The Disordered C-Terminus of Yeast Hsf1 Contains a Cryptic Low-Complexity Amyloidogenic Region


Abstract
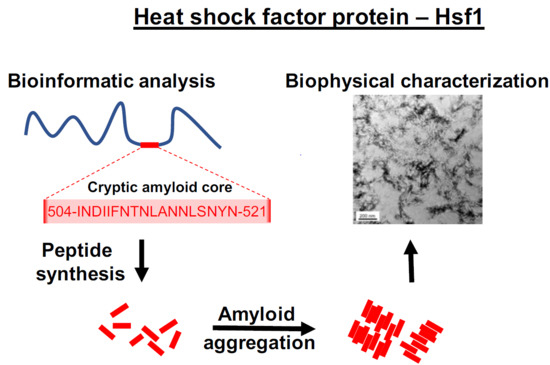
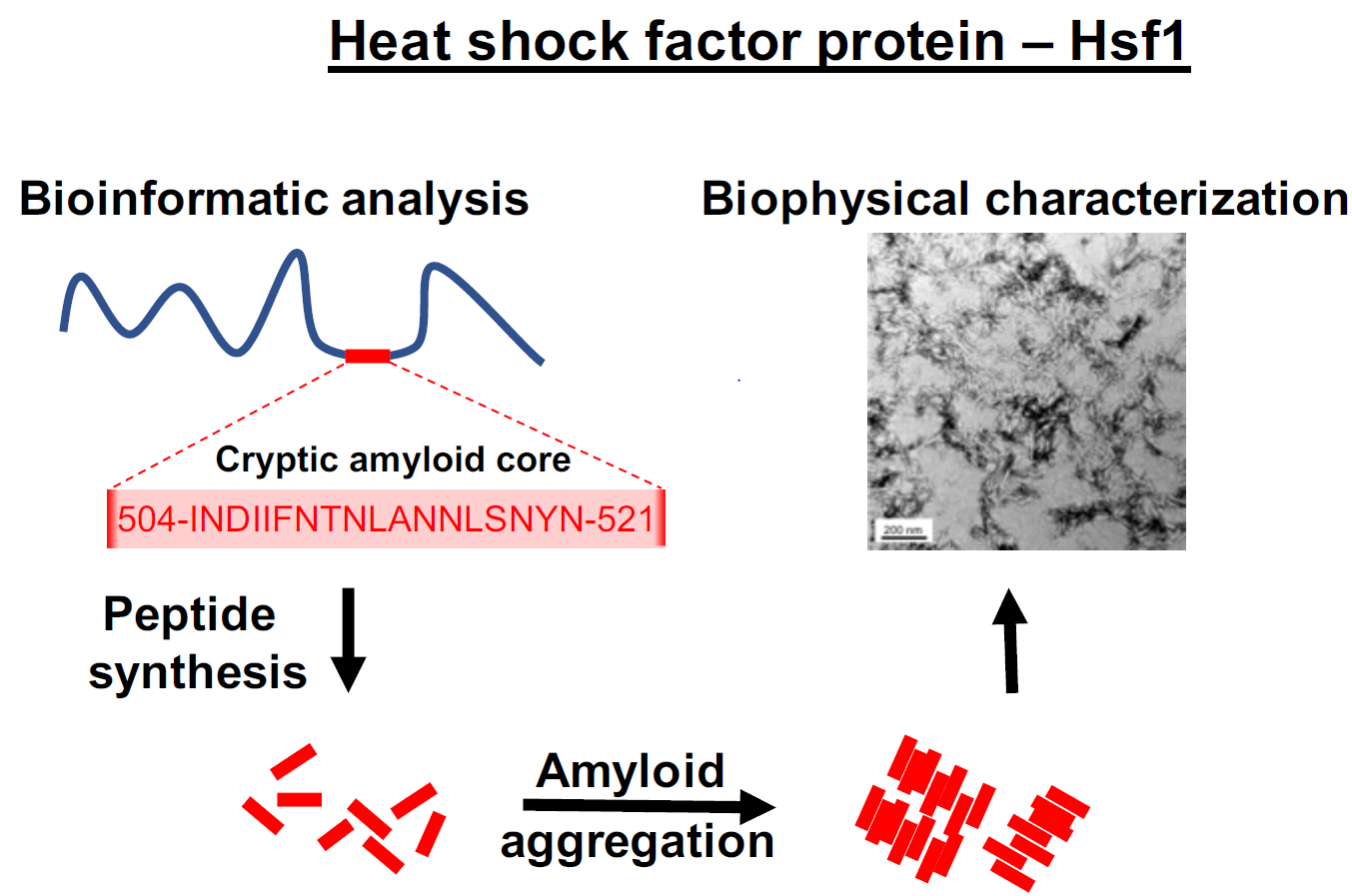
:
1. Introduction
2. Results
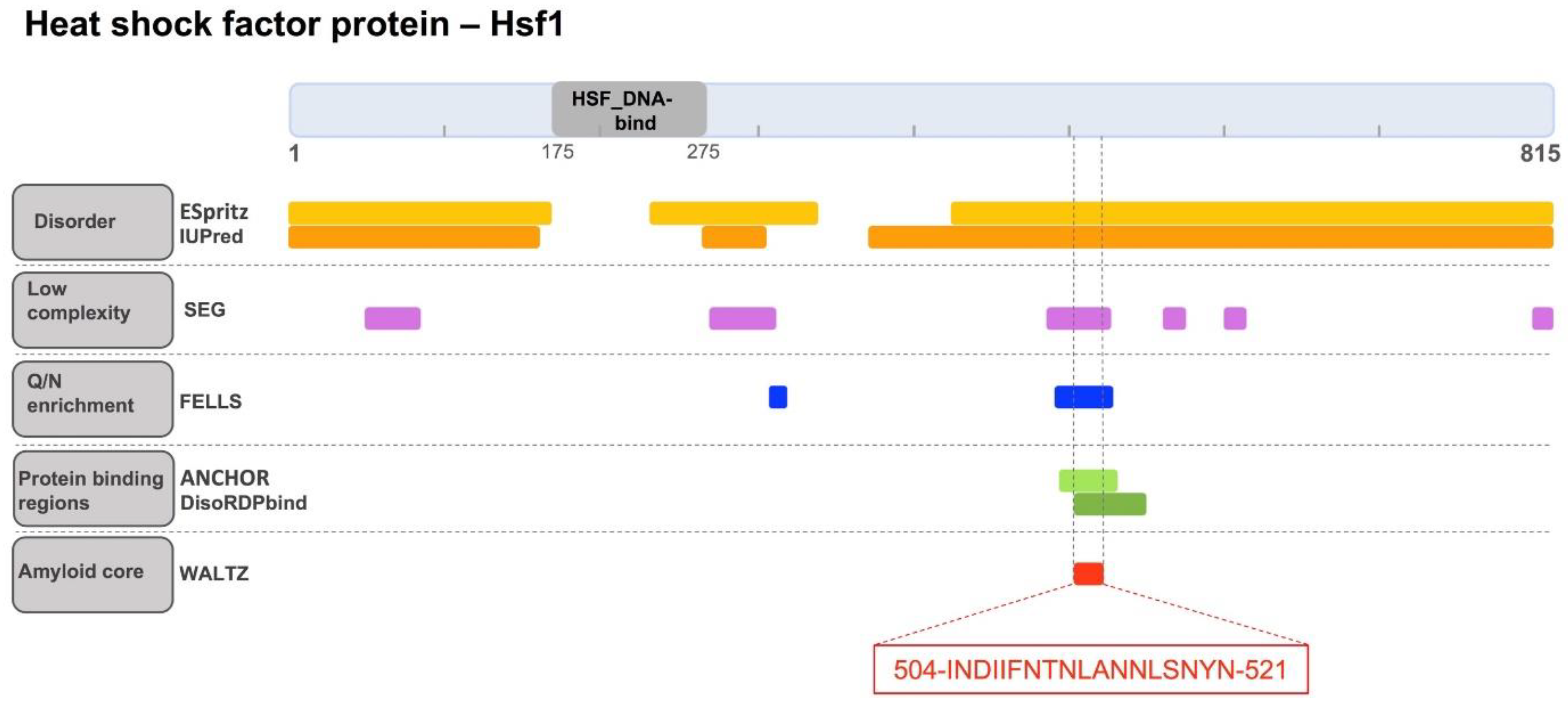
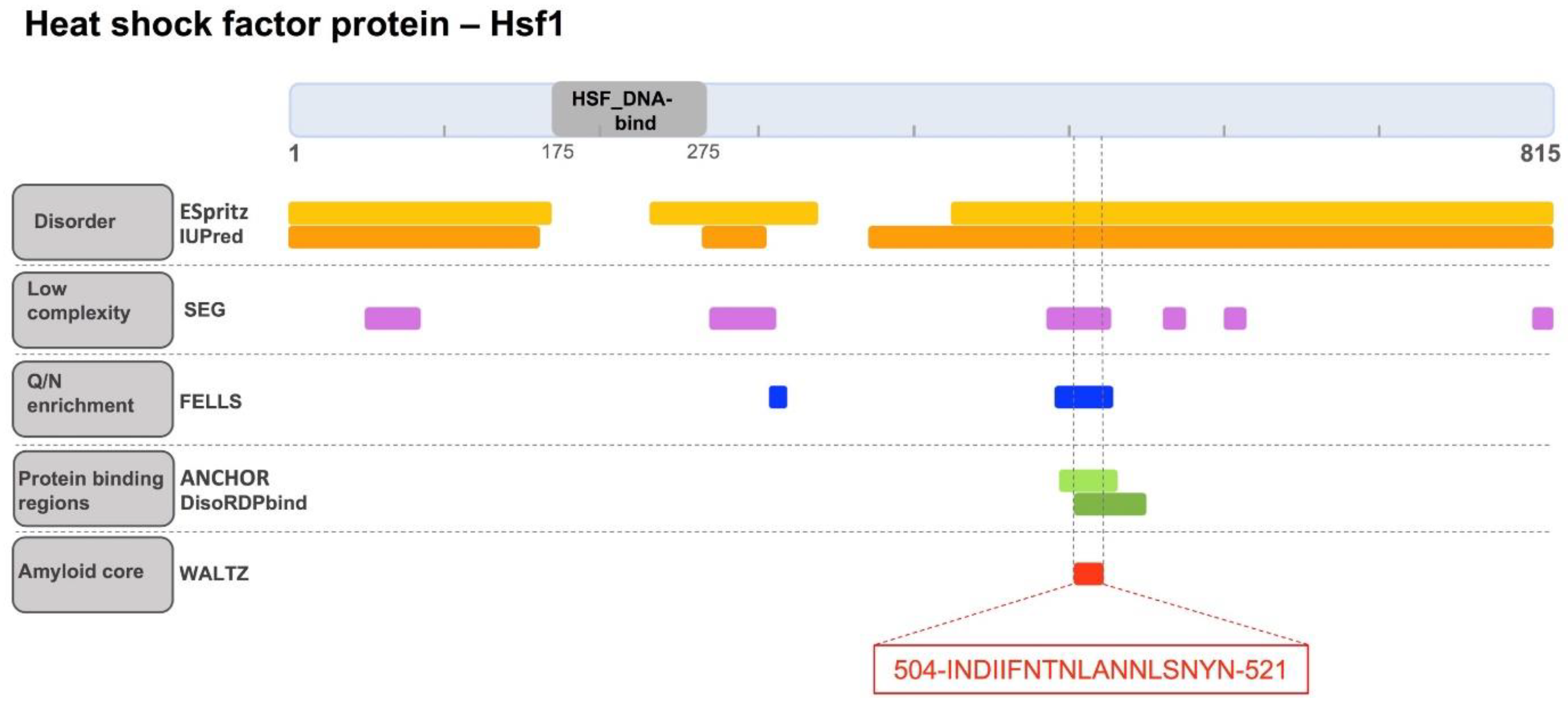
2.1. Identification of a Cryptic Amyloid Sequence Inside a Disordered and Low Complexity Region in Hsf1
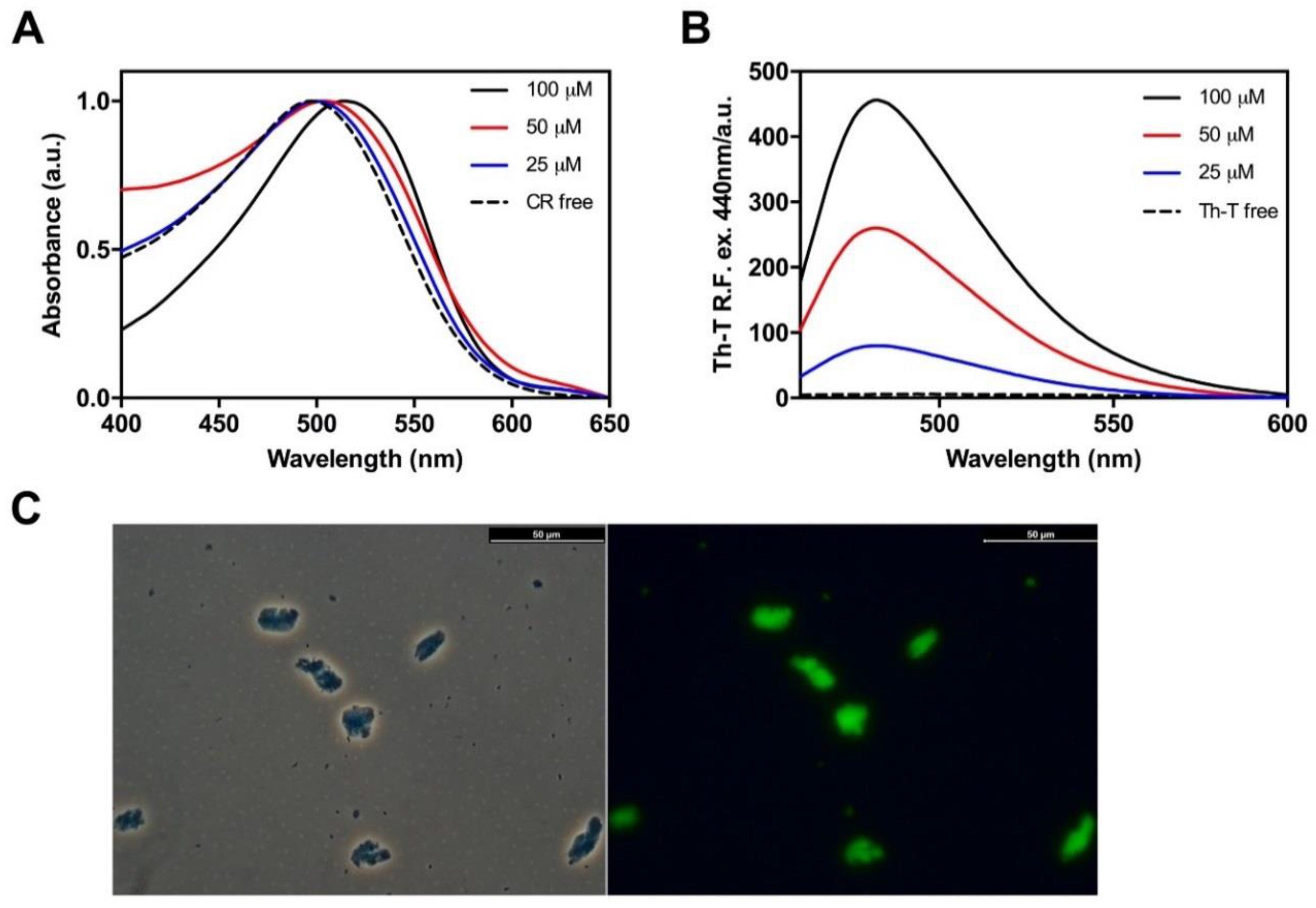
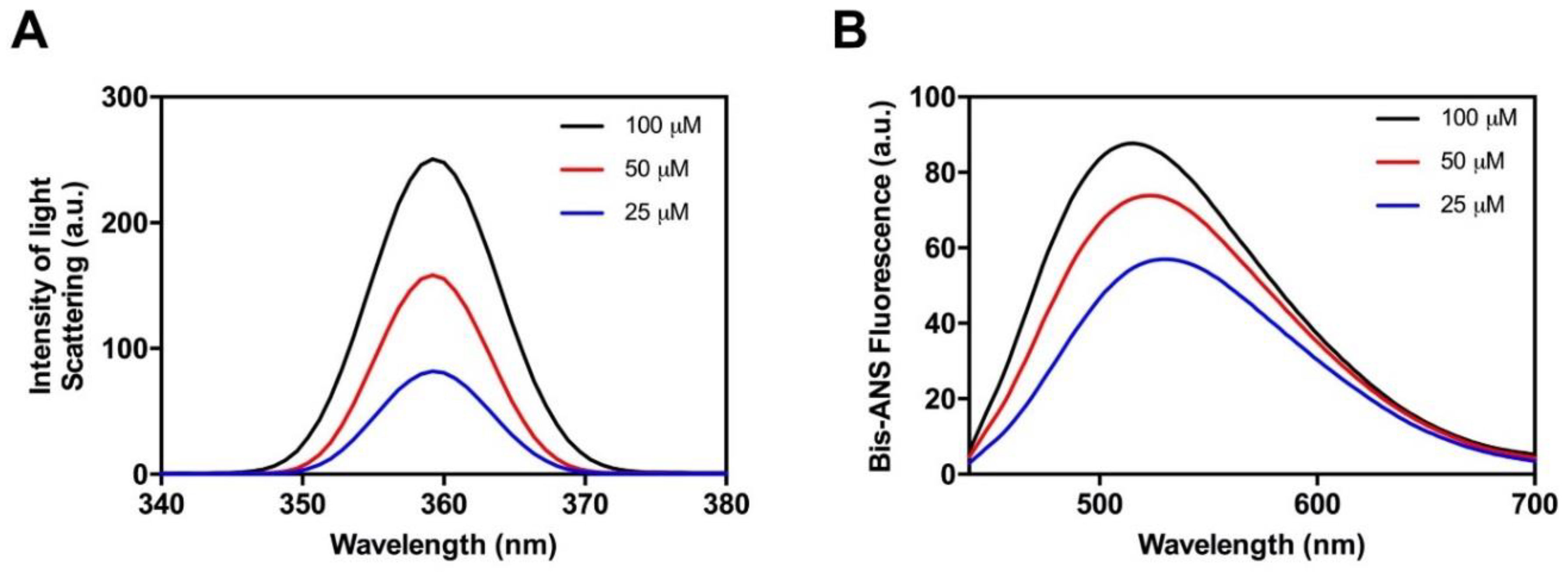
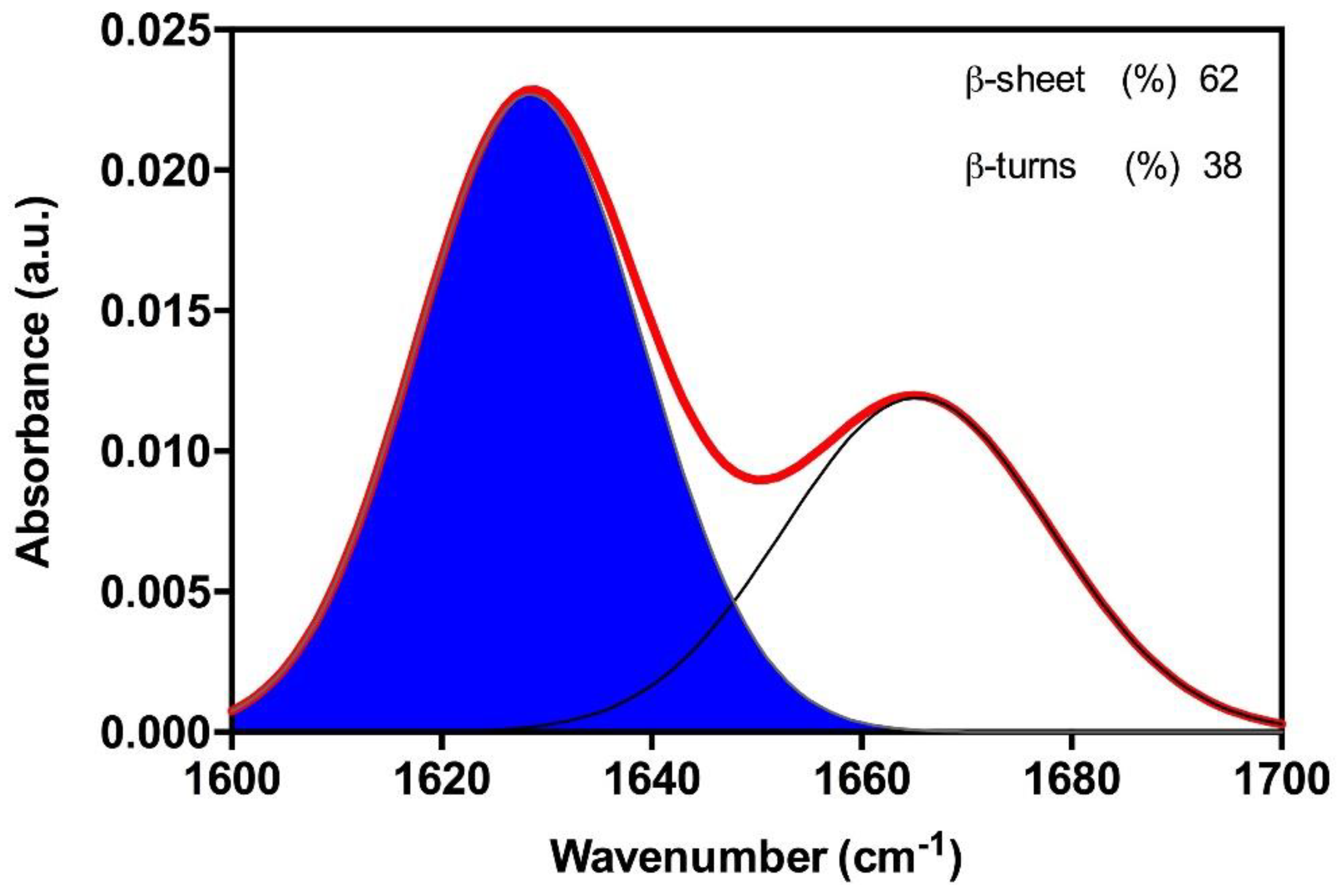
2.2. The Amyloid Core of Hsf1 Assembles into β-Sheet Enriched Aggregates
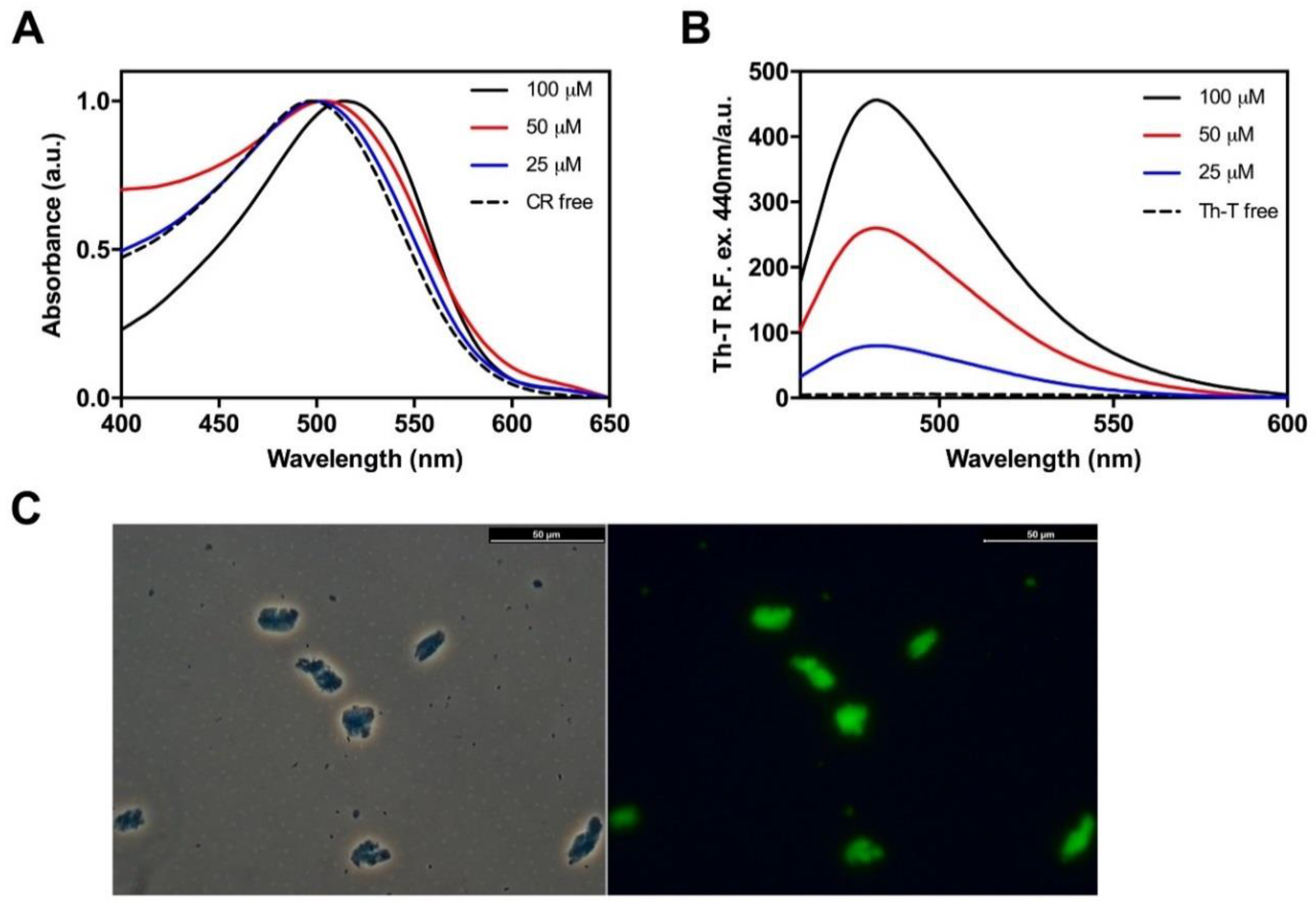
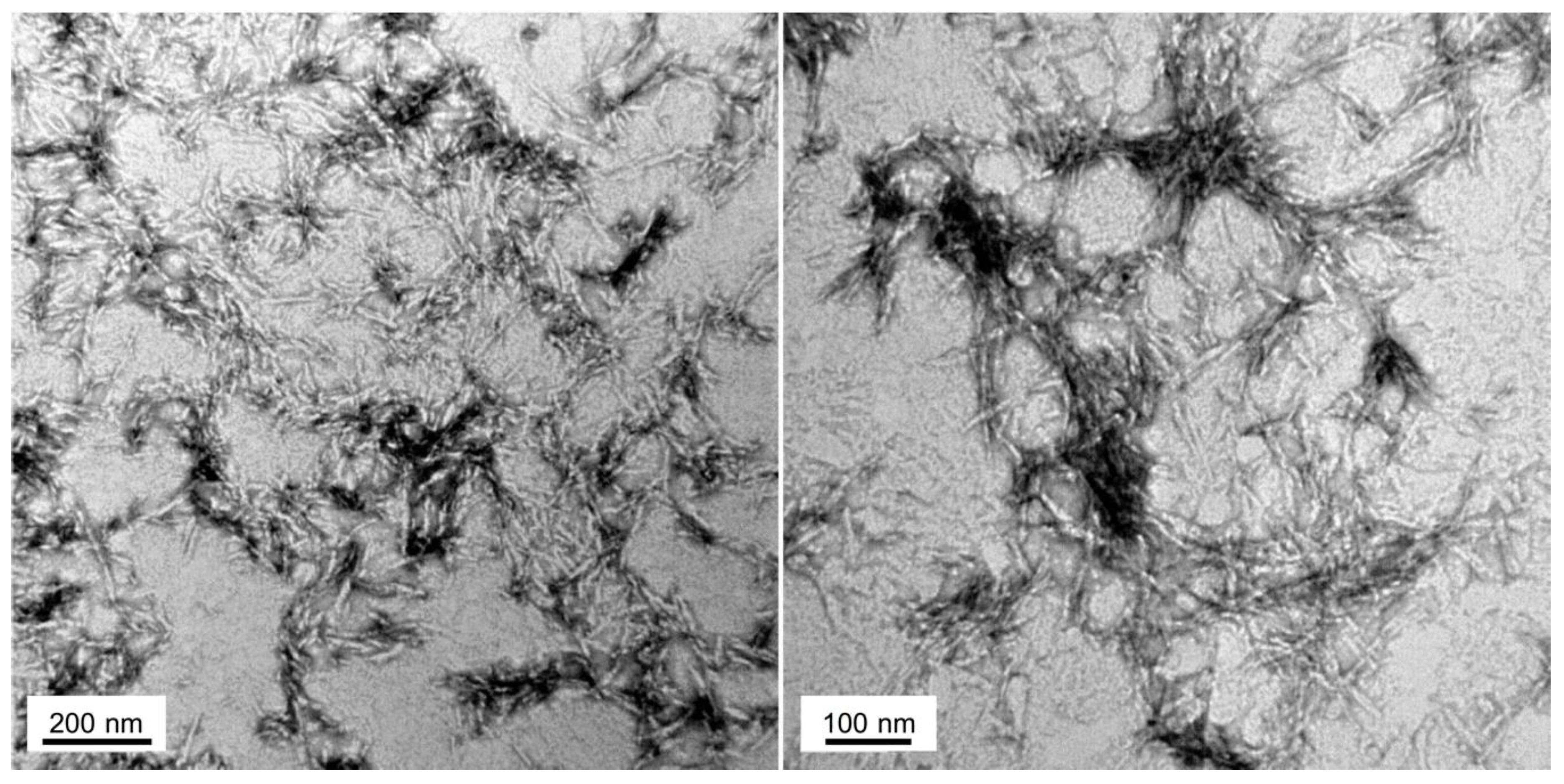
2.3. Hsf1 Amyloid Core Forms Amyloid-Like Fibrillary Structures
2.4. Hsf1 Amyloid Core Self-Assembles Very Fast
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Computational Identification of Hsf1 Soft Amyloid Cores in Low Complexity Regions
4.3. Hsf1 Soft Amyloid Core Peptide Preparation
4.4. Synchronous Light Scattering
4.5. Bis-ANS Binding
4.6. Attenuated Total Reflectance (ATR) Fourier Transform Infrared (FTIR) Spectroscopy
4.7. Binding to Amyloid Dyes
4.8. Transmission Electron Microscopy (TEM)
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Morimoto, R.I. The heat shock response: Systems biology of proteotoxic stress in aging and disease. Cold Spring Harb. Symp. Quant. Biol. 2011, 76, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Anckar, J.; Sistonen, L. Regulation of HSF1 function in the heat stress response: Implications in aging and disease. Annu. Rev. Biochem. 2011, 80, 1089–1115. [Google Scholar] [CrossRef] [PubMed]
- Baler, R.; Dahl, G.; Voellmy, R. Activation of human heat shock genes is accompanied by oligomerization, modification, and rapid translocation of heat shock transcription factor HSF1. Mol. Cell. Biol. 1993, 13, 2486–2496. [Google Scholar] [CrossRef] [PubMed]
- Anckar, J.; Sistonen, L. Heat shock factor 1 as a coordinator of stress and developmental pathways. Adv. Exp. Med. Biol. 2007, 594, 78–88. [Google Scholar] [PubMed]
- Zou, J.; Guo, Y.; Guettouche, T.; Smith, D.F.; Voellmy, R. Repression of heat shock transcription factor HSF1 activation by HSP90 (HSP90 complex) that forms a stress-sensitive complex with HSF1. Cell 1998, 94, 471–480. [Google Scholar] [CrossRef]
- Guo, Y.; Guettouche, T.; Fenna, M.; Boellmann, F.; Pratt, W.B.; Toft, D.O.; Smith, D.F.; Voellmy, R. Evidence for a mechanism of repression of heat shock factor 1 transcriptional activity by a multichaperone complex. J. Biol. Chem. 2001, 276, 45791–45799. [Google Scholar] [CrossRef] [PubMed]
- Pattaramanon, N.; Sangha, N.; Gafni, A. The carboxy-terminal domain of heat-shock factor 1 is largely unfolded but can be induced to collapse into a compact, partially structured state. Biochemistry 2007, 46, 3405–3415. [Google Scholar] [CrossRef] [PubMed]
- Ward, J.J.; Sodhi, J.S.; McGuffin, L.J.; Buxton, B.F.; Jones, D.T. Prediction and functional analysis of native disorder in proteins from the three kingdoms of life. J. Mol. Biol. 2004, 337, 635–645. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.; Vucetic, S.; Iakoucheva, L.M.; Oldfield, C.J.; Dunker, A.K.; Uversky, V.N.; Obradovic, Z. Functional anthology of intrinsic disorder. 1. Biological processes and functions of proteins with long disordered regions. J. Proteome Res. 2007, 6, 1882–1898. [Google Scholar] [CrossRef] [PubMed]
- Habchi, J.; Tompa, P.; Longhi, S.; Uversky, V.N. Introducing protein intrinsic disorder. Chem. Rev. 2014, 114, 6561–6588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cumberworth, A.; Lamour, G.; Babu, M.M.; Gsponer, J. Promiscuity as a functional trait: Intrinsically disordered regions as central players of interactomes. Biochem. J. 2013, 454, 361–369. [Google Scholar] [CrossRef] [PubMed]
- Dyson, H.J.; Wright, P.E. Coupling of folding and binding for unstructured proteins. Curr. Opin. Struct. Biol. 2002, 12, 54–60. [Google Scholar] [CrossRef]
- Yan, J.; Dunker, A.K.; Uversky, V.N.; Kurgan, L. Molecular recognition features (MoRFs) in three domains of life. Mol. BioSyst. 2016, 12, 697–710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, F.; Uversky, V.N.; Kurgan, L. Comprehensive review of methods for prediction of intrinsic disorder and its molecular functions. Cell. Mol. Life Sci. 2017, 74, 3069–3090. [Google Scholar] [CrossRef] [PubMed]
- Gemayel, R.; Chavali, S.; Pougach, K.; Legendre, M.; Zhu, B.; Boeynaems, S.; van der Zande, E.; Gevaert, K.; Rousseau, F.; Schymkowitz, J.; et al. Variable Glutamine-Rich Repeats Modulate Transcription Factor Activity. Mol. Cell 2015, 59, 615–627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pastore, A.; Temussi, P.A. The two faces of Janus: Functional interactions and protein aggregation. Curr. Opin. Struct. Biol. 2012, 22, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Ma, B.; Nussinov, R. Trp/Met/Phe hot spots in protein-protein interactions: Potential targets in drug design. Curr. Top. Med. Chem. 2007, 7, 999–1005. [Google Scholar] [CrossRef] [PubMed]
- Sabate, R.; Espargaro, A.; Grana-Montes, R.; Reverter, D.; Ventura, S. Native structure protects SUMO proteins from aggregation into amyloid fibrils. Biomacromolecules 2012, 13, 1916–1926. [Google Scholar] [CrossRef] [PubMed]
- Castillo, V.; Ventura, S. Amyloidogenic regions and interaction surfaces overlap in globular proteins related to conformational diseases. PLoS Comput. Biol. 2009, 5, e1000476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pechmann, S.; Levy, E.D.; Tartaglia, G.G.; Vendruscolo, M. Physicochemical principles that regulate the competition between functional and dysfunctional association of proteins. Proc. Natl. Acad. Sci. USA 2009, 106, 10159–10164. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.D.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Mistry, J.; Mitchell, A.L.; Potter, S.C.; Punta, M.; Qureshi, M.; Sangrador-Vegas, A.; et al. The Pfam protein families database: Towards a more sustainable future. Nucleic Acids Res. 2016, 44, D279–D285. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.S.; Liu, C.W.; Damberger, F.F.; Pelton, J.G.; Nelson, H.C.; Wemmer, D.E. Yeast heat shock transcription factor N-terminal activation domains are unstructured as probed by heteronuclear NMR spectroscopy. Protein Sci. Publ. Protein Soc. 1996, 5, 262–269. [Google Scholar] [CrossRef] [PubMed]
- Walsh, I.; Martin, A.J.; Di Domenico, T.; Tosatto, S.C. ESpritz: Accurate and fast prediction of protein disorder. Bioinformatics 2012, 28, 503–509. [Google Scholar] [CrossRef] [PubMed]
- Dosztanyi, Z.; Csizmok, V.; Tompa, P.; Simon, I. IUPred: Web server for the prediction of intrinsically unstructured regions of proteins based on estimated energy content. Bioinformatics 2005, 21, 3433–3434. [Google Scholar] [CrossRef] [PubMed]
- Wootton, J.C. Non-globular domains in protein sequences: Automated segmentation using complexity measures. Comput. Chem. 1994, 18, 269–285. [Google Scholar] [CrossRef]
- Piovesan, D.; Walsh, I.; Minervini, G.; Tosatto, S.C.E. FELLS: Fast estimator of latent local structure. Bioinformatics 2017, 33, 1889–1891. [Google Scholar] [CrossRef] [PubMed]
- Michelitsch, M.D.; Weissman, J.S. A census of glutamine/asparagine-rich regions: Implications for their conserved function and the prediction of novel prions. Proc. Natl. Acad. Sci. USA 2000, 97, 11910–11915. [Google Scholar] [CrossRef] [PubMed]
- Alberti, S.; Halfmann, R.; King, O.; Kapila, A.; Lindquist, S. A systematic survey identifies prions and illuminates sequence features of prionogenic proteins. Cell 2009, 137, 146–158. [Google Scholar] [CrossRef] [PubMed]
- Masison, D.C.; Wickner, R.B. Prion-inducing domain of yeast Ure2p and protease resistance of Ure2p in prion-containing cells. Science 1995, 270, 93–95. [Google Scholar] [CrossRef] [PubMed]
- Franzmann, T.M.; Jahnel, M.; Pozniakovsky, A.; Mahamid, J.; Holehouse, A.S.; Nuske, E.; Richter, D.; Baumeister, W.; Grill, S.W.; Pappu, R.V.; et al. Phase separation of a yeast prion protein promotes cellular fitness. Science 2018, 359. [Google Scholar] [CrossRef] [PubMed]
- Sabate, R.; Rousseau, F.; Schymkowitz, J.; Ventura, S. What makes a protein sequence a prion? PLoS Comput. Biol. 2015, 11, e1004013. [Google Scholar] [CrossRef] [PubMed]
- Sant’Anna, R.; Fernández, M.R.; Batlle, C.; Navarro, S.; de Groot, N.S.; Serpell, L.; Ventura, S. Characterization of Amyloid Cores in Prion Domains. Sci. Rep. 2016, 6, 34274. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, M.R.; Batlle, C.; Gil-Garcia, M.; Ventura, S. Amyloid cores in prion domains: Key regulators for prion conformational conversion. Prion 2017, 11, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Sabate, R.; Rousseau, F.; Schymkowitz, J.; Batlle, C.; Ventura, S. Amyloids or prions? That is the question. Prion 2015, 9, 200–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maurer-Stroh, S.; Debulpaep, M.; Kuemmerer, N.; Lopez de la Paz, M.; Martins, I.C.; Reumers, J.; Morris, K.L.; Copland, A.; Serpell, L.; Serrano, L.; et al. Exploring the sequence determinants of amyloid structure using position-specific scoring matrices. Nat. Methods 2010, 7, 237–242. [Google Scholar] [CrossRef] [PubMed]
- Conchillo-Sole, O.; de Groot, N.S.; Aviles, F.X.; Vendrell, J.; Daura, X.; Ventura, S. AGGRESCAN: A server for the prediction and evaluation of “hot spots” of aggregation in polypeptides. BMC Bioinform. 2007, 8, 65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tartaglia, G.G.; Vendruscolo, M. The Zyggregator method for predicting protein aggregation propensities. Chem. Soc. Rev. 2008, 37, 1395–1401. [Google Scholar] [CrossRef] [PubMed]
- Dosztanyi, Z.; Meszaros, B.; Simon, I. ANCHOR: Web server for predicting protein binding regions in disordered proteins. Bioinformatics 2009, 25, 2745–2746. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.; Kurgan, L. High-throughput prediction of RNA, DNA and protein binding regions mediated by intrinsic disorder. Nucleic Acids Res. 2015, 43, e121. [Google Scholar] [CrossRef] [PubMed]
- Gasteiger, E.; Gattiker, A.; Hoogland, C.; Ivanyi, I.; Appel, R.D.; Bairoch, A. ExPASy: The proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Res. 2003, 31, 3784–3788. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.; So, M.; Maat, H.; Ray, N.J.; Arisaka, F.; Goto, Y.; Carver, J.A.; Hall, D. Measurement of amyloid formation by turbidity assay-seeing through the cloud. Biophys. Rev. 2016, 8, 445–471. [Google Scholar] [CrossRef] [PubMed]
- Rosen, C.G.; Weber, G. Dimer formation from 1-amino-8-naphthalenesulfonate catalyzed by bovine serum albumin. A new fluorescent molecule with exceptional binding properties. Biochemistry 1969, 8, 3915–3920. [Google Scholar] [CrossRef] [PubMed]
- Stryer, L. The interaction of a naphthalene dye with apomyoglobin and apohemoglobin. A fluorescent probe of non-polar binding sites. J. Mol. Biol. 1965, 13, 482–495. [Google Scholar] [CrossRef]
- LeVine, H., 3rd. Thioflavine T interaction with synthetic Alzheimer’s disease beta-amyloid peptides: Detection of amyloid aggregation in solution. Protein Sci. 1993, 2, 404–410. [Google Scholar] [CrossRef] [PubMed]
- Klunk, W.E.; Pettegrew, J.W.; Abraham, D.J. Quantitative evaluation of congo red binding to amyloid-like proteins with a beta-pleated sheet conformation. J. Histochem. Cytochem. 1989, 37, 1273–1281. [Google Scholar] [CrossRef] [PubMed]
- Sunde, M.; Blake, C. The structure of amyloid fibrils by electron microscopy and X-ray diffraction. Adv. Protein Chem. 1997, 50, 123–159. [Google Scholar] [PubMed]
- Linding, R.; Schymkowitz, J.; Rousseau, F.; Diella, F.; Serrano, L. A comparative study of the relationship between protein structure and beta-aggregation in globular and intrinsically disordered proteins. J. Mol. Biol. 2004, 342, 345–353. [Google Scholar] [CrossRef] [PubMed]
- Meszaros, B.; Tompa, P.; Simon, I.; Dosztanyi, Z. Molecular principles of the interactions of disordered proteins. J. Mol. Biol. 2007, 372, 549–561. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. Intrinsic Disorder, Protein-Protein Interactions, and Disease. Adv. Protein Chem. Struct. Biol. 2018, 110, 85–121. [Google Scholar] [PubMed]
- Banani, S.F.; Lee, H.O.; Hyman, A.A.; Rosen, M.K. Biomolecular condensates: Organizers of cellular biochemistry. Nat. Rev. Mol. Cell Biol. 2017, 18, 285–298. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; McGinnis, J.P.; Si, K. Translational Control by Prion-like Proteins. Trends Cell Biol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Pallares, I.; Ventura, S. The Transcription Terminator Rho: A First Bacterial Prion. Trends Microbiol. 2017, 25, 434–437. [Google Scholar] [CrossRef] [PubMed]
- Pallares, I.; Iglesias, V.; Ventura, S. The Rho Termination Factor of Clostridium botulinum Contains a Prion-Like Domain with a Highly Amyloidogenic Core. Front. Microbiol. 2015, 6, 1516. [Google Scholar] [CrossRef] [PubMed]
- Yuan, A.H.; Hochschild, A. A bacterial global regulator forms a prion. Science 2017, 355, 198–201. [Google Scholar] [CrossRef] [PubMed]
- UniProt, C. UniProt: A hub for protein information. Nucleic Acids Res. 2015, 43, D204–D212. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | Amyloid Core | Gravy Score |
|---|---|---|
| Hsf1 | INDIIFNTNLANNLSNYN | −0.283 |
| ASYN | GVLYVG | 1.683 |
| GGAVVTGVTAVAQ | 1.238 | |
| Aβ42 | GAIIGLMVGGVVI | 2.462 |
| QKLVFFAE | 0.562 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pujols, J.; Santos, J.; Pallarès, I.; Ventura, S. The Disordered C-Terminus of Yeast Hsf1 Contains a Cryptic Low-Complexity Amyloidogenic Region. Int. J. Mol. Sci. 2018, 19, 1384. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19051384
Pujols J, Santos J, Pallarès I, Ventura S. The Disordered C-Terminus of Yeast Hsf1 Contains a Cryptic Low-Complexity Amyloidogenic Region. International Journal of Molecular Sciences. 2018; 19(5):1384. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19051384
Chicago/Turabian StylePujols, Jordi, Jaime Santos, Irantzu Pallarès, and Salvador Ventura. 2018. "The Disordered C-Terminus of Yeast Hsf1 Contains a Cryptic Low-Complexity Amyloidogenic Region" International Journal of Molecular Sciences 19, no. 5: 1384. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19051384