Estrogen and/or Estrogen Receptor α Inhibits BNIP3-Induced Apoptosis and Autophagy in H9c2 Cardiomyoblast Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
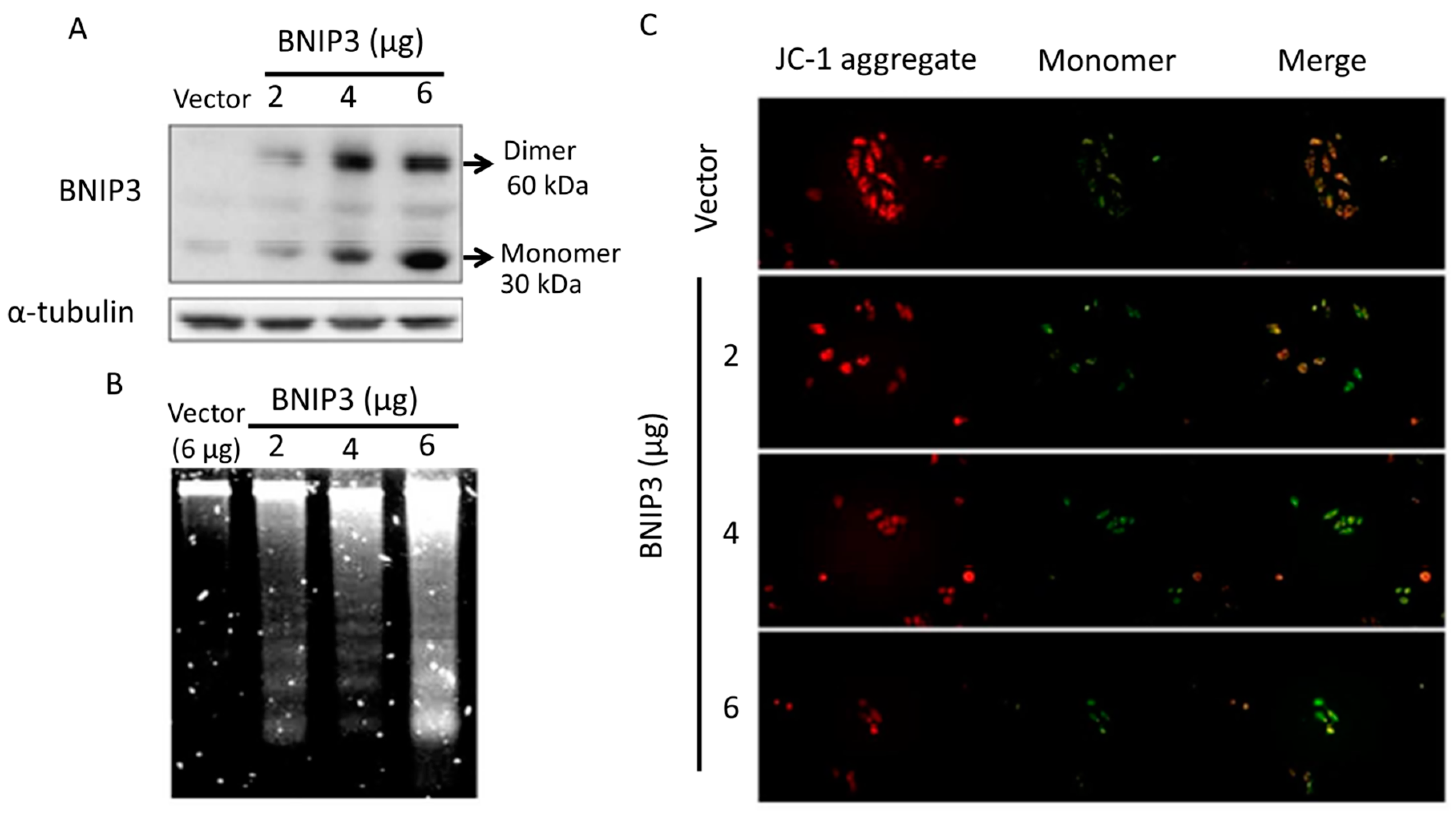
2.1. B-Cell Lymphoma 2 (BCL2)/Adenovirus E1B 19 kDa (BNIP3) Overexpression Induces Apoptosis in H9c2 Cardiomyoblast Cells
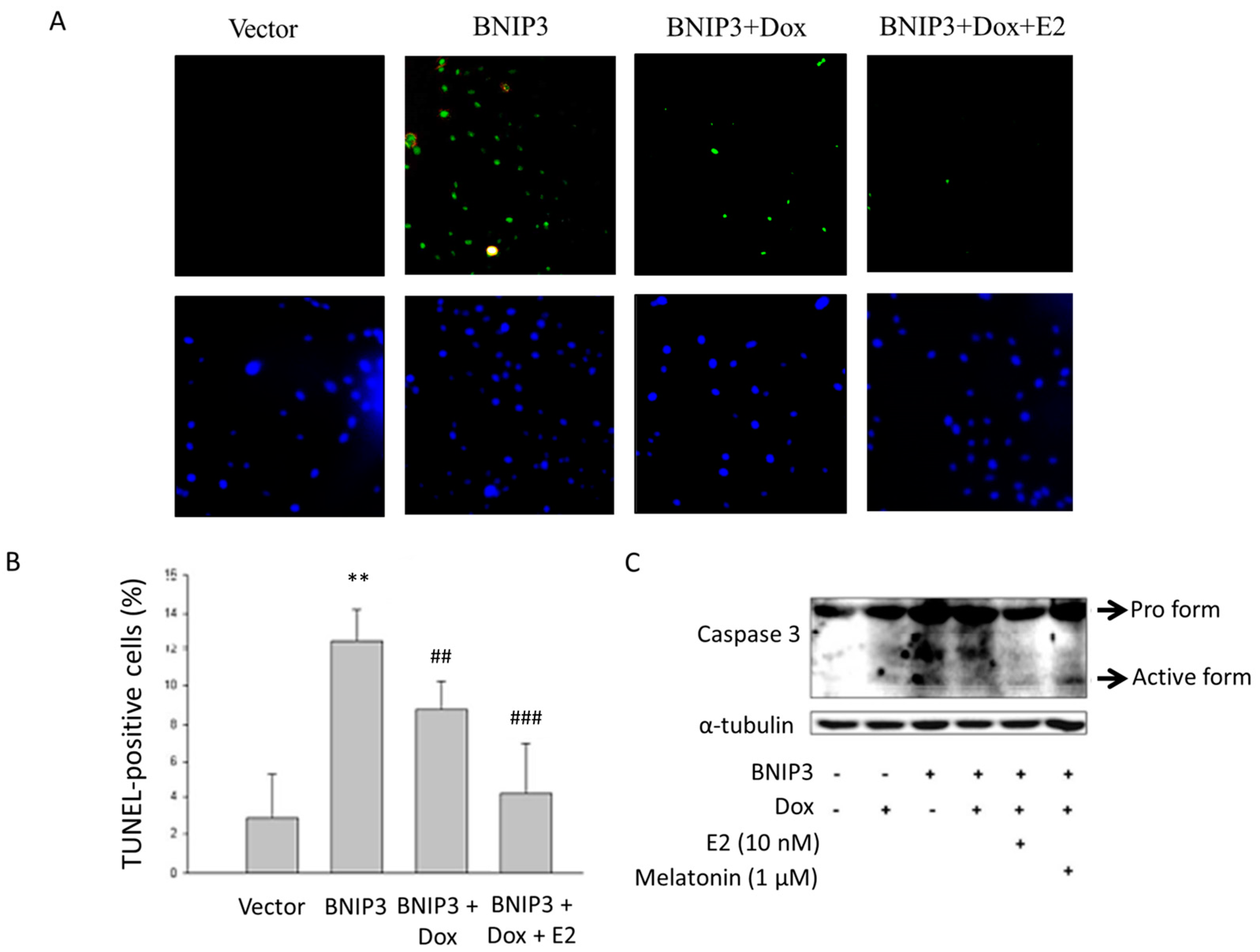
2.2. Estrogen Receptor α (ERα)/Estrogen (E2) Attenuates the Apoptotic Effect Induced by BNIP3 Overexpression
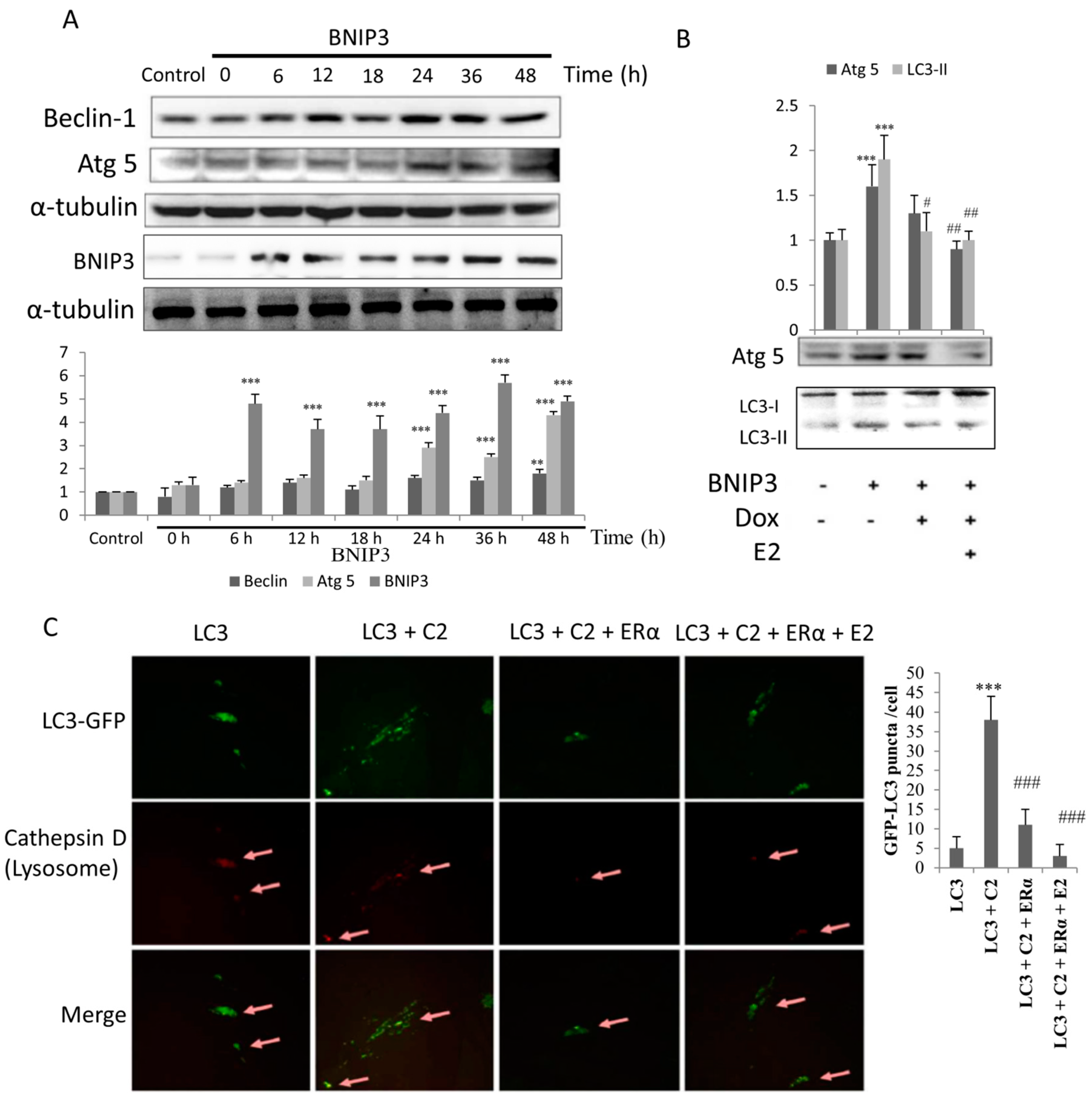
2.3. ERα/E2 Protects Against BNIP3-Induced Autophagy
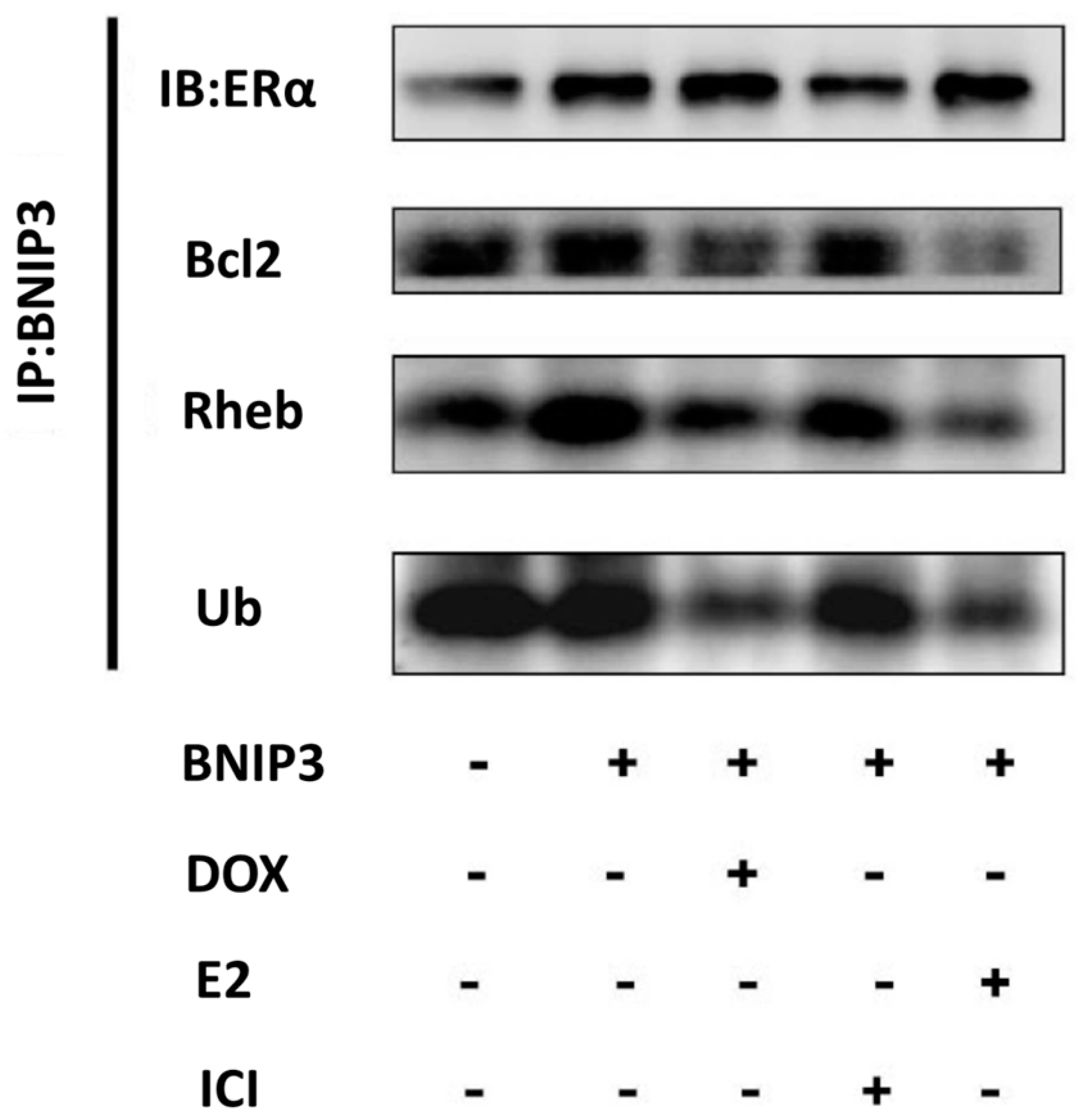
2.4. ERα/E2 Blocked Apoptosis and Autophagy by Binding with BNIP3
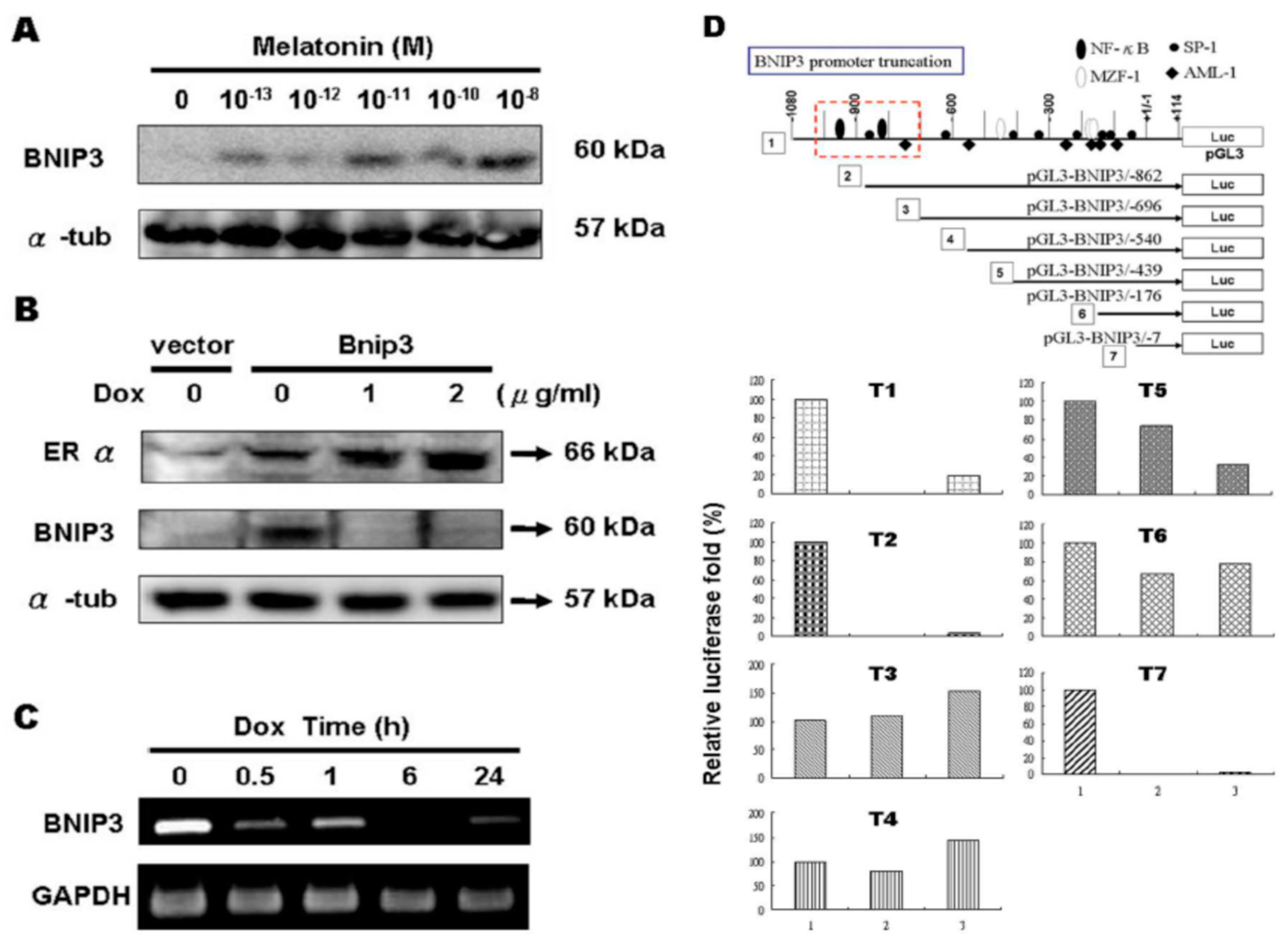
2.5. ERα Down-Regulated BNIP3 Expression
3. Discussion
4. Material and Methods
4.1. Cell Culture and Transfection
4.2. Western Blot Analysis
4.3. DNA Fragmentation
4.4. TdT-Mediated dUTP Nick End Labeling (TUNEL)
4.5. RT-PCR
4.6. Luciferase Assay
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Dhesi, P.; Tehrani, F.; Fuess, J.; Schwarz, E.R. How does the heart (not) die? The role of autophagy in cardiomyocyte homeostasis and cell death. Heart Fail. Rev. 2010, 15, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Gustafsson, A.B.; Gottlieb, R.A. Recycle or die: The role of autophagy in cardioprotection. J. Mol. Cell. Cardiol. 2008, 44, 654–661. [Google Scholar] [CrossRef] [PubMed]
- Boyd, J.M.; Malstrom, S.; Subramanian, T.; Venkatesh, L.K.; Schaeper, U.; Elangovan, B.; D’Sa-Eipper, C.; Chinnadurai, G. Adenovirus E1B 19 kDa and Bcl-2 proteins interact with a common set of cellular proteins. Cell 1994, 79, 341–351. [Google Scholar] [CrossRef]
- Bruick, R.K. Expression of the gene encoding the proapoptotic Nip3 protein is induced by hypoxia. Proc. Natl. Acad. Sci. USA 2000, 97, 9082–9087. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Ray, R.; Dubik, D.; Shi, L.F.; Cizeau, J.; Bleackley, R.C.; Saxena, S.; Gietz, R.D.; Greenberg, A.H. The E1B 19K Bcl-2-binding protein Nip3 is a dimeric mitochondrial protein that activates apoptosis. J. Exp. Med. 1997, 186, 1975–1983. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, M.; Theodorakis, P.; Subramanian, T.; Chinnadurai, G. Adenovirus E1B-19K/BCL-2 interacting protein BNIP3 contains a BH3 domain and a mitochondrial targeting sequence. J. Biol. Chem. 1998, 273, 12415–12421. [Google Scholar] [CrossRef] [PubMed]
- Ray, R.; Chen, G.; Vande Velde, C.; Cizeau, J.; Park, J.H.; Reed, J.C.; Gietz, R.D.; Greenberg, A.H. BNIP3 heterodimerizes with Bcl-2/Bcl-X-L and induces cell death independent of a Bcl-2 homology 3 (BH3) domain at both mitochondrial and nonmitochondrial sites. J. Biol. Chem. 2000, 275, 1439–1448. [Google Scholar] [CrossRef] [PubMed]
- Dorn, G.W.; Kirshenbaum, L.A. Cardiac reanimation: Targeting cardiomyocyte death by BNIP3 and NIX/BNIP3L. Oncogene 2008, 27, S158–S167. [Google Scholar] [CrossRef] [PubMed]
- Diwan, A.; Matkovich, S.J.; Yuan, Q.Y.; Zhao, W.; Yatani, A.; Brown, J.H.; Molkentin, J.D.; Kranias, E.G.; Dorn, G.W. Endoplasmic reticulum-mitochondria crosstalk in NIX-mediated murine cell death. J. Clin. Investig. 2009, 119, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, H.; Chen, X.; Baines, C.P.; Klevitsky, R.; Zhang, X.; Zhang, H.; Jaleel, N.; Chua, B.H.L.; Hewett, T.E.; Robbins, J.; et al. Ca2+- and mitochondrial-dependent cardiomyocyte necrosis as a primary mediator of heart failure. J. Clin. Investig. 2007, 117, 2431–2444. [Google Scholar] [CrossRef] [PubMed]
- Kubli, D.A.; Quinsay, M.N.; Huang, C.Q.; Lee, Y.; Gustafsson, A.B. Bnip3 functions as a mitochondrial sensor of oxidative stress during myocardial ischemia and reperfusion. Am. J. Physiol. Heart C 2008, 295, H2025–H2031. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Ai, Q.; Feng, K.; Li, Y.; Liu, X. The cardioprotective effect of dihydromyricetin prevents ischemia-reperfusion-induced apoptosis in vivo and in vitro via the PI3K/Akt and HIF-1alpha signaling pathways. Apoptosis Int. J. Program. Cell Death 2016, 21, 1366–1385. [Google Scholar] [CrossRef] [PubMed]
- Diwan, A.; Krenz, M.; Syed, F.M.; Wansapura, J.; Ren, X.P.; Koesters, A.G.; Li, H.R.; Kirshenbaum, L.A.; Hahn, H.S.; Robbins, J.; et al. Inhibition of ischemic cardiomyocyte apoptosis through targeted ablation of Bnip3 restrains postinfarction remodeling in mice. J. Clin. Investig. 2007, 117, 2825–2833. [Google Scholar] [CrossRef] [PubMed]
- Burton, T.R.; Gibson, S.B. The role of Bcl-2 family member BNIP3 in cell death and disease: NIPping at the heels of cell death. Cell Death Differ. 2009, 16, 515–523. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Ney, P.A. Role of BNIP3 and NIX in cell death, autophagy, and mitophagy. Cell Death Differ. 2009, 16, 939–946. [Google Scholar] [CrossRef] [PubMed]
- Sadoshima, J.; Izumo, S. The cellular and molecular response of cardiac myocytes to mechanical stress. Annu. Rev. Physiol. 1997, 59, 551–571. [Google Scholar] [CrossRef] [PubMed]
- Swynghedauw, B. Molecular mechanisms of myocardial remodeling. Physiol. Rev. 1999, 79, 215–262. [Google Scholar] [CrossRef] [PubMed]
- Nadal-Ginard, B.; Kajstura, J.; Leri, A.; Anversa, P. Myocyte death, growth, and regeneration in cardiac hypertrophy and failure. Circ. Res. 2003, 92, 139–150. [Google Scholar] [CrossRef] [PubMed]
- Lorell, B.H.; Carabello, B.A. Left ventricular hypertrophy: Pathogenesis, detection, and prognosis. Circulation 2000, 102, 470–479. [Google Scholar] [CrossRef] [PubMed]
- Van Empel, V.P.; De Windt, L.J. Myocyte hypertrophy and apoptosis: A balancing act. Cardiovasc. Res. 2004, 63, 487–499. [Google Scholar] [CrossRef] [PubMed]
- Sabbah, H.N. Apoptotic cell death in heart failure. Cardiovasc. Res. 2000, 45, 704–712. [Google Scholar] [CrossRef]
- Li, L.; Xu, J.; He, L.; Peng, L.; Zhong, Q.; Chen, L.; Jiang, Z. The role of autophagy in cardiac hypertrophy. Acta Biochim. Biophys. Sin. 2016, 48, 491–500. [Google Scholar] [CrossRef] [PubMed]
- Gustafsson, A.B.; Gottlieb, R.A. Autophagy in ischemic heart disease. Circ. Res. 2009, 104, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Yuan, J. Autophagy in cell death: An innocent convict? J. Clin. Investig. 2005, 115, 2679–2688. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, C.H.; You, J.Y.; Xie, Z.L. The interplay between autophagy and apoptosis in the diabetic heart. J. Mol. Cell. Cardiol. 2014, 71, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Nishida, K.; Yamaguchi, O.; Otsu, K. Crosstalk between autophagy and apoptosis in heart disease. Circ. Res. 2008, 103, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Deng, Y.; Xu, Y.; Jin, W.; Li, H. MicroRNA-223 protects neonatal rat cardiomyocytes and H9c2 cells from hypoxia-induced apoptosis and excessive autophagy via the Akt/mTOR pathway by targeting PARP-1. J. Mol. Cell. Cardiol. 2018, 118, 133–146. [Google Scholar] [CrossRef] [PubMed]
- Yaoita, H.; Ogawa, K.; Maehara, K.; Maruyama, Y. Attenuation of ischemia/reperfusion injury in rats by a caspase inhibitor. Circulation 1998, 97, 276–281. [Google Scholar] [CrossRef] [PubMed]
- Chandrashekhar, Y.; Sen, S.; Anway, R.; Shuros, A.; Anand, I. Long-term caspase inhibition ameliorates apoptosis, reduces myocardial troponin-I cleavage, protects left ventricular function, and attenuates remodeling in rats with myocardial infarction. J. Am. Coll. Cardiol. 2004, 43, 295–301. [Google Scholar] [CrossRef] [PubMed]
- Galvez, A.S.; Brunskill, E.W.; Marreez, Y.; Benner, B.J.; Regula, K.M.; Kirschenbaum, L.A.; Dorn, G.W., 2nd. Distinct pathways regulate proapoptotic Nix and BNip3 in cardiac stress. J. Biol. Chem. 2006, 281, 1442–1448. [Google Scholar] [CrossRef] [PubMed]
- Regula, K.M.; Ens, K.; Kirshenbaum, L.A. Inducible expression of BNIP3 provokes mitochondrial defects and hypoxia-mediated cell death of ventricular myocytes. Circ. Res. 2002, 91, 226–231. [Google Scholar] [CrossRef] [PubMed]
- Jousilahti, P.; Vartiainen, E.; Tuomilehto, J.; Puska, P. Sex, age, cardiovascular risk factors, and coronary heart disease: A prospective follow-up study of 14 786 middle-aged men and women in Finland. Circulation 1999, 99, 1165–1172. [Google Scholar] [CrossRef] [PubMed]
- Bhuiyan, M.S.; Shioda, N.; Fukunaga, K. Ovariectomy augments pressure overload-induced hypertrophy associated with changes in Akt and nitric oxide synthase signaling pathways in female rats. Am. J. Physiol. Endocrinol. Metabol. 2007, 293, E1606–E1614. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Arenas, I.A.; Armstrong, S.J.; Davidge, S.T. Estrogen modulation of left ventricular remodeling in the aged heart. Cardiovasc. Res. 2003, 57, 388–394. [Google Scholar] [CrossRef]
- Hsieh, D.J.; Kuo, W.W.; Lai, Y.P.; Shibu, M.A.; Shen, C.Y.; Pai, P.; Yeh, Y.L.; Lin, J.Y.; Viswanadha, V.P.; Huang, C.Y. 17beta-Estradiol and/or Estrogen Receptor beta Attenuate the Autophagic and Apoptotic Effects Induced by Prolonged Hypoxia Through HIF-1alpha-Mediated BNIP3 and IGFBP-3 Signaling Blockage. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2015, 36, 274–284. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.Y.; Kuo, W.W.; Shibu, M.A.; Lin, Y.M.; Liu, C.N.; Chen, Y.H.; Day, C.H.; Shen, C.Y.; Viswanadha, V.P.; Huang, C.Y. E2/ER beta inhibit ISO-induced cardiac cellular hypertrophy by suppressing Ca2+-calcineurin signaling. PLoS ONE 2017, 12, e0184153. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.H.; Kuo, W.W.; Shibu, M.A.; Day, C.H.; Hsieh, Y.L.; Chung, L.C.; Chen, R.J.; Wen, S.Y.; Viswanadha, V.P.; Huang, C.Y. E2/ER beta Enhances Calcineurin Protein Degradation and PI3K/Akt/MDM2 Signal Transduction to Inhibit ISO-Induced Myocardial Cell Apoptosis. Int. J. Mol. Sci. 2017, 18, 892. [Google Scholar] [CrossRef] [PubMed]
- Asokan Shibu, M.; Kuo, W.W.; Kuo, C.H.; Day, C.H.; Shen, C.Y.; Chung, L.C.; Lai, C.H.; Pan, L.F.; Vijaya Padma, V.; Huang, C.Y. Potential phytoestrogen alternatives exert cardio-protective mechanisms via estrogen receptors. BioMedicine 2017, 7, 11. [Google Scholar] [CrossRef] [PubMed]
- Ting, W.J.; Huang, C.Y.; Jiang, C.H.; Lin, Y.M.; Chung, L.C.; Shen, C.Y.; Pai, P.; Lin, K.H.; Viswanadha, V.P.; Liao, S.C. Treatment with 17beta-Estradiol Reduced Body Weight and the Risk of Cardiovascular Disease in a High-Fat Diet-Induced Animal Model of Obesity. Int. J. Mol. Sci. 2017, 18, 629. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.C.; Kuo, W.W.; Shen, C.Y.; Chen, Y.F.; Lin, Y.M.; Ho, T.J.; Padma, V.V.; Lo, J.F.; Huang, C.Y.; Huang, C.Y. Anthocyanin Attenuates Doxorubicin-Induced Cardiomyotoxicity via Estrogen Receptor-alpha/beta and Stabilizes HSF1 to Inhibit the IGF-IIR Apoptotic Pathway. Int. J. Mol. Sci. 2016, 17, 1588. [Google Scholar] [CrossRef] [PubMed]
- Huang, E.J.; Wu, C.C.; Lee, S.D.; Chen, J.H.; Liu, J.Y.; Ko, J.L.; Lin, J.A.; Lu, M.C.; Chen, L.M.; Huang, C.Y.; et al. Opposing action of estrogen receptors alpha and beta on tumor necrosis factor-alpha gene expression and caspase-8-mediated apoptotic effects in HA22T cells. Mol. Cell. Biochem. 2006, 287, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Pai, P.; Velmurugan, B.K.; Kuo, C.H.; Yen, C.Y.; Ho, T.J.; Lin, Y.M.; Chen, Y.F.; Lai, C.H.; Day, C.H.; Huang, C.Y. 17beta-Estradiol and/or estrogen receptor alpha blocks isoproterenol-induced calcium accumulation and hypertrophy via GSK3beta/PP2A/NFAT3/ANP pathway. Mol. Cell. Biochem. 2017, 434, 181–195. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.J.; Lo, J.F.; Kuo, C.H.; Chu, C.H.; Chen, L.M.; Tsai, F.J.; Tsai, C.H.; Tzang, B.S.; Kuo, W.W.; Huang, C.Y. Akt mediates 17β-estradiol and/or estrogen receptor-α inhibition of LPS-induced tumor necresis factor-α expression and myocardial cell apoptosis by suppressing the JNK1/2-NFκB pathway. J. Cell. Mol. Med. 2009, 13, 3655–3667. [Google Scholar] [CrossRef] [PubMed]
- Kostin, S.; Pool, L.; Elsasser, A.; Hein, S.; Drexler, H.C.; Arnon, E.; Hayakawa, Y.; Zimmermann, R.; Bauer, E.; Klovekorn, W.P.; et al. Myocytes die by multiple mechanisms in failing human hearts. Circ. Res. 2003, 92, 715–724. [Google Scholar] [CrossRef] [PubMed]
- Hein, S.; Arnon, E.; Kostin, S.; Schonburg, M.; Elsasser, A.; Polyakova, V.; Bauer, E.P.; Klovekorn, W.P.; Schaper, J. Progression from compensated hypertrophy to failure in the pressure-overloaded human heart: Structural deterioration and compensatory mechanisms. Circulation 2003, 107, 984–991. [Google Scholar] [CrossRef] [PubMed]
- Elsasser, A.; Vogt, A.M.; Nef, H.; Kostin, S.; Mollmann, H.; Skwara, W.; Bode, C.; Hamm, C.; Schaper, J. Human hibernating myocardium is jeopardized by apoptotic and autophagic cell death. J. Am. Coll. Cardiol. 2004, 43, 2191–2199. [Google Scholar] [CrossRef] [PubMed]
- Takemura, G.; Kanoh, M.; Minatoguchi, S.; Fujiwara, H. Cardiomyocyte apoptosis in the failing heart—A critical review from definition and classification of cell death. Int. J. Cardiol. 2013, 167, 2373–2386. [Google Scholar] [CrossRef] [PubMed]
- Yen, W.L.; Klionsky, D.J. How to Live Long and Prosper: Autophagy, Mitochondria, and Aging. Physiology 2008, 23, 248–262. [Google Scholar] [CrossRef] [PubMed]
- Ferreiro, S.F.; Vilarino, N.; Carrera, C.; Louzao, M.C.; Santamarina, G.; Cantalapiedra, A.G.; Cifuentes, J.M.; Crespo, A.; Botana, L.M. In vivo cardiomyocyte response to YTX- and AZA-1-induced damage: Autophagy versus apoptosis. Arch. Toxicol. 2016, 91, 1859–1870. [Google Scholar] [CrossRef] [PubMed]
- Wesselborg, S.; Stork, B. Autophagy signal transduction by ATG proteins: From hierarchies to networks. Cell. Mol. Life Sci. 2015, 72, 4721–4757. [Google Scholar] [CrossRef] [PubMed]
- Jung, C.H.; Ro, S.H.; Cao, J.; Otto, N.M.; Kim, D.H. mTOR regulation of autophagy. FEBS Lett. 2010, 584, 1287–1295. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.H.; Jackson, S.; Seaman, M.; Brown, K.; Kempkes, B.; Hibshoosh, H.; Levine, B. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 1999, 402, 672–676. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.H.; Kleeman, L.K.; Jiang, H.H.; Gordon, G.; Goldman, J.E.; Berry, G.; Herman, B.; Levine, B. Protection against fatal Sindbis virus encephalitis by beclin, a novel Bcl-2-interacting protein. J. Virol. 1998, 72, 8586–8596. [Google Scholar] [PubMed]
- Pattingre, S.; Tassa, A.; Qu, X.; Garuti, R.; Liang, X.H.; Mizushima, N.; Packer, M.; Schneider, M.D.; Levine, B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 2005, 122, 927–939. [Google Scholar] [CrossRef] [PubMed]
- Maejima, Y.; Isobe, M.; Sadoshima, J. Regulation of autophagy by Beclin 1 in the heart. J. Mol. Cell. Cardiol. 2016, 95, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Buja, L.M.; Vela, D. Cardiomyocyte death and renewal in the normal and diseased heart. Cardiovasc. Pathol. Off. J. Soc. Cardiovasc. Pathol. 2008, 17, 349–374. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Gustafsson, A.B. Role of apoptosis in cardiovascular disease. Apoptosis Int. J. Program. Cell Death 2009, 14, 536–548. [Google Scholar] [CrossRef] [PubMed]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef] [PubMed]
- Vande Velde, C.; Cizeau, J.; Dubik, D.; Alimonti, J.; Brown, T.; Israels, S.; Hakem, R.; Greenberg, A.H. BNIP3 and Genetic Control of Necrosis-Like Cell Death through the Mitochondrial Permeability Transition Pore. Mol. Cell. Biol. 2000, 20, 5454–5468. [Google Scholar] [CrossRef] [PubMed]
- Feng, C.C.; Lin, C.C.; Lai, Y.P.; Chen, T.S.; Asokan, S.M.; Lin, J.Y.; Lin, K.H.; Viswanadha, V.P.; Kuo, W.W.; Huang, C.Y. Hypoxia suppresses myocardial survival pathway through HIF-1 alpha-IGFBP-3-dependent signaling and enhances cardiomyocyte autophagic and apoptotic effects mainly via FoxO3a-induced BNIP3 expression. Growth Factors 2016, 34, 73–86. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.K.; Kuo, W.W.; Hsieh, D.J.; Chang, H.N.; Pai, P.Y.; Lin, K.H.; Pan, L.F.; Ho, T.J.; Viswanadha, V.P.; Huang, C.Y. CREB Negatively Regulates IGF2R Gene Expression and Downstream Pathways to Inhibit Hypoxia-Induced H9c2 Cardiomyoblast Cell Death. Int. J. Mol. Sci. 2015, 16, 27921–27930. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.H.; Kuo, C.H.; Kuo, W.W.; Ho, T.J.; Pai, P.; Chen, W.K.; Pan, L.F.; Wang, C.C.; Padma, V.V.; Huang, C.Y. NFIL3 suppresses hypoxia-induced apoptotic cell death by targeting the insulin-like growth factor 2 receptor. J. Cell. Biochem. 2015, 116, 1113–1120. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.H.; Kuo, W.W.; Jiang, A.Z.; Pai, P.; Lin, J.Y.; Chen, W.K.; Day, C.H.; Shen, C.Y.; Padma, V.V.; Huang, C.Y. Tetramethylpyrazine Ameliorated Hypoxia-Induced Myocardial Cell Apoptosis via HIF-1alpha/JNK/p38 and IGFBP3/BNIP3 Inhibition to Upregulate PI3K/Akt Survival Signaling. Cell. Physiol Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2015, 36, 334–344. [Google Scholar] [CrossRef] [PubMed]
- Yeh, Y.-L.; Ting, W.-J.; Shen, C.-Y.; Hsu, H.-H.; Chung, L.-C.; Tu, C.-C.; Chang, S.-H.; Day, C.-H.; Tsai, Y.; Huang, C.-Y. Hypoxia Augments Increased HIF-1α and Reduced Survival Protein p-Akt in Gelsolin (GSN)-Dependent Cardiomyoblast Cell Apoptosis. Cell Biochem. Biophys. 2016, 74, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Schaible, E.V.; Windschugl, J.; Bobkiewicz, W.; Kaburov, Y.; Dangel, L.; Kramer, T.; Huang, C.; Sebastiani, A.; Luh, C.; Werner, C.; et al. 2-Methoxyestradiol confers neuroprotection and inhibits a maladaptive HIF-1alpha response after traumatic brain injury in mice. J. Neurochem. 2014, 129, 940–954. [Google Scholar] [CrossRef] [PubMed]
- Gao, N.; Nester, R.A.; Sarkar, M.A. 4-Hydroxy estradiol but not 2-hydroxy estradiol induces expression of hypoxia-inducible factor 1alpha and vascular endothelial growth factor A through phosphatidylinositol 3-kinase/Akt/FRAP pathway in OVCAR-3 and A2780-CP70 human ovarian carcinoma cells. Toxicol. Appl. Pharmacol. 2004, 196, 124–135. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, S.R.; Cheng, W.C.; Su, Y.M.; Chiu, C.H.; Liou, Y.M. Molecular targets for anti-oxidative protection of green tea polyphenols against myocardial ischemic injury. BioMedicine 2014, 4, 23. [Google Scholar] [CrossRef] [PubMed]
- Fordjour, P.A.; Wang, L.; Gao, H.; Li, L.; Wang, Y.; Nyagblordzro, M.; Agyemang, K.; Fan, G. Targeting BNIP3 in inflammation-mediated heart failure: A novel concept in heart failure therapy. Heart Fail. Rev. 2016, 21, 489–497. [Google Scholar] [CrossRef] [PubMed]
- Dorn, G.W. Mitochondrial Pruning by Nix and BNip3: An Essential Function for Cardiac-Expressed Death Factors. J. Cardiovasc. Transl. 2010, 3, 374–383. [Google Scholar] [CrossRef] [PubMed]
- Gang, H.Y.; Hai, Y.; Dhingra, R.; Gordon, J.W.; Yurkova, N.; Aviv, Y.; Li, H.Z.; Aguilar, F.; Marshall, A.; Leygue, E.; et al. A Novel Hypoxia-Inducible Spliced Variant of Mitochondrial Death Gene Bnip3 Promotes Survival of Ventricular Myocytes. Circ. Res. 2011, 108, 1084–1092. [Google Scholar] [CrossRef] [PubMed]
- Quinsay, M.N.; Lee, Y.; Rikka, S.; Sayen, M.R.; Molkentin, J.D.; Gottlieb, R.A.; Gustafsson, A.B. Bnip3 mediates permeabilization of mitochondria and release of cytochrome c via a novel mechanism. J. Mol. Cell. Cardiol. 2010, 48, 1146–1156. [Google Scholar] [CrossRef] [PubMed]
- Chaanine, A.H.; Gordon, R.E.; Kohlbrenner, E.; Benard, L.; Jeong, D.; Hajjar, R.J. Potential Role of BNIP3 in Cardiac Remodeling, Myocardial Stiffness, and Endoplasmic Reticulum Mitochondrial Calcium Homeostasis in Diastolic and Systolic Heart Failure. Circ. Heart Fail. 2013, 6, 572. [Google Scholar] [CrossRef] [PubMed]
- Hulley, S.; Grady, D.; Bush, T.; Furberg, C.; Herrington, D.; Riggs, B.; Vittinghoff, E. Randomized trial of estrogen plus progestin for secondary prevention of coronary heart disease in postmenopausal women. Heart and Estrogen/progestin Replacement Study (HERS) Research Group. JAMA 1998, 280, 605–613. [Google Scholar] [CrossRef] [PubMed]
- Lobo, R.A.; Pickar, J.H.; Stevenson, J.C.; Mack, W.J.; Hodis, H.N. Back to the future: Hormone replacement therapy as part of a prevention strategy for women at the onset of menopause. Atherosclerosis 2016, 254, 282–290. [Google Scholar] [CrossRef] [PubMed]
- Clarke, S.C.; Kelleher, J.; Lloyd-Jones, H.; Slack, M.; Schofiel, P.M. A study of hormone replacement therapy in postmenopausal women with ischaemic heart disease: The Papworth HRT atherosclerosis study. BJOG Int. J. Obstet. Gynaecol. 2002, 109, 1056–1062. [Google Scholar] [CrossRef]
- Tazumi, S.; Yokota, N.; Kawakami, M.; Omoto, S.; Takamata, A.; Morimoto, K. Effects of estrogen replacement on stress-induced cardiovascular responses via renin-angiotensin system in ovariectomized rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2016, 311, R898–R905. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Sun, X.; Chou, J.; Lin, M.; Ferrario, C.M.; Zapata-Sudo, G.; Groban, L. Cardiomyocyte-specific deletion of the G protein-coupled estrogen receptor (GPER) leads to left ventricular dysfunction and adverse remodeling: A sex-specific gene profiling analysis. Biochim. Biophys. Acta 2016, 1863, 1870–1882. [Google Scholar] [CrossRef] [PubMed]
- Hale, S.L.; Birnbaum, Y.; Kloner, R.A. Estradiol, Administered Acutely, Protects Ischemic Myocardium in Both Female and Male Rabbits. J. Cardiovasc. Pharmacol. Ther. 1997, 2, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Chae, S.U.; Ha, K.C.; Piao, C.S.; Chae, S.W.; Chae, H.J. Estrogen attenuates cardiac ischemia-reperfusion injury via inhibition of calpain-mediated bid cleavage. Arch. Pharm. Res. 2007, 30, 1225–1235. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.H.; Liu, J.Y.; Wu, J.P.; Hsieh, Y.H.; Liu, C.J.; Hwang, J.M.; Lee, S.D.; Chen, L.M.; Chang, M.H.; Kuo, W.W.; et al. 17beta-estradiol reduces cardiac hypertrophy mediated through the up-regulation of PI3K/Akt and the suppression of calcineurin/NF-AT3 signaling pathways in rats. Life Sci. 2005, 78, 347–356. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.H. Anti-glycative effects of asiatic acid in human keratinocyte cells. BioMedicine 2014, 4, 19. [Google Scholar] [CrossRef] [PubMed]





© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, B.-C.; Weng, Y.-J.; Shibu, M.A.; Han, C.-K.; Chen, Y.-S.; Shen, C.-Y.; Lin, Y.-M.; Viswanadha, V.P.; Liang, H.-Y.; Huang, C.-Y. Estrogen and/or Estrogen Receptor α Inhibits BNIP3-Induced Apoptosis and Autophagy in H9c2 Cardiomyoblast Cells. Int. J. Mol. Sci. 2018, 19, 1298. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19051298
Chen B-C, Weng Y-J, Shibu MA, Han C-K, Chen Y-S, Shen C-Y, Lin Y-M, Viswanadha VP, Liang H-Y, Huang C-Y. Estrogen and/or Estrogen Receptor α Inhibits BNIP3-Induced Apoptosis and Autophagy in H9c2 Cardiomyoblast Cells. International Journal of Molecular Sciences. 2018; 19(5):1298. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19051298
Chicago/Turabian StyleChen, Bih-Cheng, Yi-Jiun Weng, Marthandam Asokan Shibu, Chien-Kuo Han, Yueh-Sheng Chen, Chia-Yao Shen, Yueh-Min Lin, Vijaya Padma Viswanadha, Hsin-Yueh Liang, and Chih-Yang Huang. 2018. "Estrogen and/or Estrogen Receptor α Inhibits BNIP3-Induced Apoptosis and Autophagy in H9c2 Cardiomyoblast Cells" International Journal of Molecular Sciences 19, no. 5: 1298. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19051298