STIM1 Knockout Enhances PDGF-Mediated Ca2+ Signaling through Upregulation of the PDGFR–PLCγ–STIM2 Cascade


Abstract
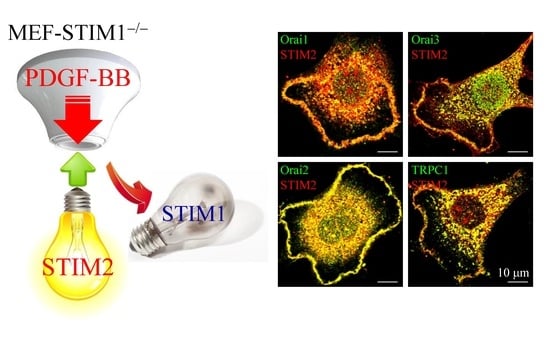
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
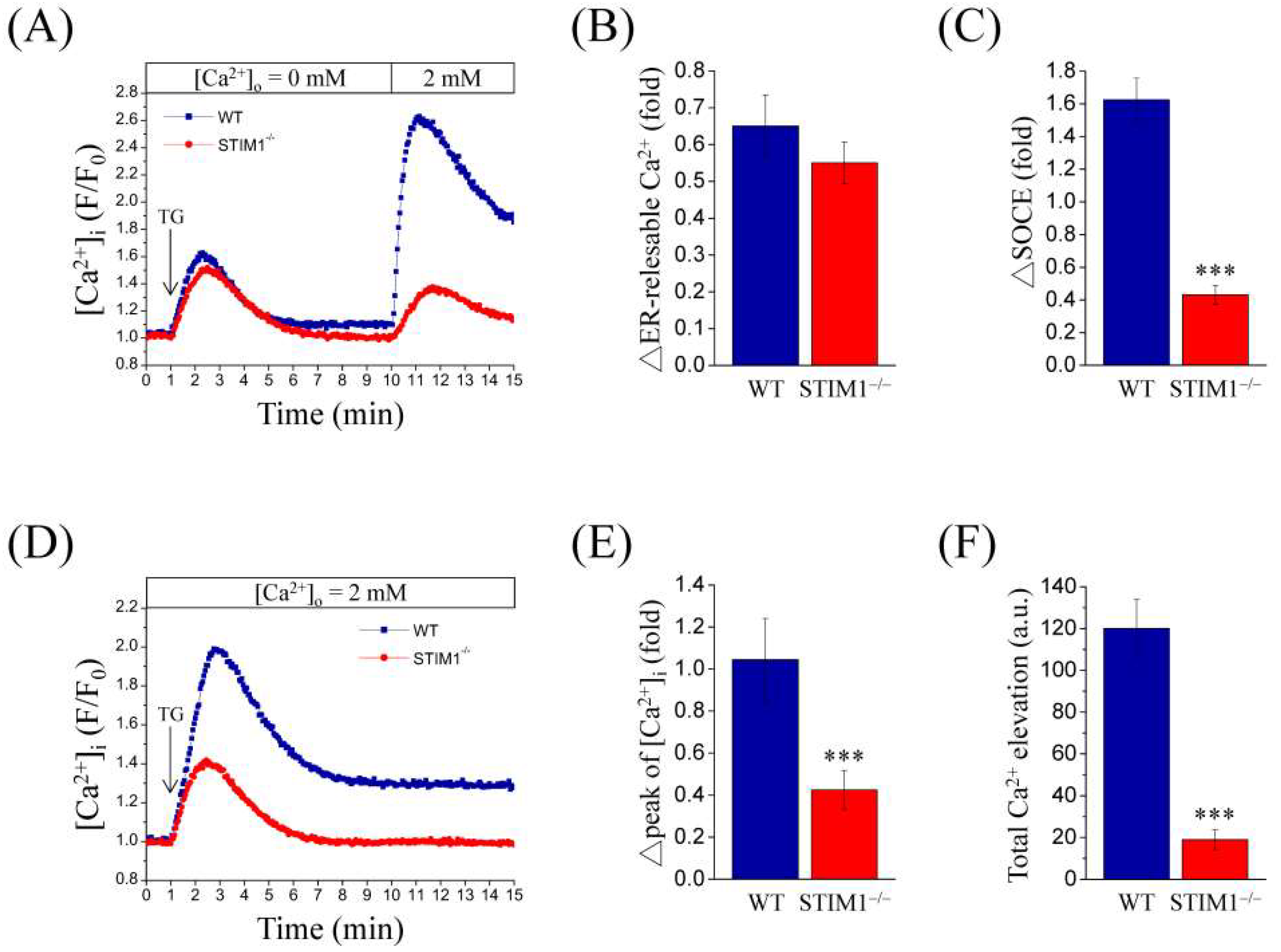
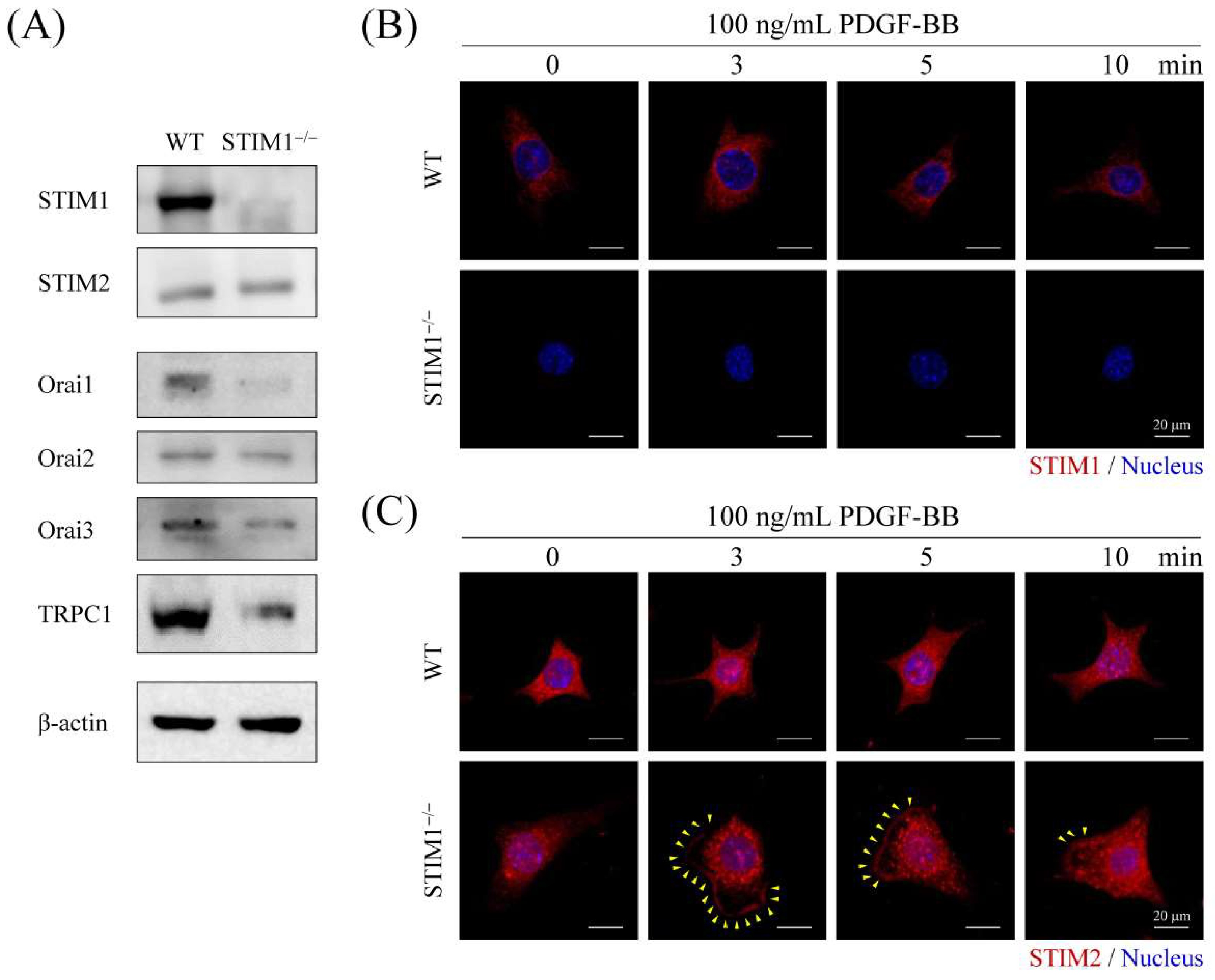
2.1. Stromal Interaction Molecule 1 (STIM1) Knockout Represses Store-Operated Ca2+ Entry (SOCE) in Mouse Embryonic Fibroblasts (MEF) Cells
2.2. PDGF-BB Induced Significant Ca2+ Elevation in STIM1 Knockout MEF Cells
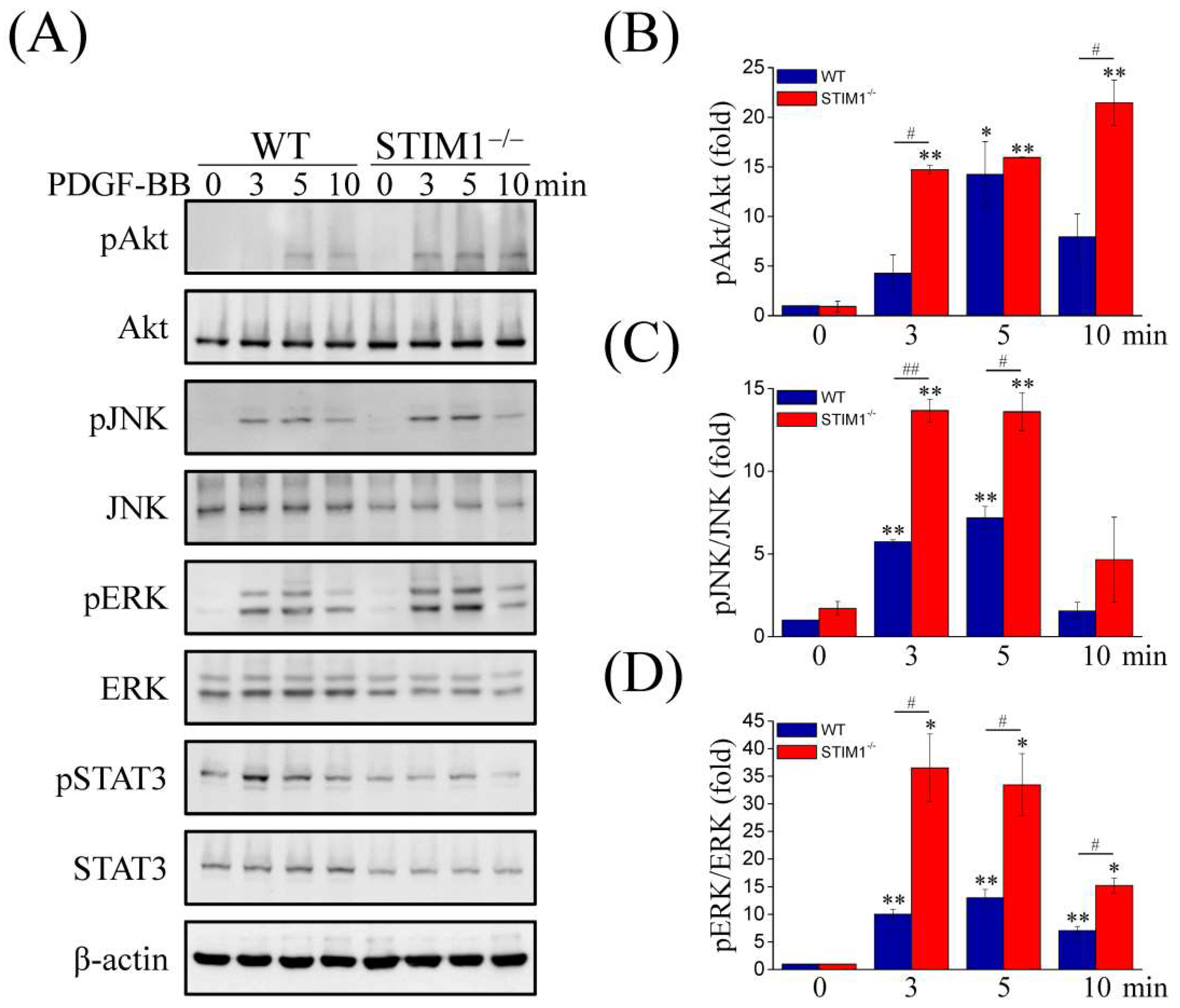
2.3. Upregulation and Activation of PDGFRα, PDGFRβ, and Phospholipase C Gamma (PLCγ) in MEF-STIM1−/− Cells
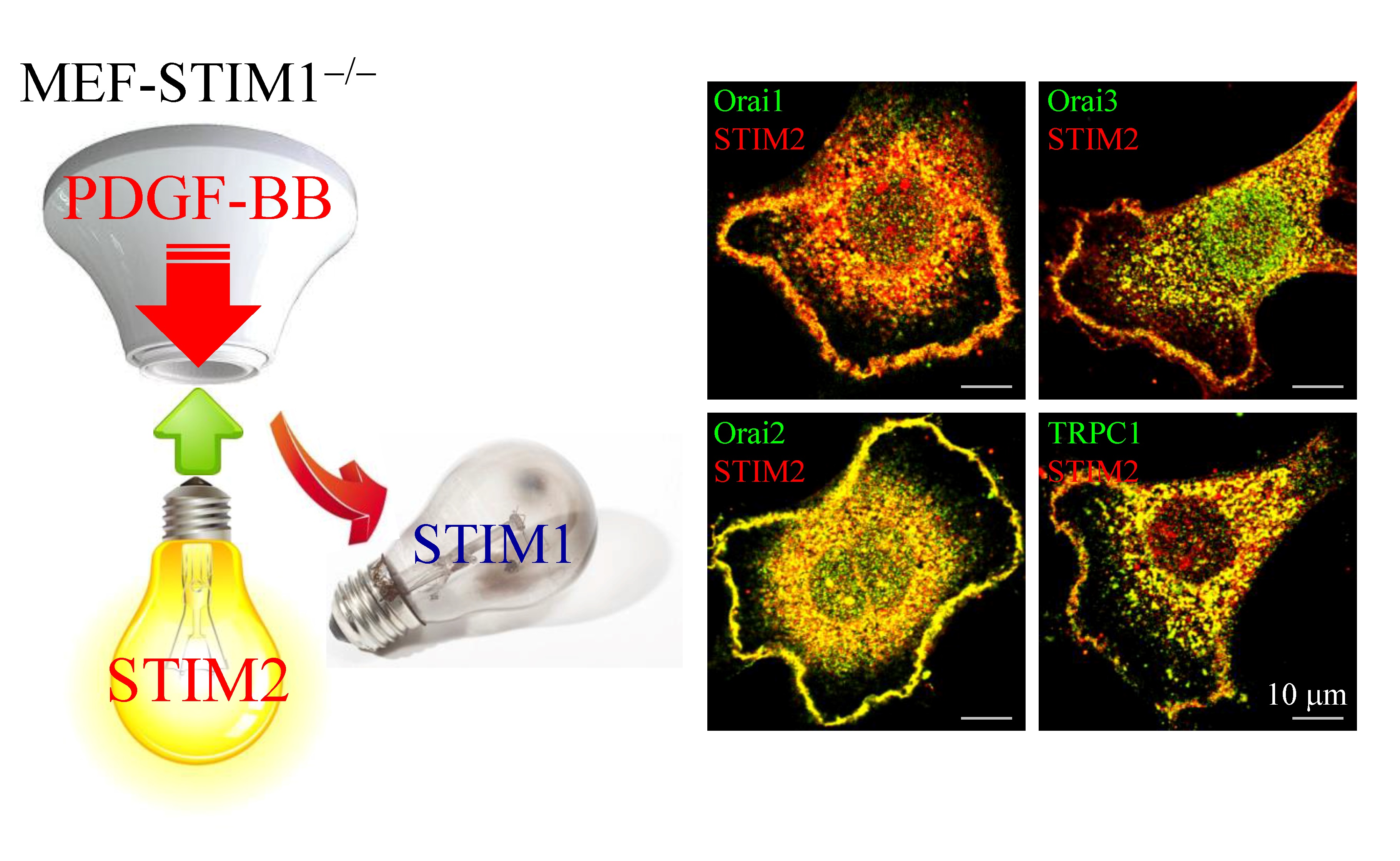
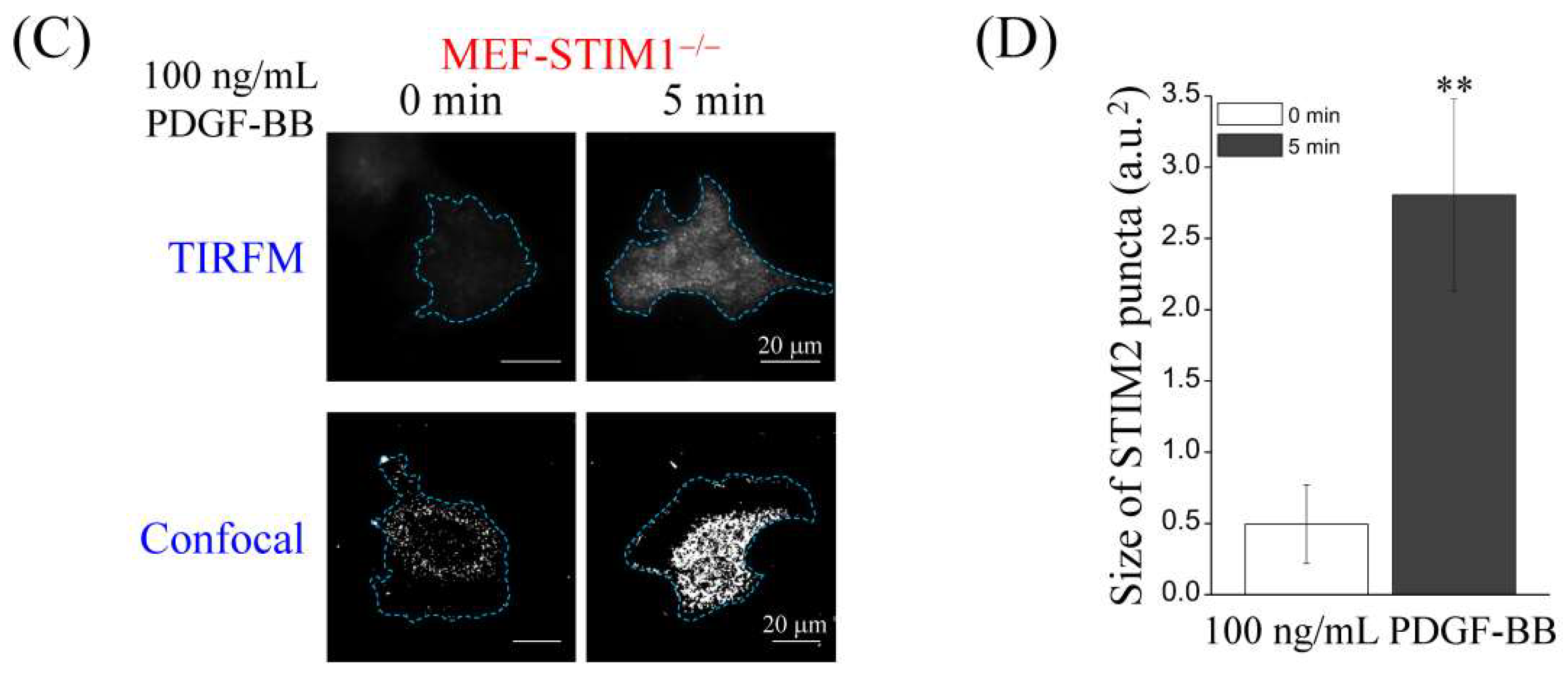
2.4. PDGF-BB-Mediated ER Store-Depletion Activates STIM2 Translocation and Puncta Formation
2.5. SOCE Inhibitors Decrease PDGF-BB-Induced Ca2+ Elevation in MEF-STIM1−/− Cells
2.6. PDGFR–PLCγ–STIM2 Signaling Induces Cell Migration in MEF-STIM1−/− Cells.
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Reagents
4.2. Single-Cell Ca2+ Measurement
4.3. Immunoblotting Analyses
4.4. Immunofluorescence Staining and Confocal Microscopy
4.5. Wound Healing Assay
4.6. Cell Proliferation Analysis
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| MEF | Mouse embryonic fibroblasts |
| PDGF | Platelet-derived growth factor |
| PDGFR | Platelet-derived growth factor receptor |
| SOCE | Store-operated Ca2+ entry |
| STIM1 | Stromal interaction molecule 1 |
| Orai1 | Ca2+ release-activated calcium modulator 1 |
| TRPC1 | Transient receptor potential canonical 1 |
| PLCγ | Phospholipase C gamma |
| RTK | Receptor tyrosine kinase |
| CREB | cAMP response element binding protein |
References
- Berchtold, M.W.; Villalobo, A. The many faces of calmodulin in cell proliferation, programmed cell death, autophagy, and cancer. Biochim. Biophys. Acta 2014, 1843, 398–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berridge, M.J.; Bootman, M.D.; Lipp, P. Calcium—A life and death signal. Nature 1998, 395, 645–648. [Google Scholar] [CrossRef] [PubMed]
- Boulware, M.J.; Marchant, J.S. Timing in cellular Ca2+ signaling. Curr. Biol. 2008, 18, R769–R776. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Stefan, M.I.; Le Novère, N. Calcium input frequency, duration and amplitude differentially modulate the relative activation of calcineurin and CaMKII. PLoS ONE 2012, 7, e43810. [Google Scholar] [CrossRef] [PubMed]
- Putney, J.W. Pharmacology of store-operated calcium channels. Mol. Interv. 2010, 10, 209. [Google Scholar] [CrossRef] [PubMed]
- Smyth, J.T.; Hwang, S.Y.; Tomita, T.; DeHaven, W.I.; Mercer, J.C.; Putney, J.W. Activation and regulation of store-operated calcium entry. J. Cell. Mol. Med. 2010, 14, 2337–2349. [Google Scholar] [CrossRef] [PubMed]
- Targos, B.; Barańska, J.; Pomorski, P. Store-operated calcium entry in physiology and pathology of mammalian cells. Acta Biochim. Pol. 2005, 52, 397–409. [Google Scholar] [PubMed]
- Cheng, K.T.; Ong, H.L.; Liu, X.; Ambudkar, I.S. Contribution of TRPC1 and Orai1 to Ca2+ entry activated by store depletion. Adv. Exp. Med. Biol. 2011, 704, 435–449. [Google Scholar] [PubMed]
- Ambudkar, I.S.; Ong, H.L.; Liu, X.; Bandyopadhyay, B.C.; Cheng, K.T. TRPC1: The link between functionally distinct store-operated calcium channels. Cell Calcium 2007, 42, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Park, C.Y.; Hoover, P.J.; Mullins, F.M.; Bachhawat, P.; Covington, E.D.; Rauser, S.; Walz, T.; Garcia, K.C.; Dolmetsch, R.E.; Lewis, R.S. STIM1 clusters and activates CRAC channels via direct binding of a cytosolic domain to Orai1. Cell 2009, 136, 876–890. [Google Scholar] [CrossRef] [PubMed]
- Daskoulidou, N.; Zeng, B.; Berglund, L.M.; Jiang, H.; Chen, G.L.; Kotova, O.; Bhandari, S.; Ayoola, J.; Griffin, S.; Atkin, S.L.; et al. High glucose enhances store-operated calcium entry by upregulating ORAI/STIM via calcineurin-NFAT signalling. J. Mol. Med. 2015, 93, 511–521. [Google Scholar] [CrossRef] [PubMed]
- Mancarella, S.; Potireddy, S.; Wang, Y.; Gao, H.; Gandhirajan, R.K.; Autieri, M.; Scalia, R.; Cheng, Z.; Wang, H.; Madesh, M.; et al. Targeted STIM deletion impairs calcium homeostasis, NFAT activation, and growth of smooth muscle. FASEB J. 2013, 27, 893–906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pulver, R.A.; Rose-Curtis, P.; Roe, M.W.; Wellman, G.C.; Lounsbury, K.M. Store-operated Ca2+ entry activates the CREB transcription factor in vascular smooth muscle. Circ. Res. 2004, 94, 1351–1358. [Google Scholar] [CrossRef] [PubMed]
- Srikanth, S.; Gwack, Y. Orai1-NFAT signalling pathway triggered by T cell receptor stimulation. Mol. Cells 2013, 35, 182–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Somasundaram, A.; Shum, A.K.; McBride, H.J.; Kessler, J.A.; Feske, S.; Miller, R.J.; Prakriya, M. Store-operated CRAC channels regulate gene expression and proliferation in neural progenitor cells. J. Neurosci. 2014, 34, 9107–9123. [Google Scholar] [CrossRef] [PubMed]
- Umemura, M.; Baljinnyam, E.; Feske, S.; De Lorenzo, M.S.; Xie, L.H.; Feng, X.; Oda, K.; Makino, A.; Fujita, T.; Yokoyama, U.; et al. Store-operated Ca2+ entry (SOCE) regulates melanoma proliferation and cell migration. PLoS ONE 2014, 9, e89292. [Google Scholar] [CrossRef] [PubMed]
- Bisaillon, J.M.; Motiani, R.K.; Gonzalez-Cobos, J.C.; Potier, M.; Halligan, K.E.; Alzawahra, W.F.; Barroso, M.; Singer, H.A.; Jourd’heuil, D.; Trebak, M. Essential role for STIM1/Orai1-mediated calcium influx in PDGF-induced smooth muscle migration. Am. J. Physiol. Cell Physiol. 2010, 298, C993–C1005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michaelis, M.; Nieswandt, B.; Stegner, D.; Eilers, J.; Kraft, R. STIM1, STIM2, and Orai1 regulate store-operated calcium entry and purinergic activation of microglia. Glia 2015, 63, 652–663. [Google Scholar] [CrossRef] [PubMed]
- Bhardwaj, R.; Müller, H.M.; Nickel, W.; Seedorf, M. Oligomerization and Ca2+/calmodulin control binding of the ER Ca2+-sensors STIM1 and STIM2 to plasma membrane lipids. Biosci. Rep. 2013, 33, e00077. [Google Scholar] [CrossRef] [PubMed]
- Rana, A.; Yen, M.; Sadaghiani, A.M.; Malmersjö, S.; Park, C.Y.; Dolmetsch, R.E.; Lewis, R.S. Alternative splicing converts STIM2 from an activator to an inhibitor of store-operated calcium channels. J. Cell Biol. 2015, 209, 653–669. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Mancarella, S.; Wang, Y.; Yue, C.; Ritchie, M.; Gill, D.L.; Soboloff, J. The short N-terminal domains of STIM1 and STIM2 control the activation kinetics of Orai1 channels. J. Biol. Chem. 2009, 284, 19164–19168. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.W.; Chen, Y.F.; Chen, Y.T.; Chiu, W.T.; Shen, M.R. The STIM1–Orai1 pathway of store-operated Ca2+ entry controls the checkpoint in cell cycle G1/S transition. Sci. Rep. 2016, 6, 22142. [Google Scholar] [CrossRef] [PubMed]
- Henke, N.; Albrecht, P.; Pfeiffer, A.; Toutzaris, D.; Zanger, K.; Methner, A. Stromal interaction molecule 1 (STIM1) is involved in the regulation of mitochondrial shape and bioenergetics and plays a role in oxidative stress. J. Biol. Chem. 2012, 287, 42042–42052. [Google Scholar] [CrossRef] [PubMed]
- Brandman, O.; Liou, J.; Park, W.S.; Meyer, T. STIM2 is a feedback regulator that stabilizes basal cytosolic and endoplasmic reticulum Ca2+ levels. Cell 2007, 131, 1327–1339. [Google Scholar] [CrossRef] [PubMed]
- DeHaven, W.I.; Smyth, J.T.; Boyles, R.R.; Putney, J.W., Jr. Calcium inhibition and calcium potentiation of Orai1, Orai2, and Orai3 calcium release-activated calcium channels. J. Biol. Chem. 2007, 282, 17548–17556. [Google Scholar] [CrossRef] [PubMed]
- Parekh, A.B. Store-operated CRAC channels: Function in health and disease. Nat. Rev. Drug Discov. 2010, 9, 399–410. [Google Scholar] [CrossRef] [PubMed]
- Soboloff, J.; Rothberg, B.S.; Madesh, M.; Gill, D.L. STIM proteins: Dynamic calcium signal transducers. Nat. Rev. Mol. Cell Biol. 2012, 13, 549–565. [Google Scholar] [CrossRef] [PubMed]
- Rosado, J.A.; Diez, R.; Smani, T.; Jardín, I. STIM and Orai1 variants in store-operated calcium entry. Front. Pharmacol. 2015, 6, 325. [Google Scholar] [CrossRef] [PubMed]
- Cheng, K.T.; Ong, H.L.; Liu, X.; Ambudkar, I.S. Contribution and regulation of TRPC channels in store-operated Ca2+ entry. Curr. Top. Membr. 2013, 71, 149–179. [Google Scholar] [PubMed]
- De Souza, L.B.; Ong, H.L.; Liu, X.; Ambudkar, I.S. Fast endocytic recycling determines TRPC1–STIM1 clustering in ER–PM junctions and plasma membrane function of channel. Biochim. Biophys. Acta 2015, 1853, 2709–2721. [Google Scholar] [CrossRef] [PubMed]
- Salido, G.M.; Jardín, I.; Rosado, J.A. The TRPC ion channels: Association with Orai1 and STIM1 proteins and participation in capacitative and non-capacitative calcium entry. Adv. Exp. Med. Biol. 2011, 704, 413–433. [Google Scholar] [PubMed]
- Ma, G.; Wei, M.; He, L.; Liu, C.; Wu, B.; Zhang, S.L.; Jing, J.; Liang, X.; Senes, A.; Tan, P.; et al. Inside-out Ca2+ signalling prompted by STIM1 conformational switch. Nat. Commun. 2015, 6, 7826. [Google Scholar] [CrossRef] [PubMed]
- Ong, H.L.; Cheng, K.T.; Liu, X.; Bandyopadhyay, B.C.; Paria, B.C.; Soboloff, J.; Pani, B.; Gwack, Y.; Srikanth, S.; Singh, B.B.; et al. Dynamic assembly of TRPC1-STIM1-Orai1 ternary complex is involved in store-operated calcium influx. Evidence for similarities in store-operated and calcium release-activated calcium channel components. J. Biol. Chem. 2007, 282, 9105–9116. [Google Scholar] [CrossRef] [PubMed]
- Bowen-Pope, D.F.; Raines, E.W. History of discovery: Platelet-derived growth factor. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 2397–2401. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y. Multifarious functions of PDGFs and PDGFRs in tumor growth and metastasis. Trends Mol. Med. 2013, 19, 460–473. [Google Scholar] [CrossRef] [PubMed]
- Raica, M.; Cimpean, A.M. Platelet-derived growth factor (PDGF)/PDGF receptors (PDGFR) axis as target for antitumor and antiangiogenic therapy. Pharmaceuticals 2010, 3, 572–599. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.S.; Wang, M.J.; Jayakumar, T.; Chou, D.S.; Ko, C.Y.; Hsu, M.J.; Hsieh, C.Y. Antiproliferative activity of hinokitiol, a tropolone derivative, is mediated via the inductions of p-JNK and p-PLCγ1 signaling in PDGF-BB-stimulated vascular smooth muscle cells. Molecules 2015, 20, 8198–8212. [Google Scholar] [CrossRef] [PubMed]
- Andrae, J.; Gallini, R.; Betsholtz, C. Role of platelet-derived growth factors in physiology and medicine. Genes Dev. 2008, 22, 1276–1312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donovan, J.; Shiwen, X.; Norman, J.; Abraham, D. Platelet-derived growth factor alpha and beta receptors have overlapping functional activities towards fibroblasts. Fibrogenesis Tissue Repair 2013, 6, 10. [Google Scholar] [CrossRef] [PubMed]
- Fretto, L.J.; Snape, A.J.; Tomlinson, J.E.; Seroogy, J.J.; Wolf, D.L.; LaRochelle, W.J.; Giese, N.A. Mechanism of platelet-derived growth factor (PDGF) AA, AB, and BB binding to alpha and beta PDGF receptor. J. Biol. Chem. 1993, 268, 3625–3631. [Google Scholar] [PubMed]
- Sachinidis, A.; Locher, R.; Hoppe, J.; Vetter, W. The platelet-derived growth factor isomers, PDGF-AA, PDGF-AB and PDGF-BB, induce contraction of vascular smooth muscle cells by different intracellular mechanisms. FEBS Lett. 1990, 275, 95–98. [Google Scholar] [CrossRef] [Green Version]
- Bornfeldt, K.E.; Raines, E.W.; Graves, L.M.; Skinner, M.P.; Krebs, E.G.; Ross, R. Platelet-derived growth factor. Distinct signal transduction pathways associated with migration versus proliferation. Ann. N. Y. Acas. Sci. 1995, 766, 416–430. [Google Scholar] [CrossRef]
- Pinzani, M. PDGF and signal transduction in hepatic stellate cells. Front. Biosci. 2002, 7, d1720–d1726. [Google Scholar] [CrossRef] [PubMed]
- Heldman, A.W.; Kandzari, D.E.; Tucker, R.W.; Crawford, L.E.; Fearon, E.R.; Koblan, K.S.; Goldschmidt-Clermont, P.J. EJ-Ras inhibits phospholipase C gamma 1 but not actin polymerization induced by platelet-derived growth factor-BB via phosphatidylinositol 3-kinase. Circ. Res. 1996, 78, 312–321. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.; Duan, M.; Yang, L.; Buch, S. Platelet-derived growth factor-BB restores human immunodeficiency virus Tat-cocaine-mediated impairment of neurogenesis: Role of TRPC1 channels. J. Neurosci. 2012, 32, 9835–9847. [Google Scholar] [CrossRef] [PubMed]
- Spinelli, A.M.; González-Cobos, J.C.; Zhang, X.; Motiani, R.K.; Rowan, S.; Zhang, W.; Garrett, J.; Vincent, P.A.; Matrougui, K.; Singer, H.A. Airway smooth muscle STIM1 and Orai1 are upregulated in asthmatic mice and mediate PDGF-activated SOCE, CRAC currents, proliferation, and migration. Pflugers Arch. 2012, 464, 482–491. [Google Scholar] [CrossRef] [PubMed]
- Suganuma, N.; Ito, S.; Aso, H.; Kondo, M.; Sato, M.; Sokabe, M.; Hasegawa, Y. STIM1 regulates platelet-derived growth factor-induced migration and Ca2+ influx in human airway smooth muscle cells. PLoS ONE 2012, 7, e45056. [Google Scholar] [CrossRef] [PubMed]
- Ma, R.; Sansom, S.C. Epidermal growth factor activates store-operated calcium channels in human glomerular mesangial cells. J. Am. Soc. Nephrol. 2001, 12, 47–53. [Google Scholar] [PubMed]
- Banai, S.; Wolf, Y.; Golomb, G.; Pearle, A.; Waltenberger, J.; Fishbein, I.; Schneider, A.; Gazit, A.; Perez, L.; Huber, R.; et al. PDGF-receptor tyrosine kinase blocker AG1295 selectively attenuates smooth muscle cell growth in vitro and reduces neointimal formation after balloon angioplasty in swine. Circulation 1998, 97, 1960–1969. [Google Scholar] [CrossRef] [PubMed]
- Cecchetti, S.; Bortolomai, I.; Ferri, R.; Mercurio, L.; Canevari, S.; Podo, F.; Miotti, S.; Iorio, E. Inhibition of phosphatidylcholine-specific phospholipase C interferes with proliferation and survival of tumor initiating cells in squamous cell carcinoma. PLoS ONE 2015, 10, e0136120. [Google Scholar] [CrossRef] [PubMed]
- Mauban, J.R.; Zacharia, J.; Fairfax, S.; Wier, W.G. PC-PLC/sphingomyelin synthase activity plays a central role in the development of myogenic tone in murine resistance arteries. Am. J. Physiol. Heart Circ. Physiol. 2015, 308, H1517–H1524. [Google Scholar] [CrossRef] [PubMed]
- Vandenberghe, M.; Raphaël, M.; Lehen’kyi, V.; Gordienko, D.; Hastie, R.; Oddos, T.; Rao, A.; Hogan, P.G.; Skryma, R.; Prevarskaya, N. Orai1 calcium channel orchestrates skin homeostasis. Proc. Natl. Acad. Sci. USA 2013, 110, E4839–E4848. [Google Scholar] [CrossRef] [PubMed]
- Numaga-Tomita, T.; Putney, J.W. Role of STIM1- and Orai1-mediated Ca2+ entry in Ca2+-induced epidermal keratinocyte differentiation. J. Cell Sci. 2013, 126, 605–612. [Google Scholar] [CrossRef] [PubMed]
- Hooper, R.; Samakai, E.; Kedra, J.; Soboloff, J. Multifaceted roles of STIM proteins. Pflugers Arch. 2013, 465, 1383–1396. [Google Scholar] [CrossRef] [PubMed]









© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, T.-Y.; Lin, Y.-H.; Chang, H.-A.; Yeh, T.-Y.; Chang, Y.-H.; Chen, Y.-F.; Chen, Y.-C.; Li, C.-C.; Chiu, W.-T. STIM1 Knockout Enhances PDGF-Mediated Ca2+ Signaling through Upregulation of the PDGFR–PLCγ–STIM2 Cascade. Int. J. Mol. Sci. 2018, 19, 1799. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19061799
Huang T-Y, Lin Y-H, Chang H-A, Yeh T-Y, Chang Y-H, Chen Y-F, Chen Y-C, Li C-C, Chiu W-T. STIM1 Knockout Enhances PDGF-Mediated Ca2+ Signaling through Upregulation of the PDGFR–PLCγ–STIM2 Cascade. International Journal of Molecular Sciences. 2018; 19(6):1799. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19061799
Chicago/Turabian StyleHuang, Tzu-Yu, Yi-Hsin Lin, Heng-Ai Chang, Tzu-Ying Yeh, Ya-Han Chang, Yi-Fan Chen, Ying-Chi Chen, Chun-Chun Li, and Wen-Tai Chiu. 2018. "STIM1 Knockout Enhances PDGF-Mediated Ca2+ Signaling through Upregulation of the PDGFR–PLCγ–STIM2 Cascade" International Journal of Molecular Sciences 19, no. 6: 1799. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19061799