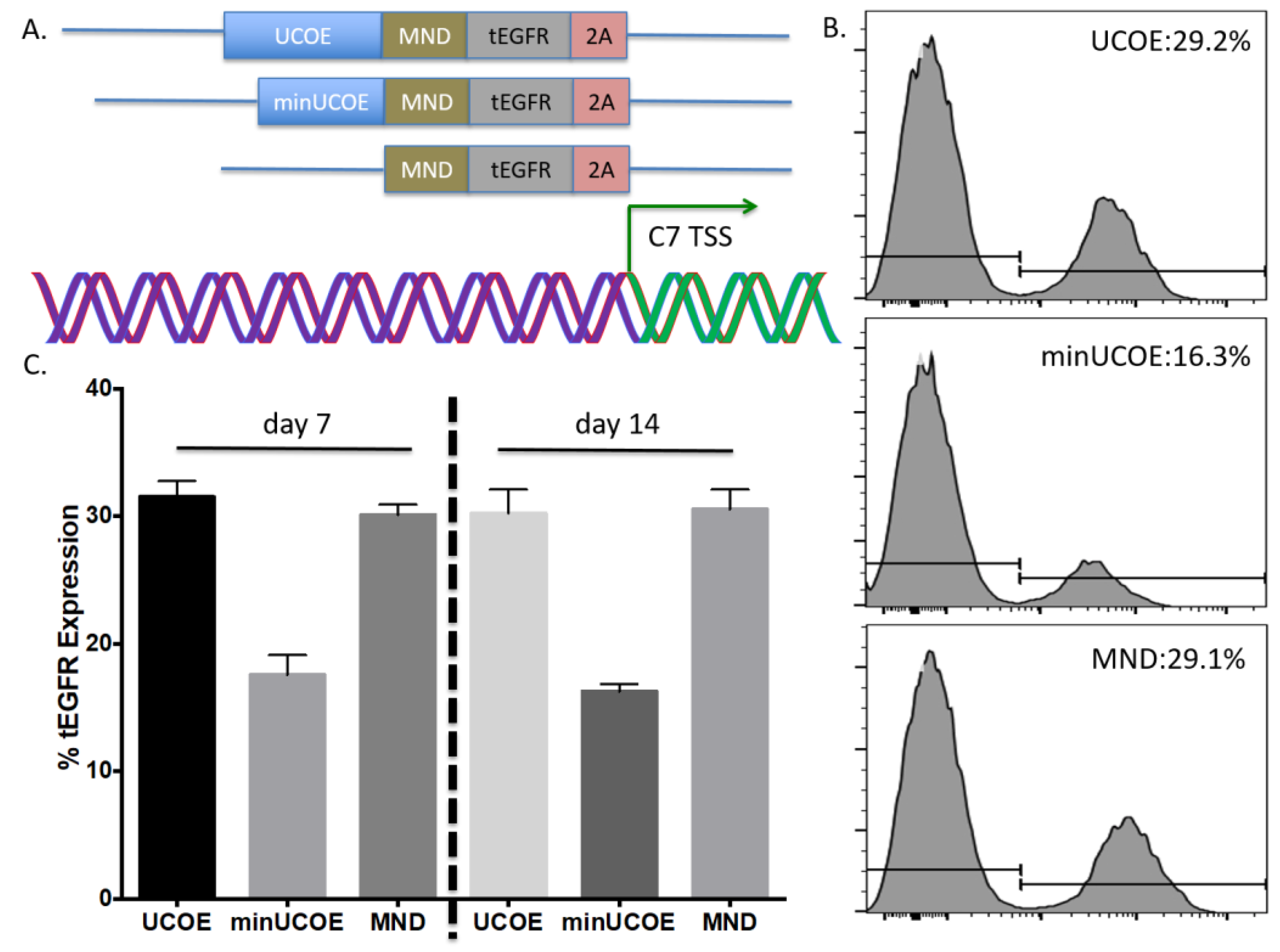
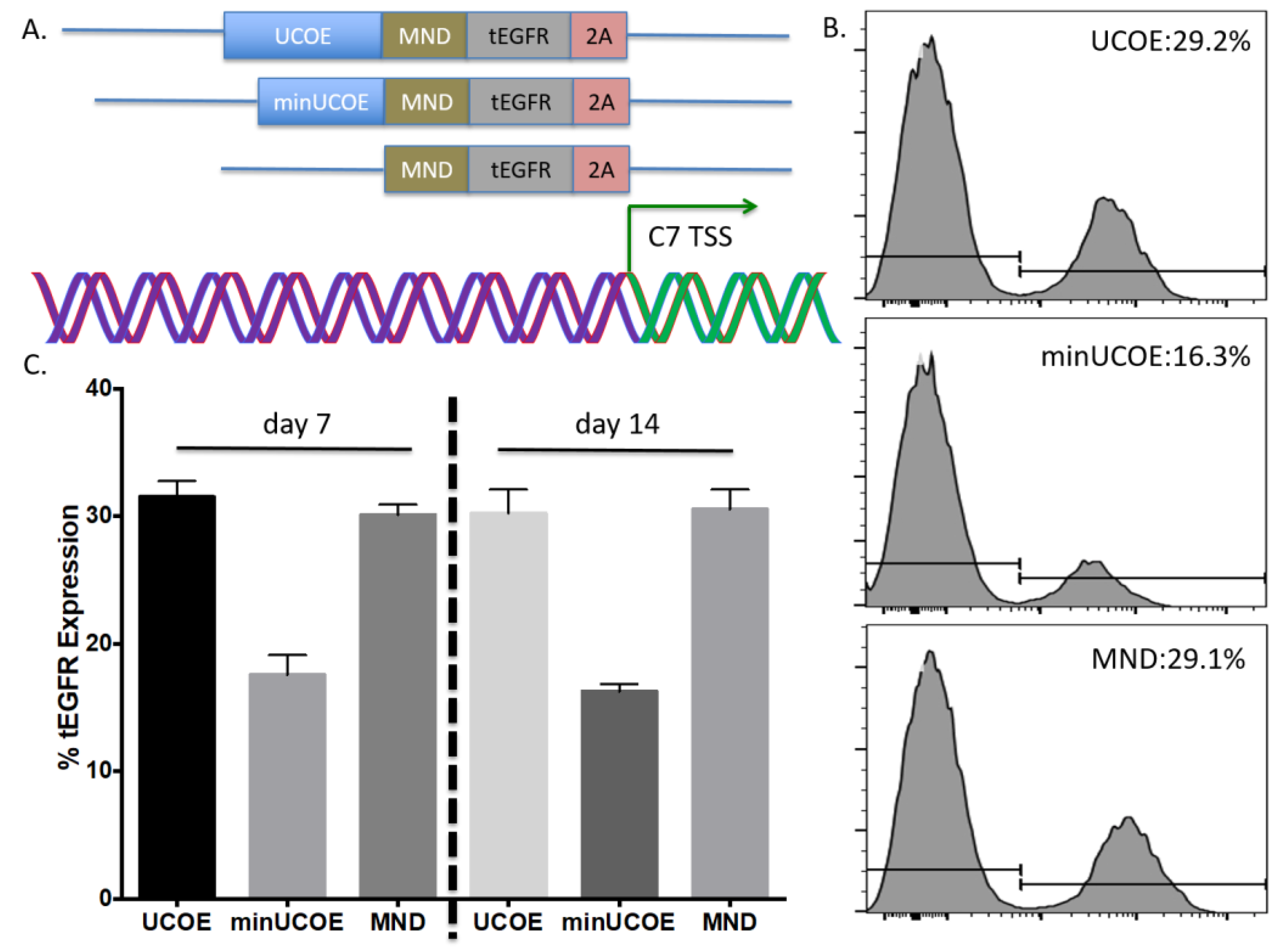
Toward further defining the optimal donor platform we generated a truncated UCOE sequence and a donor that only had a promoter (
Figure 4A). These donors were tested in primary T-cells and gene targeting frequency was measured by tEGFR expression. The lowest rates of HDR were seen in the donor with the minimal UCOE element while the MND only and UCOE MND-based donors were nearly equivalent (
Figure 4B). The levels of tEGFR expression remained stable over two weeks with significant cell expansion over that period (
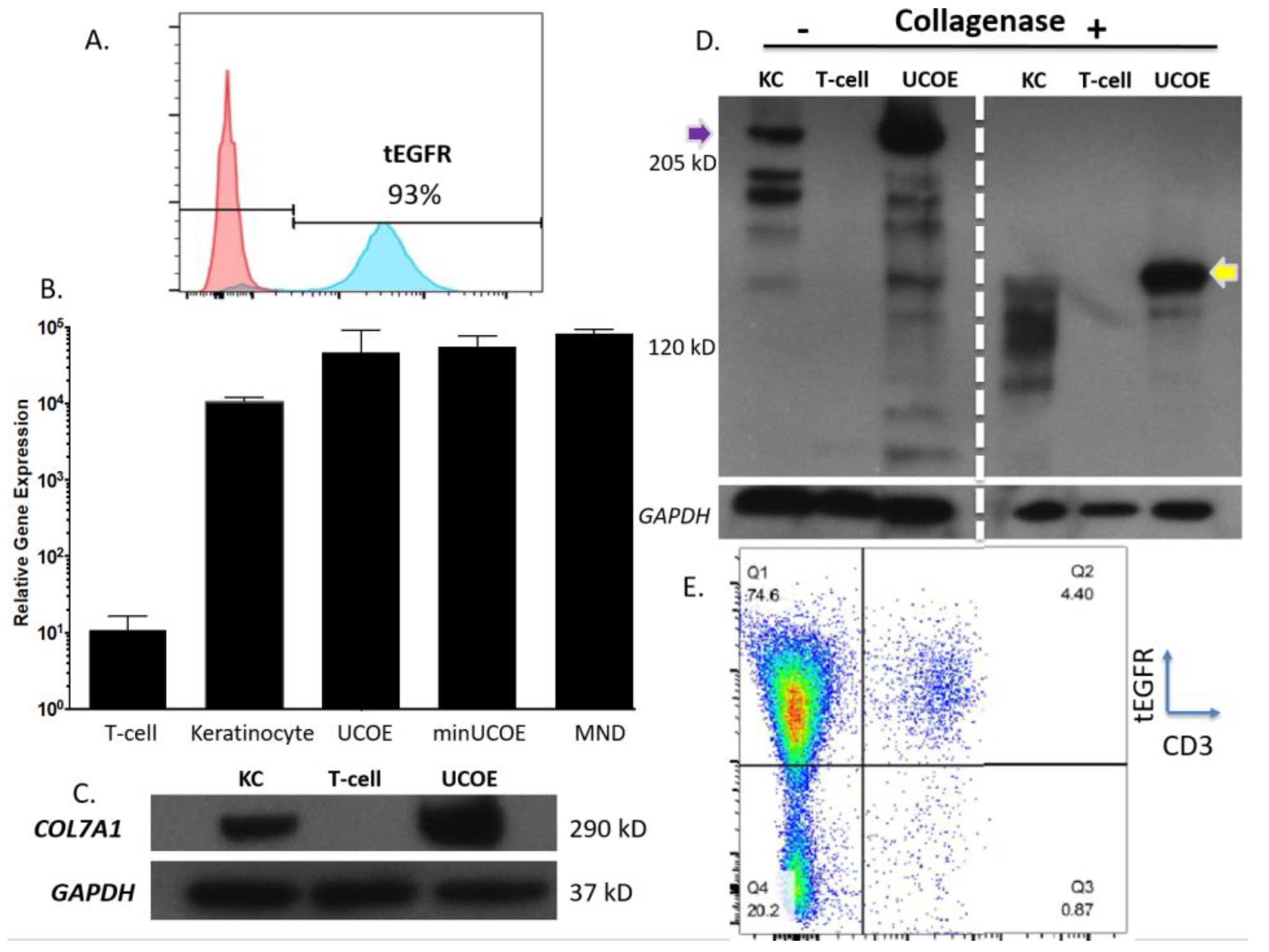
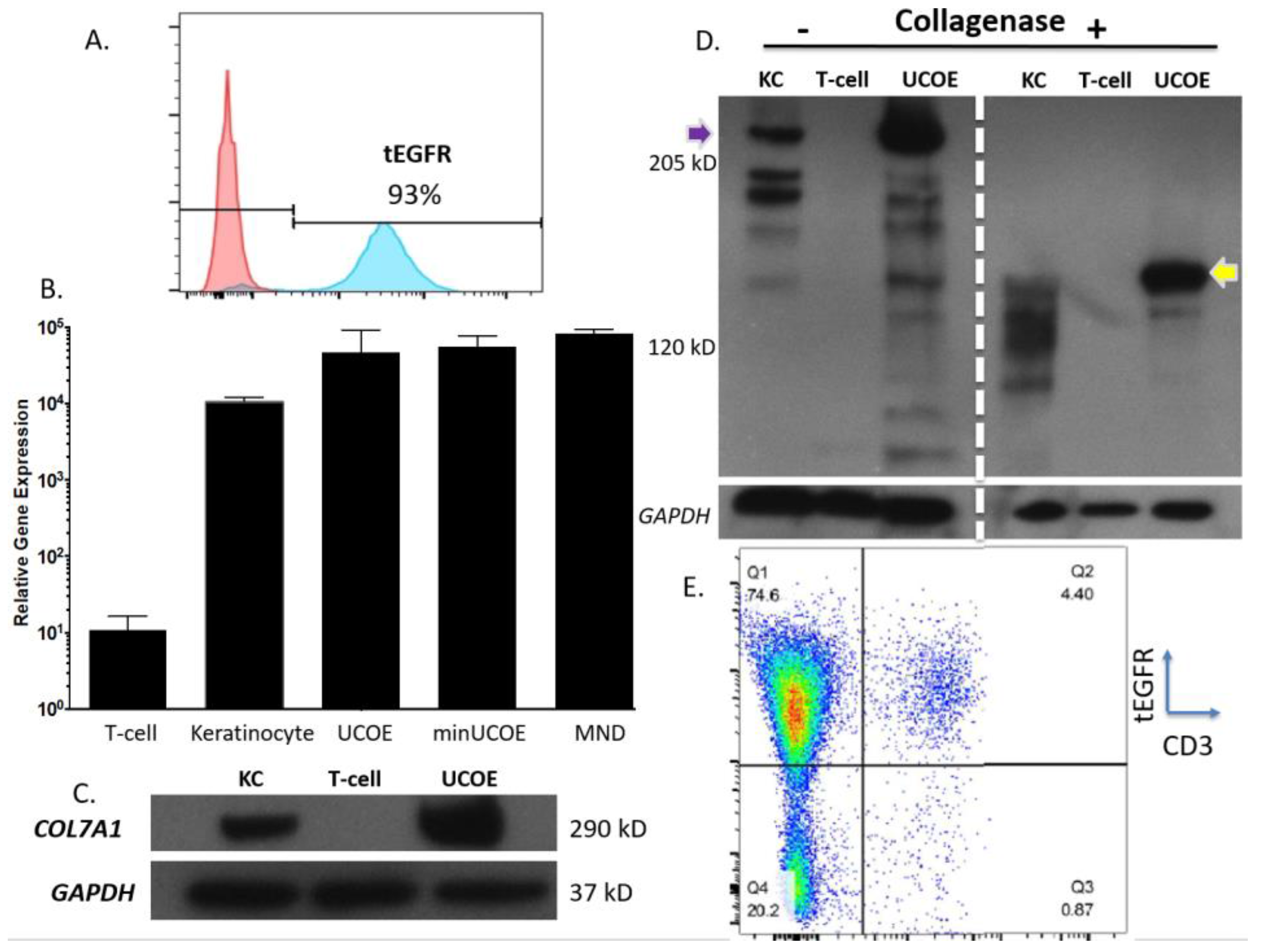
Figure 4C). To assess the properties of these modified cells we purified them by magnetic bead isolation for tEGFR expression and performed molecular and protein analysis. In highly purified cells (
Figure 5A) a fusion transcript using inside/outside PCR primers was observed showing co-expression of tEGFR and
COL7A1 (
Figure A3). Quantification of
COL7A1 mRNA expression showed that in the purified cell populations the
COL7A1 expression levels were equivalent across the donor candidates (
Figure 5B). Comparison to wild type keratinocytes showed that the engineered T-cells produce more
COL7A1 mRNA than this major
COL7A1 producing cell population. Western blot analysis demonstrated that the T-cells were capable of producing higher levels of
COL7A1 protein than keratinocytes and that the peptide adopted the proper architecture as evidenced by collagenase digestion (
Figure 5C,D). Next, in order to make the T-cells broadly applicable we ablated the T-cell receptor such that they lose their ability to cause GVHD (
Figure 5E). Importantly, this strategy has been used clinically for immunotherapy [
33].
The cell choices for our study are carefully considered: UCB HSCs represent a potential lifelong source of COL7A1 and represent a putative advance over current bone marrow transplant that infuses and engrafts cells with physiological levels of COL7A1. T-cells could be considered for augmentative or shorter-term treatment options given that they may circulate for a short time, deliver the payload, and then slowly decline due to effector cell exhaustion. To realize the potential for this approach our T-cell engineering design included TCR disruption such that they cannot initiate GVHD. As such, both of the cell populations have a favorable immunological profile with a potential as an off the shelf product for delivery to multiple patients.
Our strategy is reliant on AAV-based delivery of the donor and this size restricted vector is ideal for facilitating HDR. A recent human genome level screen showed that Cas9 mediated upregulation of target genes occurs within a ~200 bp window proximal to the transcriptional start site [
25]. This is highly significant given that our UMET (UCOE.MND.tEGFR.t2A) construct allows for 500 bp of homology arm sequence making our operational capacity within range of every gene in the human genome. We also observed that the MND promoter only version mediated similar expression levels to UMET over a 14-day time course (
Figure 4). This adds to the amount of donor sequence that can be included in the targeting arms and provides expanded flexibility under circumstances/in models where the targeting window may exceed 500 bp.
Our studies provide proof of principle in cells with high translational impact. Importantly, the design is intended to be flexible and expansive in its application with potential for cellular engineering in support of translation, production, and manufacturing. In conclusion, our robust platform represents a broadly applicable tool for gene upregulation with direct application in diverse models, organisms, and gene and cellular expression systems.
4. Materials and Methods
4.1. Human Cell Sample Purification
Primary T-cells and umbilical cord blood derived CD34 cells were obtained in accordance with the Declaration of Helsinki requirements for research on human subjects with approval of the University of Minnesota Institutional Review Board IRB# 0305M47681. T-cells were isolated using the RosetteSep Human T Cell Enrichment Cocktail (STEMCELL Technologies, Cambridge, MA, USA) and the CD34 MicroBead kit (Miltenyi Biotec, Auburn, CA, USA) was used to enrich for CD34+ cells from cord blood. EGFR positive selection was accomplished by adding 3.0 µg/mL of phycoerythrin (PE) labeled anti-human EGFR antibody (Biolegend, San Diego, CA, USA; Clone AY13) and the EasySep PE positive selection kit (STEMCELL Technologies, Cambridge, MA, USA) Human embryonic kidney 293T cell line were purchased from ThermoFisher (Waltham, MA, USA).
4.2. Culture Conditions
293T cells were maintained in Dulbecco’s Modification of Eagle’s Medium supplemented with glutamax, non-essential amino acids, penicillin/streptomycin and 10% fetal bovine serum ThermoFisher (Waltham, MA, USA). Cells were maintained at 37 °C and 5% CO2.
T-cells were grown in X-VIVO-20 (Lonza, Allendale, NJ, USA) with 10% AB serum (Valley Biomedical, Winchester, VA, USA), 300 IU of IL-2 and 5 ng/mL each of IL-7 and IL-15 (PeproTech, Rocky Hill, NJ, USA), N-Acetyl-l-Cysteine, penicillin/streptomycin, and Gluta-MAX-I each from ThermoFisher (Waltham, MA, USA).
CD34+ hematopoietic stem cells were cultured in StemSpan SFMII media containing 1 µM SR1 each from STEMCELL Technologies, Cambridge, MA, USA and human cytokines Flt-3 ligand (100 ng/mL), SCF (100 ng/mL), TPO (100 ng/mL), IL-6 (100 ng/mL) all from Biolegend, San Diego, CA, USA.
4.3. CRISPR/Cas9
Guide RNAs were obtained from Synthego (Redwood City, CA, USA) and were used at a concentration of 1 μg with 10 μg of Cas9 protein (Aldevron, Fargo, ND, USA) or 1 μg of mRNA (TriLink, San Diego, CA, USA). Guide RNA target sequences (5′-3′) were:
COL7A1-1: GGCAGUAAAAGCCGUCAGCU
COL7A1-2: GCGGACGCGCAGGCAAGACC
COL7A1-3: AGAAAAGUCCCUGAUCUCGG
TRAC: GAGAAUCAAAAUCGGUGAAU
AAVS1: GUCACCAAUCCUGUCCCUAG
4.4. Gene Transfer
Plasmids for guide RNA candidate testing were delivered via Lipofectamine 2000 (ThermoFisher, Waltham, MA, USA) to 293 cells in cis using the Guide-IT CRISPR/Cas9 system (Clontech, Mountain View, CA, USA) T-cells were activated with CD3/CD28 Dynabeads (ThermoFisher, Waltham, MA, USA) at a 3:1 bead to cell ratio. Six hours prior to gene transfer the beads were removed. Electroporation was performed using the Neon Electroporation System (ThermoFisher, Waltham, MA, USA) with 10 μL tips using buffer T. T cell Neon settings: 1400 volts, 10 ms, 3 pulses. HSC Neon settings: 1450 volts, 10 ms, 3 pulses. Cells were plated in anti-biotic free media for 12–24 h.
GFP mRNA was obtained from TriLink BioTechnologies (San Diego, CA, USA). AAV-6 particles were produced by Vigene (Rockville, MD, USA) and were added at the indicated MOI and AAV particle units are genome copies (GC)/mL. COL7A1 upregulation testing was performed using gRNA 3 and in 293T cells using dCAS9-VP64-GFP that was a gift from Feng Zhang (Addgene plasmid # 61422).
4.5. Molecular Analysis
293T cells treated for 72 h with the candidate nuclease and the COL7A1 target locus sequence was amplified with F: 5′-TGGTCACTGTGATTGACCTAAA-3′ and R: 5′-GGAGTTGGCTGGGTTGT-3′ at 94C 2 min and 40 rounds of 94C 40 s, 58C 40 s, 68C 1 min, and a final extension of 68C for 10 min. Surveyor assay was performed using the Surveyor Mutation Detection Kit (Integrated DNA Technologies, Coralville, IA, USA) with resolution on a 10% polyacrylamide gel.
Inside/out HDR PCR. Genomic DNA was amplified using AAVS1 F: 5′-GGACGAGCTGTACAAGTAACG-3′ and R: 5′-GAGACAGTGACCAACCATCC-3′ or tEGFR F: 5′-CAGTGTGCCCACTACATTGA-3′ and COL7A1 R: 5′-TGAGGAGCCATCCAGTAAGA-3′ using Phusion High-Fidelity DNA Polymerase (New England BioLabs, MA, USA) with the following conditions: 98C × 30 s and 35 cycles of 98C × 10 s, 63C × 15 s, and 72C × 30 s.
Amplicons were either directly sequenced (Sequetech, Mountain View, CA, USA) or TA cloned (ThermoFisher, Waltham, MA, USA) and then sequenced.
Inference of CRISPR Edits (ICE). Sanger files were analyzed for insertions and deletions using the ICE algorithim (
https://ice.synthego.com/#/; Synthego, Redwood City, CA, USA).
Quantitative reverse transcription PCR. Total RNA was reverse transcribed with SuperScript VILO (ThermoFisher) and analyzed by TaqMan gene expression assay using the 2−∆∆CT method. The COL7A1 probe was Hs00164310_m1 and the normalization control was GAPDH Hs99999905_m1 (ThermoFisher).
4.6. Western Blot
Cell lysates were resuspended in RIPA buffer (MilliporeSigma, Burlington, MA, USA). Undigested or collagen digested (via 37C incubation for 2 h at 37 °C with collagenase from Worthington Biochemical Co., Freehold, NJ, USA) were resolved on a Tris-Acetate gel under reducing conditions. GAPDH was the loading control (MilliporeSigma, Burlington, MA, USA) and collagenase samples were probed with an anti-C7 antibody that was a kind gift from Dr. David Woodley and Dr. Mei Chen. Full length C7 protein was analyzed with an anti-human antibody (Abnova, Walnut, CA, USA). Image densitometry was performed with ImageJ, (National Institutes of Health, Bethesda, MD, USA).
4.7. Colony Forming Assay
Ten thousand tEGFR purified HSCs were placed in MethoCult semi-solid media (STEMCELL Technologies, Cambridge, MA, USA). At day 14 the colonies were enumerated and scored for morphology by an experienced, blinded reviewer.
4.8. Flow Cytometry
FACS data was acquired on a BD LSRII Cytometer (Becton Dickinson, Franklin Lakes, NJ, USA) and data was analyzed with FlowJo 10.4.2 (FlowJo, LLC, Ashland, OR, USA). The following antibodies were used: CD34: Alexa Fluor 488 clone 581; CD133 PE-Dazzle Clone clone 7; CD3 APC/Cy7 clone: OKT3; Zombie VioletTM Fixable Viability Kit all from BioLegend (San Diego, CA, USA).
4.9. Images
Design images are power point templates from Motiflio (Ellicott City, MD, USA)

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}