Prognostic Value of RNASEH2A-, CDK1-, and CD151-Related Pathway Gene Profiling for Kidney Cancers

Abstract
:1. Background
2. Results
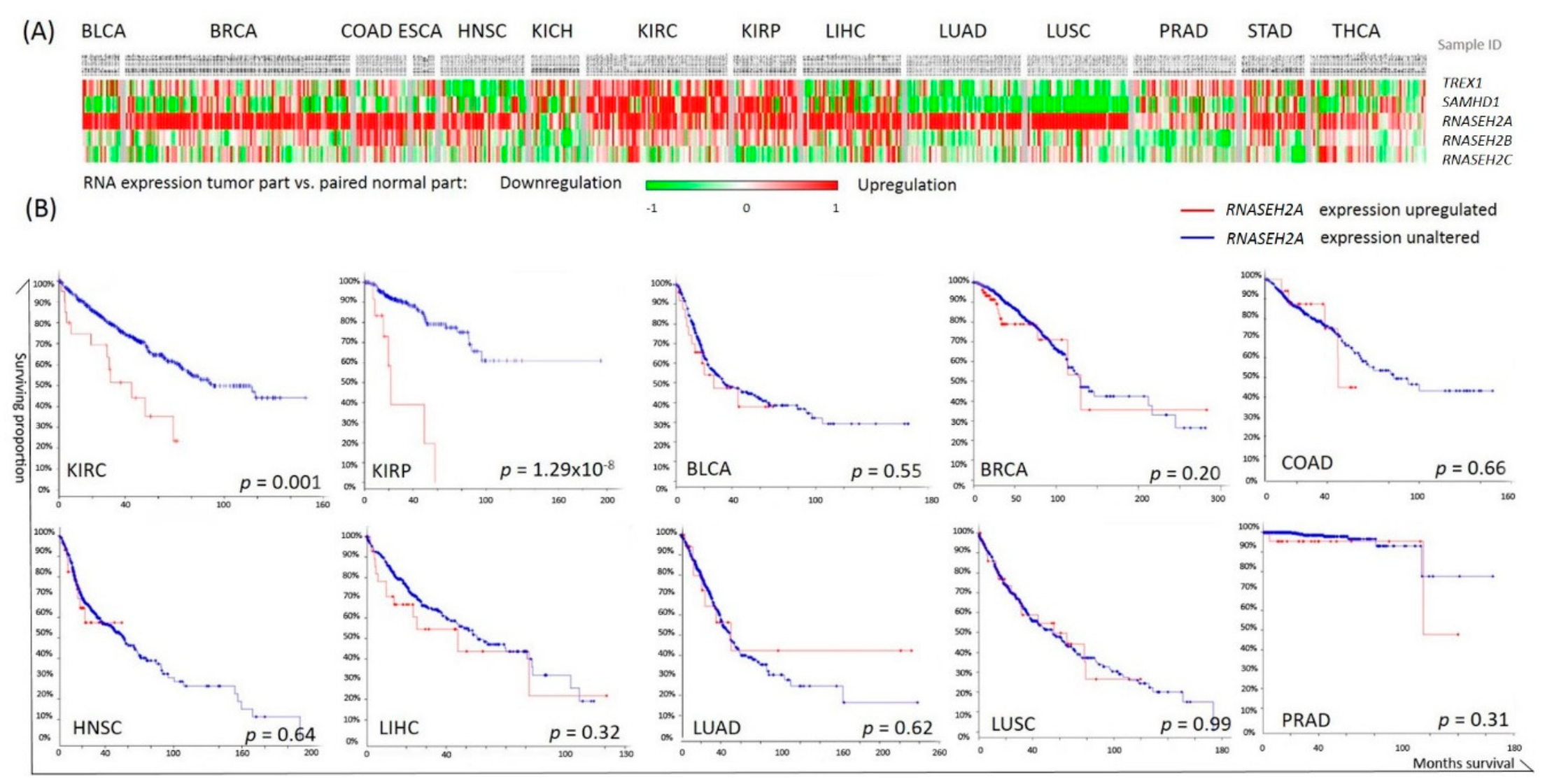
2.1. Pan-Cancer Analysis of Nucleotide Degrading Enzyme Genes
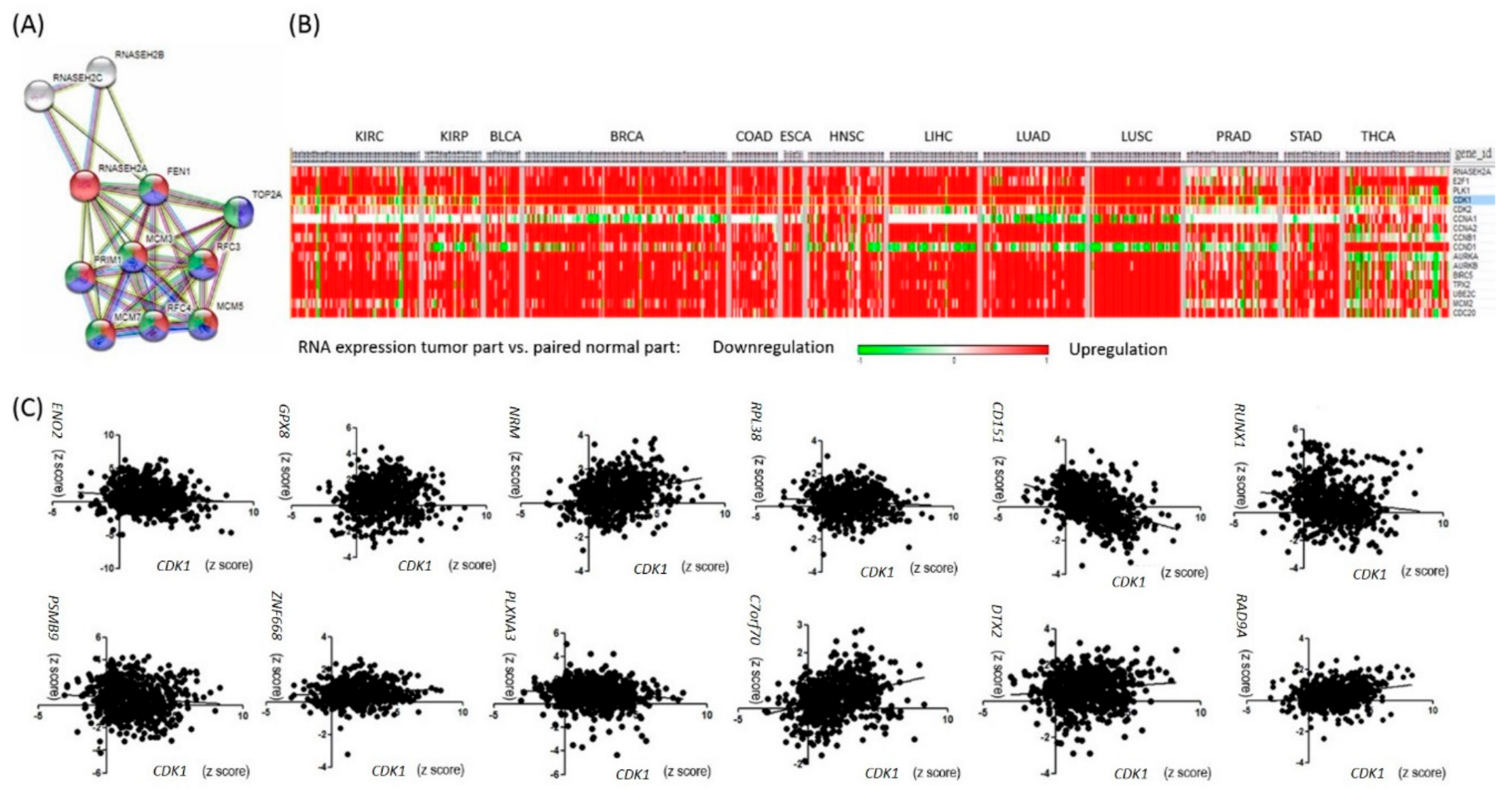
2.2. RNASEH2A Network Analysis and Identification of an Alternative Pathway Promoting Tumor Growth in CDK1-Low Tumors
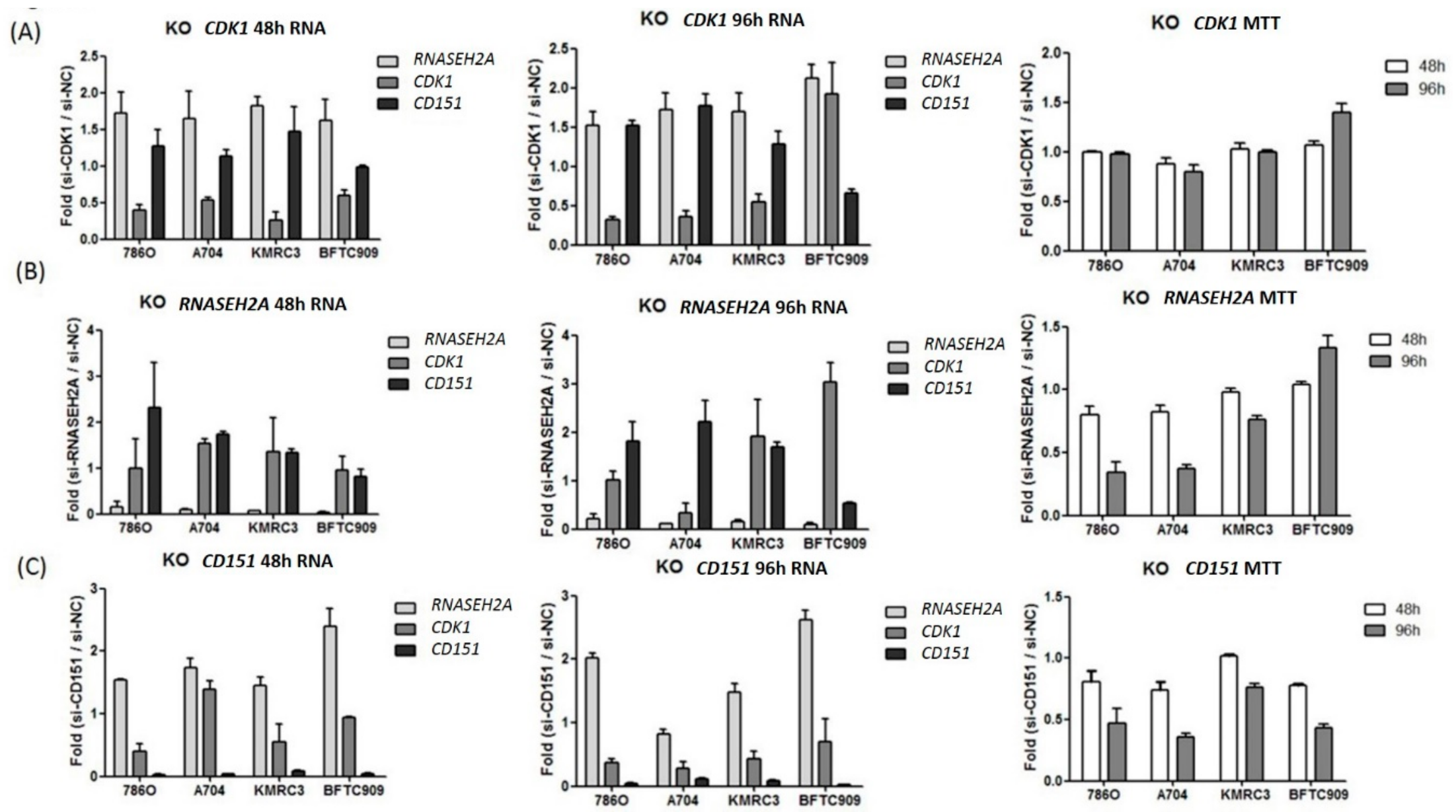
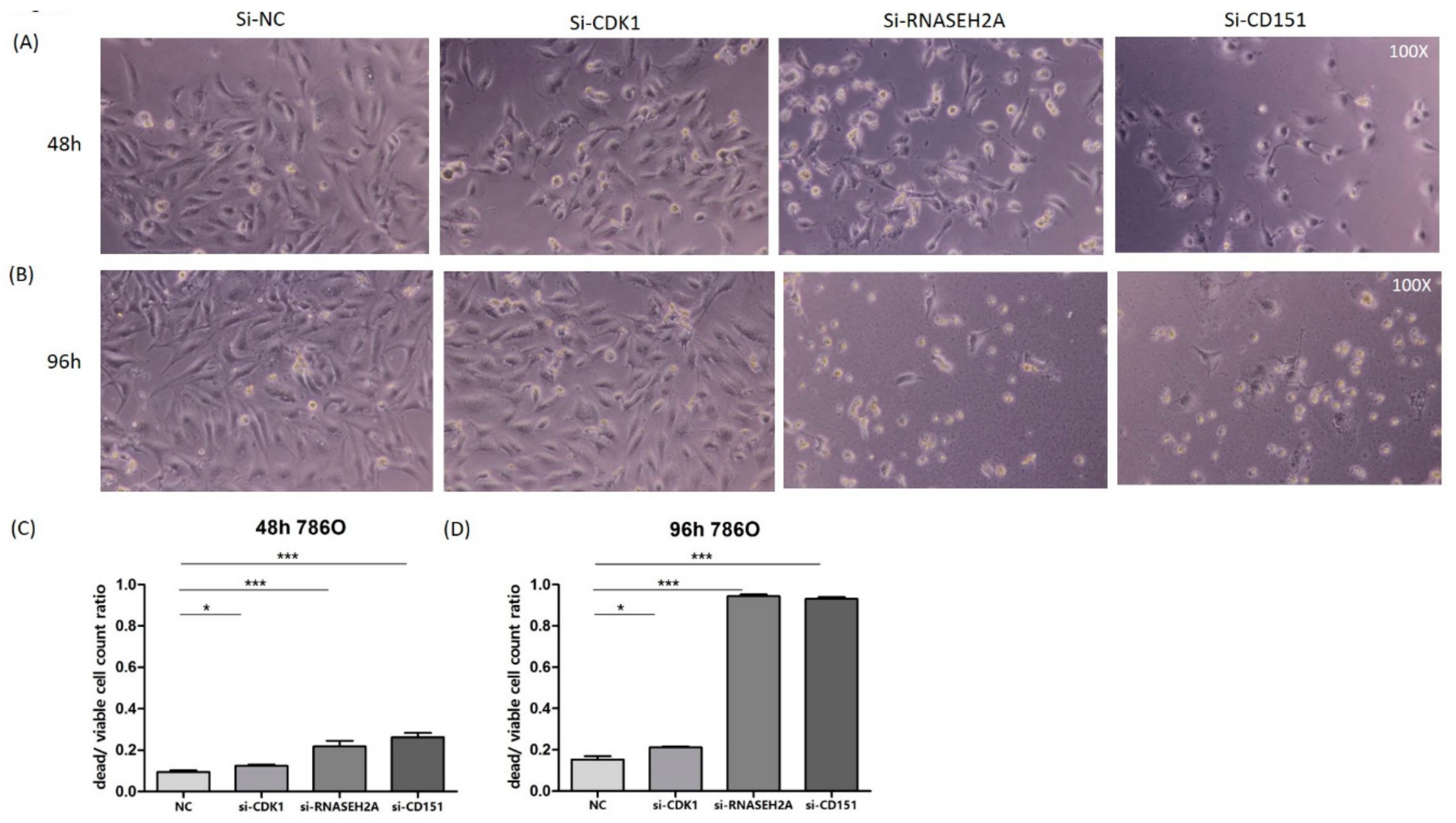
2.3. Analyses of the Interactions of RNASEH2A, CDK1, and CD151 Using Knockdown Studies in Four Kidney Cancer Cell Lines
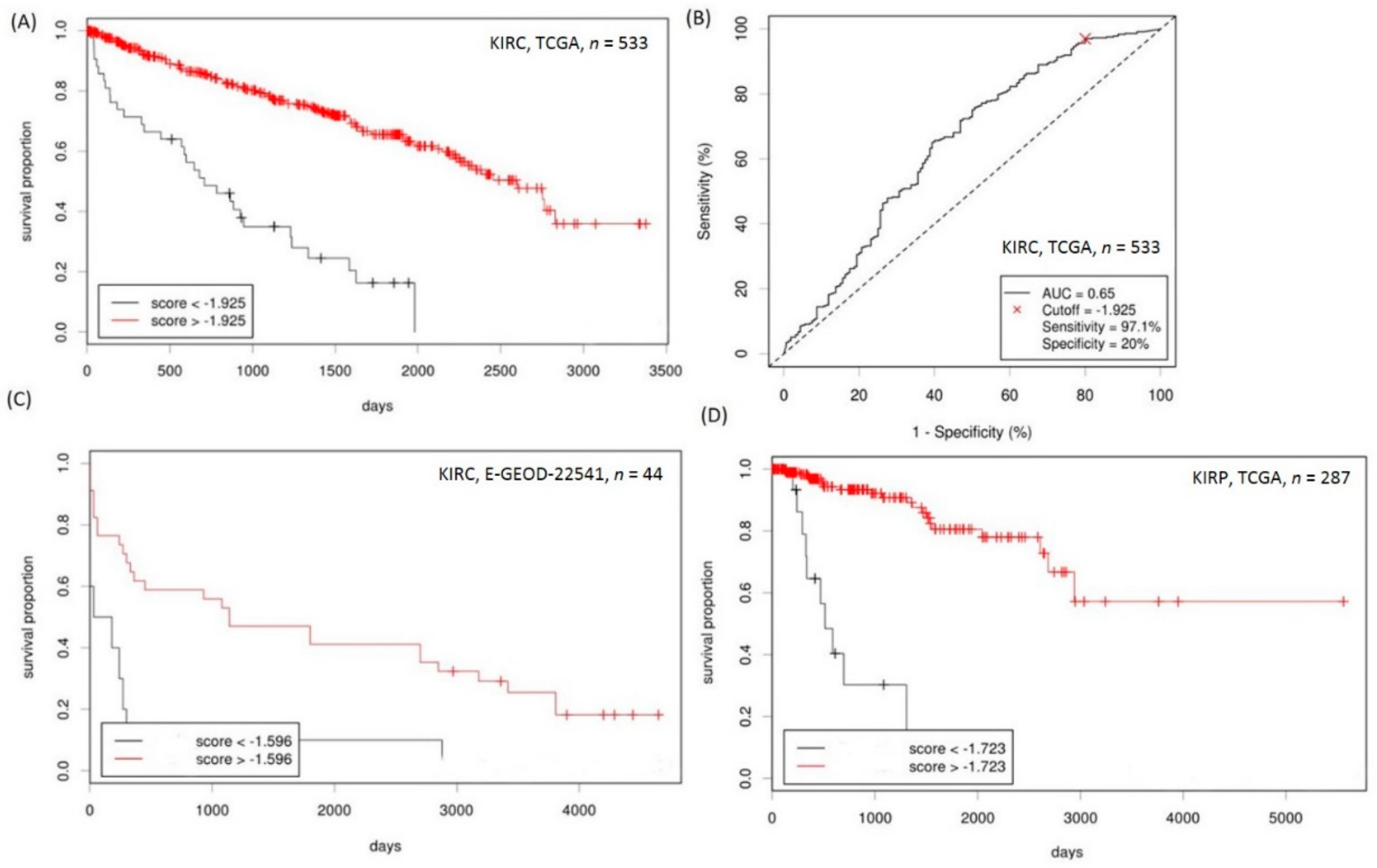
2.4. Analyses of the Prognostic Values of CD151-Related Genes in RCC
2.5. Establishment of a Prognostication Scoring System for Survival Prediction in RCC
3. Discussion
4. Methods
4.1. Study Populations
4.2. Cell Line Studies
4.3. RNA Isolation and Real-Time PCR Analyses
4.4. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Kohnken, R.; Kodigepalli, K.M.; Wu, L. Regulation of deoxynucleotide metabolism in cancer: Novel mechanisms and therapeutic implications. Mol. Cancer 2015, 14, 176. [Google Scholar] [CrossRef] [PubMed]
- Merati, M.; Buethe, D.J.; Cooper, K.D.; Honda, K.S.; Wang, H.; Gerstenblith, M.R. Aggressive CD8(+) epidermotropic cutaneous T-cell lymphoma associated with homozygous mutation in SAMHD1. JAAD Case Rep. 2015, 1, 227–229. [Google Scholar] [CrossRef] [PubMed]
- Volkman, H.E.; Stetson, D.B. The enemy within: Endogenous retroelements and autoimmune disease. Nat. Immunol. 2014, 15, 415–422. [Google Scholar] [CrossRef] [PubMed]
- Pokatayev, V.; Hasin, N.; Chon, H.; Cerritelli, S.M.; Sakhuja, K.; Ward, J.M.; Morris, H.D.; Yan, N.; Crouch, R.J. RNase H2 catalytic core Aicardi-Goutieres syndrome-related mutant invokes cGAS-STING innate immune-sensing pathway in mice. J. Exp. Med. 2016, 213, 329–336. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.A.; Huang, H.Y.; Chang, Y.S.; Lin, C.L.; Lai, I.L.; Chang, J.G. DNA-Sensing and Nuclease Gene Expressions as Markers for Colorectal Cancer Progression. Oncology 2016, 92, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; Cao, Z. Is the role of human RNase H2 restricted to its enzyme activity? Prog. Biophys. Mol. Biol. 2016, 121, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Sparks, J.L.; Chon, H.; Cerritelli, S.M.; Kunkel, T.A.; Johansson, E.; Crouch, R.J.; Burgers, P.M. RNase H2-initiated ribonucleotide excision repair. Mol. Cell 2012, 47, 980–986. [Google Scholar] [CrossRef] [PubMed]
- Williams, K.A.; Lee, M.; Hu, Y.; Andreas, J.; Patel, S.J.; Zhang, S.; Chines, P.; Elkahloun, A.; Chandrasekharappa, S.; Gutkind, J.S.; et al. A systems genetics approach identifies CXCL14, ITGAX, and LPCAT2 as novel aggressive prostate cancer susceptibility genes. PLoS Genet. 2014, 10, e1004809. [Google Scholar] [CrossRef] [PubMed]
- Megchelenbrink, W.; Katzir, R.; Lu, X.; Ruppin, E.; Notebaart, R.A. Synthetic dosage lethality in the human metabolic network is highly predictive of tumor growth and cancer patient survival. Proc. Natl. Acad. Sci. USA 2015, 112, 12217–12222. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Morris, J.H.; Cook, H.; Kuhn, M.; Wyder, S.; Simonovic, M.; Santos, A.; Doncheva, N.T.; Roth, A.; Bork, P.; et al. The STRING database in 2017: Quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res. 2017, 45, D362–D368. [Google Scholar] [CrossRef] [PubMed]
- Xi, Q.; Huang, M.; Wang, Y.; Zhong, J.; Liu, R.; Xu, G.; Jiang, L.; Wang, J.; Fang, Z.; Yang, S. The expression of CDK1 is associated with proliferation and can be a prognostic factor in epithelial ovarian cancer. Tumour Biol. 2015, 36, 4939–4948. [Google Scholar] [CrossRef] [PubMed]
- Hongo, F.; Takaha, N.; Oishi, M.; Ueda, T.; Nakamura, T.; Naitoh, Y.; Naya, Y.; Kamoi, K.; Okihara, K.; Matsushima, T.; et al. CDK1 and CDK2 activity is a strong predictor of renal cell carcinoma recurrence. Urol. Oncol. 2014, 32, 1240–1246. [Google Scholar] [CrossRef] [PubMed]
- Gan, W.; Zhao, H.; Li, T.; Liu, K.; Huang, J. CDK1 interacts with iASPP to regulate colorectal cancer cell proliferation through p53 pathway. Oncotarget 2017, 8, 71618–71629. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Masuda, N.; Tsukamoto, F.; Inaji, H.; Akiyama, F.; Sonoo, H.; Kurebayashi, J.; Yoshidome, K.; Tsujimoto, M.; Takei, H.; et al. The cell cycle profiling-risk score based on CDK1 and 2 predicts early recurrence in node-negative, hormone receptor-positive breast cancer treated with endocrine therapy. Cancer Lett. 2014, 355, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Asghar, U.; Witkiewicz, A.K.; Turner, N.C.; Knudsen, E.S. The history and future of targeting cyclin-dependent kinases in cancer therapy. Nat. Rev. Drug Discov. 2015, 14, 130–146. [Google Scholar] [CrossRef] [PubMed]
- Sadej, R.; Grudowska, A.; Turczyk, L.; Kordek, R.; Romanska, H.M. CD151 in cancer progression and metastasis: A complex scenario. Lab. Investig. 2014, 94, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Yoo, S.H.; Lee, K.; Chae, J.Y.; Moon, K.C. CD151 expression can predict cancer progression in clear cell renal cell carcinoma. Histopathology 2011, 58, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Juengel, E.; Maxeiner, S.; Rutz, J.; Justin, S.; Roos, F.; Khoder, W.; Tsaur, I.; Nelson, K.; Bechstein, W.O.; Haferkamp, A.; et al. Sulforaphane inhibits proliferation and invasive activity of everolimus-resistant kidney cancer cells in vitro. Oncotarget 2016, 7, 85208–85219. [Google Scholar] [CrossRef] [PubMed]
- Ridge, C.A.; Pua, B.B.; Madoff, D.C. Epidemiology and staging of renal cell carcinoma. Semin. Intervent. Radiol. 2014, 31, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Bacik, J.; Murphy, B.A.; Russo, P.; Mazumdar, M. Interferon-alfa as a comparative treatment for clinical trials of new therapies against advanced renal cell carcinoma. J. Clin. Oncol. 2002, 20, 289–296. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Park, W.S.; Park, E.Y.; Park, B.; Joo, J.; Joung, J.Y.; Seo, H.K.; Lee, K.H.; Chung, J. The prognostic value of BAP1, PBRM1, pS6, PTEN, TGase2, PD-L1, CA9, PSMA, and Ki-67 tissue markers in localized renal cell carcinoma: A retrospective study of tissue microarrays using immunohistochemistry. PLoS ONE 2017, 12, e0179610. [Google Scholar] [CrossRef] [PubMed]
- Giridhar, K.V.; Sosa, C.P.; Hillman, D.W.; Sanhueza, C.; Dalpiaz, C.L.; Costello, B.A.; Quevedo, F.J.; Pitot, H.C.; Dronca, R.S.; Ertz, D.; et al. Whole Blood mRNA Expression-Based Prognosis of Metastatic Renal Cell Carcinoma. Int. J. Mol. Sci. 2017, 18, 2326. [Google Scholar] [CrossRef] [PubMed]
- Wuttig, D.; Baier, B.; Fuessel, S.; Meinhardt, M.; Herr, A.; Hoefling, C.; Toma, M.; Grimm, M.O.; Meye, A.; Rolle, A.; et al. Gene signatures of pulmonary metastases of renal cell carcinoma reflect the disease-free interval and the number of metastases per patient. Int. J. Cancer 2009, 125, 474–482. [Google Scholar] [CrossRef] [PubMed]
- Budczies, J.; Klauschen, F.; Sinn, B.V.; Gyorffy, B.; Schmitt, W.D.; Darb-Esfahani, S.; Denkert, C. Cutoff Finder: A comprehensive and straightforward Web application enabling rapid biomarker cutoff optimization. PLoS ONE 2012, 7, e51862. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable | Group | Unadjusted HR (95% CI) | p Value | Adjusted HR (95% CI) | p Value |
|---|---|---|---|---|---|
| KIRC (n = 533) | |||||
| Age (per year) | 1.02 (1.02–1.04) | <0.0001 | - | - | |
| Sex | Male | 0.95 (0.69–1.31) | 0.77 | - | - |
| Female | 1 | ||||
| Stage | High (stage III–IV) | 4.28 (3.06–5.99) | <0.0001 | - | - |
| Low (stage I–II) | 1 | ||||
| Distant metastasis | M1 | 4.59 (3.32–6.33) | <0.0001 | - | - |
| M0 | 1 | ||||
| Fuhrman grade | High (Gr 3–4) | 2.85 (1.98–4.11) | <0.0001 | - | - |
| Low (Gr 1–2) | 1 | ||||
| Prediction score | Low (<−1.925) | 4.53 (3.05–6.73) | <0.0001 | 2.68 (1.75–4.11) | <0.0001 |
| High (≥−1.925) | 1 | ||||
| KIRP (n = 287) | |||||
| Age (per year) | 1.01 (0.98–1.04) | 0.56 | - | - | |
| Sex | Male | 0.72 (0.32–1.62) | 0.43 | - | - |
| Female | 1 | ||||
| Stage | High (stage (III–IV) | 4.86 (2.22–10.66) | <0.0001 | - | - |
| Low (stage I–II) | 1 | ||||
| Prediction score | Low (<−1.723) | 13.19 (6.03–28.83) | <0.0001 | 7.44 (3.01–18.41) | <0.0001 |
| High (≥−1.723) | 1 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, C.-A.; Huang, H.-Y.; Yen, J.-C.; Chang, J.-G. Prognostic Value of RNASEH2A-, CDK1-, and CD151-Related Pathway Gene Profiling for Kidney Cancers. Int. J. Mol. Sci. 2018, 19, 1586. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19061586
Yang C-A, Huang H-Y, Yen J-C, Chang J-G. Prognostic Value of RNASEH2A-, CDK1-, and CD151-Related Pathway Gene Profiling for Kidney Cancers. International Journal of Molecular Sciences. 2018; 19(6):1586. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19061586
Chicago/Turabian StyleYang, Chin-An, Hsi-Yuan Huang, Ju-Chen Yen, and Jan-Gowth Chang. 2018. "Prognostic Value of RNASEH2A-, CDK1-, and CD151-Related Pathway Gene Profiling for Kidney Cancers" International Journal of Molecular Sciences 19, no. 6: 1586. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19061586