Molecular Markers of Therapy-Resistant Glioblastoma and Potential Strategy to Combat Resistance


Abstract
:
1. Introduction
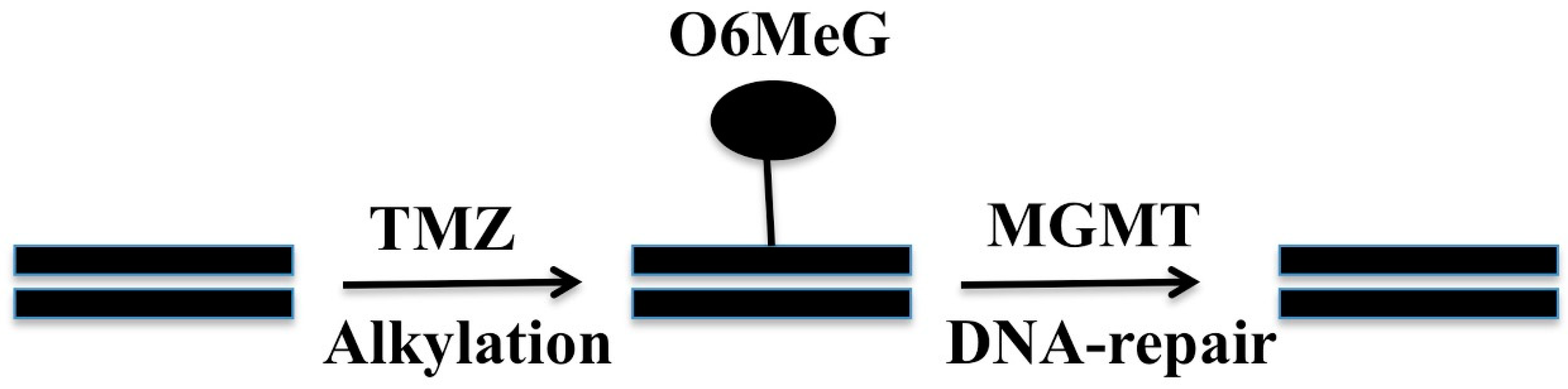
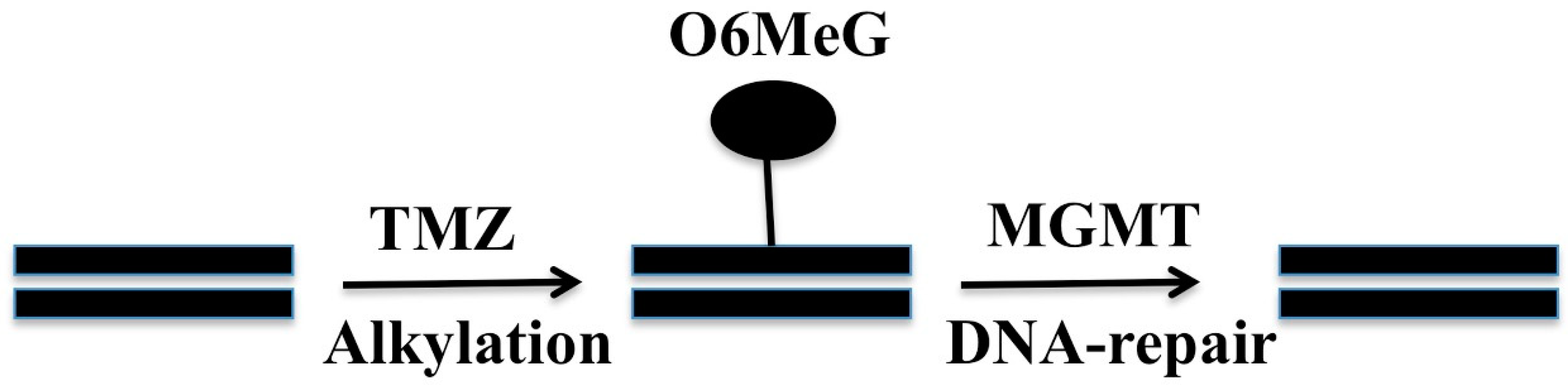
2. O6-Methylguanine Methyltransferase (MGMT)
3. Epidermal Growth Factor Receptor (EGFR)
4. Isocitrate Dehydrogenase (IDH)1/2
5. 1p19q Co-Deletion
6. α-Thalassemia/Mental Retardation Syndrome X-Linked (ATRX)
7. Telomerase Reverse Transcriptase (TERT)
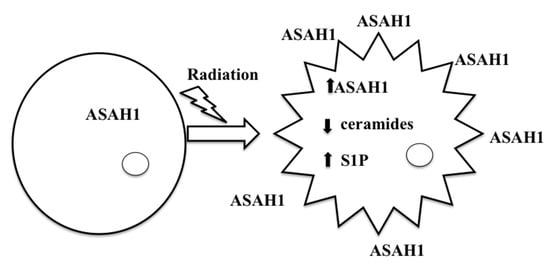
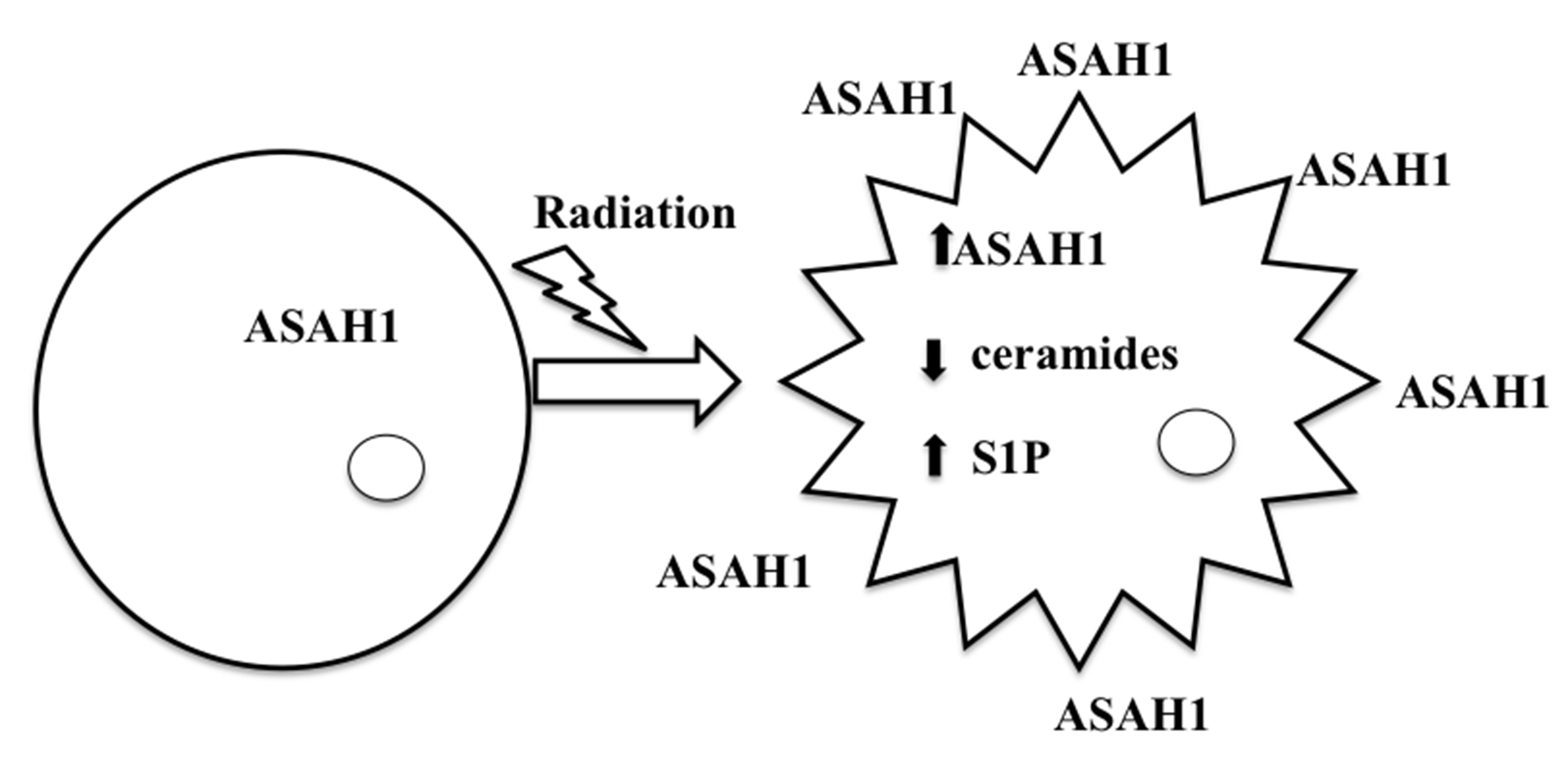
8. Acid Ceramidase (ASAH1) as a Druggable Target to Combat Multiple Therapy-Resistant Cancers
9. ASAH1-Induced Radioresistance in GBM
10. Identification of Novel Drug Targets to Combat Radioresistant GBM
Acknowledgments
Conflicts of Interest
Abbreviations
| 5-ALA | 5-aminolevulinic acid |
| ASAH1 | acid ceramidase |
| AML | acute myeloid leukemia |
| CerS | ceramide synthases |
| DMG | N-dimethyl glycine |
| GBM | glioblastoma |
| IDH | isocitrate dehydrogenase |
| MGMT | O6-methylguanine-methyltransferase |
| MRI | magnetic resonance imaging |
| S1P | sphingosine-1-phosphate |
| SPHK1 | sphingosine kinase 1 |
| SPHK2 | sphingosine kinase 2 |
| TTF | tumor-treating field |
References
- Ostrom, Q.T.; Gittleman, H.; Farah, P.; Ondracek, A.; Chen, Y.; Wolinsky, Y.; Stroup, N.E.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS statistical report: Primary brain and central nervous system tumors diagnosed in the United States in 2006–2010. Neuro Oncol. 2013, 15, ii1–ii56. [Google Scholar] [CrossRef] [PubMed]
- Ostrom, Q.T.; Gittleman, H.; Fulop, J.; Liu, M.; Blanda, R.; Kromer, C.; Wolinsky, Y.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Central Nervous System Tumors Diagnosed in the United States in 2008–2012. Neuro Oncol. 2015, 17, iv1–iv62. [Google Scholar] [CrossRef] [PubMed]
- Ostrom, Q.T.; Gittleman, H.; Liao, P.; Rouse, C.; Chen, Y.; Dowling, J.; Wolinsky, Y.; Kruchko, C.; Barnholtz-Sloan, J. CBTRUS Statistical Report: Primary Brain and Central Nervous System Tumors Diagnosed in the United States in 2007–2011. Neuro Oncol. 2014, 16, 1–63. [Google Scholar] [CrossRef] [PubMed]
- Fisher, J.L.; Schwartzbaum, J.A.; Wrensch, M.; Wiemels, J.L. Epidemiology of brain tumors. Neurol. Clin. 2007, 25, 867–890. [Google Scholar] [CrossRef] [PubMed]
- Goodenberger, M.L.; Jenkins, R.B. Genetics of adult glioma. Cancer Genet. 2012, 205, 613–621. [Google Scholar] [CrossRef] [PubMed]
- Farrell, C.J.; Plotkin, S.R. Genetic causes of brain tumors: Neurofibromatosis, tuberous sclerosis, von Hippel-Lindau, and other syndromes. Neurol. Clin. 2007, 25, 925–946. [Google Scholar] [CrossRef] [PubMed]
- Chaichana, K.L.; Parker, S.L.; Olivi, A.; Quinones-Hinojosa, A. Long-term seizure outcomes in adult patients undergoing primary resection of malignant brain astrocytomas. Clinical article. J. Neurosurg. 2009, 111, 282–292. [Google Scholar] [CrossRef] [PubMed]
- Chandra, V.; Rock, A.K.; Opalak, C.; Stary, J.M.; Sima, A.P.; Carr, M.; Vega, R.A.; Broaddus, W.C. A systematic review of perioperative seizure prophylaxis during brain tumor resection: The case for a multicenter randomized clinical trial. Neurosurg. Focus 2017, 43, E18. [Google Scholar] [CrossRef] [PubMed]
- Wychowski, T.; Wang, H.; Buniak, L.; Henry, J.C.; Mohile, N. Considerations in prophylaxis for tumor-associated epilepsy: Prevention of status epilepticus and tolerability of newer generation AEDs. Clin. Neurol. Neurosurg. 2013, 115, 2365–2369. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.M.; Parney, I.F.; Huang, W.; Anderson, F.A., Jr.; Asher, A.L.; Bernstein, M.; Lillehei, K.O.; Brem, H.; Berger, M.S.; Laws, E.R.; et al. Patterns of care for adults with newly diagnosed malignant glioma. JAMA 2005, 293, 557–564. [Google Scholar] [CrossRef] [PubMed]
- Kalpathy-Cramer, J.; Gerstner, E.R.; Emblem, K.E.; Andronesi, O.; Rosen, B. Advanced magnetic resonance imaging of the physical processes in human glioblastoma. Cancer Res. 2014, 74, 4622–4637. [Google Scholar] [CrossRef] [PubMed]
- Drappatz, J.; Schiff, D.; Kesari, S.; Norden, A.D.; Wen, P.Y. Medical management of brain tumor patients. Neurol. Clin. 2007, 25, 1035–1071, ix. [Google Scholar] [CrossRef] [PubMed]
- Kostaras, X.; Cusano, F.; Kline, G.A.; Roa, W.; Easaw, J. Use of dexamethasone in patients with high-grade glioma: A clinical practice guideline. Curr. Oncol. 2014, 21, e493–e503. [Google Scholar] [CrossRef] [PubMed]
- Chinot, O.L.; Wick, W.; Mason, W.; Henriksson, R.; Saran, F.; Nishikawa, R.; Carpentier, A.F.; Hoang-Xuan, K.; Kavan, P.; Cernea, D.; et al. Bevacizumab plus radiotherapy-temozolomide for newly diagnosed glioblastoma. N. Engl. J. Med. 2014, 370, 709–722. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, M.R.; Dignam, J.J.; Armstrong, T.S.; Wefel, J.S.; Blumenthal, D.T.; Vogelbaum, M.A.; Colman, H.; Chakravarti, A.; Pugh, S.; Won, M.; et al. A randomized trial of bevacizumab for newly diagnosed glioblastoma. N. Engl. J. Med. 2014, 370, 699–708. [Google Scholar] [CrossRef] [PubMed]
- Lai, A.; Kharbanda, S.; Pope, W.B.; Tran, A.; Solis, O.E.; Peale, F.; Forrest, W.F.; Pujara, K.; Carrillo, J.A.; Pandita, A.; et al. Evidence for sequenced molecular evolution of IDH1 mutant glioblastoma from a distinct cell of origin. J. Clin. Oncol. 2011, 29, 4482–4490. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, K.; Hiwatashi, A.; Togao, O.; Kikuchi, K.; Hatae, R.; Yoshimoto, K.; Mizoguchi, M.; Suzuki, S.O.; Yoshiura, T.; Honda, H. MR Imaging-Based Analysis of Glioblastoma Multiforme: Estimation of IDH1 Mutation Status. Am. J. Neuroradiol. 2016, 37, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Aldape, K.; Nejad, R.; Louis, D.N.; Zadeh, G. Integrating molecular markers into the World Health Organization classification of CNS tumors: A survey of the neuro-oncology community. Neuro Oncol. 2017, 19, 336–344. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef] [PubMed]
- Dean, M.; Fojo, T.; Bates, S. Tumour stem cells and drug resistance. Nat. Rev. Cancer 2005, 5, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Christensen, K.; Schroder, H.D.; Kristensen, B.W. CD133 identifies perivascular niches in grade II-IV astrocytomas. J. Neurooncol. 2008, 90, 157–170. [Google Scholar] [CrossRef] [PubMed]
- Zeppernick, F.; Ahmadi, R.; Campos, B.; Dictus, C.; Helmke, B.M.; Becker, N.; Lichter, P.; Unterberg, A.; Radlwimmer, B.; Herold-Mende, C.C. Stem cell marker CD133 affects clinical outcome in glioma patients. Clin. Cancer Res. 2008, 14, 123–129. [Google Scholar] [CrossRef] [PubMed]
- Osuka, S.; Van Meir, E.G. Overcoming therapeutic resistance in glioblastoma: The way forward. J. Clin. Investig. 2017, 127, 415–426. [Google Scholar] [CrossRef] [PubMed]
- Lefranc, F.; Kiss, R. The sodium pump alpha1 subunit as a potential target to combat apoptosis-resistant glioblastomas. Neoplasia 2008, 10, 198–206. [Google Scholar] [CrossRef] [PubMed]
- Barbagallo, D.; Caponnetto, A.; Cirnigliaro, M.; Brex, D.; Barbagallo, C.; D’Angeli, F.; Morrone, A.; Caltabiano, R.; Barbagallo, G.M.; Ragusa, M.; et al. CircSMARCA5 Inhibits Migration of Glioblastoma Multiforme Cells by Regulating a Molecular Axis Involving Splicing Factors SRSF1/SRSF3/PTB. Int. J. Mol. Sci. 2018, 19, 480. [Google Scholar] [CrossRef] [PubMed]
- Karsy, M.; Neil, J.A.; Guan, J.; Mahan, M.A.; Colman, H.; Jensen, R.L. A practical review of prognostic correlations of molecular biomarkers in glioblastoma. Neurosurg. Focus 2015, 38, E4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hegi, M.E.; Diserens, A.C.; Gorlia, T.; Hamou, M.F.; de Tribolet, N.; Weller, M.; Kros, J.M.; Hainfellner, J.A.; Mason, W.; Mariani, L.; et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N. Engl. J. Med. 2005, 352, 997–1003. [Google Scholar] [CrossRef] [PubMed]
- Gorlia, T.; van den Bent, M.J.; Hegi, M.E.; Mirimanoff, R.O.; Weller, M.; Cairncross, J.G.; Eisenhauer, E.; Belanger, K.; Brandes, A.A.; Allgeier, A.; et al. Nomograms for predicting survival of patients with newly diagnosed glioblastoma: Prognostic factor analysis of EORTC and NCIC trial 26981-22981/CE.3. Lancet Oncol. 2008, 9, 29–38. [Google Scholar] [CrossRef]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Zarnett, O.J.; Sahgal, A.; Gosio, J.; Perry, J.; Berger, M.S.; Chang, S.; Das, S. Treatment of elderly patients with glioblastoma: A systematic evidence-based analysis. JAMA Neurol. 2015, 72, 589–596. [Google Scholar] [CrossRef] [PubMed]
- Felsberg, J.; Thon, N.; Eigenbrod, S.; Hentschel, B.; Sabel, M.C.; Westphal, M.; Schackert, G.; Kreth, F.W.; Pietsch, T.; Loffler, M.; et al. Promoter methylation and expression of MGMT and the DNA mismatch repair genes MLH1, MSH2, MSH6 and PMS2 in paired primary and recurrent glioblastomas. Int. J. Cancer 2011, 129, 659–670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weller, M.; Tabatabai, G.; Kastner, B.; Felsberg, J.; Steinbach, J.P.; Wick, A.; Schnell, O.; Hau, P.; Herrlinger, U.; Sabel, M.C.; et al. MGMT Promoter Methylation Is a Strong Prognostic Biomarker for Benefit from Dose-Intensified Temozolomide Rechallenge in Progressive Glioblastoma: The DIRECTOR Trial. Clin. Cancer Res. 2015, 21, 2057–2064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wick, W.; Weller, M.; van den Bent, M.; Sanson, M.; Weiler, M.; von Deimling, A.; Plass, C.; Hegi, M.; Platten, M.; Reifenberger, G. MGMT testing--the challenges for biomarker-based glioma treatment. Nat. Rev. Neurol. 2014, 10, 372–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bush, N.A.; Butowski, N. The Effect of Molecular Diagnostics on the Treatment of Glioma. Curr. Oncol. Rep. 2017, 19, 26. [Google Scholar] [CrossRef] [PubMed]
- Quillien, V.; Lavenu, A.; Karayan-Tapon, L.; Carpentier, C.; Labussiere, M.; Lesimple, T.; Chinot, O.; Wager, M.; Honnorat, J.; Saikali, S.; et al. Comparative assessment of 5 methods (methylation-specific polymerase chain reaction, MethyLight, pyrosequencing, methylation-sensitive high-resolution melting, and immunohistochemistry) to analyze O6-methylguanine-DNA-methyltranferase in a series of 100 glioblastoma patients. Cancer 2012, 118, 4201–4211. [Google Scholar] [PubMed]
- Thorne, A.H.; Zanca, C.; Furnari, F. Epidermal growth factor receptor targeting and challenges in glioblastoma. Neuro Oncol. 2016, 18, 914–918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ludwig, K.; Kornblum, H.I. Molecular markers in glioma. J. Neurooncol. 2017, 134, 505–512. [Google Scholar] [CrossRef] [PubMed]
- Reardon, D.A.; Wen, P.Y.; Mellinghoff, I.K. Targeted molecular therapies against epidermal growth factor receptor: Past experiences and challenges. Neuro Oncol. 2014, 16 (Suppl. 8), viii7–viii13. [Google Scholar] [CrossRef] [PubMed]
- Patel, M.; Vogelbaum, M.A.; Barnett, G.H.; Jalali, R.; Ahluwalia, M.S. Molecular targeted therapy in recurrent glioblastoma: Current challenges and future directions. Expert Opin. Investig. Drugs 2012, 21, 1247–1266. [Google Scholar] [CrossRef] [PubMed]
- Parsons, D.W.; Jones, S.; Zhang, X.; Lin, J.C.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Siu, I.M.; Gallia, G.L.; et al. An integrated genomic analysis of human glioblastoma multiforme. Science 2008, 321, 1807–1812. [Google Scholar] [CrossRef] [PubMed]
- Stancheva, G.; Goranova, T.; Laleva, M.; Kamenova, M.; Mitkova, A.; Velinov, N.; Poptodorov, G.; Mitev, V.; Kaneva, R.; Gabrovsky, N. IDH1/IDH2 but not TP53 mutations predict prognosis in Bulgarian glioblastoma patients. Biomed. Res. Int. 2014, 2014, 654727. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Parsons, D.W.; Jin, G.; McLendon, R.; Rasheed, B.A.; Yuan, W.; Kos, I.; Batinic-Haberle, I.; Jones, S.; Riggins, G.J.; et al. IDH1 and IDH2 mutations in gliomas. N. Engl. J. Med. 2009, 360, 765–773. [Google Scholar] [CrossRef] [PubMed]
- Labussiere, M.; Boisselier, B.; Mokhtari, K.; Di Stefano, A.L.; Rahimian, A.; Rossetto, M.; Ciccarino, P.; Saulnier, O.; Paterra, R.; Marie, Y.; et al. Combined analysis of TERT, EGFR, and IDH status defines distinct prognostic glioblastoma classes. Neurology 2014, 83, 1200–1206. [Google Scholar] [CrossRef] [PubMed]
- Zou, P.; Xu, H.; Chen, P.; Yan, Q.; Zhao, L.; Zhao, P.; Gu, A. IDH1/IDH2 mutations define the prognosis and molecular profiles of patients with gliomas: A meta-analysis. PLoS ONE 2013, 8, e68782. [Google Scholar] [CrossRef] [PubMed]
- Houillier, C.; Wang, X.; Kaloshi, G.; Mokhtari, K.; Guillevin, R.; Laffaire, J.; Paris, S.; Boisselier, B.; Idbaih, A.; Laigle-Donadey, F.; et al. IDH1 or IDH2 mutations predict longer survival and response to temozolomide in low-grade gliomas. Neurology 2010, 75, 1560–1566. [Google Scholar] [CrossRef] [PubMed]
- Leu, S.; von Felten, S.; Frank, S.; Vassella, E.; Vajtai, I.; Taylor, E.; Schulz, M.; Hutter, G.; Hench, J.; Schucht, P.; et al. IDH/MGMT-driven molecular classification of low-grade glioma is a strong predictor for long-term survival. Neuro Oncol. 2013, 15, 469–479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beiko, J.; Suki, D.; Hess, K.R.; Fox, B.D.; Cheung, V.; Cabral, M.; Shonka, N.; Gilbert, M.R.; Sawaya, R.; Prabhu, S.S.; et al. IDH1 mutant malignant astrocytomas are more amenable to surgical resection and have a survival benefit associated with maximal surgical resection. Neuro Oncol. 2014, 16, 81–91. [Google Scholar] [CrossRef] [PubMed]
- Dang, L.; Yen, K.; Attar, E.C. IDH mutations in cancer and progress toward development of targeted therapeutics. Ann. Oncol. 2016, 27, 599–608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riemenschneider, M.J.; Jeuken, J.W.; Wesseling, P.; Reifenberger, G. Molecular diagnostics of gliomas: State of the art. Acta Neuropathol. 2010, 120, 567–584. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.S.; Perry, A.; Borell, T.J.; Lee, H.K.; O’Fallon, J.; Hosek, S.M.; Kimmel, D.; Yates, A.; Burger, P.C.; Scheithauer, B.W.; et al. Alterations of chromosome arms 1p and 19q as predictors of survival in oligodendrogliomas, astrocytomas, and mixed oligoastrocytomas. J. Clin. Oncol. 2000, 18, 636–645. [Google Scholar] [CrossRef] [PubMed]
- Cairncross, G.; Wang, M.; Shaw, E.; Jenkins, R.; Brachman, D.; Buckner, J.; Fink, K.; Souhami, L.; Laperriere, N.; Curran, W.; et al. Phase III trial of chemoradiotherapy for anaplastic oligodendroglioma: Long-term results of RTOG 9402. J. Clin. Oncol. 2013, 31, 337–343. [Google Scholar] [CrossRef] [PubMed]
- Cairncross, G.; Berkey, B.; Shaw, E.; Jenkins, R.; Scheithauer, B.; Brachman, D.; Buckner, J.; Fink, K.; Souhami, L.; Laperierre, N.; et al. Phase III trial of chemotherapy plus radiotherapy compared with radiotherapy alone for pure and mixed anaplastic oligodendroglioma: Intergroup Radiation Therapy Oncology Group Trial 9402. J. Clin. Oncol. 2006, 24, 2707–2714. [Google Scholar] [CrossRef] [PubMed]
- Kaloshi, G.; Benouaich-Amiel, A.; Diakite, F.; Taillibert, S.; Lejeune, J.; Laigle-Donadey, F.; Renard, M.A.; Iraqi, W.; Idbaih, A.; Paris, S.; et al. Temozolomide for low-grade gliomas: Predictive impact of 1p/19q loss on response and outcome. Neurology 2007, 68, 1831–1836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boots-Sprenger, S.H.; Sijben, A.; Rijntjes, J.; Tops, B.B.; Idema, A.J.; Rivera, A.L.; Bleeker, F.E.; Gijtenbeek, A.M.; Diefes, K.; Heathcock, L.; et al. Significance of complete 1p/19q co-deletion, IDH1 mutation and MGMT promoter methylation in gliomas: Use with caution. Mod. Pathol. 2013, 26, 922–929. [Google Scholar] [CrossRef] [PubMed]
- Laxton, R.C.; Popov, S.; Doey, L.; Jury, A.; Bhangoo, R.; Gullan, R.; Chandler, C.; Brazil, L.; Sadler, G.; Beaney, R.; et al. Primary glioblastoma with oligodendroglial differentiation has better clinical outcome but no difference in common biological markers compared with other types of glioblastoma. Neuro Oncol. 2013, 15, 1635–1643. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Ma, W.; Zhao, H. Loss of heterozygosity 1p/19q and survival in glioma: A meta-analysis. Neuro Oncol. 2014, 16, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Wirsching, H.G.; Weller, M. The Role of Molecular Diagnostics in the Management of Patients with Gliomas. Curr. Treat. Options Oncol. 2016, 17, 51. [Google Scholar] [CrossRef] [PubMed]
- Nandakumar, P.; Mansouri, A.; Das, S. The Role of ATRX in Glioma Biology. Front. Oncol. 2017, 7, 236. [Google Scholar] [CrossRef] [PubMed]
- Wiestler, B.; Capper, D.; Holland-Letz, T.; Korshunov, A.; von Deimling, A.; Pfister, S.M.; Platten, M.; Weller, M.; Wick, W. ATRX loss refines the classification of anaplastic gliomas and identifies a subgroup of IDH mutant astrocytic tumors with better prognosis. Acta Neuropathol. 2013, 126, 443–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koschmann, C.; Calinescu, A.A.; Nunez, F.J.; Mackay, A.; Fazal-Salom, J.; Thomas, D.; Mendez, F.; Kamran, N.; Dzaman, M.; Mulpuri, L.; et al. ATRX loss promotes tumor growth and impairs nonhomologous end joining DNA repair in glioma. Sci. Transl. Med. 2016, 8, 328ra28. [Google Scholar] [CrossRef] [PubMed]
- Killela, P.J.; Reitman, Z.J.; Jiao, Y.; Bettegowda, C.; Agrawal, N.; Diaz, L.A., Jr.; Friedman, A.H.; Friedman, H.; Gallia, G.L.; Giovanella, B.C.; et al. TERT promoter mutations occur frequently in gliomas and a subset of tumors derived from cells with low rates of self-renewal. Proc. Natl. Acad. Sci. USA 2013, 110, 6021–6026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eckel-Passow, J.E.; Lachance, D.H.; Molinaro, A.M.; Walsh, K.M.; Decker, P.A.; Sicotte, H.; Pekmezci, M.; Rice, T.; Kosel, M.L.; Smirnov, I.V.; et al. Glioma Groups Based on 1p/19q, IDH, and TERT Promoter Mutations in Tumors. N. Engl. J. Med. 2015, 372, 2499–2508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koelsche, C.; Sahm, F.; Capper, D.; Reuss, D.; Sturm, D.; Jones, D.T.; Kool, M.; Northcott, P.A.; Wiestler, B.; Bohmer, K.; et al. Distribution of TERT promoter mutations in pediatric and adult tumors of the nervous system. Acta Neuropathol. 2013, 126, 907–915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shankar, G.M.; Francis, J.M.; Rinne, M.L.; Ramkissoon, S.H.; Huang, F.W.; Venteicher, A.S.; Akama-Garren, E.H.; Kang, Y.J.; Lelic, N.; Kim, J.C.; et al. Rapid Intraoperative Molecular Characterization of Glioma. JAMA Oncol. 2015, 1, 662–667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernardo, K.; Hurwitz, R.; Zenk, T.; Desnick, R.J.; Ferlinz, K.; Schuchman, E.H.; Sandhoff, K. Purification, characterization, and biosynthesis of human acid ceramidase. J. Biol. Chem. 1995, 270, 11098–11102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gatt, S. Enzymic Hydrolysis and Synthesis of Ceramides. J. Biol. Chem. 1963, 238, 3131–3133. [Google Scholar] [PubMed]
- Zeidan, Y.H.; Jenkins, R.W.; Korman, J.B.; Liu, X.; Obeid, L.M.; Norris, J.S.; Hannun, Y.A. Molecular targeting of acid ceramidase: Implications to cancer therapy. Curr. Drug Targets 2008, 9, 653–661. [Google Scholar] [CrossRef] [PubMed]
- Ogretmen, B.; Hannun, Y.A. Biologically active sphingolipids in cancer pathogenesis and treatment. Nat. Rev. Cancer 2004, 4, 604–616. [Google Scholar] [CrossRef] [PubMed]
- Pettus, B.J.; Chalfant, C.E.; Hannun, Y.A. Ceramide in apoptosis: An overview and current perspectives. Biochim. Biophys. Acta 2002, 1585, 114–125. [Google Scholar] [CrossRef]
- Taha, T.A.; Mullen, T.D.; Obeid, L.M. A house divided: Ceramide, sphingosine, and sphingosine-1-phosphate in programmed cell death. Biochim. Biophys. Acta 2006, 1758, 2027–2036. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.S.; Awad, A.J.; Shabani, S.; Doan, N. Molecular Targeting of Acid Ceramidase in Glioblastoma: A Review of Its Role, Potential Treatment, and Challenges. Pharmaceutics 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- Young, N.; Pearl, D.K.; Van Brocklyn, J.R. Sphingosine-1-phosphate regulates glioblastoma cell invasiveness through the urokinase plasminogen activator system and CCN1/Cyr61. Mol. Cancer Res. 2009, 7, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Young, N.; Van Brocklyn, J.R. Roles of sphingosine-1-phosphate (S1P) receptors in malignant behavior of glioma cells. Differential effects of S1P2 on cell migration and invasiveness. Exp. Cell Res. 2007, 313, 1615–1627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mullen, T.D.; Hannun, Y.A.; Obeid, L.M. Ceramide synthases at the centre of sphingolipid metabolism and biology. Biochem. J. 2012, 441, 789–802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mullen, T.D.; Obeid, L.M. Ceramide and apoptosis: Exploring the enigmatic connections between sphingolipid metabolism and programmed cell death. Anticancer Agents Med. Chem. 2012, 12, 340–363. [Google Scholar] [CrossRef] [PubMed]
- White-Gilbertson, S.; Mullen, T.; Senkal, C.; Lu, P.; Ogretmen, B.; Obeid, L.; Voelkel-Johnson, C. Ceramide synthase 6 modulates TRAIL sensitivity and nuclear translocation of active caspase-3 in colon cancer cells. Oncogene 2009, 28, 1132–1141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bieberich, E. Ceramide signaling in cancer and stem cells. Future Lipidol. 2008, 3, 273–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sawada, M.; Nakashima, S.; Banno, Y.; Yamakawa, H.; Hayashi, K.; Takenaka, K.; Nishimura, Y.; Sakai, N.; Nozawa, Y. Ordering of ceramide formation, caspase activation, and Bax/Bcl-2 expression during etoposide-induced apoptosis in C6 glioma cells. Cell Death Differ. 2000, 7, 761–772. [Google Scholar] [CrossRef] [PubMed]
- Pitson, S.M.; Moretti, P.A.; Zebol, J.R.; Lynn, H.E.; Xia, P.; Vadas, M.A.; Wattenberg, B.W. Activation of sphingosine kinase 1 by ERK1/2-mediated phosphorylation. EMBO J. 2003, 22, 5491–5500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saad, A.F.; Meacham, W.D.; Bai, A.; Anelli, V.; Elojeimy, S.; Mahdy, A.E.; Turner, L.S.; Cheng, J.; Bielawska, A.; Bielawski, J.; et al. The functional effects of acid ceramidase overexpression in prostate cancer progression and resistance to chemotherapy. Cancer Biol. Ther. 2007, 6, 1455–1460. [Google Scholar] [CrossRef] [PubMed]
- Seelan, R.S.; Qian, C.; Yokomizo, A.; Bostwick, D.G.; Smith, D.I.; Liu, W. Human acid ceramidase is overexpressed but not mutated in prostate cancer. Genes Chromosom. Cancer 2000, 29, 137–146. [Google Scholar] [CrossRef]
- Lai, M.; Realini, N.; La Ferla, M.; Passalacqua, I.; Matteoli, G.; Ganesan, A.; Pistello, M.; Mazzanti, C.M.; Piomelli, D. Complete Acid Ceramidase ablation prevents cancer-initiating cell formation in melanoma cells. Sci. Rep. 2017, 7, 7411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Musumarra, G.; Barresi, V.; Condorelli, D.F.; Scire, S. A bioinformatic approach to the identification of candidate genes for the development of new cancer diagnostics. Biol. Chem. 2003, 384, 321–327. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.F.; Pearson, J.M.; Feith, D.J.; Loughran, T.P., Jr. The emergence of acid ceramidase as a therapeutic target for acute myeloid leukemia. Expert Opin. Ther. Targets 2017, 21, 583–590. [Google Scholar] [CrossRef] [PubMed]
- Mahdy, A.E.; Cheng, J.C.; Li, J.; Elojeimy, S.; Meacham, W.D.; Turner, L.S.; Bai, A.; Gault, C.R.; McPherson, A.S.; Garcia, N.; et al. Acid ceramidase upregulation in prostate cancer cells confers resistance to radiation: AC inhibition, a potential radiosensitizer. Mol. Ther. 2009, 17, 430–438. [Google Scholar] [CrossRef] [PubMed]
- Samsel, L.; Zaidel, G.; Drumgoole, H.M.; Jelovac, D.; Drachenberg, C.; Rhee, J.G.; Brodie, A.M.; Bielawska, A.; Smyth, M.J. The ceramide analog, B13, induces apoptosis in prostate cancer cell lines and inhibits tumor growth in prostate cancer xenografts. Prostate 2004, 58, 382–393. [Google Scholar] [CrossRef] [PubMed]
- Selzner, M.; Bielawska, A.; Morse, M.A.; Rudiger, H.A.; Sindram, D.; Hannun, Y.A.; Clavien, P.A. Induction of apoptotic cell death and prevention of tumor growth by ceramide analogues in metastatic human colon cancer. Cancer Res. 2001, 61, 1233–1240. [Google Scholar] [PubMed]
- Vethakanraj, H.S.; Sesurajan, B.P.; Padmanaban, V.P.; Jayaprakasam, M.; Murali, S.; Sekar, A.K. Anticancer effect of acid ceramidase inhibitor ceranib-2 in human breast cancer cell lines MCF-7, MDA MB-231 by the activation of SAPK/JNK, p38 MAPK apoptotic pathways, inhibition of the Akt pathway, downregulation of ERalpha. Anticancer Drugs 2018, 29, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Ma, S.; Liu, H.; Han, H.; Wang, S. HCFU inhibits cervical cancer cells growth and metastasis by inactivating Wnt/beta-catenin pathway. J. Cell Biochem. 2017. [Google Scholar] [CrossRef] [PubMed]
- Realini, N.; Solorzano, C.; Pagliuca, C.; Pizzirani, D.; Armirotti, A.; Luciani, R.; Costi, M.P.; Bandiera, T.; Piomelli, D. Discovery of highly potent acid ceramidase inhibitors with in vitro tumor chemosensitizing activity. Sci. Rep. 2013, 3, 1035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morimoto, K.; Koh, M. Postoperative adjuvant use of carmofur for early breast cancer. Osaka City Med. J. 2003, 49, 77–83. [Google Scholar] [PubMed]
- Abuhusain, H.J.; Matin, A.; Qiao, Q.; Shen, H.; Kain, N.; Day, B.W.; Stringer, B.W.; Daniels, B.; Laaksonen, M.A.; Teo, C.; et al. A metabolic shift favoring sphingosine 1-phosphate at the expense of ceramide controls glioblastoma angiogenesis. J. Biol. Chem. 2013, 288, 37355–37364. [Google Scholar] [CrossRef] [PubMed]
- Annabi, B.; Lachambre, M.P.; Plouffe, K.; Sartelet, H.; Beliveau, R. Modulation of invasive properties of CD133+ glioblastoma stem cells: A role for MT1-MMP in bioactive lysophospholipid signaling. Mol. Carcinog. 2009, 48, 910–919. [Google Scholar] [CrossRef] [PubMed]
- Doan, N.B.; Alhajala, H.; Al-Gizawiy, M.M.; Mueller, W.M.; Rand, S.D.; Connelly, J.M.; Cochran, E.J.; Chitambar, C.R.; Clark, P.; Kuo, J.; et al. Acid ceramidase and its inhibitors: A de novo drug target and a new class of drugs for killing glioblastoma cancer stem cells with high efficiency. Oncotarget 2017, 8, 112662–112674. [Google Scholar] [CrossRef] [PubMed]
- Doan, N.B.; Nguyen, H.S.; Al-Gizawiy, M.M.; Mueller, W.M.; Sabbadini, R.A.; Rand, S.D.; Connelly, J.M.; Chitambar, C.R.; Schmainda, K.M.; Mirza, S.P. Acid ceramidase confers radioresistance to glioblastoma cells. Oncol. Rep. 2017, 38, 1932–1940. [Google Scholar] [CrossRef] [PubMed]
- Doan, N.B.; Nguyen, H.S.; Montoure, A.; Al-Gizawiy, M.M.; Mueller, W.M.; Kurpad, S.; Rand, S.D.; Connelly, J.M.; Chitambar, C.R.; Schmainda, K.M.; et al. Acid ceramidase is a novel drug target for pediatric brain tumors. Oncotarget 2017, 8, 24753–24761. [Google Scholar] [CrossRef] [PubMed]
- Hara, S.; Nakashima, S.; Kiyono, T.; Sawada, M.; Yoshimura, S.; Iwama, T.; Banno, Y.; Shinoda, J.; Sakai, N. p53-Independent ceramide formation in human glioma cells during gamma-radiation-induced apoptosis. Cell Death Differ. 2004, 11, 853–861. [Google Scholar] [CrossRef] [PubMed]
- Kubota, T.; Fujita, S.; Kodaira, S.; Yamamoto, T.; Josui, K.; Arisawa, Y.; Suto, A.; Ishibiki, K.; Abe, O.; Mabuchi, K.; et al. Antitumor activity of fluoropyrimidines and thymidylate synthetase inhibition. Jpn. J. Cancer Res. 1991, 82, 476–482. [Google Scholar] [CrossRef] [PubMed]
- Sato, S.; Ueyama, T.; Fukui, H.; Miyazaki, K.; Kuwano, M. Anti-tumor effects of carmofur on human 5-FU resistant cells. Gan Kagaku Ryoho Cancer Chemother. 1999, 26, 1613–1616. [Google Scholar]
- Watanabe, M.; Kodaira, S.; Takahashi, T.; Tominaga, T.; Hojo, K.; Kato, T.; Kunitomo, K.; Isomoto, H.; Ohashi, Y.; Yasutomi, M. Randomized trial of the efficacy of adjuvant chemotherapy for colon cancer with combination therapy incorporating the oral pyrimidine 1-hexylcarbamoyl-5-fluorouracil. Langenbeck’s Arch. Surg. 2006, 391, 330–337. [Google Scholar] [CrossRef] [PubMed]
- Gebai, A.; Gorelik, A.; Li, Z.; Illes, K.; Nagar, B. Structural basis for the activation of acid ceramidase. Nat. Commun. 2018, 9, 1621. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; He, J.; Xie, X.; Su, G.; Teitz-Tennenbaum, S.; Sabel, M.S.; Lubman, D.M. Serum autoantibody profiling using a natural glycoprotein microarray for the prognosis of early melanoma. J. Proteome Res. 2010, 9, 6044–6051. [Google Scholar] [CrossRef] [PubMed]
- Alexander, B.M.; Cloughesy, T.F. Adult Glioblastoma. J. Clin. Oncol. 2017, 35, 2402–2409. [Google Scholar] [CrossRef] [PubMed]
- Hou, L.C.; Veeravagu, A.; Hsu, A.R.; Tse, V.C. Recurrent glioblastoma multiforme: A review of natural history and management options. Neurosurg. Focus 2006, 20, E5. [Google Scholar] [CrossRef] [PubMed]
- Wong, E.T.; Hess, K.R.; Gleason, M.J.; Jaeckle, K.A.; Kyritsis, A.P.; Prados, M.D.; Levin, V.A.; Yung, W.K. Outcomes and prognostic factors in recurrent glioma patients enrolled onto phase II clinical trials. J. Clin. Oncol. 1999, 17, 2572–2578. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Rao, L.; Wang, H.L.; Mao, Z.W.; Lei, R.H.; Yang, Z.Y.; Qing, H.; Deng, Y.L. Transcriptome analysis of glioma cells for the dynamic response to gamma-irradiation and dual regulation of apoptosis genes: A new insight into radiotherapy for glioblastomas. Cell Death Dis. 2013, 4, e895. [Google Scholar] [CrossRef] [PubMed]
- Doan, N.B.; Nguyen, H.S.; Alhajala, H.S.; Jaber, B.; Al-Gizawiy, M.M.; Erin Ahn, E.-Y.; Mueller, W.M.; Chitambar, C.R.; Mirza, S.P.; Schmainda, K.M. Identification of radiation responsive genes and transcriptome profiling via complete RNA sequencing in a stable radioresistant U87 glioblastoma model. Oncotarget 2018, 9, 23532–23542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, G.T.; Murray, G.I. Current mechanistic insights into the roles of matrix metalloproteinases in tumour invasion and metastasis. J. Pathol. 2015, 237, 273–281. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.L.; Kuo, F.H.; Chen, P.N.; Hsieh, Y.H.; Yu, N.Y.; Yang, W.E.; Hsieh, M.J.; Yang, S.F. Andrographolide suppresses the migratory ability of human glioblastoma multiforme cells by targeting ERK1/2-mediated matrix metalloproteinase-2 expression. Oncotarget 2017, 8, 105860–105872. [Google Scholar] [CrossRef] [PubMed]
- Iwadate, Y. Epithelial-mesenchymal transition in glioblastoma progression. Oncol. Lett. 2016, 11, 1615–1620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, X.Z.; Xu, S.L.; Xin, Y.H.; Yu, S.C.; Ping, Y.F.; Chen, L.; Xiao, H.L.; Wang, B.; Yi, L.; Wang, Q.L.; et al. Tumor-associated microglia/macrophages enhance the invasion of glioma stem-like cells via TGF-beta1 signaling pathway. J. Immunol. 2012, 189, 444–453. [Google Scholar] [CrossRef] [PubMed]
- Monteiro, A.R.; Hill, R.; Pilkington, G.J.; Madureira, P.A. The Role of Hypoxia in Glioblastoma Invasion. Cells 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Proescholdt, M.A.; Merrill, M.J.; Stoerr, E.M.; Lohmeier, A.; Pohl, F.; Brawanski, A. Function of carbonic anhydrase IX in glioblastoma multiforme. Neuro Oncol. 2012, 14, 1357–1366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.; Li, Y.; Wang, R.; Li, Y.; Shi, P.; Kan, Z.; Pang, X. Inhibition of REST Suppresses Proliferation and Migration in Glioblastoma Cells. Int. J. Mol. Sci. 2016, 17. [Google Scholar] [CrossRef] [PubMed]
- Nakatani, K.; Yoshimi, N.; Mori, H.; Sakai, H.; Shinoda, J.; Andoh, T.; Sakai, N. The significance of the expression of tumor suppressor gene DCC in human gliomas. J. Neurooncol. 1998, 40, 237–242. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Yoon, J.G.; Li, L.; Yu, W.; Shao, J.; Hua, D.; Zheng, S.; Hood, L.; Goodlett, D.R.; Foltz, G.; et al. The SOX2 response program in glioblastoma multiforme: An integrated ChIP-seq, expression microarray, and microRNA analysis. BMC Genom. 2011, 12, 11. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Yan, P.F.; Zhao, H.Y.; Zhang, F.C.; Zhao, W.H.; Feng, M. Inhibitor of Nicotinamide Phosphoribosyltransferase Sensitizes Glioblastoma Cells to Temozolomide via Activating ROS/JNK Signaling Pathway. Biomed. Res. Int. 2016, 2016, 1450843. [Google Scholar] [CrossRef] [PubMed]
- Baysan, M.; Woolard, K.; Cam, M.C.; Zhang, W.; Song, H.; Kotliarova, S.; Balamatsias, D.; Linkous, A.; Ahn, S.; Walling, J.; et al. Detailed longitudinal sampling of glioma stem cells in situ reveals Chr7 gain and Chr10 loss as repeated events in primary tumor formation and recurrence. Int. J. Cancer 2017, 141, 2002–2013. [Google Scholar] [CrossRef] [PubMed]
- Stricker, S.H.; Feber, A.; Engstrom, P.G.; Caren, H.; Kurian, K.M.; Takashima, Y.; Watts, C.; Way, M.; Dirks, P.; Bertone, P.; et al. Widespread resetting of DNA methylation in glioblastoma-initiating cells suppresses malignant cellular behavior in a lineage-dependent manner. Genes Dev. 2013, 27, 654–669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Razavi, S.M.; Lee, K.E.; Jin, B.E.; Aujla, P.S.; Gholamin, S.; Li, G. Immune Evasion Strategies of Glioblastoma. Front. Surg. 2016, 3, 11. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
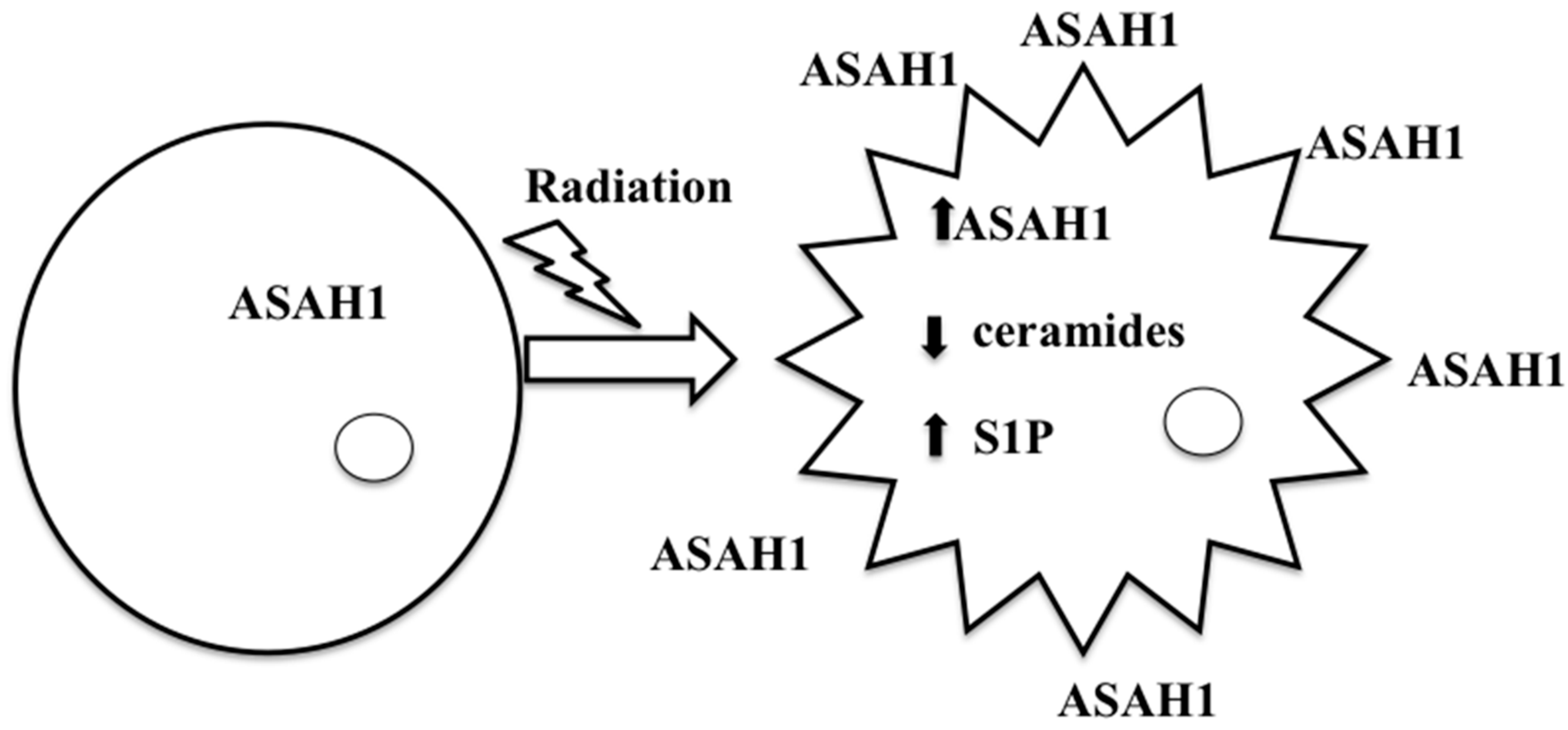{kind=link}
| GO:0006954: Inflammatory Response | GO:0000187: Activation of MAPK Activity | |
| CCL26 | C–C motif chemokine ligand 26 (CCL26) | anaplastic lymphoma receptor tyrosine kinase (ALK) |
| CCL3 | C–C motif chemokine ligand 3 (CCL3) | chondroitin sulfate proteoglycan 4 (CSPG4) |
| CXCL8 | C–X–C motif chemokine ligand 8 (CXCL8) | dual specificity phosphatase 7 (DUSP7) |
| GPR68 | G protein-coupled receptor 68 (GPR68) | formyl peptide receptor 1 (FPR1) |
| NFKBID | NFKB inhibitor delta (NFKBID) | transforming growth factor beta 3 (TGFB3) |
| TNFAIP3 | TNF alpha-induced protein 3 (TNFAIP3) | tumor protein p73 (TP73) |
| TNFRSF10D | TNF receptor superfamily member 10d (TNFRSF10D) | |
| TNIP3 | TNFAIP3 interacting protein 3 (TNIP3) | GO:0004222: Metalloendopeptidase Activity |
| XCR1 | X–C motif chemokine receptor 1 (XCR1) | ADAM metallopeptidase domain 12 (ADAM12) |
| BDKRB1 | bradykinin receptor B1 (BDKRB1) | ADAM metallopeptidase domain 19 (ADAM19) |
| BDKRB2 | bradykinin receptor B2 (BDKRB2) | ADAM metallopeptidase with thrombospondin type 1 motif 1 (ADAMTS1) |
| CHST4 | carbohydrate sulfotransferase 4 (CHST4) | ADAM metallopeptidase with thrombospondin type 1 motif 14 (ADAMTS14) |
| C3 | complement C3 (C3) | bone morphogenetic protein 1 (BMP1) |
| FPR1 | formyl peptide receptor 1 (FPR1) | matrix metallopeptidase 12 (MMP12) |
| GBP5 | guanylate binding protein 5 (GBP5) | matrix metallopeptidase 3 (MMP3) |
| IL24 | interleukin 24 (IL24) | matrix metallopeptidase 7 (MMP7) |
| IL36B | interleukin 36, beta (IL36B) | membrane metalloendopeptidase (MME) |
| NFATC4 | nuclear factor of activated T-cells 4 (NFATC4) | teashirt zinc finger homeobox 2 (TSHZ2) |
| PTGER2 | prostaglandin E receptor 2 (PTGER2) | |
| SDC1 | syndecan 1 (SDC1) | GO:0071356: Cellular Response to Tumor Necrosis Factor |
| ZC3H12A | zinc finger CCCH-type-containing 12A (ZC3H12A) | C–C motif chemokine ligand 26 (CCL26) |
| C–C motif chemokine ligand 3 (CCL3) | ||
| GO:0010718: Positive Regulation of Epithelial to Mesenchymal Transition | C–X–C motif chemokine ligand 8 (CXCL8) | |
| BAMBI | BMP and activin membrane-bound inhibitor (BAMBI) | ankyrin repeat domain 1 (ANKRD1) |
| GLIPR2 | GLI pathogenesis related 2 (GLIPR2) | collagen type I alpha 1 chain (COL1A1) |
| AXIN2 | axin 2 (AXIN2) | endothelin 1 (EDN1) |
| COL1A1 | collagen type I alpha 1 chain (COL1A1) | hyaluronan synthase 2 (HAS2) |
| TGFB3 | transforming growth factor beta 3 (TGFB3) | periostin (POSTN) |
| GO:0016477: Cell Migration | ||
| BAMBI | BMP and activin membrane-bound inhibitor (BAMBI) | GO:0044344: Cellular Response to Fibroblast Growth Factor |
| EPHA3 | EPH receptor A3 (EPHA3) | C–X–C motif chemokine ligand 8 (CXCL8) |
| EPHB3 | EPH receptor B3 (EPHB3) | collagen type I alpha 1 chain (COL1A1) |
| ERG | ERG, ETS transcription factor (ERG) | periostin (POSTN) |
| WWC1 | WW and C2 domain containing 1 (WWC1) | snail family transcriptional repressor 2 (SNAI2) |
| BDKRB1 | bradykinin receptor B1 (BDKRB1) | |
| CSPG4 | chondroitin sulfate proteoglycan 4 (CSPG4) | GO:0071560: Cellular Response to Transforming Growth Factor Beta |
| COL5A1 | collagen type V alpha 1 chain (COL5A1) | ankyrin repeat domain 1 (ANKRD1) |
| FSCN1 | fascin actin-bundling protein 1 (FSCN1) | collagen type I alpha 1 chain (COL1A1) |
| LCP1 | lymphocyte cytosolic protein 1 (LCP1) | endothelin 1 (EDN1) |
| PODXL | podocalyxin like (PODXL) | periostin (POSTN) |
| PSG2 | pregnancy specific beta-1-glycoprotein 2 (PSG2) | phosphodiesterase 3A (PDE3A) |
| SDC1 | syndecan 1 (SDC1) | |
| GO:0001525: Angiogenesis | ribonuclease A family member 1, pancreatic (RNASE1) | |
| CXCL8 | C–X–C motif chemokine ligand 8 (CXCL8) | ribonuclease A family member 2 (RNASE2) |
| EPHB3 | EPH receptor B3 (EPHB3) | |
| EPHB4 | EPH receptor B4 (EPHB4) | GO:0090263: Positive Regulation of Canonical Wnt Signaling Pathway |
| ACKR3 | atypical chemokine receptor 3 (ACKR3) | BMP and activin membrane bound inhibitor (BAMBI) |
| CSPG4 | chondroitin sulfate proteoglycan 4 (CSPG4) | R-spondin 3 (RSPO3) |
| COL8A1 | collagen type VIII alpha 1 chain (COL8A1) | SRY-box 4 (SOX4) |
| NRXN3 | neurexin 3 (NRXN3) | axin 2 (AXIN2) |
| NDNF | neuron-derived neurotrophic factor (NDNF) | collagen type I alpha 1 chain (COL1A1) |
| NRP2 | neuropilin 2 (NRP2) | distal-less homeobox 5 (DLX5) |
| SERPINE1 | serpin family E member 1 (SERPINE1) | leucine rich repeat containing G protein-coupled receptor 4 (LGR4) |
| ZC3H12A | zinc finger CCCH-type containing 12A (ZC3H12A) | |
| GO:0008283: Cell Proliferation | 4-aminobutyrate aminotransferase (ABAT) | |
| E2F8 | E2F transcription factor 8 (E2F8) | BCL2 interacting protein 3 (BNIP3) |
| ERG | ERG, ETS transcription factor (ERG) | carbonic anhydrase 9 (CA9) |
| ROS1 | ROS proto-oncogene 1, receptor tyrosine kinase (ROS1) | cytochrome P450 family 1 subfamily A member 1 (CYP1A1) |
| ALK | anaplastic lymphoma receptor tyrosine kinase (ALK) | egl-9 family hypoxia inducible factor 3 (EGLN3) |
| AXIN2 | axin 2 (AXIN2) | lysyl oxidase like 2 (LOXL2) |
| CDC25A | cell division cycle 25A (CDC25A) | mucin 1, cell surface associated (MUC1) |
| CSPG4 | chondroitin sulfate proteoglycan 4 (CSPG4) | periostin (POSTN) |
| CYP1A1 | cytochrome P450 family 1 subfamily A member 1 (CYP1A1) | transforming growth factor beta 3 (TGFB3) |
| DLX5 | distal-less homeobox 5 (DLX5) | |
| FSCN1 | fascin actin-bundling protein 1 (FSCN1) | |
| FGF5 | fibroblast growth factor 5 (FGF5) | |
| GRPR | gastrin releasing peptide receptor (GRPR) | |
| MYH10 | myosin heavy chain 10 (MYH10) | |
| PDK1 | pyruvate dehydrogenase kinase 1 (PDK1) | |
| UHRF1 | ubiquitin like with PHD and ring finger domains 1 (UHRF1) | |
| GO:0016049: Cell Growth | ||
| ROS1 | ROS proto-oncogene 1, receptor tyrosine kinase (ROS1) | |
| EDN1 | endothelin 1 (EDN1) | |
| IL7R | interleukin 7 receptor (IL7R) | |
| NDNF | neuron-derived neurotrophic factor (NDNF) | |
| TGFB3 | transforming growth factor beta 3 (TGFB3) | |
| GO:0006915: Apoptotic Process | GO:0008152: Metabolic Process |
| BCL2 binding component 3 (BBC3) | 1-acylglycerol-3-phosphate O-acyltransferase 2 (AGPAT2) |
| DCC netrin 1 receptor (DCC) | UDP glucuronosyltransferase family 1 member A1 (UGT1A1) |
| PYD and CARD domain-containing (PYCARD) | UDP glucuronosyltransferase family 1 member A10 (UGT1A10) |
| TNF receptor-associated factor 5 (TRAF5) | UDP glucuronosyltransferase family 1 member A3 (UGT1A3) |
| XIAP-associated factor 1 (XAF1) | UDP glucuronosyltransferase family 1 member A4 (UGT1A4) |
| Brain-expressed X-linked 2 (BEX2) | UDP glucuronosyltransferase family 1 member A5 (UGT1A5) |
| caspase 1 (CASP1) | UDP glucuronosyltransferase family 1 member A6 (UGT1A6) |
| cathepsin H (CTSH) | UDP glucuronosyltransferase family 1 member A7 (UGT1A7) |
| complement C5a receptor 1 (C5AR1) | UDP glucuronosyltransferase family 1 member A8 (UGT1A8) |
| engulfment and cell motility 1 (ELMO1) | UDP glucuronosyltransferase family 1 member A9 (UGT1A9) |
| interleukin 1 beta (IL1B) | acyl-CoA synthetase medium-chain family member 5 (ACSM5) |
| mitogen-activated protein kinase kinase 6 (MAP2K6) | acyl-CoA synthetase short-chain family member 1 (ACSS1) |
| nuclear receptor subfamily 4 group A member 1 (NR4A1) | acyl-CoA synthetase short-chain family member 3 (ACSS3) |
| phorbol-12-myristate-13-acetate-induced protein 1 (PMAIP1) | glutathione S-transferase mu 5 (GSTM5) |
| secreted frizzled related protein 2 (SFRP2) | haloacid dehalogenase-like hydrolase domain-containing 3 (HDHD3) |
| tyrosyl-tRNA synthetase (YARS) | lipase E, hormone sensitive type (LIPE) |
| mannosidase alpha class 1C member 1 (MAN1C1) | |
| GO:0072332: Intrinsic Apoptotic Signaling Pathway by p53 Class Mediator | |
| PERP, TP53 apoptosis effector (PERP) | GO:0007155: Cell Adhesion |
| PYD and CARD domain-containing (PYCARD) | CD22 molecule (CD22) |
| phorbol-12-myristate-13-acetate-induced protein 1 (PMAIP1) | CD9 molecule (CD9) |
| zinc finger matrin-type 1 (ZMAT1) | EPH receptor A4 (EPHA4) |
| zinc finger protein 385D (ZNF385D) | adhesion G protein-coupled receptor G1 (ADGRG1) |
| amelotin (AMTN) | |
| GO:2000406: Positive Regulation of T Cell Migration | basal cell adhesion molecule (Lutheran blood group) (BCAM) |
| PYD and CARD domain-containing (PYCARD) | brevican (BCAN) |
| TNF receptor superfamily member 14 (TNFRSF14) | cadherin 11 (CDH11) |
| integrin subunit alpha 4 (ITGA4) | collagen type IV alpha 6 chain (COL4A6) |
| fasciculation and elongation protein zeta 1 (FEZ1) | |
| GO:0002457: T Cell Antigen Processing and Presentation | fibulin 7 (FBLN7) |
| intercellular adhesion molecule 1 (ICAM1) | hemicentin 2 (HMCN2) |
| raftlin, lipid raft linker 1 (RFTN1) | hyaluronan synthase 1 (HAS1) |
| integrin subunit alpha 11 (ITGA11) | |
| GO:0002282: Microglial Cell Activation Involved in Immune Response | integrin subunit alpha 2 (ITGA2) |
| interleukin 33 (IL33) | integrin subunit alpha 4 (ITGA4) |
| toll like receptor 3 (TLR3) | integrin subunit alpha L (ITGAL) |
| integrin subunit beta 8 (ITGB8) | |
| GO:0007165: Signal Transduction | intercellular adhesion molecule 1 (ICAM1) |
| ArfGAP with RhoGAP domain, ankyrin repeat and PH domain 2 (ARAP2) | junction plakoglobin (JUP) |
| G kinase anchoring protein 1 (GKAP1) | laminin subunit alpha 2 (LAMA2) |
| G protein subunit gamma 11 (GNG11) | laminin subunit alpha 3 (LAMA3) |
| GULP, engulfment adaptor PTB domain-containing 1 (GULP1) | ninjurin 1 (NINJ1) |
| KIT proto-oncogene receptor tyrosine kinase (KIT) | protein kinase C epsilon (PRKCE) |
| MX dynamin like GTPase 1 (MX1) | protein kinase, X-linked (PRKX) |
| NDP, norrin cystine knot growth factor (NDP) | protocadherin 17 (PCDH17) |
| NLR family pyrin domain-containing 12 (NLRP12) | sphingosine-1-phosphate receptor 1 (S1PR1) |
| NLR family pyrin domain-containing 3 (NLRP3) | trophoblast glycoprotein (TPBG) |
| PYD and CARD domain-containing (PYCARD) | versican (VCAN) |
| Ras association domain family member 9 (RASSF9) | |
| Rho family GTPase 2 (RND2) | GO:0045746: Negative Regulation Of Notch Signaling Pathway |
| SPARC-related modular calcium binding 1 (SMOC1) | ChaC glutathione-specific gamma-glutamylcyclotransferase 1 (CHAC1) |
| TNF receptor-associated factor 5 (TRAF5) | MAGE family member A1 (MAGEA1) |
| TNF receptor superfamily member 11b (TNFRSF11B) | Hes-related family bHLH transcription factor with YRPW motif 1 (HEY1) |
| amyloid beta precursor protein binding family B member 1 interacting protein (APBB1IP) | maternally expressed 3 (non-protein coding) (MEG3) |
| androgen receptor (AR) | |
| basal cell adhesion molecule (Lutheran blood group) (BCAM) | GO:0010759: Positive Regulation of Macrophage Chemotaxis |
| calcitonin-related polypeptide beta (CALCB) | chemerin chemokine-like receptor 1 (CMKLR1) |
| caspase 1 (CASP1) | complement C5a receptor 1 (C5AR1) |
| chimerin 1 (CHN1) | tumor necrosis factor superfamily member 18 (TNFSF18) |
| complement C5a receptor 1 (C5AR1) | |
| fibroblast growth factor 18 (FGF18) | GO:0006351: Transcription, DNA-Templated |
| fibroblast growth factor 7 (FGF7) | CREB3 regulatory factor (CREBRF) |
| growth differentiation factor 15 (GDF15) | DNA damage inducible transcript 3 (DDIT3) |
| inositol-trisphosphate 3-kinase A (ITPKA) | E2F transcription factor 7 (E2F7) |
| insulin like growth factor binding protein 1 (IGFBP1) | HKR1, GLI-Kruppel zinc finger family member (HKR1) |
| insulin like growth factor binding protein 5 (IGFBP5) | Kruppel-like factor 2 (KLF2) |
| integrin subunit alpha L (ITGAL) | Kruppel-like factor 9 (KLF9) |
| interleukin 1 beta (IL1B) | MAGE family member A1 (MAGEA1) |
| interleukin 15 receptor subunit alpha (IL15RA) | MAX dimerization protein 1 (MXD1) |
| junction plakoglobin (JUP) | MLX interacting protein like (MLXIPL) |
| mitogen-activated protein kinase 10 (MAPK10) | NLR family pyrin domain-containing 3 (NLRP3) |
| mitogen-activated protein kinase kinase 6 (MAP2K6) | RAR related orphan receptor B (RORB) |
| nuclear receptor subfamily 2 group F member 1 (NR2F1) | SATB homeobox 1 (SATB1) |
| nuclear receptor subfamily 4 group A member 1 (NR4A1) | T-box 3 (TBX3) |
| nuclear receptor subfamily 4 group A member 2 (NR4A2) | TGFB-induced factor homeobox 2 like, X-linked (TGIF2LX) |
| phosphodiesterase 10A (PDE10A) | ZFP14 zinc finger protein (ZFP14) |
| phosphodiesterase 1A (PDE1A) | androgen receptor (AR) |
| phosphodiesterase 4D (PDE4D) | endoplasmic reticulum to nucleus signaling 1 (ERN1) |
| placental growth factor (PGF) | forkhead box P2 (FOXP2) |
| plasminogen activator, urokinase (PLAU) | hair growth associated (HR) |
| protein kinase AMP-activated catalytic subunit alpha 2 (PRKAA2) | hes-related family bHLH transcription factor with YRPW motif 1 (HEY1) |
| protein kinase C epsilon (PRKCE) | homeobox B7 (HOXB7) |
| protein kinase C zeta (PRKCZ) | homeobox B8 (HOXB8) |
| ras-related dexamethasone induced 1 (RASD1) | homeobox B9 (HOXB9) |
| ribosomal protein S6 kinase A2 (RPS6KA2) | interleukin 33 (IL33) |
| ribosomal protein S6 kinase A6 (RPS6KA6) | iroquois homeobox 5 (IRX5) |
| secreted and transmembrane 1 (SECTM1) | leucine zipper tumor suppressor 1 (LZTS1) |
| single Ig and TIR domain-containing (SIGIRR) | mitogen-activated protein kinase kinase 6 (MAP2K6) |
| thrombomodulin (THBD) | myelin expression factor 2 (MYEF2) |
| toll like receptor 3 (TLR3) | neuronal PAS domain protein 2 (NPAS2) |
| transducin-like enhancer of split 1 (TLE1) | nuclear factor I B (NFIB) |
| tumor necrosis factor superfamily member 10 (TNFSF10) | nuclear protein 1, transcriptional regulator (NUPR1) |
| tumor necrosis factor superfamily member 13b (TNFSF13B) | nuclear receptor coactivator 7 (NCOA7) |
| tumor necrosis factor superfamily member 18 (TNFSF18) | nuclear receptor subfamily 2 group F member 1 (NR2F1) |
| tyrosyl-tRNA synthetase (YARS) | nuclear receptor subfamily 4 group A member 1 (NR4A1) |
| unc-5 netrin receptor B (UNC5B) | nuclear receptor subfamily 4 group A member 2 (NR4A2) |
| unc-5 netrin receptor C (UNC5C) | protein kinase AMP-activated catalytic subunit alpha 2 (PRKAA2) |
| very low density lipoprotein receptor (VLDLR) | thyroid hormone receptor beta (THRB) |
| transducin-like enhancer of split 1 (TLE1) | |
| tribbles pseudokinase 3 (TRIB3) | |
| tumor protein p63 (TP63) | |
| twist family bHLH transcription factor 2 (TWIST2) | |
| vestigial-like family member 2 (VGLL2) | |
| visual system homeobox 1 (VSX1) | |
| zinc finger and SCAN domain containing 16 (ZSCAN16) | |
| zinc finger family member 788 (ZNF788) | |
| zinc finger protein 117 (ZNF117) | |
| zinc finger protein 138 (ZNF138) | |
| zinc finger protein 20 (ZNF20) | |
| zinc finger protein 273 (ZNF273) | |
| zinc finger protein 28 (ZNF28) | |
| zinc finger protein 30 (ZNF30) | |
| zinc finger protein 320 (ZNF320) | |
| zinc finger protein 354B (ZNF354B) | |
| zinc finger protein 396 (ZNF396) | |
| zinc finger protein 415 (ZNF415) | |
| zinc finger protein 419 (ZNF419) | |
| zinc finger protein 433 (ZNF433) | |
| zinc finger protein 44 (ZNF44) | |
| zinc finger protein 442 (ZNF442) | |
| zinc finger protein 443 (ZNF443) | |
| zinc finger protein 468 (ZNF468) | |
| zinc finger protein 521 (ZNF521) | |
| zinc finger protein 525 (ZNF525) | |
| zinc finger protein 528 (ZNF528) | |
| zinc finger protein 549 (ZNF549) | |
| zinc finger protein 563 (ZNF563) | |
| zinc finger protein 572 (ZNF572) | |
| zinc finger protein 577 (ZNF577) | |
| zinc finger protein 625 (ZNF625) | |
| zinc finger protein 649 (ZNF649) | |
| zinc finger protein 674 (ZNF674) | |
| zinc finger protein 680 (ZNF680) | |
| zinc finger protein 71 (ZNF71) | |
| zinc finger protein 761 (ZNF761) | |
| zinc finger protein 765 (ZNF765) | |
| zinc finger protein 792 (ZNF792) | |
| zinc finger protein 799 (ZNF799) | |
| zinc finger protein 806 (ZNF806) | |
| zinc finger protein 816 (ZNF816) | |
| zinc finger protein 83 (ZNF83) | |
| zinc finger protein 845 (ZNF845) | |
| zinc finger protein 85 (ZNF85) | |
| zinc finger protein 883 (ZNF883) | |
| zinc finger protein 888 (ZNF888) | |
| zinc finger with KRAB and SCAN domains 7 (ZKSCAN7) |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nguyen, H.S.; Shabani, S.; Awad, A.J.; Kaushal, M.; Doan, N. Molecular Markers of Therapy-Resistant Glioblastoma and Potential Strategy to Combat Resistance. Int. J. Mol. Sci. 2018, 19, 1765. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19061765
Nguyen HS, Shabani S, Awad AJ, Kaushal M, Doan N. Molecular Markers of Therapy-Resistant Glioblastoma and Potential Strategy to Combat Resistance. International Journal of Molecular Sciences. 2018; 19(6):1765. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19061765
Chicago/Turabian StyleNguyen, Ha S., Saman Shabani, Ahmed J. Awad, Mayank Kaushal, and Ninh Doan. 2018. "Molecular Markers of Therapy-Resistant Glioblastoma and Potential Strategy to Combat Resistance" International Journal of Molecular Sciences 19, no. 6: 1765. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19061765