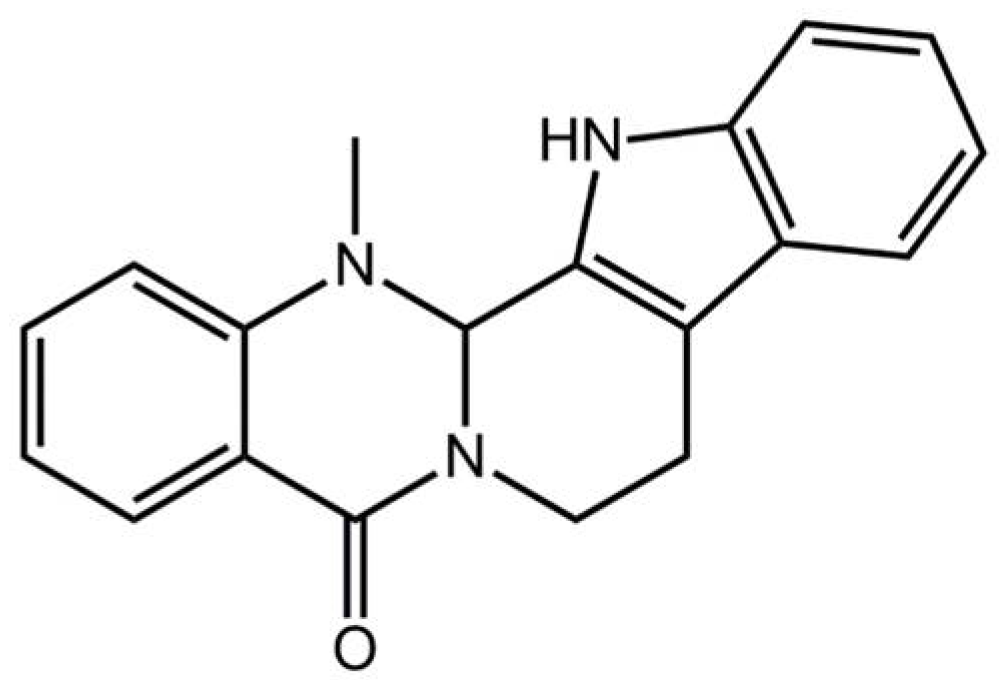
Pharmacological Basis for the Use of Evodiamine in Alzheimer’s Disease: Antioxidation and Antiapoptosis


Abstract
:1. Introduction
2. Results
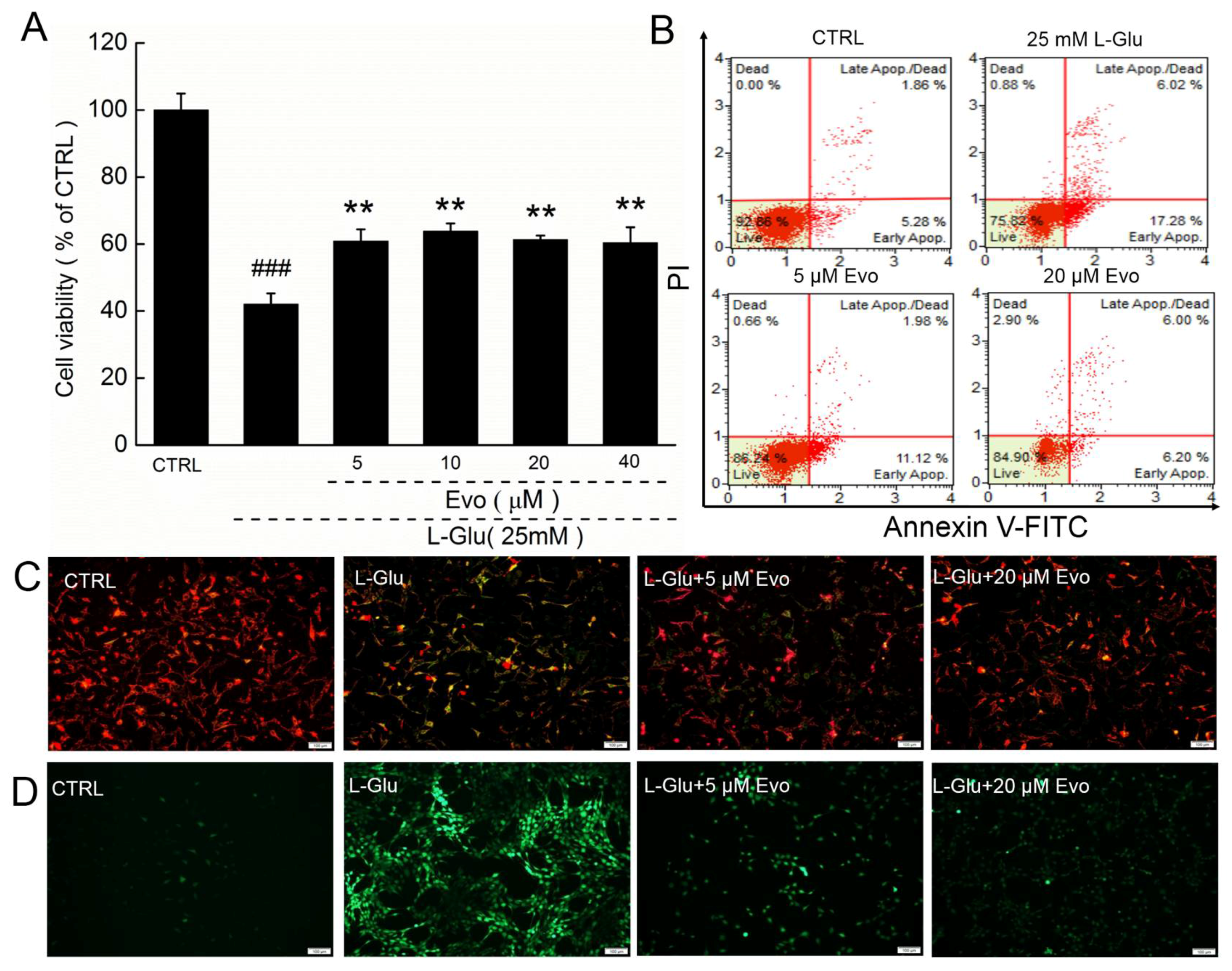
2.1. Evo Protects HT22 Cells against l-Glu Damage via Regulation of Mitochondrial Function
2.2. Evo Ameliorates the Expression Levels of Apoptosis-Related Proteins in HT22 Cells
2.3. Evo Improved AD-Like Behavior in d-gal and AlCl3-Induced AD Mice
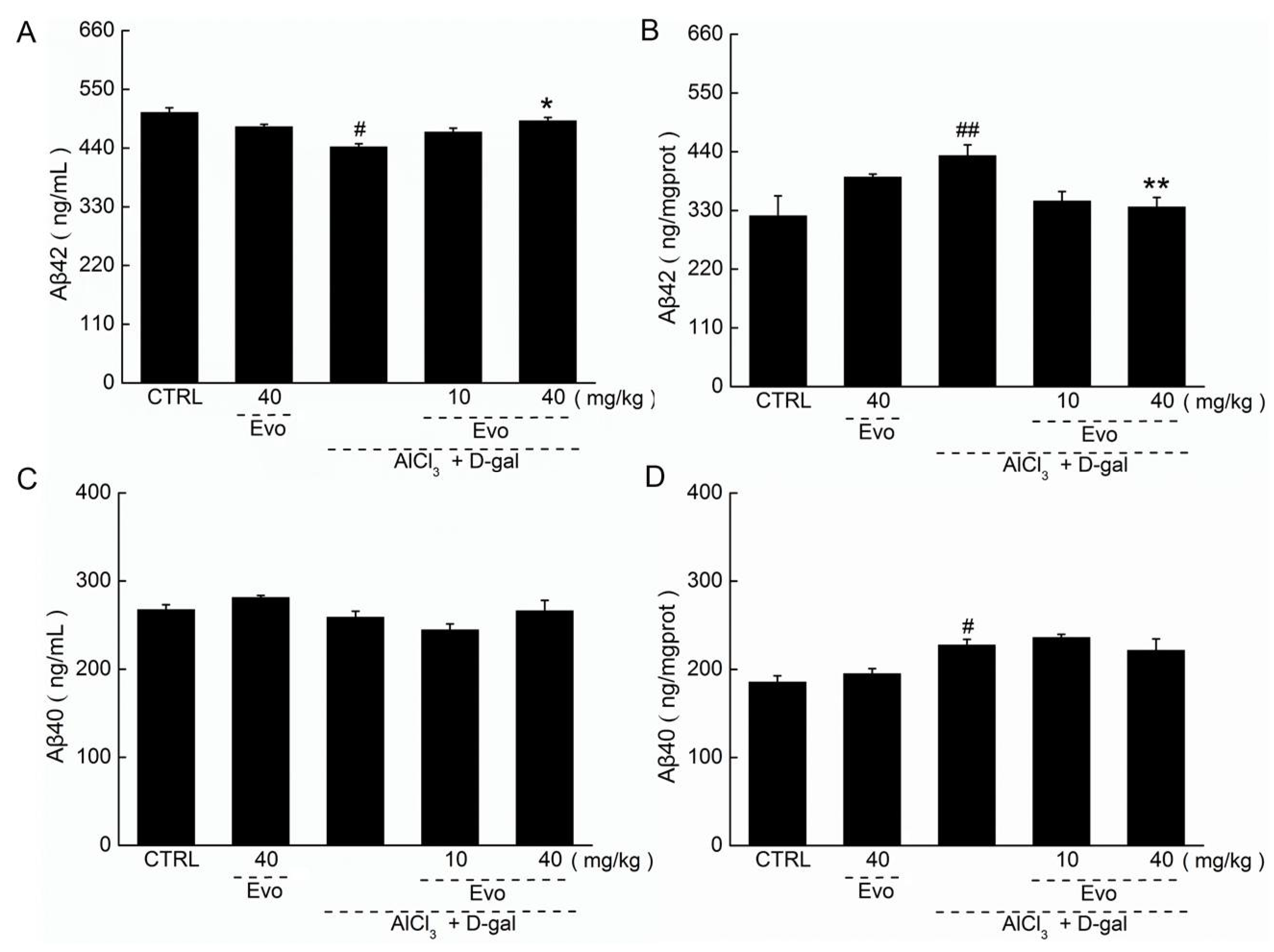
2.4. The Effect of Evo on the Levels of Aβ42 and Aβ40 in Serum and Cerebral Cortex of AD Mice
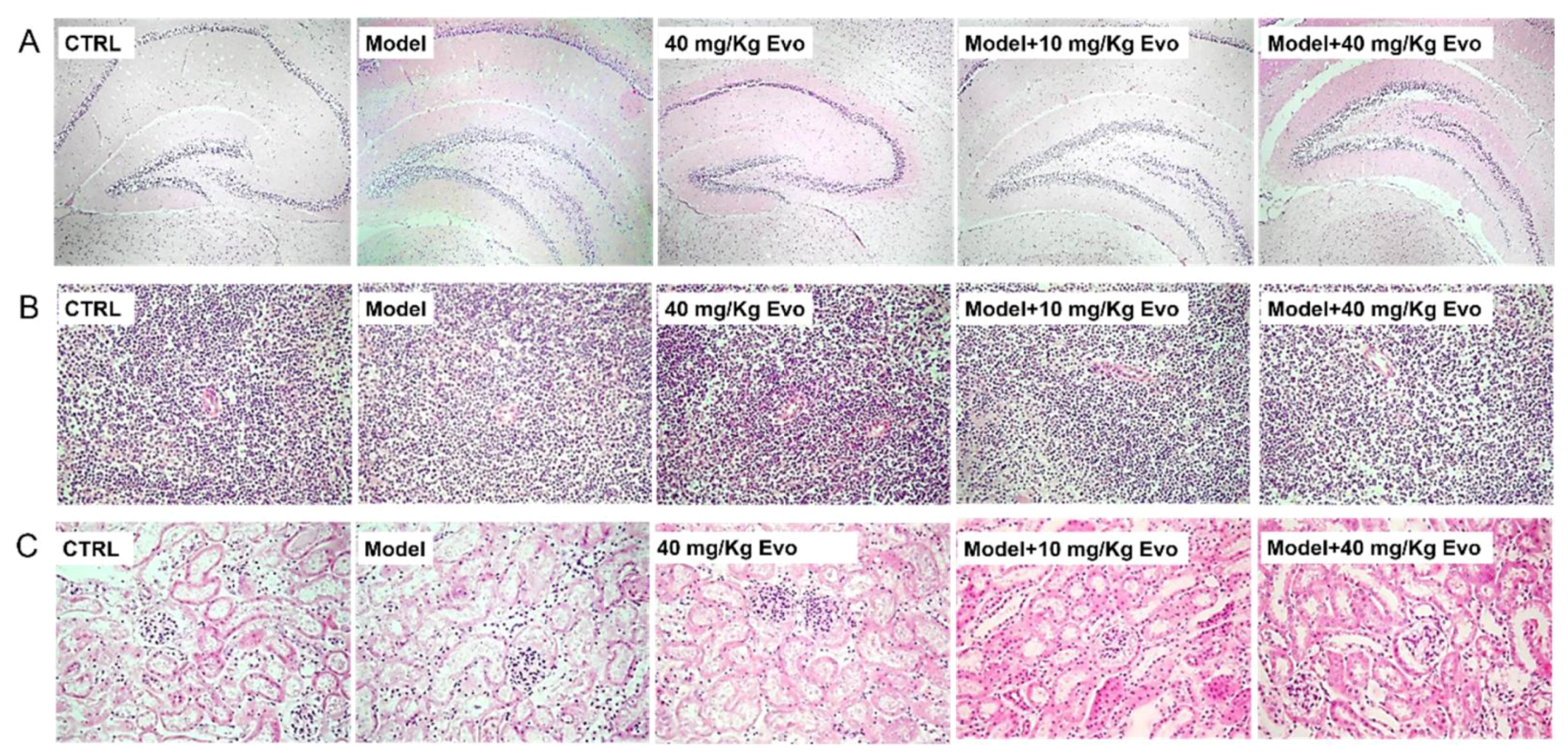
2.5. The Effect of Evo on the Pathology of the Brain, Spleen, and Kidney in AD Mice
2.6. Evo Regulated the Levels of Cholinergic Neurotransmitters in AD Mice
2.7. Evo Displayed Anti-Oxidative Effects in AD Mice
3. Discussion
4. Materials and Methods
4.1. Chemicals and Antibodies
4.2. Cell Culture
4.3. Cell Viability Assay
4.4. Cell Apoptosis Assay
4.5. Measurement of MMP and Intracellular ROS Levels
4.6. Western Blot
4.7. The Development of AD Mouse Model and Agent Administration Process
4.8. Behavioral Tests
4.8.1. Open Field Experiment Test
4.8.2. Morris Water Maze Test
4.9. Enzyme-Linked Immunosorbent Assay
4.10. Histological Examination
4.11. Statistical Analysis
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
Abbreviations
| Ach | acetylcholine |
| AchE | acetylcholine esterase |
| AD | Alzheimer’s disease |
| Akt | protein kinase B |
| AlCl3 | aluminum trichloride |
| ANOVA | one-way analysis of variance |
| Aβ | amyloid beta |
| Bcl-2 | B-cell lymphoma-2 |
| ChAT | choline acetyltransferase |
| d-gal | d-galactose |
| Evo | Evodiamine |
| ELISA | enzyme-linked immunosorbent assay |
| GAPDH | glyceraldehyde-3-phosphate dehydrogenase |
| GSH-Px | glutathione peroxidase |
| H&E | hematoxylin–eosin staining |
| l-Glu | l-glutamic acid |
| MAPK | mitogen-activated protein kinase |
| MMP | mitochondrial membrane potential |
| mTOR | mammalian target of rapamycin |
| MWM | Morris water maze test |
| ROS | reactive oxygen species |
| SOD | superoxide dismutase |
References
- Mattson, M.P. Pathways towards and away from Alzheimer’s disease. Nature 2004, 430, 631–639. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Huh, J.W.; Eom, T.; Na, N.; Lee, Y.; Kim, J.S.; Kim, S.U.; Shim, I.; Lee, S.R.; Kim, E. Effects of Newly Synthesized Recombinant Human Amyloid-β Complexes and Poly-Amyloid-β Fibers on Cell Apoptosis and Cognitive Decline. J. Microbiol. Biotechnol. 2017, 27, 2044–2051. [Google Scholar] [CrossRef] [PubMed]
- Sohn, E.J.; Shin, M.J.; Eum, W.S.; Kim, D.W.; Yong, J.I.; Ryu, E.J.; Park, J.H.; Cho, S.B.; Cha, H.J.; Kim, S.J.; et al. Tat-NOL3 protects against hippocampal neuronal cell death induced by oxidative stress through the regulation of apoptotic pathways. Int. J. Mol. Med. 2016, 38, 225–235. [Google Scholar] [CrossRef] [PubMed]
- Cheignon, C.; Tomas, M.; Bonnefont-Rousselot, D.; Faller, P.; Hureau, C.; Collin, F. Oxidative stress and the amyloid beta peptide in Alzheimer’s disease. Redox Biol. 2018, 14, 450–464. [Google Scholar] [CrossRef] [PubMed]
- Foidl, B.M.; Do-Dinh, P.; Hutter-Schmid, B.; Bliem, H.R.; Humpel, C. Cholinergic neurodegeneration in an Alzheimer mouse model overexpressing amyloid-precursor protein with the Swedish-Dutch-Iowa mutations. Neurobiol. Learn. Mem. 2016, 136, 86–96. [Google Scholar] [CrossRef] [PubMed]
- Schliebs, R.; Arendt, T. The significance of the cholinergic system in the brain during aging and in Alzheimer’s disease. J. Neural Transm. 2006, 113, 1625–1644. [Google Scholar] [CrossRef] [PubMed]
- Craig, L.A.; Hong, N.S.; McDonald, R.J. Revisiting the cholinergic hypothesis in the development of Alzheimer’s disease. Neurosci. Biobehav. Rev. 2011, 35, 1397–1409. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y.; Ahn, S.M.; Wang, Z.; Choi, Y.W.; Shin, H.K.; Choi, B.T. Neuroprotective effects of 2,3,5,4′-tetrahydoxystilbene-2-O-β-d-glucoside from Polygonum multiflorum against glutamate-induced oxidative toxicity in HT22 cells. J. Ethnopharmacol. 2017, 195, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Liu, D.; Zheng, Y.; Li, H.; Hao, C.; Ouyang, W. Protective effects of kinetin against aluminum chloride and d-galactose induced cognitive impairment and oxidative damage in mouse. Brain Res. Bull. 2017, 134, 262–272. [Google Scholar] [CrossRef] [PubMed]
- Qu, Z.; Zhang, J.; Yang, H.; Huo, L.; Gao, J.; Chen, H.; Gao, W. Protective effect of tetrahydropalmatine against d-galactose induced memory impairment in rat. Physiol. Behav. 2016, 154, 114–125. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Chen, X.; Lu, W.; Zhang, S.; Guan, X.; Li, Z.; Wang, D. Anti-Oxidative Stress Activity Is Essential for Amanita caesarea Mediated Neuroprotection on Glutamate-Induced Apoptotic HT22 Cells and an Alzheimer’s Disease Mouse Model. Int. J. Mol. Sci. 2017, 18, 1623. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.Y.; Liu, J.G.; Li, H.; Yang, H.M. Pharmacological Effects of Active Components of Chinese Herbal Medicine in the Treatment of Alzheimer’s Disease: A Review. Am. J. Chin. Med. 2016, 44, 1525–1541. [Google Scholar] [CrossRef] [PubMed]
- Gavaraskar, K.; Dhulap, S.; Hirwani, R.R. Therapeutic and cosmetic applications of Evodiamine and its derivatives—A patent review. Fitoterapia 2015, 106, 22–35. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.M.; Gao, K.; Wang, D.M.; Quan, X.Z.; Liu, J.N.; Ma, C.M.; Qin, C.; Zhang, L.F. Evodiamine improves congnitive abilities in SAMP8 and APPswe/PS1ΔE9 transgenic mouse models of Alzheimer’s disease. Acta Pharmacol. Sin. 2011, 32, 295–302. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Deng, D.; Shao, N.; Xu, Y.; Xue, L.; Peng, Y.; Liu, Y.; Zhi, F. Evodiamine activates cellular apoptosis through suppressing PI3K/AKT and activating MAPK in glioma. OncoTargets Ther. 2018, 11, 1183–1192. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Wu, S.; Zhang, Y.; Wang, L.; Guo, Y. Antitumor effect of triptolide in T-cell lymphoblastic lymphoma by inhibiting cell viability, invasion, and epithelial-mesenchymal transition via regulating the PI3K/AKT/mTOR pathway. OncoTargets Ther. 2018, 11, 769–779. [Google Scholar] [CrossRef] [PubMed]
- Souza, L.C.; Jesse, C.R.; Del Fabbro, L.; de Gomes, M.G.; Gomes, N.S.; Filho, C.B.; Goes, A.T.R.; Wilhelm, E.A.; Luchese, C.; Roman, S.S.; et al. Aging exacerbates cognitive and anxiety alterations induced by an intracerebroventricular injection of amyloid-β1–42 peptide in mice. Mol. Cell. Neurosci. 2018, 88, 93–106. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Jiao, J.J.; Holscher, C.; Wu, M.N.; Zhang, J.; Tong, J.Q.; Dong, X.F.; Qu, X.S.; Cao, Y.; Cai, H.Y.; et al. A novel GLP-1/GIP/Gcg triagonist reduces cognitive deficits and pathology in the 3xTg mouse model of Alzheimer’s disease. Hippocampus 2018, 28, 358–372. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; An, S.; Hu, W.; Teng, M.; Wang, X.; Qu, Y.; Liu, Y.; Yuan, Y.; Wang, D. The Neuroprotective Properties of Hericium erinaceus in Glutamate-Damaged Differentiated PC12 Cells and an Alzheimer’s Disease Mouse Model. Int. J. Mol. Sci. 2016, 17, 1810. [Google Scholar] [CrossRef] [PubMed]
- Frozza, R.L.; Lourenco, M.V.; De Felice, F.G. Challenges for Alzheimer’s Disease Therapy: Insights from Novel Mechanisms Beyond Memory Defects. Front. Neurosci. 2018, 12, 37. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Shang, Q.; Han, M.; Chen, K.; Xu, H. Traditional Chinese medicine injection for angina pectoris: An overview of systematic reviews. Am. J. Chin. Med. 2014, 42, 37–59. [Google Scholar] [CrossRef] [PubMed]
- Sheng, Z.H.; Cai, Q. Mitochondrial transport in neurons: Impact on synaptic homeostasis and neurodegeneration. Nat. Rev. Neurosci. 2012, 13, 77–93. [Google Scholar] [CrossRef] [PubMed]
- Shigenaga, M.K.; Hagen, T.M.; Ames, B.N. Oxidative damage and mitochondrial decay in aging. Proc. Natl. Acad. Sci. USA 1994, 91, 10771–10778. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, P.; Yadav, R.S.; Chandravanshi, L.P.; Shukla, R.K.; Dhuriya, Y.K.; Chauhan, L.K.; Dwivedi, H.N.; Pant, A.B.; Khanna, V.K. Unraveling the mechanism of neuroprotection of curcumin in arsenic induced cholinergic dysfunctions in rats. Toxicol. Appl. Pharmacol. 2014, 279, 428–440. [Google Scholar] [CrossRef] [PubMed]
- Romero-Leguizamon, C.R.; Ramirez-Latorre, J.A.; Mora-Munoz, L.; Guerrero-Naranjo, A. Signaling pathways mTOR and AKT in epilepsy. Rev. Neurol. 2016, 63, 33–41. [Google Scholar] [PubMed]
- Datta, S.R.; Ranger, A.M.; Lin, M.Z.; Sturgill, J.F.; Ma, Y.C.; Cowan, C.W.; Dikkes, P.; Korsmeyer, S.J.; Greenberg, M.E. Survival factor-mediated BAD phosphorylation raises the mitochondrial threshold for apoptosis. Dev. Cell 2002, 3, 631–643. [Google Scholar] [CrossRef]
- Bivik, C.A.; Larsson, P.K.; Kagedal, K.M.; Rosdahl, I.K.; Ollinger, K.M. UVA/B-induced apoptosis in human melanocytes involves translocation of cathepsins and Bcl-2 family members. J. Investig. Dermatol. 2006, 126, 1119–1127. [Google Scholar] [CrossRef] [PubMed]
- Walsh, R.N.; Cummins, R.A. The Open-Field Test: A critical review. Psychol. Bull. 1976, 83, 482–504. [Google Scholar] [CrossRef] [PubMed]
- Choleris, E.; Thomas, A.W.; Kavaliers, M.; Prato, F.S. A detailed ethological analysis of the mouse open field test: Effects of diazepam, chlordiazepoxide and an extremely low frequency pulsed magnetic field. Neurosci. Biobehav. Rev. 2001, 25, 235–260. [Google Scholar] [CrossRef]
- Pratico, D.; Uryu, K.; Sung, S.; Tang, S.; Trojanowski, J.Q.; Lee, V.M. Aluminum modulates brain amyloidosis through oxidative stress in APP transgenic mice. FASEB J. 2002, 16, 1138–1140. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; Zuo, P.; Zhang, Q.; Li, X.; Hu, Y.; Long, J.; Packer, L.; Liu, J. Chronic systemic d-galactose exposure induces memory loss, neurodegeneration, and oxidative damage in mice: Protective effects of R-alpha-lipoic acid. J. Neurosci. Res. 2006, 84, 647–654. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Prakash, A.; Dogra, S. Naringin alleviates cognitive impairment, mitochondrial dysfunction and oxidative stress induced by d-galactose in mice. Food Chem. Toxicol. 2010, 48, 626–632. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.M.; Lu, J.; Zheng, Y.L.; Zhou, Z.; Shan, Q.; Ma, D.F. Purple sweet potato color repairs d-galactose-induced spatial learning and memory impairment by regulating the expression of synaptic proteins. Neurobiol Learn. Mem. 2008, 90, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, D.A.; Griffin, S.; Munch, G.; Pasinetti, G.M. Amyloid β-peptide and amyloid pathology are central to the oxidative stress and inflammatory cascades under which Alzheimer’s disease brain exists. J. Alzheimers Dis. 2002, 4, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Cai, Q.; Tammineni, P. Mitochondrial Aspects of Synaptic Dysfunction in Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 57, 1087–1103. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.; Zhao, Y.; Zhang, T.; Lan, J.; Yang, J.; Yuan, L.; Zhang, Q.; Pan, K.; Zhang, K. Galantamine inhibits β-amyloid-induced cytostatic autophagy in PC12 cells through decreasing ROS production. Cell Prolif. 2018. [Google Scholar] [CrossRef] [PubMed]
- Fossati, S.; Giannoni, P.; Solesio, M.E.; Cocklin, S.L.; Cabrera, E.; Ghiso, J.; Rostagno, A. The carbonic anhydrase inhibitor methazolamide prevents amyloid beta-induced mitochondrial dysfunction and caspase activation protecting neuronal and glial cells in vitro and in the mouse brain. Neurobiol. Dis. 2016, 86, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Abulfadl, Y.S.; El-Maraghy, N.N.; Ahmed, A.A.E.; Nofal, S.; Badary, O.A. Protective effects of thymoquinone on d-galactose and aluminum chloride induced neurotoxicity in rats: Biochemical, histological and behavioral changes. Neurol. Res. 2018, 40, 324–333. [Google Scholar] [CrossRef] [PubMed]
- Melo, J.B.; Agostinho, P.; Oliveira, C.R. Involvement of oxidative stress in the enhancement of acetylcholinesterase activity induced by amyloid beta-peptide. Neurosci. Res. 2003, 45, 117–127. [Google Scholar] [CrossRef]
- Park, S.J.; Jung, J.M.; Lee, H.E.; Lee, Y.W.; Kim, D.H.; Kim, J.M.; Hong, J.G.; Lee, C.H.; Jung, I.H.; Cho, Y.B.; et al. The memory ameliorating effects of INM-176, an ethanolic extract of Angelica gigas, against scopolamine- or Aβ1–42-induced cognitive dysfunction in mice. J. Ethnopharmacol. 2012, 143, 611–620. [Google Scholar] [CrossRef] [PubMed]
- Auld, D.S.; Kornecook, T.J.; Bastianetto, S.; Quirion, R. Alzheimer’s disease and the basal forebrain cholinergic system: Relations to β-amyloid peptides, cognition, and treatment strategies. Prog. Neurobiol. 2002, 68, 209–245. [Google Scholar] [CrossRef]
- Ferreira-Vieira, T.H.; Guimaraes, I.M.; Silva, F.R.; Ribeiro, F.M. Alzheimer’s disease: Targeting the Cholinergic System. Curr. Neuropharmacol. 2016, 14, 101–115. [Google Scholar] [CrossRef] [PubMed]
- Tabet, N. Acetylcholinesterase inhibitors for Alzheimer’s disease: Anti-inflammatories in acetylcholine clothing! Age Ageing 2006, 35, 336–338. [Google Scholar] [CrossRef] [PubMed]
- Wilcock, G.K.; Esiri, M.M.; Bowen, D.M.; Smith, C.C. Alzheimer’s disease. Correlation of cortical choline acetyltransferase activity with the severity of dementia and histological abnormalities. J. Neurol. Sci. 1982, 57, 407–417. [Google Scholar] [CrossRef]
- Swaab, D.F. Chapter II Neurobiology and neuropathology of the human hypothalamus. Handb. Chem. Neuroanat. 1997, 13, 39–137. [Google Scholar]
- Ishii, M.; Iadecola, C. Metabolic and Non-Cognitive Manifestations of Alzheimer’s Disease: The Hypothalamus as Both Culprit and Target of Pathology. Cell Metab. 2015, 22, 761–776. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Chen, Y.; Cai, G.; Li, X.; Wang, D. Carnosic acid induces apoptosis of hepatocellular carcinoma cells via ROS-mediated mitochondrial pathway. Chem.-Biol. Interact. 2017, 277, 91–100. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Items | Neurotransmitters | CTRL | Evo (40 mg/kg) | AlCl3 + d-gal | ||
|---|---|---|---|---|---|---|
| Model | Evo (mg/kg) | |||||
| 10 | 40 | |||||
| Serum | Ach (μg/mL) | 886.8 ± 22.6 | 895.0 ± 31.4 | 773.3 ± 14.2 ## | 845.0 ± 28.2 ** | 916.7 ± 26.7 ** |
| AchE (nmol/L) | 146.8 ± 4.0 | 138.7 ± 10.0 | 174.7 ± 7.8 ## | 132.0 ± 4.8 ** | 152.7 ± 4.2 * | |
| ChAT(pmol/L) | 275.7 ± 11.0 | 259.3 ± 6.2 | 231.4 ± 10.7 # | 260.2 ± 7.2 * | 264.3 ± 4.3 * | |
| Hypothalamus | Ach (μg/mgprot) | 271.2 ± 27.2 | 249.2 ± 22.5 | 176.5 ± 18.5 ## | 209.3 ± 14.5 | 258.6 ± 13.1 ** |
| AchE (nmol/gprot) | 56.7 ± 4.2 | 60.3 ± 4.4 | 72.4 ± 4.4 ## | 66.1 ± 5.3 | 58.7 ± 1.8 * | |
| ChAT (pmol/gprot) | 126.3 ± 12.8 | 123.3 ± 14.5 | 75.8 ± 6.4 ### | 87.0 ± 7.7 | 145.9 ± 16.7 *** | |
| Cerebral Cortex | Ach (μg/mgprot) | 765.9 ± 23.1 | 748.7 ± 23.8 | 546.3 ± 10.7 # | 615.2 ± 17.5 * | 626.4 ± 21.5 * |
| AchE (nmol/gprot) | 58.5 ± 6.8 | 66.5 ± 5.0 | 103.5 ± 5.1 ### | 84.9 ± 2.9 ** | 73.6 ± 3.2 *** | |
| ChAT (pmol/gprot) | 240.0 ± 15.8 | 232.5 ± 4.3 | 165.1 ± 3.1 # | 243.0 ± 14.0 ** | 212.2 ± 12.8 * | |
| Items | Factors | CTRL | Evo (40 mg/kg) | AlCl3 + d-gal | ||
|---|---|---|---|---|---|---|
| Model | Evo (mg/kg) | |||||
| 10 | 40 | |||||
| Serum | ROS (U/mL) | 458.8 ± 6.0 | 465.5 ± 6.3 | 535.2 ± 6.2 # | 515.6 ± 13.3 | 482.5 ± 16.5 * |
| SOD (U/mL) | 322.1 ± 3.7 | 322.9 ± 7.3 | 282.5 ± 18.3 # | 310.8 ± 7.2 * | 330.8 ± 18.7 * | |
| GSH-Px (U/mL) | 748.5 ± 25.2 | 792.5 ± 55.2 | 655.4 ± 23.0 # | 711.7 ± 6.7 | 765.0 ± 10.2 * | |
| Hypothalamus | ROS (U/mgprot) | 123.5 ± 9.9 | 103.8 ± 8.4 | 160.8 ± 10.5 ## | 137.6 ± 10.3 | 122.4 ± 6.0 ** |
| SOD (U/mgprot) | 123.2 ± 12.6 | 112.6 ± 14.7 | 70.8 ± 7.5 ### | 98.4 ± 3.7 * | 116.2 ± 15.5 ** | |
| GSH-Px (U/mgprot) | 198.5 ± 18.7 | 191.9 ± 25.1 | 136.0 ± 13.6 ## | 199.5 ± 11.9 ** | 236.4 ± 26.3 ** | |
| Cerebral Cortex | ROS(U/mgprot) | 208.2 ± 15.8 | 220.1 ± 14.2 | 375.7 ± 20.2 ### | 363.1 ± 15.9 | 313.4 ± 13.9 * |
| SOD(U/mgprot) | 164.7 ± 15.2 | 189.6 ± 17.9 | 121.4 ± 12.2 # | 164.1 ± 4.7 * | 183.9 ± 6.0 * | |
| GSH-Px (U/mgprot) | 335.7 ± 20.6 | 349.3 ± 9.2 | 265.6 ± 3.5 # | 321.7 ± 11.3 * | 339.3 ± 21. 1* | |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Y.; Wang, J.; Wang, C.; Li, Z.; Liu, X.; Zhang, J.; Lu, J.; Wang, D. Pharmacological Basis for the Use of Evodiamine in Alzheimer’s Disease: Antioxidation and Antiapoptosis. Int. J. Mol. Sci. 2018, 19, 1527. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19051527
Zhang Y, Wang J, Wang C, Li Z, Liu X, Zhang J, Lu J, Wang D. Pharmacological Basis for the Use of Evodiamine in Alzheimer’s Disease: Antioxidation and Antiapoptosis. International Journal of Molecular Sciences. 2018; 19(5):1527. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19051527
Chicago/Turabian StyleZhang, Yongfeng, Jiaqi Wang, Chunyue Wang, Zhiping Li, Xin Liu, Jun Zhang, Jiahui Lu, and Di Wang. 2018. "Pharmacological Basis for the Use of Evodiamine in Alzheimer’s Disease: Antioxidation and Antiapoptosis" International Journal of Molecular Sciences 19, no. 5: 1527. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19051527