Fewer Functional Deficits and Reduced Cell Death after Ranibizumab Treatment in a Retinal Ischemia Model


Abstract
:1. Introduction
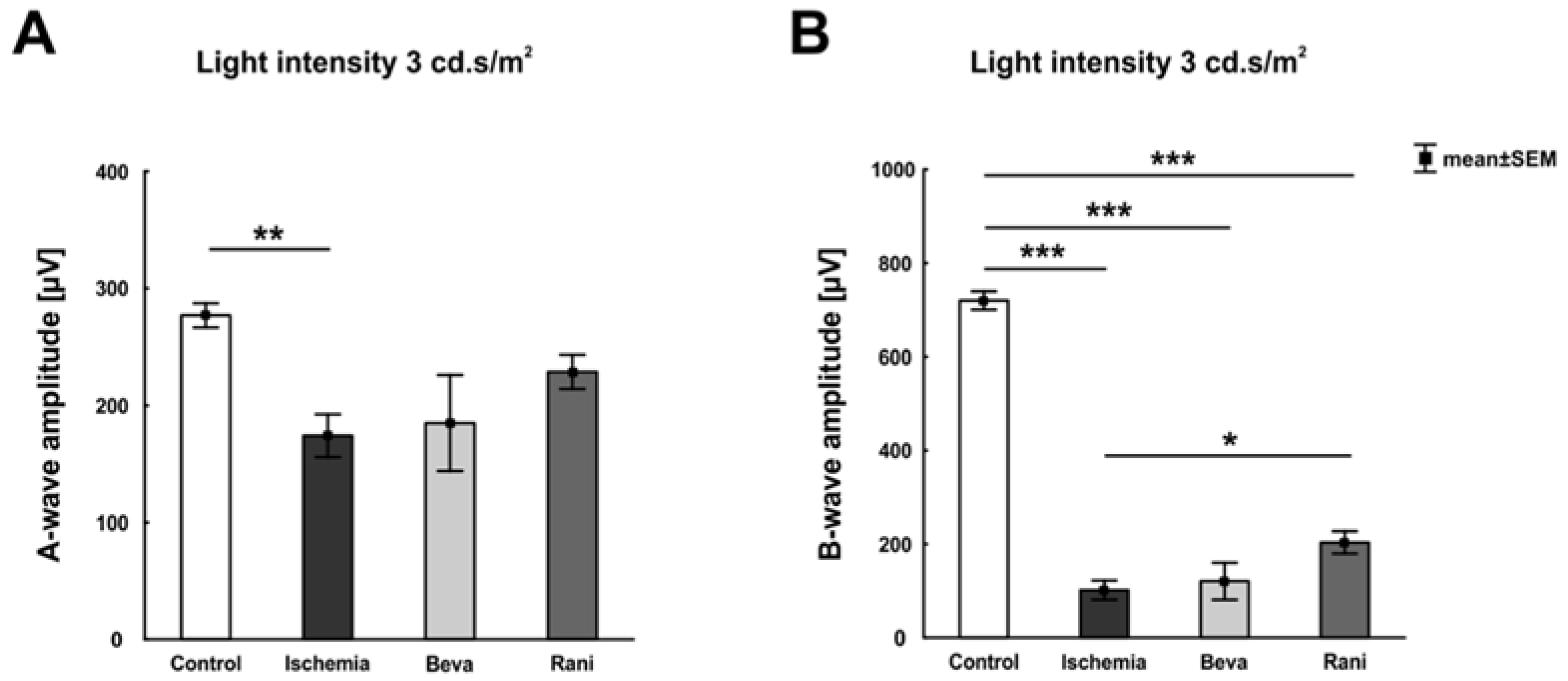
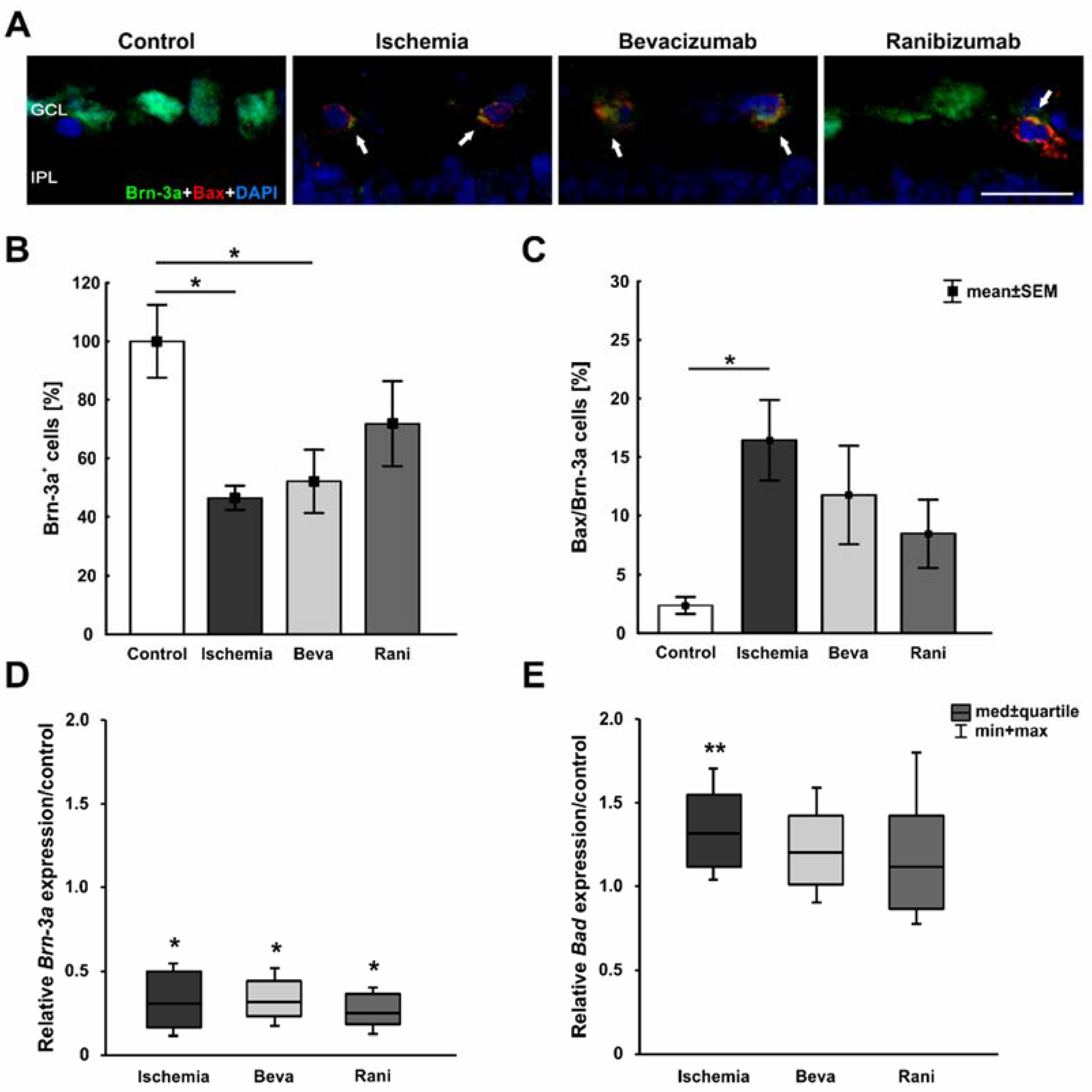
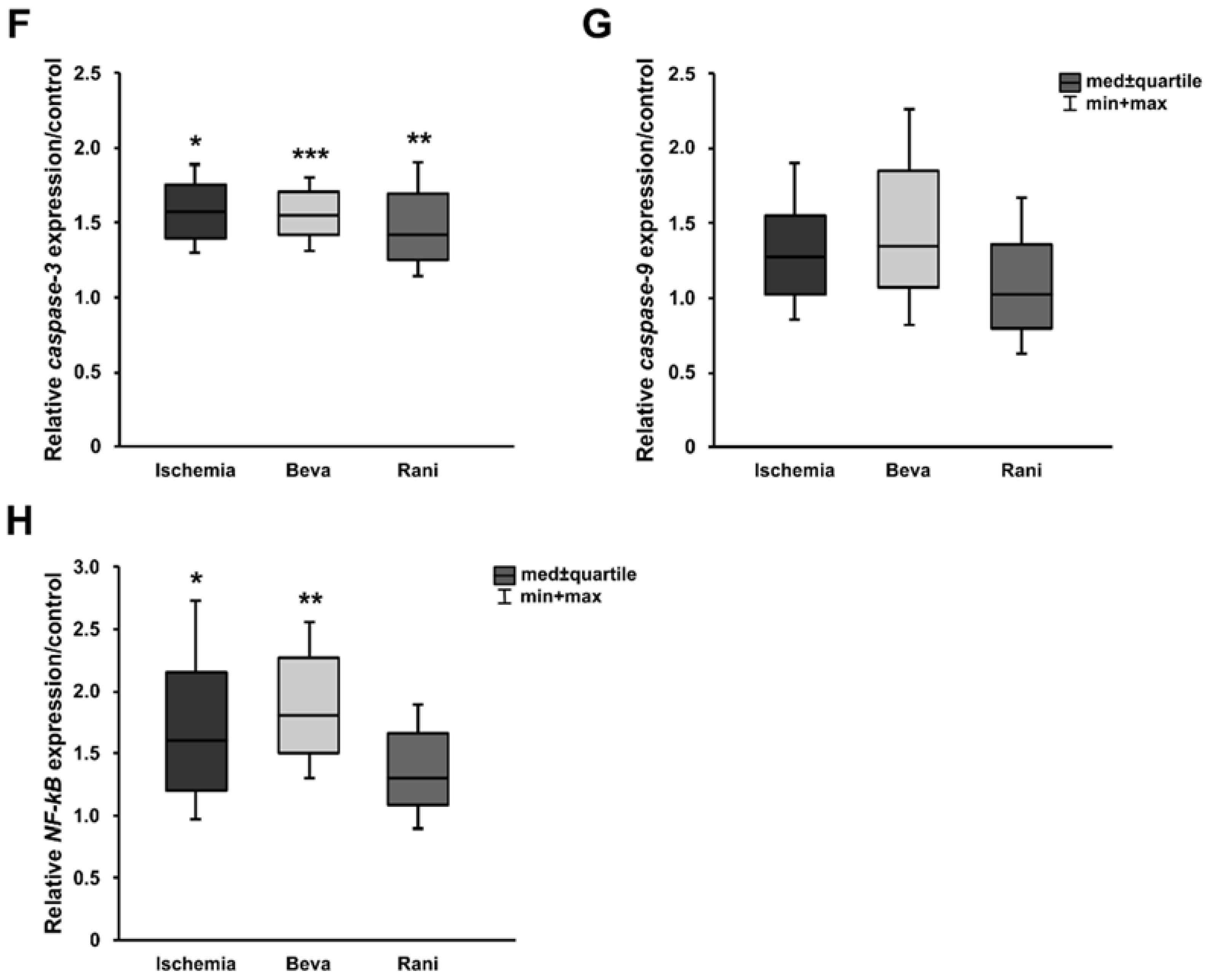
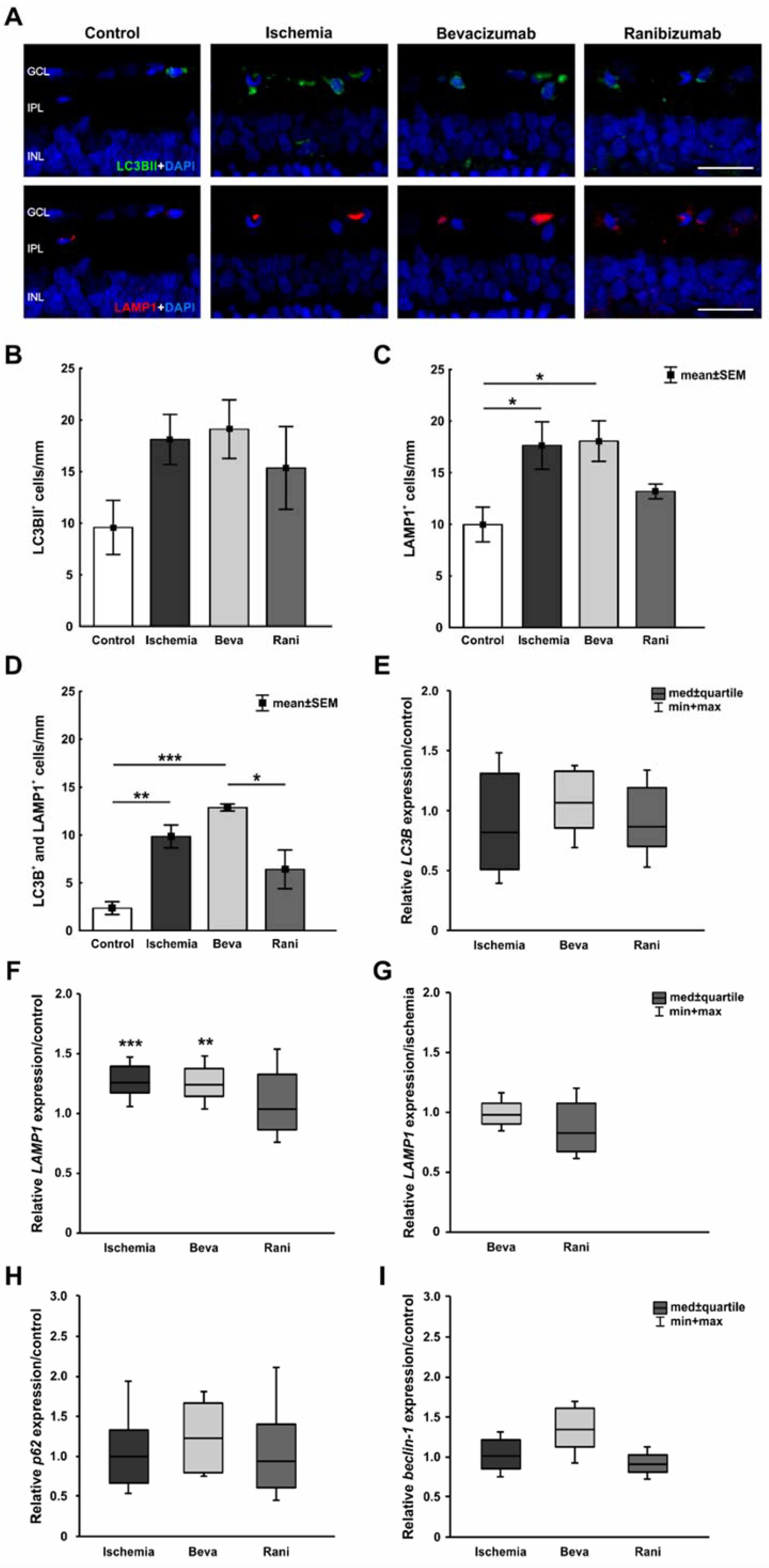
2. Results
3. Discussion
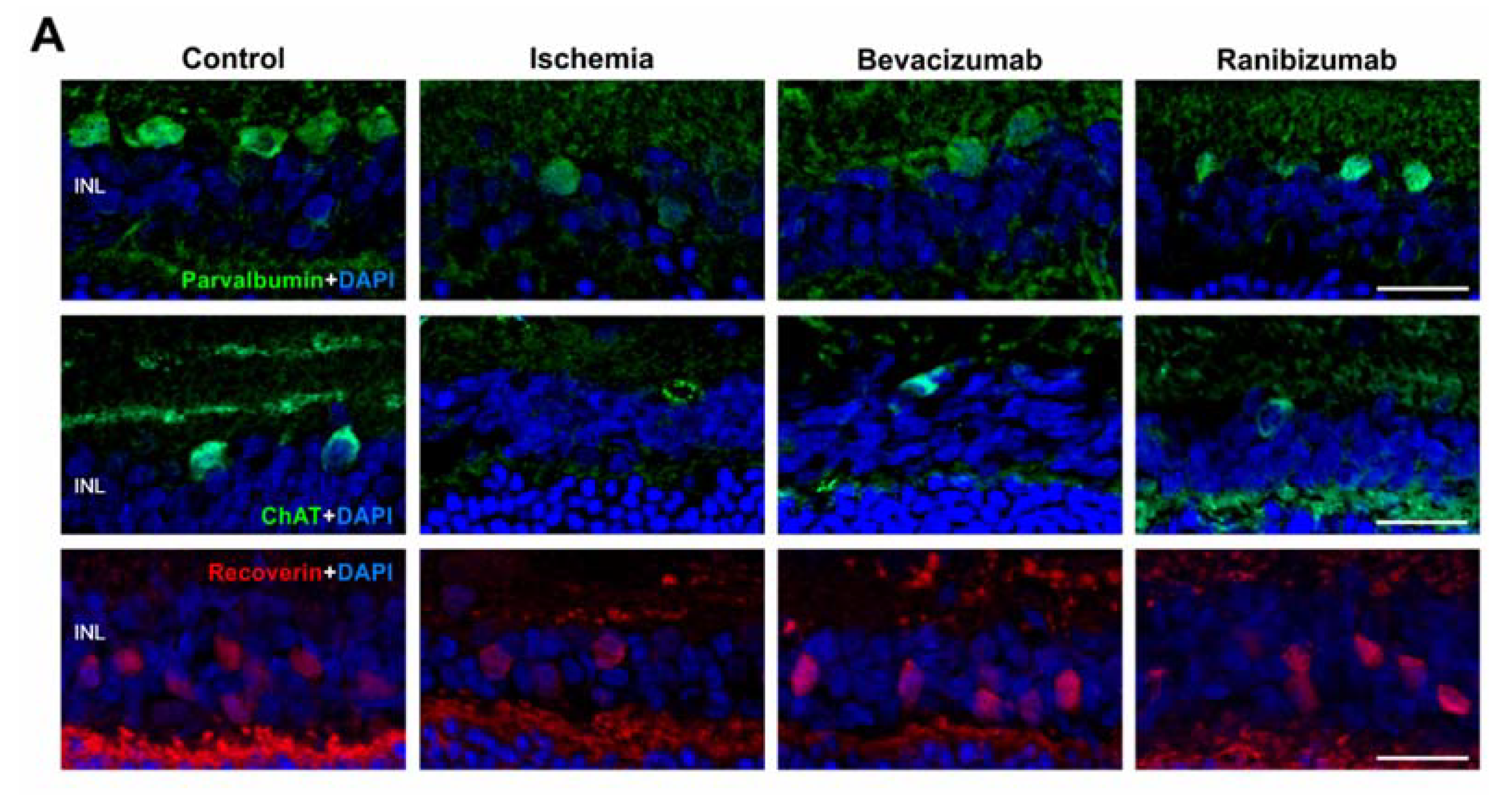
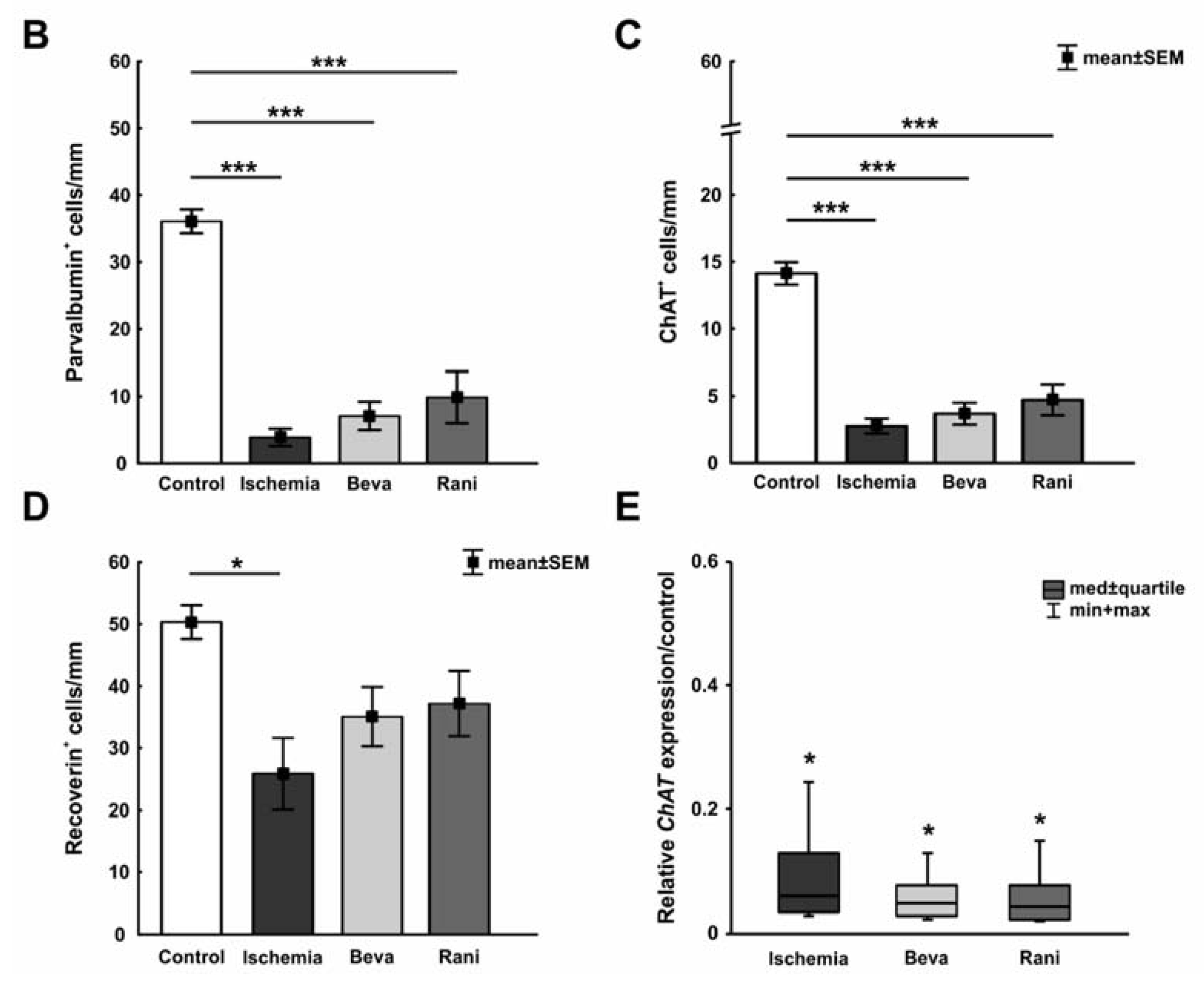
3.1. Certain Preservation of the Neuronal Function
3.2. Some Prevention of Cell Death and Loss of RGCs after Ranibizumab Treatment
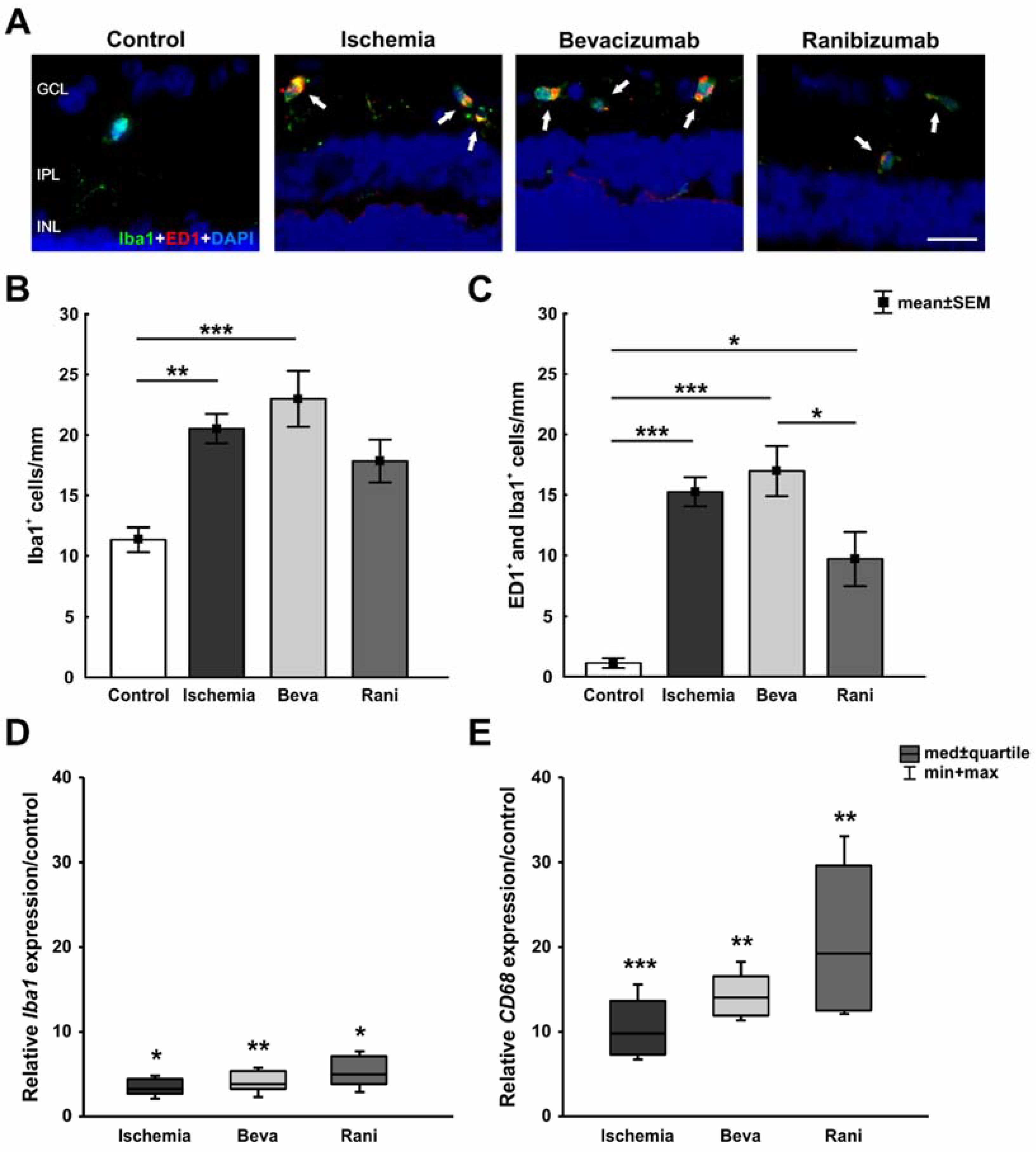
3.3. Positive Impact of the Anti-VEGF Therapy on the Microglia and Cone Bipolar Cells
4. Materials and Methods
4.1. Animals
4.2. Induction of Ischemia/Reperfusion
4.3. Treatment with Bevacizumab and Ranibizumab
4.4. Electroretinogram Measurements
4.5. Tissue Collection and Preparation
4.6. Immunohistology of Retinal Sections
4.7. Quantitative Real-Time PCR Analysis of Retinal Tissue
4.8. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Conflicts of Interest
References
- Cai, M.; Zhang, X.; Li, Y.; Xu, H. Toll-like receptor 3 activation drives the inflammatory response in oxygen-induced retinopathy in rats. Br. J. Ophthalmol. 2015, 99, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.J.; Braun, T.A.; Wordinger, R.J.; Clark, A.F. Progressive morphological changes and impaired retinal function associated with temporal regulation of gene expression after retinal ischemia/reperfusion injury in mice. Mol. Neurodegener. 2013, 8, 21. [Google Scholar] [CrossRef] [PubMed]
- Bek, T. Inner retinal Ischaemia: Current understanding and needs for further investigations. Acta Ophthalmol. 2009, 87, 362–367. [Google Scholar] [CrossRef] [PubMed]
- Hardy, P.; Beauchamp, M.; Sennlaub, F.; Gobeil, F., Jr.; Tremblay, L.; Mwaikambo, B.; Lachapelle, P.; Chemtob, S. New insights into the retinal circulation: Inflammatory lipid mediators in ischemic retinopathy. Prostaglandins Leukot. Essent. Fat. Acids 2005, 72, 301–325. [Google Scholar] [CrossRef] [PubMed]
- Minhas, G.; Morishita, R.; Anand, A. Preclinical models to investigate retinal ischemia: Advances and drawbacks. Front. Neurol. 2012, 3, 75. [Google Scholar] [CrossRef] [PubMed]
- Kaur, C.; Foulds, W.S.; Ling, E.A. Hypoxia-ischemia and retinal ganglion cell damage. Clin. Ophthalmol. 2008, 2, 879–889. [Google Scholar] [CrossRef] [PubMed]
- Osborne, N.N.; Casson, R.J.; Wood, J.P.; Chidlow, G.; Graham, M.; Melena, J. Retinal ischemia: Mechanisms of damage and potential therapeutic strategies. Prog. Retin. Eye Res. 2004, 23, 91–147. [Google Scholar] [CrossRef] [PubMed]
- Belforte, N.; Sande, P.H.; de Zavalia, N.; Fernandez, D.C.; Silberman, D.M.; Chianelli, M.S.; Rosenstein, R.E. Ischemic tolerance protects the rat retina from glaucomatous damage. PLoS ONE 2011, 6, e23763. [Google Scholar] [CrossRef] [PubMed]
- Dijk, F.; Kraal-Muller, E.; Kamphuis, W. Ischemia-induced changes of AMPA-type glutamate receptor subunit expression pattern in the rat retina: A real-time quantitative pcr study. Investig. Ophthalmol. Vis. Sci. 2004, 45, 330–341. [Google Scholar] [CrossRef]
- Joachim, S.C.; Jehle, T.; Boehm, N.; Gramlich, O.W.; Lagreze, W.A.; Pfeiffer, N.; Grus, F.H. Effect of ischemia duration on autoantibody response in rats undergoing retinal ischemia-reperfusion. Ophthalmic Res. 2012, 48, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Schmid, H.; Renner, M.; Dick, H.B.; Joachim, S.C. Loss of inner retinal neurons after retinal ischemia in rats. Investig. Ophthalmol. Vis. Sci. 2014, 55, 2777–2787. [Google Scholar] [CrossRef] [PubMed]
- Szabo, M.E.; Droy-Lefaix, M.T.; Doly, M.; Braquet, P. Free radical-mediated effects in reperfusion injury: A histologic study with superoxide dismutase and egb 761 in rat retina. Ophthalmic Res. 1991, 23, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Tsujikawa, A.; Ogura, Y.; Hiroshiba, N.; Miyamoto, K.; Kiryu, J.; Tojo, S.J.; Miyasaka, M.; Honda, Y. Retinal ischemia-reperfusion injury attenuated by blocking of adhesion molecules of vascular endothelium. Investig. Ophthalmol. Vis. Sci. 1999, 40, 1183–1190. [Google Scholar]
- Lam, T.T.; Abler, A.S.; Tso, M.O. Apoptosis and caspases after ischemia-reperfusion injury in rat retina. Investig. Ophthalmol. Vis. Sci. 1999, 40, 967–975. [Google Scholar]
- Zheng, G.Y.; Zhang, C.; Li, Z.G. Early activation of caspase-1 after retinal ischemia and reperfusion injury in mice. Chin. Med. J. 2004, 117, 717–721. [Google Scholar] [PubMed]
- Fortes Filho, J.B.; Maia, M.; Tartarella, M.B.; Meyer, F.S.; Fortes, B.G.; Kliemann, L.M. Experimental histopathological study on retinal and renal cellular response to intravitreous antiangiogenic drugs. Int. J. Ophthalmol. 2014, 7, 437–440. [Google Scholar] [PubMed]
- Pavlidis, E.T.; Pavlidis, T.E. Role of bevacizumab in colorectal cancer growth and its adverse effects: A review. World J. Gastroenterol. 2013, 19, 5051–5060. [Google Scholar] [CrossRef] [PubMed]
- Rosenfeld, P.J.; Moshfeghi, A.A.; Puliafito, C.A. Optical coherence tomography findings after an intravitreal injection of bevacizumab (avastin) for neovascular age-related macular degeneration. Ophthalmic Surg. Lasers Imaging 2005, 36, 331–335. [Google Scholar] [PubMed]
- Schmucker, C.; Ehlken, C.; Agostini, H.T.; Antes, G.; Ruecker, G.; Lelgemann, M.; Loke, Y.K. A safety review and meta-analyses of bevacizumab and ranibizumab: Off-label versus goldstandard. PLoS ONE 2012, 7, e42701. [Google Scholar] [CrossRef] [PubMed]
- Nadal-Nicolas, F.M.; Jimenez-Lopez, M.; Salinas-Navarro, M.; Sobrado-Calvo, P.; Alburquerque-Bejar, J.J.; Vidal-Sanz, M.; Agudo-Barriuso, M. Whole number, distribution and co-expression of brn3 transcription factors in retinal ganglion cells of adult albino and pigmented rats. PLoS ONE 2012, 7, e49830. [Google Scholar] [CrossRef] [PubMed]
- Cervia, D.; Catalani, E.; Dal Monte, M.; Casini, G. Vascular endothelial growth factor in the ischemic retina and its regulation by somatostatin. J. Neurochem. 2012, 120, 818–829. [Google Scholar] [CrossRef] [PubMed]
- Bates, D.O.; Jones, R.O. The role of vascular endothelial growth factor in wound healing. Int. J. Low Extremity Wounds 2003, 2, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Abcouwer, S.F.; Lin, C.M.; Wolpert, E.B.; Shanmugam, S.; Schaefer, E.W.; Freeman, W.M.; Barber, A.J.; Antonetti, D.A. Effects of ischemic preconditioning and bevacizumab on apoptosis and vascular permeability following retinal ischemia-reperfusion injury. Investig. Ophthalmol. Vis. Sci. 2010, 51, 5920–5933. [Google Scholar] [CrossRef] [PubMed]
- Funk, M.; Karl, D.; Georgopoulos, M.; Benesch, T.; Sacu, S.; Polak, K.; Zlabinger, G.J.; Schmidt-Erfurth, U. Neovascular age-related macular degeneration: Intraocular cytokines and growth factors and the influence of therapy with ranibizumab. Ophthalmology 2009, 116, 2393–2399. [Google Scholar] [CrossRef] [PubMed]
- Funatsu, H.; Yamashita, H.; Ikeda, T.; Mimura, T.; Eguchi, S.; Hori, S. Vitreous levels of interleukin-6 and vascular endothelial growth factor are related to diabetic macular edema. Ophthalmology 2003, 110, 1690–1696. [Google Scholar] [CrossRef]
- Ehlken, C.; Rennel, E.S.; Michels, D.; Grundel, B.; Pielen, A.; Junker, B.; Stahl, A.; Hansen, L.L.; Feltgen, N.; Agostini, H.T.; et al. Levels of vegf but not VEGF(165b) are increased in the vitreous of patients with retinal vein occlusion. Am. J. Ophthalmol. 2011, 152, 298–303. [Google Scholar] [CrossRef] [PubMed]
- Campochiaro, P.A.; Channa, R.; Berger, B.B.; Heier, J.S.; Brown, D.M.; Fiedler, U.; Hepp, J.; Stumpp, M.T. Treatment of diabetic macular edema with a designed ankyrin repeat protein that binds vascular endothelial growth factor: A phase I/II study. Am. J. Ophthalmol. 2013, 155, 697–704. [Google Scholar] [CrossRef] [PubMed]
- Thaler, S.; Fiedorowicz, M.; Choragiewicz, T.J.; Bolz, S.; Tura, A.; Henke-Fahle, S.; Yoeruek, E.; Zrenner, E.; Bartz-Schmidt, K.U.; Ziemssen, F.; et al. Toxicity testing of the vegf inhibitors bevacizumab, ranibizumab and pegaptanib in rats both with and without prior retinal ganglion cell damage. Acta Ophthalmol. 2010, 88, e170–e176. [Google Scholar] [CrossRef] [PubMed]
- Saenz-de-Viteri, M.; Fernandez-Robredo, P.; Hernandez, M.; Bezunartea, J.; Reiter, N.; Recalde, S.; Garcia-Layana, A. Single- and repeated-dose toxicity study of bevacizumab, ranibizumab, and aflibercept in arpe-19 cells under normal and oxidative stress conditions. Biochem. Pharmacol. 2016, 103, 129–139. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P.; Ruiz de Almodovar, C. VEGF ligands and receptors: Implications in neurodevelopment and neurodegeneration. Cell. Mol. Life Sci. 2013, 70, 1763–1778. [Google Scholar] [CrossRef] [PubMed]
- Iwona, B.S. Growth factors in the pathogenesis of retinal neurodegeneration in diabetes mellitus. Curr. Neuropharmacol. 2016, 14, 792–804. [Google Scholar] [CrossRef] [PubMed]
- Famiglietti, E.V.; Stopa, E.G.; McGookin, E.D.; Song, P.; LeBlanc, V.; Streeten, B.W. Immunocytochemical localization of vascular endothelial growth factor in neurons and glial cells of human retina. Brain Res. 2003, 969, 195–204. [Google Scholar] [CrossRef]
- Lee, I.; Lee, H.; Kim, J.M.; Chae, E.H.; Kim, S.J.; Chang, N. Short-term hyperhomocysteinemia-induced oxidative stress activates retinal glial cells and increases vascular endothelial growth factor expression in rat retina. Biosci. Biotechnol. Biochem. 2007, 71, 1203–1210. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.F.; Maguire, M.G.; Ying, G.S.; Grunwald, J.E.; Fine, S.L.; Jaffe, G.J. Ranibizumab and bevacizumab for neovascular age-related macular degeneration. N. Engl. J. Med. 2011, 364, 1897–1908. [Google Scholar] [CrossRef] [PubMed]
- Chakravarthy, U.; Harding, S.P.; Rogers, C.A.; Downes, S.M.; Lotery, A.J.; Wordsworth, S.; Reeves, B.C. Ranibizumab versus bevacizumab to treat neovascular age-related macular degeneration: One-year findings from the ivan randomized trial. Ophthalmology 2012, 119, 1399–1411. [Google Scholar] [CrossRef] [PubMed]
- Saint-Geniez, M.; Maharaj, A.S.; Walshe, T.E.; Tucker, B.A.; Sekiyama, E.; Kurihara, T.; Darland, D.C.; Young, M.J.; D’Amore, P.A. Endogenous vegf is required for visual function: Evidence for a survival role on muller cells and photoreceptors. PLoS ONE 2008, 3, e3554. [Google Scholar] [CrossRef] [PubMed]
- Myers, A.C.; Lovestam Adrian, M.; Bruun, A.; Ghosh, F.; Andreasson, S.; Ponjavic, V. Retinal function and morphology in rabbit after intravitreal injection of VEGF inhibitors. Curr. Eye Res. 2012, 37, 399–407. [Google Scholar] [CrossRef] [PubMed]
- Joachim, S.C.; Renner, M.; Reinhard, J.; Theiss, C.; May, C.; Lohmann, S.; Reinehr, S.; Stute, G.; Faissner, A.; Marcus, K.; et al. Protective effects on the retina after ranibizumab treatment in an ischemia model. PLoS ONE 2017, 12, e0182407. [Google Scholar] [CrossRef] [PubMed]
- Adachi, M.; Takahashi, K.; Nishikawa, M.; Miki, H.; Uyama, M. High intraocular pressure-induced ischemia and reperfusion injury in the optic nerve and retina in rats. Graefes Arch. Clin. Exp. Ophthalmol. 1996, 234, 445–451. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Gong, B.; Hatala, D.A.; Kern, T.S. Retinal ischemia and reperfusion causes capillary degeneration: Similarities to diabetes. Investig. Ophthalmol. Vis. Sci. 2007, 48, 361–367. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Li, C.; Guo, H.; Kern, T.S.; Huang, K.; Zheng, L. Curcumin inhibits neuronal and vascular degeneration in retina after ischemia and reperfusion injury. PLoS ONE 2011, 6, e23194. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Klionsky, D.J. Eaten alive: A history of macroautophagy. Nat. Cell Biol. 2010, 12, 814–822. [Google Scholar] [CrossRef] [PubMed]
- Marino, G.; Niso-Santano, M.; Baehrecke, E.H.; Kroemer, G. Self-consumption: The interplay of autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 2014, 15, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Wei, T.; Kang, Q.; Ma, B.; Gao, S.; Li, X.; Liu, Y. Activation of autophagy and paraptosis in retinal ganglion cells after retinal ischemia and reperfusion injury in rats. Exp. Ther. Med. 2015, 9, 476–482. [Google Scholar] [CrossRef] [PubMed]
- Piras, A.; Gianetto, D.; Conte, D.; Bosone, A.; Vercelli, A. Activation of autophagy in a rat model of retinal ischemia following high intraocular pressure. PLoS ONE 2011, 6, e22514. [Google Scholar] [CrossRef] [PubMed]
- Kornek, B.; Storch, M.K.; Weissert, R.; Wallstroem, E.; Stefferl, A.; Olsson, T.; Linington, C.; Schmidbauer, M.; Lassmann, H. Multiple sclerosis and chronic autoimmune encephalomyelitis: A comparative quantitative study of axonal injury in active, inactive, and remyelinated lesions. Am. J. Pathol. 2000, 157, 267–276. [Google Scholar] [CrossRef]
- Langmann, T. Microglia activation in retinal degeneration. J. Leukoc. Biol. 2007, 81, 1345–1351. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Parlier, R.; Shen, J.K.; Lutty, G.A.; Vinores, S.A. VEGF receptor blockade markedly reduces retinal microglia/macrophage infiltration into laser-induced cnv. PLoS ONE 2013, 8, e71808. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Yu, B.; Xiang, Y.H.; Han, X.J.; Xu, Y.; So, K.F.; Xu, A.D.; Ruan, Y.W. Changes in retinal morphology, electroretinogram and visual behavior after transient global ischemia in adult rats. PLoS ONE 2013, 8, e65555. [Google Scholar] [CrossRef] [PubMed]
- Renner, M.; Stute, G.; Alzureiqi, M.; Reinhard, J.; Wiemann, S.; Schmid, H.; Faissner, A.; Dick, H.B.; Joachim, S.C. Optic nerve degeneration after retinal ischemia/reperfusion in a rodent model. Front. Cell. Neurosci. 2017, 11, 254. [Google Scholar] [CrossRef] [PubMed]
- Reinehr, S.; Kuehn, S.; Casola, C.; Koch, D.; Stute, G.; Grotegut, P.; Dick, H.B.; Joachim, S.C. Hsp27 immunization reinforces AII amacrine cell and synapse damage induced by s100 in an autoimmune glaucoma model. Cell Tissue Res. 2018, 371, 237–249. [Google Scholar] [CrossRef] [PubMed]
- Kuehn, S.; Rodust, C.; Stute, G.; Grotegut, P.; Meissner, W.; Reinehr, S.; Dick, H.B.; Joachim, S.C. Concentration-dependent inner retina layer damage and optic nerve degeneration in a nmda model. J. Mol. Neurosci. 2017, 63, 283–299. [Google Scholar] [CrossRef] [PubMed]
- Reinehr, S.; Reinhard, J.; Wiemann, S.; Stute, G.; Kuehn, S.; Woestmann, J.; Dick, H.B.; Faissner, A.; Joachim, S.C. Early remodelling of the extracellular matrix proteins tenascin-c and phosphacan in retina and optic nerve of an experimental autoimmune glaucoma model. J. Cell. Mol. Med. 2016, 20, 2122–2137. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Type/Mechanism | Primary Antibody with Dilution | Company | Secondary Anibody with Dilution | Company |
|---|---|---|---|---|
| RGCs | Anti-Brn-3a, 1:100 | Santa Cruz Biotechnology, Heidelberg, Germany | Alexa 488, 1:500 | Dianova, Hamburg, Germany |
| Apoptotic cells | Anti-Bax, 1:100 | Abcam, Cambridge, UK | Alexa 555, 1:400 | Invitrogen, Darmstadt, Germany |
| Early autophagy | Anti-LC3BII, 1:100 | Cell Signaling, Danvers, MA, USA | Alexa 488, 1:500 | Invitrogen |
| Late autophagy | Anti-LAMP1, 1:100 | Abcam | Alexa 555, 1:400 | Invitrogen |
| Microglia | Anti-Iba1, 1:400 | Wako Chemicals, Neuss, Germany | Alexa 488, 1:500 | Invitrogen |
| Activated microglia and macrophages | Anti-ED1, 1:200 | Millipore, Darmstadt, Germany | Alexa 555, 1:500 | Invitrogen |
| Amacrine cells | Anti-parvalbumin, 1:100 | Swant, Marly, Switzerland | Alexa 488, 1:500 | Invitrogen |
| Cholinergic amacrine cells | Anti-ChAT, 1:250 | Millipore | Alexa 488, 1:500 | Jackson Immuno Research, Cambridgeshire, UK |
| Cone bipolar cells | Anti-recoverin, 1:1000 | Millipore | Alexa 555, 1:400 | Invitrogen |
| Gene | Primer Sequence (5′–3′) | Amplicon Size | Primer Efficiency |
|---|---|---|---|
| β-Actin-F | cccgcgagtacaaccttct | 72 bp | 1.000 |
| β-Actin-R | cgtcatccatggcgaact | ||
| Cyclophilin-F | tgctggaccaaacacaaatg | 88 bp | 1.000 |
| Cyclophilin-R | cttcccaaagaccacatgct | ||
| Bad-F | caggcagccaataacagtca | 68 bp | 1.000 |
| Bad-R | tacgaactgtggcgactcc | ||
| Beclin-1-F | aggatggtgtctctcgaagatt | 77 bp | 0.969 |
| Beclin-1-R | gatcagagtgaagctattagcactttc | ||
| Brn-3a (Pou4f1)-F | ctggccaacctcaagatcc | 72 bp | 1.000 |
| Brn-3a (Pou4f1)-R | cgtgagcgactcgaacct | ||
| Caspase-3-F | ccgacttcctgtatgcttactcta | 70 bp | 1.000 |
| Caspase-3-R | catgacccgtcccttgaa | ||
| Caspase-9-F | cgtggtggtcatcctctctc | 81 bp | 1.000 |
| Caspase-9-R | gagcatccatctgtgccata | ||
| CD68-F | ctcacaaaaaggctgccact | 60 bp | 1.000 |
| CD68-R | ttccggtggttgtaggtgtc | ||
| ChAT-F | gcctcatctctggtgtgctt | 62 bp | 1.000 |
| ChAT-R | gtcagtgggaagggagtgg | ||
| Iba1-F | ctccgaggagacgttcagtt | 96 bp | 0.855 |
| Iba1-R | tttttctcctcatacatcagaatcatcagaat | ||
| LAMP1-F | ctgaaggtggggaacaagag | 66 bp | 1.000 |
| LAMP1-R | caggctagaagtggcattca | ||
| LC3B-F | tgaatatgagcgaactcatcaag | 92 bp | 1.000 |
| LC3B-R | catgctgtgcccattcac | ||
| NFκB-F | ctggcagctcttctcaaagc | 70 bp | 1.000 |
| NFκB-R | ccaggtcatagagaggctcaa | ||
| P62-F | ggggaccctgataacatcaa | 73 bp | 0.976 |
| P62-R | ccttgccggctaagatgag |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Palmhof, M.; Lohmann, S.; Schulte, D.; Stute, G.; Wagner, N.; Dick, H.B.; Joachim, S.C. Fewer Functional Deficits and Reduced Cell Death after Ranibizumab Treatment in a Retinal Ischemia Model. Int. J. Mol. Sci. 2018, 19, 1636. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19061636
Palmhof M, Lohmann S, Schulte D, Stute G, Wagner N, Dick HB, Joachim SC. Fewer Functional Deficits and Reduced Cell Death after Ranibizumab Treatment in a Retinal Ischemia Model. International Journal of Molecular Sciences. 2018; 19(6):1636. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19061636
Chicago/Turabian StylePalmhof, Marina, Stephanie Lohmann, Dustin Schulte, Gesa Stute, Natalie Wagner, H. Burkhard Dick, and Stephanie C. Joachim. 2018. "Fewer Functional Deficits and Reduced Cell Death after Ranibizumab Treatment in a Retinal Ischemia Model" International Journal of Molecular Sciences 19, no. 6: 1636. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19061636