Systemic Inflammatory Response and Atherosclerosis: The Paradigm of Chronic Inflammatory Rheumatic Diseases

Abstract
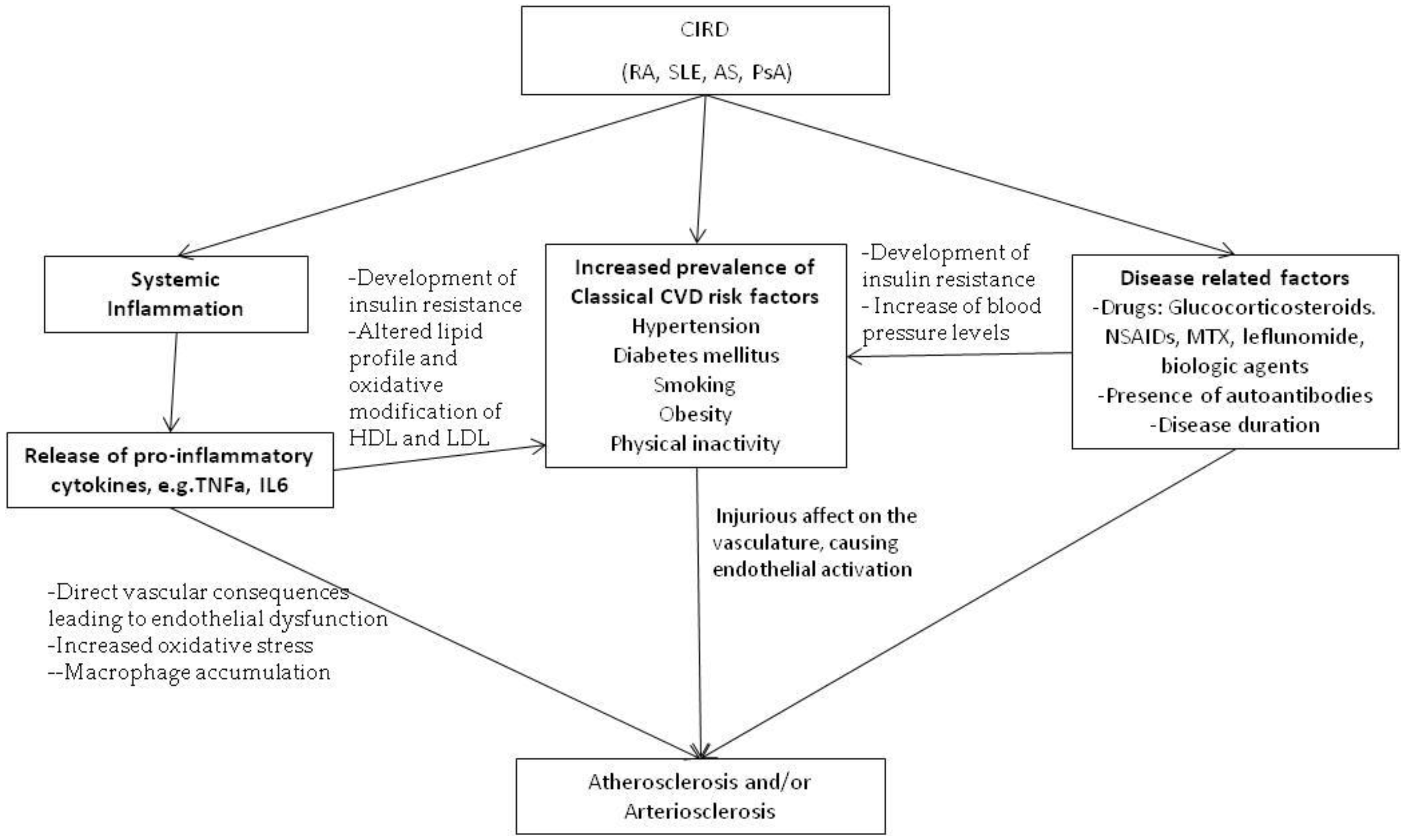
:1. Introduction
2. Search Strategy
3. Thematic Sections
3.1. Epidemiology
3.1.1. Rheumatoid Arthritis
3.1.2. Systemic Lupus Erythematosus and Antiphospholipid Syndrome
3.1.3. Ankylosing Spondylitis
3.2. Classical Cardiovascular Risk Factors and Metabolic Syndrome (MetS)
4. Immunological Mechanisms
4.1. Endothelial Dysfunction
4.1.1. Oxidative Stress
4.1.2. Innate Immunity, Toll Like Receptor (TLR) Signaling, and NLRP3 Inflammasome Activation
4.1.3. Macrophage Accumulation
4.1.4. Pro-Inflammatory Cytokines
5. What It All Means for Treatment of Patients with CIRD
6. Practical Implications
Author Contributions
Conflicts of Interest
Abbreviation
| ACPA | Anti-citrullinated protein/peptide antibodies |
| Aix | Augmentation index |
| APS | Antiphospholipid syndrome |
| AS | Ankylosing spondylitis |
| ASDAS | Ankylosing Spondylitis Disease Activity Score |
| BASDAI | Bath Ankylosing Spondylitis Disease Activity Index |
| BMI | Body mass index |
| cDMARDs | Conventional disease modifying antirheumatic drugs |
| CHD | Coronary heart disease |
| cIMT | Carotid intima-media thickness |
| CIRD | Chronic inflammatory rheumatic diseases |
| CRP | C-reactive protein |
| CV | Cardiovascular |
| CVD | Cardiovascular disease |
| DAMPs | Damage-associated molecular patterns |
| DM | Diabetes Mellitus |
| DR3 | Death-domain receptor 3 |
| ds-DNA | Double stranded deoxyribonucleic acid |
| e.g., | For example |
| EC | Endothelial cell |
| ESR | Erythrocyte sedimantation rate |
| EULAR | European League Against Rheumatism |
| FMD | Flow-mediated vasodilation |
| HDL | High-density lipoprotein |
| i.e., | Id est (that is) |
| ICAM | Intercellular adhension molecule |
| IFN | Interferon |
| IL | Interleukin |
| IR | Insulin resistance |
| LDL | Low-density lipoprotein |
| Lp(a) | Lipoprotein a |
| MetS | Metabolic syndrome |
| MI | Myocardial infarction |
| MTX | Methotrexate |
| NFkB | Nuclear Factor Kappa B |
| NLRP3 | NOD-like receptor family pyrin domain containing 3 |
| NO | Nitric oxide |
| NSAIDs | Non-steroid antiinflammatory drugs |
| oxLDL | Oxidised low-density lipoprotein |
| PAMPs | Pathogen-associated molecular patterns |
| PGI2 | Prostacyclin |
| PRRs | Pattern-recognition receptors |
| PsA | Psoriatic arthritis |
| PWV | Pulse wave velocity |
| RA | Rhaumatoid arthritis |
| RNA | Ribonucleic acid |
| ROS | Reactive oxygen species |
| SLE | Systemic Lupus Erythematosus |
| SLEDAI | Systemic Lupus Erythematosus Disease Activity Index |
| SLICC | Systemic Lupus International Collaborating Clinics/American College of Rheumatology |
| SpA | Spondyloarthropathies |
| TC | Triglycerides |
| TGF | Transforming growth factor |
| Th | T-helper |
| TLR | Toll like receptor |
| TM | Thrombomodulin |
| TNFa | Tumor necrosis factor-a |
| VCAM | Vascular endothelial adhension molecule |
References
- Peters, M.J.; Symmons, D.P.M.; McCarey, D.; Dijkmans, B.A.C.; Nicola, P.; Kvien, T.K.; McInnes, I.B.; Haentzschel, H.; Gonzalez-Gay, M.A.; Provan, S.; et al. EULAR evidence-based recommendations for cardiovascular risk management in patients with rheumatoid arthritis and other forms of inflammatory arthritis. Ann. Rheum. Dis. 2010, 69, 325–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aviña-Zubieta, J.A.; Choi, H.K.; Sadatsafavi, M.; Etminan, M.; Esdaile, J.M.; Lacaille, D. Risk of cardiovascular mortality in patients with rheumatoid arthritis: A meta-analysis of observational studies. Arthritis Care Res. 2008, 59, 1690–1697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dadoun, S.; Zeboulon-Ktorza, N.; Combescure, C.; Elhai, M.; Rozenberg, S.; Gossec, L.; Fautrel, B. Mortality in rheumatoid arthritis over the last fifty years: Systematic review and meta-analysis. Jt. Bone Spine 2013, 80, 29–33. [Google Scholar] [CrossRef] [PubMed]
- Agca, R.; Heslinga, S.C.; Rollefstad, S.; Heslinga, M.; McInnes, I.B.; Peters, M.J.L.; Kvien, T.K.; Dougados, M.; Radner, H.; Atzeni, F.; et al. Nurmohamed, EULAR recommendations for cardiovascular disease risk management in patients with rheumatoid arthritis and other forms of inflammatory joint disorders: 2015/2016 update. Ann. Rheum. Dis. 2017, 76, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Liao, K.P.; Solomon, D.H. Traditional cardiovascular risk factors, inflammation and cardiovascular risk in rheumatoid arthritis. Rheumatology 2013, 52, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Solomon, D.H.; Kremer, J.; Curtis, J.R.; Hochberg, M.C.; Reed, G.; Tsao, P.; Farkouh, M.E.; Setoguchi, S.; Greenberg, J.D. Explaining the cardiovascular risk associated with rheumatoid arthritis: Traditional risk factors versus markers of rheumatoid arthritis severity. Ann. Rheum. Dis. 2010, 69, 1920–1925. [Google Scholar] [CrossRef] [PubMed]
- Sfikakis, P.P.; Bournia, V.-K.; Kitas, G. Do non-steroidal anti-inflammatory drugs increase or decrease cardiovascular risk in patients with rheumatoid arthritis? Clin. Exp. Rheumatol. 2014, 73, 1515–1521. [Google Scholar]
- Van Halm, V.P.; Peters, M.J.L.; Voskuyl, A.E.; Boers, M.; Lems, W.F.; Visser, M.; Stehouwer, C.D.A.; Spijkerman, A.M.W.; Dekker, J.M.; Nijpels, G.; et al. Rheumatoid arthritis versus diabetes as a risk factor for cardiovascular disease: A cross-sectional study, the CARRE Investigation. Ann. Rheum. Dis. 2009, 68, 1395–1400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stamatelopoulos, K.S.; Kitas, G.D.; Papamichael, C.M.; Chryssohoou, E.; Kyrkou, K.; Georgiopoulos, G.; Protogerou, A.; Panoulas, V.F.; Sandoo, A.; Tentolouris, N.; et al. Atherosclerosis in Rheumatoid Arthritis Versus Diabetes: A Comparative Study. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 1702–1708. [Google Scholar] [CrossRef] [PubMed]
- Lindhardsen, J.; Ahlehoff, O.; Gislason, G.H.; Madsen, O.R.; Olesen, J.B.; Torp-Pedersen, C.; Hansen, P.R. The risk of myocardial infarction in rheumatoid arthritis and diabetes mellitus: A Danish nationwide cohort study. Ann. Rheum. Dis. 2011, 70, 929–934. [Google Scholar] [CrossRef] [PubMed]
- Esdaile, J.M.; Abrahamowicz, M.; Grodzicky, T.; Li, Y.; Panaritis, C.; Berger, R.D.; Côté, R.; Grover, S.A.; Fortin, P.R.; Clarke, A.E.; et al. Traditional Framingham risk factors fail to fully account for accelerated atherosclerosis in systemic lupus erythematosus. Arthritis Rheumatol. 2001, 44, 2331–2337. [Google Scholar] [CrossRef] [Green Version]
- Bartels, C.M.; Buhr, K.A.; Goldberg, J.W.; Bell, C.L.; Visekruna, M.; Nekkanti, S.; Greenlee, R.T. Mortality and Cardiovascular Burden of Systemic Lupus Erythematosus in a US Population-based Cohort. J. Rheumatol. 2014, 41, 680–687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schoenfeld, S.R.; Kasturi, S.; Costenbader, K.H. The epidemiology of atherosclerotic cardiovascular disease among patients with SLE: A systematic review. Semin. Arthritis Rheum. 2013, 43, 77–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, G.C.; Liu, H.R.; Leng, R.X.; Li, X.P.; Li, X.M.; Pan, H.F.; Ye, D.Q. Subclinical atherosclerosis in patients with systemic lupus erythematosus: A systemic review and meta-analysis. Autoimmun. Rev. 2016, 15, 22–37. [Google Scholar] [CrossRef] [PubMed]
- Tektonidou, M.G.; Kravvariti, E.; Konstantonis, G.; Tentolouris, N.; Sfikakis, P.P.; Protogerou, A. Subclinical atherosclerosis in Systemic Lupus Erythematosus: Comparable risk with Diabetes Mellitus and Rheumatoid Arthritis. Autoimmun. Rev. 2017, 16, 308–312. [Google Scholar] [CrossRef] [PubMed]
- Henrot, P.; Foret, J.; Barnetche, T.; Lazaro, E.; Duffau, P.; Seneschal, J.; Schaeverbeke, T.; Truchetet, M.E.; Richez, C. Assessment of subclinical atherosclerosis in systemic lupus erythematosus: A systematic review and meta-analysis. Jt. Bone Spine 2018, 85, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Schieir, O.; Tosevski, C.; Glazier, R.H.; Hogg-Johnson, S.; Badley, E.M. Incident myocardial infarction associated with major types of arthritis in the general population: A systematic review and meta-analysis. Ann. Rheum. Dis. 2017, 76, 1396–1404. [Google Scholar] [CrossRef] [PubMed]
- Mathieu, S.; Soubrier, M. Cardiovascular events in ankylosing spondylitis: A 2018 meta-analysis. Ann. Rheum. Dis. 2018. [Google Scholar] [CrossRef] [PubMed]
- Papagoras, C.; Markatseli, T.E.; Saougou, I.; Alamanos, Y.; Zikou, A.K.; Voulgari, P.V.; Kiortsis, D.N.; Drosos, A.A. Cardiovascular risk profile in patients with spondyloarthritis. Jt. Bone Spine 2014, 81, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Han, C.; Robinson, D.W.; Hackett, M.V.; Paramore, L.C.; Fraeman, K.H.; Bala, M.V. Cardiovascular disease and risk factors in patients with rheumatoid arthritis, psoriatic arthritis, and ankylosing spondylitis. J. Rheumatol. 2006, 33, 2167–2172. [Google Scholar] [PubMed]
- Puig, L. Cardiometabolic Comorbidities in Psoriasis and Psoriatic Arthritis. Int. J. Mol. Sci. 2017, 19, 58. [Google Scholar] [CrossRef] [PubMed]
- Bournia, V.-K.; Kitas, G.; Protogerou, A.D.; Sfikakis, P.P. Impact of non-steroidal anti-inflammatory drugs on cardiovascular risk: Is it the same in osteoarthritis and rheumatoid arthritis? Mod. Rheumatol. 2017, 27, 559–569. [Google Scholar] [CrossRef] [PubMed]
- Arida, A.; Protogerou, A.D.; Konstantonis, G.; Konsta, M.; Delicha, E.M.; Kitas, G.D.; Sfikakis, P.P. Subclinical Atherosclerosis Is Not Accelerated in Patients with Ankylosing Spondylitis with Low Disease Activity: New Data and Metaanalysis of Published Studies. J. Rheumatol. 2015, 42, 2098–2105. [Google Scholar] [CrossRef] [PubMed]
- Bakland, G.; Gran, J.T.; Nossent, J.C. Increased mortality in ankylosing spondylitis is related to disease activity. Ann. Rheum. Dis. 2011, 70, 1921–1925. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Hennekens, C.H.; Buring, J.E.; Rifai, N. C-Reactive Protein and Other Markers of Inflammation in the Prediction of Cardiovascular Disease in Women. N. Engl. J. Med. 2000, 342, 836–843. [Google Scholar] [CrossRef] [PubMed]
- Kaptoge, S.; Thompson, S.G.; Danesh, J. Emerging Risk Factors Collaboration, C-Reactive Protein, Fibrinogen, and Cardiovascular Risk. N. Engl. J. Med. 2013, 368, 84–86. [Google Scholar] [CrossRef]
- Ridker, P.M.; Luscher, T.F. Anti-inflammatory therapies for cardiovascular disease. Eur. Heart J. 2014, 35, 1782–1791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, M.Y.; Li, C.J.; Hou, M.F.; Chu, P.Y. New Insights into the Role of Inflammation in the Pathogenesis of Atherosclerosis. Int. J. Mol. Sci. 2017, 18, 2034. [Google Scholar] [CrossRef] [PubMed]
- Peters, M.J.; Van Halm, V.P.; Voskuyl, A.E.; Smulders, Y.M.; Boers, M.; Lems, W.F.; Visser, M.; Stehouwer, C.D.; Dekker, J.M.; Nijpels, G.; et al. Does rheumatoid arthritis equal diabetes mellitus as an independent risk factor for cardiovascular disease? A prospective study. Arthritis Care Res. 2009, 61, 1571–1579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lévy, L.; Fautrel, B.; Barnetche, T.; Schaeverbeke, T. Incidence and risk of fatal myocardial infarction and stroke events in rheumatoid arthritis patients. A systematic review of the literature. Clin. Exp. Rheumatol. 2008, 26, 673–679. [Google Scholar] [PubMed]
- Meune, C.; Touzé, E.; Trinquart, L.; Allanore, Y. High risk of clinical cardiovascular events in rheumatoid arthritis: Levels of associations of myocardial infarction and stroke through a systematic review and meta-analysis. Arch. Cardiovasc. Dis. 2010, 103, 253–261. [Google Scholar] [CrossRef] [PubMed]
- Mok, C.C.; Kwok, C.L.; Ho, L.Y.; Chan, P.T.; Yip, S.F. Life expectancy, standardized mortality ratios, and causes of death in six rheumatic diseases in Hong Kong, China. Arthritis Rheumatol. 2011, 63, 1182–1189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Exarchou, S.; Lie, E.; Lindström, U.; Askling, J.; Forsblad-d’Elia, H.; Turesson, C.; Kristensen, L.E.; Jacobsson, L.T. Mortality in ankylosing spondylitis: Results from a nationwide population-based study. Ann. Rheum. Dis. 2016, 75, 1466–1472. [Google Scholar] [CrossRef] [PubMed]
- Haroon, N.N.; Paterson, J.M.; Li, P.; Inman, R.D.; Haroon, N. Patients with Ankylosing Spondylitis Have Increased Cardiovascular and Cerebrovascular Mortality. Ann. Intern. Med. 2015, 163, 409–416. [Google Scholar] [CrossRef] [PubMed]
- Jamnitski, A.; Visman, I.M.; Peters, M.J.L.; Boers, M.; Dijkmans, B.A.C.; Nurmohamed, M.T. Prevalence of cardiovascular diseases in psoriatic arthritis resembles that of rheumatoid arthritis. Ann. Rheum. Dis. 2011, 70, 875–876. [Google Scholar] [CrossRef] [PubMed]
- Nurmohamed, M.T.; van der Horst-Bruinsma, I.; Maksymowych, W.P. Cardiovascular and Cerebrovascular Diseases in Ankylosing Spondylitis: Current Insights. Curr. Rheumatol. Rep. 2012, 14, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Gladman, D.D.; Ang, M.; Su, L.; Tom, B.D.; Schentag, C.T.; Farewell, V.T. Cardiovascular morbidity in psoriatic arthritis. Ann. Rheum. Dis. 2009, 68, 1131–1135. [Google Scholar] [CrossRef] [PubMed]
- Ambrosino, P.; Lupoli, R.; Di Minno, A.; Tasso, M.; Peluso, R.; Di Minno, M.N.D. Subclinical atherosclerosis in patients with rheumatoid arthritis. Thromb. Haemost. 2015, 113, 916–930. [Google Scholar] [CrossRef] [PubMed]
- Di Minno, M.N.D.; Ambrosino, P.; Lupoli, R.; Di Minno, A.; Tasso, M.; Peluso, R.; Tremoli, E. Cardiovascular risk markers in patients with psoriatic arthritis: A meta-analysis of literature studies. Ann. Med. 2015, 47, 346–353. [Google Scholar] [CrossRef] [PubMed]
- Ambrosino, P.; Tasso, M.; Lupoli, R.; Di Minno, A.; Baldassarre, D.; Tremoli, E.; Di Minno, M.N.D. Non-invasive assessment of arterial stiffness in patients with rheumatoid arthritis: A systematic review and meta-analysis of literature studies. Ann. Med. 2015, 47, 457–467. [Google Scholar] [CrossRef] [PubMed]
- Sacre, K.; Escoubet, B.; Pasquet, B.; Chauveheid, M.P.; Zennaro, M.C.; Tubach, F.; Papo, T. Increased arterial stiffness in systemic lupus erythematosus (SLE) patients at low risk for cardiovascular disease: A cross-sectional controlled study. PLoS ONE 2014, 9, e94511. [Google Scholar] [CrossRef] [PubMed]
- Shang, Q.; Tam, L.; Li, E.; Yip, G.; Yu, C. Increased arterial stiffness correlated with disease activity in systemic lupus erythematosus. Lupus 2008, 17, 1096–1102. [Google Scholar] [CrossRef] [PubMed]
- Cypiene, A.; Kovaite, M.; Venalis, A.; Dadoniene, J.; Rugiene, R.; Petrulioniene, Z.; Ryliskyte, L.; Laucevicius, A. Arterial wall dysfunction in systemic lupus erythematosus. Lupus 2009, 18, 522–529. [Google Scholar] [CrossRef] [PubMed]
- Capkin, E.; Kiris, A.; Karkucak, M.; Durmus, I.; Gokmen, F.; Cansu, A.; Tosun, M.; Ayar, A. Investigation of effects of different treatment modalities on structural and functional vessel wall properties in patients with ankylosing spondylitis. Jt. Bone Spine 2011, 78, 378–382. [Google Scholar] [CrossRef] [PubMed]
- Bodnár, N.; Kerekes, G.; Seres, I.; Paragh, G.; Kappelmayer, J.; Némethné, Z.G.; Szegedi, G.; Shoenfeld, Y.; Sipka, S.; Soltész, P.; et al. Assessment of Subclinical Vascular Disease Associated with Ankylosing Spondylitis. J. Rheumatol. 2011, 38, 723–729. [Google Scholar] [CrossRef] [PubMed]
- Costa, L.; Caso, F.; D’Elia, L.; Atteno, M.; Peluso, R.; Del Puente, A.; Strazzullo, P.; Scarpa, R. Psoriatic arthritis is associated with increased arterial stiffness in the absence of known cardiovascular risk factors: A case control study. Clin. Rheumatol. 2012, 31, 711–715. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Shang, Q.; Li, E.K.; Leung, Y.Y.; Kun, E.W.; Kwok, L.W.; Li, M.; Li, T.K.; Zhu, T.Y.; Yu, C.M.; et al. Cumulative inflammatory burden is independently associated with increased arterial stiffness in patients with psoriatic arthritis: A prospective study. Arthritis Res. Ther. 2015, 17, 75. [Google Scholar] [CrossRef] [PubMed]
- Di Minno, M.N.D.; Ambrosino, P.; Lupoli, R.; Di Minno, A.; Tasso, M.; Peluso, R.; Tremoli, E. Clinical assessment of endothelial function in patients with rheumatoid arthritis: A meta-analysis of literature studies. Eur. J. Intern. Med. 2015, 26, 835–842. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.Z.; Wang, P.; Guan, S.Y.; Li, H.M.; Leng, R.X.; Pan, H.F.; Ye, D.Q. Decreased flow-mediated dilatation in patients with rheumatoid arthritis: A meta-analysis. Postgrad. Med. J. 2017, 93, 260–265. [Google Scholar] [CrossRef] [PubMed]
- Sabio, J.M.; Vargas-Hitos, J.; Zamora-Pasadas, M.; Mediavilla, J.D.; Navarrete, N.; Ramirez, Á.; Hidalgo-Tenorio, C.; Jáimez, L.; Martín, J.; Jiménez-Alonso, J. Metabolic syndrome is associated with increased arterial stiffness and biomarkers of subclinical atherosclerosis in patients with systemic lupus erythematosus. J. Rheumatol. 2009, 36, 2204–2211. [Google Scholar] [CrossRef] [PubMed]
- Valero-Gonzalez, S.; Castejon, R.; Jimenez-Ortiz, C.; Rosado, S.; Tutor-Ureta, P.; Vargas, J.A.; Yebra-Bango, M. Increased arterial stiffness is independently associated with metabolic syndrome and damage index in systemic lupus erythematosus patients. Scand. J. Rheumatol. 2014, 43, 54–58. [Google Scholar] [CrossRef] [PubMed]
- Protogerou, A.; Zampeli, E.; Tentolouris, N.; Makrilakis, K.; Kitas, G.; Sfikakis, P.P. Subclinical femoral atheromatosis in rheumatoid arthritis: Comparable prevalence to diabetes mellitus in a case-control study. Ann. Rheum. Dis. 2012, 71, 1534–1536. [Google Scholar] [CrossRef] [PubMed]
- Del Rincón, I.; Polak, J.F.; O’leary, D.H.; Battafarano, D.F.; Erikson, J.M.; Restrepo, J.F.; Molina, E.; Escalante, A. Systemic inflammation and cardiovascular risk factors predict rapid progression of atherosclerosis in rheumatoid arthritis. Ann. Rheum. Dis. 2015, 74, 1118–1123. [Google Scholar] [CrossRef] [PubMed]
- Myasoedova, E.; Chandran, A.; Ilhan, B.; Major, B.T.; Michet, C.J.; Matteson, E.L.; Crowson, C.S. The role of rheumatoid arthritis (RA) flare and cumulative burden of RA severity in the risk of cardiovascular disease. Ann. Rheum. Dis. 2016, 75, 560–565. [Google Scholar] [CrossRef] [PubMed]
- Arida, A.; Protogerou, A.D.; Konstantonis, G.; Fragiadaki, K.; Kitas, G.D.; Sfikakis, P.P. Atherosclerosis is not accelerated in rheumatoid arthritis of low activity or remission, regardless of antirheumatic treatment modalities. Rheumatology 2017, 56, 934–939. [Google Scholar] [CrossRef] [PubMed]
- Kitas, G.D.; Gabriel, S.E. Cardiovascular disease in rheumatoid arthritis: State of the art and future perspectives. Ann. Rheum. Dis. 2011, 70, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Arida, A.; Zampeli, E.; Konstantonis, G.; Fragiadaki, K.; Kitas, G.D.; Protogerou, A.D.; Sfikakis, P.P. Rheumatoid arthritis is sufficient to cause atheromatosis but not arterial stiffness or hypertrophy in the absence of classical cardiovascular risk factors. Clin. Rheumatol. 2015, 34, 853–859. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Chen, L.; Delzell, E.; Muntner, P.; Hillegass, W.B.; Safford, M.M.; Millan, I.Y.N.; Crowson, C.S.; Curtis, J.R. The association between inflammatory markers, serum lipids and the risk of cardiovascular events in patients with rheumatoid arthritis. Ann. Rheum. Dis. 2014, 73, 1301–1308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernatsky, S.; Boivin, J.F.; Joseph, L.; Manzi, S.; Ginzler, E.; Gladman, D.D.; Urowitz, M.; Fortin, P.R.; Petri, M.; Barr, S.; et al. Mortality in systemic lupus erythematosus. Arthritis Rheumatol. 2006, 54, 2550–2557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tektonidou, M.G.; Wang, Z.; Ward, M.M. Brief Report: Trends in Hospitalizations Due to Acute Coronary Syndromes and Stroke in Patients with Systemic Lupus Erythematosus, 1996 to 2012. Arthritis Rheumatol. 2016, 68, 2680–2685. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, P.; Kumar, A.; Kumar, S.; Aggarwal, A.; Sinha, N.; Misra, R. Subclinical atherosclerosis and endothelial dysfunction in young South-Asian patients with systemic lupus erythematosus. Clin. Rheumatol. 2009, 28, 1259–1265. [Google Scholar] [CrossRef] [PubMed]
- Kiss, E.; Soltesz, P.; Der, H.; Kocsis, Z.; Tarr, T.; Bhattoa, H.; Shoenfeld, Y.; Szegedi, G. Reduced flow-mediated vasodilation as a marker for cardiovascular complications in lupus patients. J. Autoimmun. 2006, 27, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Selzer, F.; Sutton-Tyrrell, K.; Fitzgerald, S.; Tracy, R.; Kuller, L.; Manzi, S. Vascular stiffness in women with systemic lupus erythematosus. Hypertension 2001, 37, 1075–1082. [Google Scholar] [CrossRef] [PubMed]
- Battista, F.; Pucci, G.; Bocci, E.B.; Anastasio, F.; Crapa, M.; Sanesi, L.; Gerli, R.; Schillaci, G. 2B.09: Arterial stiffness and disease-related organ damage in systemic lupus erythematosus. J. Hypertens. 2015, 33, e24. [Google Scholar] [CrossRef] [PubMed]
- Chou, C.H.; Lin, M.C.; Peng, C.L.; Wu, Y.C.; Sung, F.C.; Kao, C.H.; Liu, S.H. A nationwide population-based retrospective cohort study: Increased risk of acute coronary syndrome in patients with ankylosing spondylitis. Scand. J. Rheumatol. 2014, 43, 132–136. [Google Scholar] [CrossRef] [PubMed]
- Keller, J.J.; Hsu, J.L.; Lin, S.M.; Chou, C.C.; Wang, L.H.; Wang, J.; Bai, C.H.; Chiou, H.Y. Increased risk of stroke among patients with ankylosing spondylitis: A population-based matched-cohort study. Rheumatol. Int. 2014, 34, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Demiralp, E.; Kardesoglu, E.; Kiralp, M.Z.; Cebeci, B.S.; Keskin, İ.; Ozmen, N.; Dursun, H. Aortic elasticity in patients with ankylosing spondylitis. Acta Cardiol. 2004, 59, 630–634. [Google Scholar] [CrossRef] [PubMed]
- Berg, I.J.; Semb, A.G.; van der Heijde, D.; Kvien, T.K.; Olsen, I.C.; Dagfinrud, H.; Provan, S.A. CRP and ASDAS are associated with future elevated arterial stiffness, a risk marker of cardiovascular disease, in patients with ankylosing spondylitis: Results after 5-year follow-up. Ann. Rheum. Dis. 2015, 74, 1562–1566. [Google Scholar] [CrossRef] [PubMed]
- Di Minno, M.N.D.; Iervolino, S.; Peluso, R.; Scarpa, R.; Di Minno, G. CaRRDs Study Group. Carotid Intima-Media Thickness in Psoriatic Arthritis: Differences between Tumor Necrosis Factor-Blockers and Traditional Disease-Modifying Antirheumatic Drugs. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 705–712. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Juanatey, C.; Llorca, J.; Miranda-Filloy, J.A.; Amigo-Diaz, E.; Testa, A.; Garcia-Porrua, C.; Martin, J.; Gonzalez-Gay, M.A. Endothelial dysfunction in psoriatic arthritis patients without clinically evident cardiovascular disease or classic atherosclerosis risk factors. Arthritis Care Res. 2007, 57, 287–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez-juanatey, C.; Llorca, J.; Amigo-Diaz, E.; Dierssen, T.; Martin, J.; Gonzalez-Gay, M.A. High prevalence of subclinical atherosclerosis in psoriatic arthritis patients without clinically evident cardiovascular disease or classic atherosclerosis risk factors. Arthritis Care Res. 2007, 57, 1074–1080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Odegaard, J.I.; Chawla, A. Pleiotropic Actions of Insulin Resistance and Inflammation in Metabolic Homeostasis. Science 2013, 339, 172–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baghdadi, L.R.; Woodman, R.J.; Shanahan, E.M.; Mangoni, A.A. The impact of traditional cardiovascular risk factors on cardiovascular outcomes in patients with rheumatoid arthritis: A systematic review and meta-analysis. PLoS ONE 2015, 10, e0117952. [Google Scholar] [CrossRef] [PubMed]
- Chung, C.P.; Giles, J.T.; Petri, M.; Szklo, M.; Post, W.; Blumenthal, R.S.; Gelber, A.C.; Ouyang, P.; Jenny, N.S.; Bathon, J.M. Prevalence of Traditional Modifiable Cardiovascular Risk Factors in Patients with Rheumatoid Arthritis: Comparison with Control Subjects from the Multi-Ethnic Study of Atherosclerosis. Semin. Arthritis Rheum. 2012, 41, 535–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stavropoulos-Kalinoglou, A.; Metsios, G.S.; Panoulas, V.F.; Douglas, K.M.; Nevill, A.M.; Jamurtas, A.Z.; Kita, M.; Koutedakis, Y.; Kitas, G.D. Associations of obesity with modifiable risk factors for the development of cardiovascular disease in patients with rheumatoid arthritis. Ann. Rheum. Dis. 2009, 68, 242–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panoulas, V.F.; Douglas, K.M.; Milionis, H.J.; Stavropoulos-Kalinglou, A.; Nightingale, P.; Kita, M.D.; Tselios, A.L.; Metsios, G.S.; Elisaf, M.S.; Kitas, G.D. Prevalence and associations of hypertension and its control in patients with rheumatoid arthritis. Rheumatology 2007, 46, 1477–1482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panoulas, V.F.; Metsios, G.S.; Pace, A.V.; John, H.; Treharne, G.J.; Banks, M.J.; Kitas, G.D. Hypertension in rheumatoid arthritis. Rheumatology 2008, 47, 1286–1298. [Google Scholar] [CrossRef] [PubMed]
- Protogerou, A.D.; Panagiotakos, D.B.; Zampeli, E.; Argyris, A.A.; Arida, K.; Konstantonis, G.D.; Pitsavos, C.; Kitas, G.D.; Sfikakis, P.P. Arterial hypertension assessed “out-of-office” in a contemporary cohort of rheumatoid arthritis patients free of cardiovascular disease is characterized by high prevalence, low awareness, poor control and increased vascular damage-associated “white coat” phenomenon. Arthritis Res. Ther. 2013, 15, R142. [Google Scholar] [CrossRef] [PubMed]
- Bartels, C.M.; Johnson, H.; Voelker, K.; Thorpe, C.; McBride, P.; Jacobs, E.A.; Pandhi, N.; Smith, M. Impact of Rheumatoid Arthritis on Receiving a Diagnosis of Hypertension Among Patients With Regular Primary Care. Arthritis Care Res. 2014, 66, 1281–1288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, C.P.; Oeser, A.; Solus, J.F.; Avalos, I.; Gebretsadik, T.; Shintani, A.; Raggi, P.; Sokka, T.; Pincus, T.; Stein, C.M. Prevalence of the metabolic syndrome is increased in rheumatoid arthritis and is associated with coronary atherosclerosis. Atherosclerosis 2008, 196, 756–763. [Google Scholar] [CrossRef] [PubMed]
- Dessein, P.H.; Tobias, M.; Veller, M.G. Metabolic syndrome and subclinical atherosclerosis in rheumatoid arthritis. J. Rheumatol. 2006, 33, 2425–2432. [Google Scholar] [PubMed]
- Rostom, S.; Mengat, M.; Lahlou, R.; Hari, A.; Bahiri, R.; Hajjaj-Hassouni, N. Metabolic syndrome in rheumatoid arthritis: Case control study. BMC Musculoskelet. Disord. 2013, 14, 147. [Google Scholar] [CrossRef] [PubMed]
- Rask-Madsen, C.; Domínguez, H.; Ihlemann, N.; Hermann, T.; Køber, L.; Torp-Pedersen, C. Tumor Necrosis Factor-Inhibits Insulin’s Stimulating Effect on Glucose Uptake and Endothelium-Dependent Vasodilation in Humans. Circulation 2003, 108, 1815–1821. [Google Scholar] [CrossRef] [PubMed]
- Carey, A.L.; Febbraio, M.A. Interleukin-6 and insulin sensitivity: Friend or foe? Diabetologia 2004, 47, 1135–1142. [Google Scholar] [CrossRef] [PubMed]
- Kiortsis, D.N.; Mavridis, A.K.; Vasakos, S.; Nikas, S.N.; Drosos, A.A. Effects of infliximab treatment on insulin resistance in patients with rheumatoid arthritis and ankylosing spondylitis. Ann. Rheum. Dis. 2005, 64, 765–766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huvers, F.C.; Popa, C.; Netea, M.G.; van den Hoogen, F.H.; Tack, C.J. Improved insulin sensitivity by anti-TNFalpha antibody treatment in patients with rheumatic diseases. Ann. Rheum. Dis. 2007, 66, 558–559. [Google Scholar] [CrossRef] [PubMed]
- Toms, T.E.; Panoulas, V.F.; John, H.; Douglas, K.M.; Kitas, G.D. Methotrexate therapy associates with reduced prevalence of the metabolic syndrome in rheumatoid arthritis patients over the age of 60—More than just an anti-inflammatory effect? A cross sectional study. Arthritis Res. Ther. 2009, 11, R110. [Google Scholar] [CrossRef] [PubMed]
- Toms, T.E.; Panoulas, V.F.; Douglas, K.M.; Griffiths, H.R.; Kitas, G.D. Lack of association between glucocorticoid use and presence of the metabolic syndrome in patients with rheumatoid arthritis: A cross-sectional study. Arthritis Res. Ther. 2008, 10, R145. [Google Scholar] [CrossRef] [PubMed]
- Van Sijl, A.M.; Peters, M.J.; Knol, D.L.; de Vet, R.H.; Sattar, N.; Dijkmans, B.A.; Smulders, Y.M.; Nurmohamed, M.T. The Effect of TNF-alpha Blocking Therapy on Lipid Levels in Rheumatoid Arthritis: A Meta-Analysis. Semin. Arthritis Rheum. 2011, 41, 393–400. [Google Scholar] [CrossRef] [PubMed]
- Liao, K.P.; Cai, T.; Gainer, V.S.; Cagan, A.; Murphy, S.N.; Liu, C.; Churchill, S.; Shaw, S.Y.; Kohane, I.; Solomon, D.H.; et al. Lipid and Lipoprotein Levels and Trend in Rheumatoid Arthritis Compared to the General Population. Arthritis Care Res. 2013, 65, 2046–2050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choy, E.; Sattar, N. Interpreting lipid levels in the context of high-grade inflammatory states with a focus on rheumatoid arthritis: A challenge to conventional cardiovascular risk actions. Ann. Rheum. Dis. 2009, 68, 460–469. [Google Scholar] [CrossRef] [PubMed]
- Charles-Schoeman, C.; Watanabe, J.; Lee, Y.Y.; Furst, D.E.; Amjadi, S.; Elashoff, D.; Park, G.; McMahon, M.; Paulus, H.E.; Fogelman, A.M.; et al. Abnormal function of high-density lipoprotein is associated with poor disease control and an altered protein cargo in rheumatoid arthritis. Arthritis Rheumatol. 2009, 60, 2870–2879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filippatos, T.D.; Derdemezis, C.S.; Voulgari, P.V.; Tsimihodimos, V.; Elisaf, M.S.; Tselepis, A.D.; Drosos, A.A. Effects of 12 months of treatment with disease-modifying anti-rheumatic drugs on low and high density lipoprotein subclass distribution in patients with early rheumatoid arthritis: A pilot study. Scand. J. Rheumatol. 2013, 42, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Popa, C.; van den Hoogen, F.H.; Radstake, T.R.; Netea, M.G.; Eijsbouts, A.E.; Den Heijer, M.; van der Meer, J.W.; van Riel, P.L.; Stalenhoef, A.F.; Barrera, P. Modulation of lipoprotein plasma concentrations during long-term anti-TNF therapy in patients with active rheumatoid arthritis. Ann. Rheum. Dis. 2007, 66, 1503–1507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schimmel, E.K.; Yazici, Y. Increased lipid levels but unchanged atherogenic index in rheumatoid arthritis patients treated with biologic disease modifying antirheumatic drugs: Published experience. Clin. Exp. Rheumatol. 2009, 27, 446–451. [Google Scholar] [PubMed]
- Semb, A.G.; Kvien, T.K.; DeMicco, D.A.; Fayyad, R.; Wun, C.C.; LaRosa, J.C.; Betteridge, J.; Pedersen, T.R.; Holme, I. Effect of intensive lipid-lowering therapy on cardiovascular outcome in patients with and those without inflammatory joint disease. Arthritis Rheumatol. 2012, 64, 2836–2846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soulaidopoulos, S.; Nikiphorou, E.; Dimitroulas, T.; Kitas, G.D. The Role of Statins in Disease Modification and Cardiovascular Risk in Rheumatoid Arthritis. Front. Med. 2018, 5, 24. [Google Scholar] [CrossRef] [PubMed]
- Rontoyanni, V.G.; Sfikakis, P.P.; Kitas, G.D.; Protogerou, A.D. Marine n-3 fatty acids for cardiovascular risk reduction and disease control in rheumatoid arthritis: “kill two birds with one stone”? Curr. Pharm. Des. 2012, 18, 1531–1542. [Google Scholar] [CrossRef] [PubMed]
- Elkan, A.C.; Håkansson, N.; Frostegård, J.; Hafström, I. Low level of physical activity in women with rheumatoid arthritis is associated with cardiovascular risk factors but not with body fat mass—A cross sectional study. BMC Musculoskelet. Disord. 2011, 12, 13. [Google Scholar] [CrossRef] [PubMed]
- Padyukov, L.; Silva, C.; Stolt, P.; Alfredsson, L.; Klareskog, L. A gene-environment interaction between smoking and shared epitope genes in HLA-DR provides a high risk of seropositive rheumatoid arthritis. Arthritis Rheumatol. 2004, 50, 3085–3092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szodoray, P.; Szabó, Z.; Kapitány, A.; Gyetvai, Á.; Lakos, G.; Szántó, S.; Szücs, G.; Szekanecz, Z. Anti-citrullinated protein/peptide autoantibodies in association with genetic and environmental factors as indicators of disease outcome in rheumatoid arthritis. Autoimmun. Rev. 2010, 9, 140–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, C.P.; Avalos, I.; Oeser, A.; Gebretsadik, T.; Shintani, A.; Raggi, P.; Stein, C.M. High prevalence of the metabolic syndrome in patients with systemic lupus erythematosus: Association with disease characteristics and cardiovascular risk factors. Ann. Rheum. Dis. 2006, 66, 208–214. [Google Scholar] [CrossRef] [PubMed]
- Chung, C.P.; Oeser, A.; Solus, J.F.; Gebretsadik, T.; Shintani, A.; Avalos, I.; Sokka, T.; Raggi, P.; Pincus, T.; Stein, C.M. Inflammation-associated insulin resistance: Differential effects in rheumatoid arthritis and systemic lupus erythematosus define potential mechanisms. Arthritis Rheumatol. 2008, 58, 2105–2112. [Google Scholar] [CrossRef] [PubMed]
- Chung, C.P.; Oeser, A.; Solus, J.; Avalos, I.; Gebretsadik, T.; Shintani, A.; Linton, M.F.; Fazio, S.; Stein, C.M. Inflammatory mechanisms affecting the lipid profile in patients with systemic lupus erythematosus. J. Rheumatol. 2007, 34, 1849–1854. [Google Scholar] [PubMed]
- Borba, E.F.; Bonfá, E. Dyslipoproteinemias in systemic lupus erythematosus: Influence of disease, activity, and anticardiolipin antibodies. Lupus 1997, 6, 533–539. [Google Scholar] [CrossRef] [PubMed]
- Nuttall, S.L.; Heaton, S.; Piper, M.K.; Martin, U.; Gordon, C. Cardiovascular risk in systemic lupus erythematosus—Evidence of increased oxidative stress and dyslipidaemia. Rheumatology 2003, 42, 758–762. [Google Scholar] [CrossRef] [PubMed]
- McMahon, M.; Grossman, J.; FitzGerald, J.; Dahlin-Lee, E.; Wallace, D.J.; Thong, B.Y.; Badsha, H.; Kalunian, K.; Charles, C.; Navab, M.; et al. Proinflammatory high-density lipoprotein as a biomarker for atherosclerosis in patients with systemic lupus erythematosus and rheumatoid arthritis. Arthritis Rheumatol. 2006, 54, 2541–2549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papadakis, J.A.; Sidiropoulos, P.I.; Karvounaris, S.A.; Vrentzos, G.E.; Spanakis, E.K.; Ganotakis, E.S.; Kritikos, H.D.; Mikhailidis, D.P.; Boumpas, D.T. High prevalence of metabolic syndrome and cardiovascular risk factors in men with ankylosing spondylitis on anti-TNFalpha treatment: Correlation with disease activity. Clin. Exp. Rheumatol. 2009, 27, 292–298. [Google Scholar] [PubMed]
- Mathieu, S.; Gossec, L.; Dougados, M.; Soubrier, M. Cardiovascular profile in ankylosing spondylitis: A systematic review and meta-analysis. Arthritis Care Res. 2011, 63, 557–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Labitigan, M.; Bahče-Altuntas, A.; Kremer, J.M.; Reed, G.; Greenberg, J.D.; Jordan, N.; Putterman, C.; Broder, A. Higher rates and clustering of abnormal lipids, obesity, and diabetes mellitus in psoriatic arthritis compared with rheumatoid arthritis. Arthritis Care Res. 2014, 66, 600–607. [Google Scholar] [CrossRef] [PubMed]
- Haroon, M.; Gallagher, P.; Heffernan, E.; FitzGerald, O. High Prevalence of Metabolic Syndrome and of Insulin Resistance in Psoriatic Arthritis is Associated with the Severity of Underlying Disease. J. Rheumatol. 2014, 41, 1357–1365. [Google Scholar] [CrossRef] [PubMed]
- Costa, L.; Caso, F.; Atteno, M.; Del Puente, A.; Darda, M.A.; Caso, P.; Ortolan, A.; Fiocco, U.; Ramonda, R.; Punzi, L.; et al. Impact of 24-month treatment with etanercept, adalimumab, or methotrexate on metabolic syndrome components in a cohort of 210 psoriatic arthritis patients. Clin. Rheumatol. 2013, 33, 833–839. [Google Scholar] [CrossRef] [PubMed]
- Mehta, N.N.; Gelfand, J.M. High-density lipoprotein cholesterol function improves after successful treatment of psoriasis: A step forward in the right direction. J. Investig. Dermatol. 2014, 134, 592–595. [Google Scholar] [CrossRef] [PubMed]
- Davignon, J.; Ganz, P. Role of Endothelial Dysfunction in Atherosclerosis. Circulation 2004, 109, III-27–III-32. [Google Scholar] [CrossRef] [PubMed]
- Bordy, R.; Totoson, P.; Prati, C.; Marie, C.; Wendling, D.; Demougeot, C. Microvascular endothelial dysfunction in rheumatoid arthritis. Nat. Rev. Rheumatol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Tabas, I.; Williams, K.J.; Boren, J. Subendothelial Lipoprotein Retention as the Initiating Process in Atherosclerosis: Update and Therapeutic Implications. Circulation 2007, 116, 1832–1844. [Google Scholar] [CrossRef] [PubMed]
- Murdaca, G.; Colombo, B.M.; Cagnati, P.; Gulli, R.; Spanò, F.; Puppo, F. Endothelial dysfunction in rheumatic autoimmune diseases. Atherosclerosis 2012, 224, 309–317. [Google Scholar] [CrossRef] [PubMed]
- Libby, P.; Ridker, P.M.; Maseri, A. Inflammation and atherosclerosis. Circulation 2002, 105, 1135–1143. [Google Scholar] [CrossRef] [PubMed]
- Steyers, C.; Miller, F. Endothelial Dysfunction in Chronic Inflammatory Diseases. Int. J. Mol. Sci. 2014, 15, 11324–11349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dessein, P.H.; Joffe, B.I.; Singh, S. Biomarkers of endothelial dysfunction, cardiovascular risk factors and atherosclerosis in rheumatoid arthritis. Arthritis Res. Ther. 2005, 7, R634–R643. [Google Scholar] [CrossRef] [PubMed]
- Dessein, P.H.; Solomon, A.; Woodiwiss, A.J.; Norton, G.R.; Tsang, L.; Gonzalez-Gay, M.A. Marked Independent Relationship between Circulating Interleukin-6 Concentrations and Endothelial Activation in Rheumatoid Arthritis. Mediat. Inflamm. 2013, 2013, 510243. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Gay, M.A.; Gonzalez-Juanatey, C.; Miranda-Filloy, J.A.; Garcia-Unzueta, M.T.; Llorca, J. Lack of association between carotid intima-media wall thickness and carotid plaques and markers of endothelial cell activation in rheumatoid arthritis patients undergoing anti-TNF therapy. Acta Reumatol. Port. 2012, 37, 155–159. [Google Scholar] [PubMed]
- Gonzalez-Gay, M.A.; Garcia-Unzueta, M.T.; De Matias, J.M.; Gonzalez-Juanatey, C.; Garcia-Porrua, C.; Sanchez-Andrade, A.; Martin, J.; Llorca, J. Influence of anti-TNF-alpha infliximab therapy on adhesion molecules associated with atherogenesis in patients with rheumatoid arthritis. Clin. Exp. Rheumatol. 2006, 24, 373–379. [Google Scholar] [PubMed]
- Kerekes, G.; Szekanecz, Z.; Dér, H.; Sándor, Z.; Lakos, G.; Muszbek, L.; Csipö, I.; Sipka, S.; Seres, I.; Paragh, G.; et al. Endothelial dysfunction and atherosclerosis in rheumatoid arthritis: A multiparametric analysis using imaging techniques and laboratory markers of inflammation and autoimmunity. J. Rheumatol. 2008, 35, 398–406. [Google Scholar] [PubMed]
- Dimitroulas, T.; Hodson, J.; Sandoo, A.; Smith, J.; Kitas, G.D. Endothelial injury in rheumatoid arthritis: A crosstalk between dimethylarginines and systemic inflammation. Arthritis Res. Ther. 2017, 19, 32. [Google Scholar] [CrossRef] [PubMed]
- Yao, G.H.; Liu, Z.H.; Zhang, X.; Zheng, C.X.; Chen, H.P.; Zeng, C.H.; Li, L.S. Circulating thrombomodulin and vascular cell adhesion molecule-1 and renal vascular lesion in patients with lupus nephritis. Lupus 2008, 17, 720–726. [Google Scholar] [CrossRef] [PubMed]
- Santos, M.J.; Carmona-Fernandes, D.; Canhão, H.; da Silva, J.C.; Fonseca, J.E.; Gil, V. Early vascular alterations in SLE and RA patients—A step towards understanding the associated cardiovascular risk. PLoS ONE 2012, 7, e44668. [Google Scholar] [CrossRef] [PubMed]
- Surdacki, A.; Sulicka, J.; Korkosz, M.; Mikołajczyk, T.; Telesińska-Jasiówka, D.; Klimek, E.; Kierzkowska, I.; Guzik, T.; Grodzicki, T.K. Blood Monocyte Heterogeneity and Markers of Endothelial Activation in Ankylosing Spondylitis. J. Rheumatol. 2014, 41, 481–489. [Google Scholar] [CrossRef] [PubMed]
- Genre, F.; López-Mejías, R.; Miranda-Filloy, J.A.; Carnero-López, B.; Gómez-Acebo, I.; Blanco, R.; Ochoa, R.; Rueda, J.; Gonzalez-Juanatey, C.; Llorca, J.; et al. Asymmetric dimethylarginine serum levels in non-diabetic ankylosing spondylitis patients undergoing TNF-α antagonist therapy. Clin. Exp. Rheumatol. 2013, 31, 749–755. [Google Scholar] [PubMed]
- Erre, G.L.; Sanna, P.; Zinellu, A.; Ponchietti, A.; Fenu, P.; Sotgia, S.; Carru, C.; Ganau, A.; Passiu, G. Plasma asymmetric dimethylarginine (ADMA) levels and atherosclerotic disease in ankylosing spondylitis: A cross-sectional study. Clin. Rheumatol. 2011, 30, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Garg, N.; Krishan, P.; Syngle, A. Atherosclerosis in Psoriatic Arthritis: A Multiparametric Analysis Using Imaging Technique and Laboratory Markers of Inflammation and Vascular Function. Int. J. Angiol. 2016, 25, 222–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolliker Frers, R.A.; Cosentino, V.; Tau, J.; Kerzberg, E.M.; Urdapilleta, A.; Chiocconi, M.; Kogan, N.; Otero-Losada, M.; Capani, F. Immune-Mediated Inflammation Promotes Subclinical Atherosclerosis in Recent-Onset Psoriatic Arthritis Patients without Conventional Cardiovascular Risk Factors. Front. Immunol. 2018, 9, 139. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Li, Y.; Li, Y.; Ren, X.; Zhang, X.; Hu, D.; Gao, Y.; Xing, Y.; Shang, H. Oxidative Stress-Mediated Atherosclerosis: Mechanisms and Therapies. Front. Physiol. 2017, 8, 600. [Google Scholar] [CrossRef] [PubMed]
- Hassan, S.Z.; Gheita, T.A.; Kenawy, S.A.; Fahim, A.T.; El-Sorougy, I.M.; Abdou, M.S. Oxidative stress in systemic lupus erythematosus and rheumatoid arthritis patients: Relationship to disease manifestations and activity. Int. J. Rheum. Dis. 2011, 14, 325–331. [Google Scholar] [CrossRef] [PubMed]
- Vasanthi, P.; Nalini, G.; Rajasekhar, G. Status of oxidative stress in rheumatoid arthritis. Int. J. Rheum. Dis. 2009, 12, 29–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-González, A.; Gaxiola-Robles, R.; Zenteno-Savín, T. Oxidative stress in patients with rheumatoid arthritis. Rev. Investig. Clin. 2015, 67, 46–53. [Google Scholar]
- Solmaz, D.; Kozacı, D.; Sarı, İ.; Taylan, A.; Önen, F.; Akkoç, N.; Akar, S. Oxidative stress and related factors in patients with ankylosing spondylitis. Eur. J. Rheumatol. 2016, 3, 20–24. [Google Scholar] [CrossRef] [PubMed]
- Stanek, A.; Cholewka, A.; Wielkoszyński, T.; Romuk, E.; Sieroń, K.; Sieroń, A. Increased Levels of Oxidative Stress Markers, Soluble CD40 Ligand, and Carotid Intima-Media Thickness Reflect Acceleration of Atherosclerosis in Male Patients with Ankylosing Spondylitis in Active Phase and without the Classical Cardiovascular Risk Factors. Oxid. Med. Cell. Longev. 2017, 2017, 9712536. [Google Scholar] [CrossRef] [PubMed]
- Coaccioli, S.; Panaccione, A.; Biondi, R.; Sabatini, C.; Landucci, P.; Del, R.G.; Fantera, M.; Monno, A.M.; Di, L.C.; Paladini, A.; et al. Evaluation of oxidative stress in rheumatoid and psoriatic arthritis and psoriasis. Clin. Ter. 2009, 160, 467–472. [Google Scholar] [PubMed]
- Cacciapaglia, F.; Anelli, M.G.; Rizzo, D.; Morelli, E.; Scioscia, C.; Mazzotta, D.; Iannone, F.; Lapadula, G. Influence of TNF-α inhibition on oxidative stress of rheumatoid arthritis patients. Reumatismo 2016, 67, 97–102. [Google Scholar] [CrossRef] [PubMed]
- Lundberg, A.M.; Hansson, G.K. Innate immune signals in atherosclerosis. Clin. Immunol. 2010, 134, 5–24. [Google Scholar] [CrossRef] [PubMed]
- Witztum, J.L.; Lichtman, A.H. The Influence of Innate and Adaptive Immune Responses on Atherosclerosis. Annu. Rev. Pathol. Mech. Dis. 2014, 9, 73–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adamczak, D.M. The Role of Toll-Like Receptors and Vitamin D in Cardiovascular Diseases—A Review. Int. J. Mol. Sci. 2017, 18, 2252. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Pope, R.M. Toll-like receptor signaling: A potential link among rheumatoid arthritis, systemic lupus, and atherosclerosis. J. Leukoc. Biol. 2010, 88, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Ospelt, C.; Brentano, F.; Rengel, Y.; Stanczyk, J.; Kolling, C.; Tak, P.P.; Gay, R.E.; Gay, S.; Kyburz, D. Overexpression of toll-like receptors 3 and 4 in synovial tissue from patients with early rheumatoid arthritis: Toll-like receptor expression in early and longstanding arthritis. Arthritis Rheumatol. 2008, 58, 3684–3692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radstake, T.R.; Roelofs, M.F.; Jenniskens, Y.M.; Oppers-Walgreen, B.; van Riel, P.L.; Barrera, P.; Joosten, L.A.; van den Berg, W.B. Expression of Toll-like receptors 2 and 4 in rheumatoid synovial tissue and regulation by proinflammatory cytokines interleukin-12 and interleukin-18 via interferon-γ? Arthritis Rheumatol. 2004, 50, 3856–3865. [Google Scholar] [CrossRef] [PubMed]
- Christensen, S.R.; Shupe, J.; Nickerson, K.; Kashgarian, M.; Flavell, R.A.; Shlomchik, M.J. Toll-like receptor 7 and TLR9 dictate autoantibody specificity and have opposing inflammatory and regulatory roles in a murine model of lupus. Immunity 2006, 25, 417–428. [Google Scholar] [CrossRef] [PubMed]
- De Rycke, L.; Vandooren, B.; Kruithof, E.; De Keyser, F.; Veys, E.M.; Baeten, D. Tumor necrosis factor alpha blockade treatment down-modulates the increased systemic and local expression of Toll-like receptor 2 and Toll-like receptor 4 in spondylarthropathy. Arthritis Rheumatol. 2005, 52, 2146–2158. [Google Scholar] [CrossRef] [PubMed]
- Assassi, S.; Reveille, J.D.; Arnett, F.C.; Weisman, M.H.; Ward, M.M.; Agarwal, S.K.; Gourh, P.; Bhula, J.; Sharif, R.; Sampat, K.; et al. Whole-blood gene expression profiling in ankylosing spondylitis shows upregulation of toll-like receptor 4 and 5. J. Rheumatol. 2011, 38, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Candia, L.; Marquez, J.; Hernandez, C.; Zea, A.H.; Espinoza, L.R. Toll-like receptor-2 expression is upregulated in antigen-presenting cells from patients with psoriatic arthritis: A pathogenic role for innate immunity? J. Rheumatol. 2007, 34, 374–379. [Google Scholar] [PubMed]
- Schroder, K.; Zhou, R.; Tschopp, J. The NLRP3 Inflammasome: A Sensor for Metabolic Danger? Science 2010, 327, 296–300. [Google Scholar] [CrossRef] [PubMed]
- Tedgui, A.; Mallat, Z. Cytokines in Atherosclerosis: Pathogenic and Regulatory Pathways. Physiol. Rev. 2006, 86, 515–581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altaf, A.; Qu, P.; Zhao, Y.; Wang, H.; Lou, D.; Niu, N. NLRP3 inflammasome in peripheral blood monocytes of acute coronary syndrome patients and its relationship with statins. Coron. Artery Dis. 2015, 26, 409–421. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Xie, W.L.; Kong, W.W.; Chen, D.; Qu, P. Expression of the NLRP3 Inflammasome in Carotid Atherosclerosis. J. Stroke Cerebrovasc. Dis. 2015, 24, 2455–2466. [Google Scholar] [CrossRef] [PubMed]
- Zheng, F.; Xing, S.; Gong, Z.; Mu, W.; Xing, Q. Silence of NLRP3 Suppresses Atherosclerosis and Stabilizes Plaques in Apolipoprotein E-Deficient Mice. Mediat. Inflamm. 2014, 2014, 507208. [Google Scholar] [CrossRef] [PubMed]
- Duewell, P.; Kono, H.; Rayner, K.J.; Sirois, C.M.; Vladimer, G.; Bauernfeind, F.G.; Abela, G.S.; Franchi, L.; Nunez, G.; Schnurr, M.; et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 2010, 464, 1357–1361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajamäki, K.; Lappalainen, J.; Öörni, K.; Välimäki, E.; Matikainen, S.; Kovanen, P.T.; Eklund, K.K. Cholesterol crystals activate the NLRP3 inflammasome in human macrophages: A novel link between cholesterol metabolism and inflammation. PLoS ONE 2010, 5, e11765. [Google Scholar] [CrossRef] [PubMed]
- Mathews, R.J.; Robinson, J.I.; Battellino, M.; Wong, C.; Taylor, J.C.; Eyre, S.; Churchman, S.M.; Wilson, A.G.; Isaacs, J.D.; Hyrich, K.; et al. Evidence of NLRP3-inflammasome activation in rheumatoid arthritis (RA); genetic variants within the NLRP3-inflammasome complex in relation to susceptibility to RA and response to anti-TNF treatment. Ann. Rheum. Dis. 2014, 73, 1202–1210. [Google Scholar] [CrossRef] [PubMed]
- Choulaki, C.; Papadaki, G.; Repa, A.; Kampouraki, E.; Kambas, K.; Ritis, K.; Bertsias, G.; Boumpas, D.T.; Sidiropoulos, P. Enhanced activity of NLRP3 inflammasome in peripheral blood cells of patients with active rheumatoid arthritis. Arthritis Res. Ther. 2015, 17, 257. [Google Scholar] [CrossRef] [PubMed]
- Kastbom, A.; Klingberg, E.; Verma, D.; Carlsten, H.; Forsblad-d’Elia, H.; Wesamaa, J.; Cedergren, J.; Eriksson, P.; Söderkvist, P. Genetic variants in CARD8 but not in NLRP3 are associated with ankylosing spondylitis. Scand. J. Rheumatol. 2013, 42, 465–468. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Fu, R.; Guo, C.; Huang, Y.; Wang, H.; Wang, S.; Zhao, J.; Yang, N. Anti-dsDNA antibodies bind to TLR4 and activate NLRP3 inflammasome in lupus monocytes/macrophages. J. Transl. Med. 2016, 14, 156. [Google Scholar] [CrossRef] [PubMed]
- Kahlenberg, J.M.; Kaplan, M.J. The inflammasome and lupus. Curr. Opin. Rheumatol. 2014, 26, 475–481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, M.S.; Kang, Y.; Lee, N.; Wahl, E.R.; Kim, S.H.; Kang, K.S.; Lazova, R.; Kang, I. Self double-stranded (ds)DNA induces IL-1β production from human monocytes by activating NLRP3 inflammasome in the presence of anti-dsDNA antibodies. J. Immunol. 2013, 190, 1407–1415. [Google Scholar] [CrossRef] [PubMed]
- Asgari, E.; Le Friec, G.; Yamamoto, H.; Perucha, E.; Sacks, S.S.; Köhl, J.; Cook, H.T.; Kemper, C. C3a modulates IL-1β secretion in human monocytes by regulating ATP efflux and subsequent NLRP3 inflammasome activation. Blood 2013, 122, 3473–3481. [Google Scholar] [CrossRef] [PubMed]
- Kahlenberg, J.M.; Thacker, S.G.; Berthier, C.C.; Cohen, C.D.; Kretzler, M.; Kaplan, M.J. Inflammasome Activation of IL-18 Results in Endothelial Progenitor Cell Dysfunction in Systemic Lupus Erythematosus. J. Immunol. 2011, 187, 6143–6156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samstad, E.O.; Niyonzima, N.; Nymo, S.; Aune, M.H.; Ryan, L.; Bakke, S.S.; Lappegård, K.T.; Brekke, O.L.; Lambris, J.D.; Damås, J.K.; et al. Cholesterol Crystals Induce Complement-Dependent Inflammasome Activation and Cytokine Release. J. Immunol. 2014, 192, 2837–2845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, S.; Chen, H.; Wu, G.; Zhao, C. The association of NLRP3 and TNFRSF1A polymorphisms with risk of ankylosing spondylitis and treatment efficacy of etanercept. J. Clin. Lab. Anal. 2017, 31, e22138. [Google Scholar] [CrossRef] [PubMed]
- Hansson, G.K. Inflammation, atherosclerosis, and coronary artery disease. N. Engl. J. Med. 2005, 352, 1685–1695. [Google Scholar] [CrossRef] [PubMed]
- Weiss, G.; Willeit, J.; Kiechl, S.; Fuchs, D.; Jarosch, E.; Oberhollenzer, F.; Reibnegger, G.; Tilz, G.P.; Gerstenbrand, F.; Wachter, H. Increased concentrations of neopterin in carotid atherosclerosis. Atherosclerosis 1994, 106, 263–271. [Google Scholar] [CrossRef]
- Ray, K.K.; Morrow, D.A.; Sabatine, M.S.; Shui, A.; Rifai, N.; Cannon, C.P.; Braunwald, E. Long-term prognostic value of neopterin: A novel marker of monocyte activation in patients with acute coronary syndrome. Circulation 2007, 115, 3071–3078. [Google Scholar] [CrossRef] [PubMed]
- Rho, Y.H.; Solus, J.; Raggi, P.; Oeser, A.; Gebretsadik, T.; Shintani, A.; Stein, C.M. Macrophage activation and coronary atherosclerosis in systemic lupus erythematosus and rheumatoid arthritis. Arthritis Care Res. 2011, 63, 535–541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voloshyna, I.; Modayil, S.; Littlefield, M.J.; Belilos, E.; Belostocki, K.; Bonetti, L.; Rosenblum, G.; Carsons, S.E.; Reiss, A.B. Plasma from rheumatoid arthritis patients promotes pro-atherogenic cholesterol transport gene expression in THP-1 human macrophages. Exp. Biol. Med. 2013, 238, 1192–1197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Fu, Q.; Cui, H.; Qu, B.; Pan, W.; Shen, N.; Bao, C. Interferon-α priming promotes lipid uptake and macrophage-derived foam cell formation: A novel link between interferon-α and atherosclerosis in lupus. Arthritis Rheumatol. 2011, 63, 492–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korman, B.D.; Huang, C.C.; Skamra, C.; Wu, P.; Koessler, R.; Yao, D.; Huang, Q.Q.; Pearce, W.; Sutton-Tyrrell, K.; Kondos, G.; et al. Inflammatory expression profiles in monocyte-to-macrophage differentiation in patients with systemic lupus erythematosus and relationship with atherosclerosis. Arthritis Res. Ther. 2014, 16, R147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghazizadeh, R.; Shimizu, H.; Tosa, M.; Ghazizadeh, M. Pathogenic mechanisms shared between psoriasis and cardiovascular disease. Int. J. Med. Sci. 2010, 7, 284–289. [Google Scholar] [CrossRef] [PubMed]
- Ait-Oufella, H.; Taleb, S.; Mallat, Z.; Tedgui, A. Recent advances on the role of cytokines in atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 969–979. [Google Scholar] [CrossRef] [PubMed]
- Ramji, D.P.; Davies, T.S. Cytokines in atherosclerosis: Key players in all stages of disease and promising therapeutic targets. Cytokine Growth Factor Rev. 2015, 26, 673–685. [Google Scholar] [CrossRef] [PubMed]
- Svenungsson, E.; Fei, G.Z.; Jensen-Urstad, K.; De Faire, U.; Hamsten, A.; Frostegård, J. TNF-α: A link between hypertriglyceridaemia and inflammation in SLE patients with cardiovascular disease. Lupus 2003, 12, 454–461. [Google Scholar] [CrossRef] [PubMed]
- Ley, K.; Laudanna, C.; Cybulsky, M.I.; Nourshargh, S. Getting to the site of inflammation: The leukocyte adhesion cascade updated. Nat. Rev. Immunol. 2007, 7, 678–689. [Google Scholar] [CrossRef] [PubMed]
- Folcik, V.A.; Aamir, R.; Cathcart, M.K. Cytokine modulation of LDL oxidation by activated human monocytes. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 1954–1961. [Google Scholar] [CrossRef] [PubMed]
- Didion, S. Cellular and Oxidative Mechanisms Associated with Interleukin-6 Signaling in the Vasculature. Int. J. Mol. Sci. 2017, 18, 2563. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.J.; Kim, W.J.; Bae, H.U.; Kim, D.I.; Park, Y.B.; Park, J.E.; Kwon, B.S.; Lee, W.H. Involvement of TL1A and DR3 in induction of pro-inflammatory cytokines and matrix metalloproteinase-9 in atherogenesis. Cytokine 2005, 29, 229–235. [Google Scholar] [CrossRef] [PubMed]
- McLaren, J.E.; Calder, C.J.; McSharry, B.P.; Sexton, K.; Salter, R.C.; Singh, N.N.; Wilkinson, G.W.; Wang, E.C.; Ramji, D.P. The TNF-Like Protein 1A-Death Receptor 3 Pathway Promotes Macrophage Foam Cell Formation In Vitro. J. Immunol. 2010, 184, 5827–5834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steiner, G.; Tohidast-Akrad, M.; Witzmann, G.; Vesely, M.; Studnicka-Benke, A.; Gal, A.; Kunaver, M.; Zenz, P.; Smolen, J.S. Cytokine production by synovial T cells in rheumatoid arthritis. Rheumatology 1999, 38, 202–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cavagna, L.; Boffini, N.; Cagnotto, G.; Inverardi, F.; Grosso, V.; Caporali, R. Atherosclerosis and Rheumatoid Arthritis: More Than a Simple Association. Mediat. Inflamm. 2012, 2012, 147354. [Google Scholar] [CrossRef] [PubMed]
- Pasceri, V.; Yeh, E.T. A tale of two diseases: Atherosclerosis and rheumatoid arthritis. Circulation 1999, 100, 2124–2126. [Google Scholar] [CrossRef] [PubMed]
- Skeoch, S.; Bruce, I.N. Atherosclerosis in rheumatoid arthritis: Is it all about inflammation? Nat. Rev. Rheumatol. 2015, 11, 390–400. [Google Scholar] [CrossRef] [PubMed]
- Cassatella, M.A.; da Silva, G.P.; Tinazzi, I.; Facchetti, F.; Scapini, P.; Calzetti, F.; Tamassia, N.; Wei, P.; Nardelli, B.; Roschke, V.; et al. Soluble TNF-like cytokine (TL1A) production by immune complexes stimulated monocytes in rheumatoid arthritis. J. Immunol. 2007, 178, 7325–7333. [Google Scholar] [CrossRef] [PubMed]
- Bamias, G.; Siakavellas, S.I.; Stamatelopoulos, K.S.; Chryssochoou, E.; Papamichael, C.; Sfikakis, P.P. Circulating levels of TNF-like cytokine 1A (TL1A) and its decoy receptor 3 (DcR3) in rheumatoid arthritis. Clin. Immunol. 2008, 129, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Bamias, G.; Stamatelopoulos, K.; Zampeli, E.; Protogerou, A.; Sigala, F.; Papamichael, C.; Christopoulos, P.; Kitas, G.D.; Sfikakis, P.P. Circulating levels of TNF-like cytokine 1A correlate with the progression of atheromatous lesions in patients with rheumatoid arthritis. Clin. Immunol. 2013, 147, 144–150. [Google Scholar] [CrossRef] [PubMed]
- McGrath, C.M.; Young, S.P. Lipid and Metabolic Changes in Rheumatoid Arthritis. Curr. Rheumatol. Rep. 2015, 17, 57. [Google Scholar] [CrossRef] [PubMed]
- Daïen, C.I.; Duny, Y.; Barnetche, T.; Daurès, J.P.; Combe, B.; Morel, J. Effect of TNF inhibitors on lipid profile in rheumatoid arthritis: A systematic review with meta-analysis. Ann. Rheum. Dis. 2012, 71, 862–868. [Google Scholar] [CrossRef] [PubMed]
- Obermoser, G.; Pascual, V. The interferon-α signature of systemic lupus erythematosus. Lupus 2010, 19, 1012–1019. [Google Scholar] [CrossRef] [PubMed]
- Wigren, M.; Nilsson, J.; Kaplan, M.J. Pathogenic immunity in systemic lupus erythematosus and atherosclerosis: Common mechanisms and possible targets for intervention. J. Intern. Med. 2015, 278, 494–506. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.Y.; Qu, B.; Ye, P.; Li, J.; Bao, C.D. Vulnerability of atherosclerotic plaques is associated with type I interferon in a murine model of lupus and atherosclerosis. Genet. Mol. Res. 2015, 14, 14871–14881. [Google Scholar] [CrossRef] [PubMed]
- Sumarac-Dumanovic, M.; Stevanovic, D.; Ljubic, A.; Jorga, J.; Simic, M.; Stamenkovic-Pejkovic, D.; Starcevic, V.; Trajkovic, V.; Micic, D. Increased activity of interleukin-23/interleukin-17 proinflammatory axis in obese women. Int. J. Obes. 2009, 33, 151–156. [Google Scholar] [CrossRef] [PubMed]
- Fabbrini, E.; Cella, M.; Mccartney, S.A.; Fuchs, A.; Abumrad, N.A.; Pietka, T.A.; Chen, Z.; Finck, B.N.; Han, D.H.; Magkos, F.; et al. Association between specific adipose tissue CD4+ T-cell populations and insulin resistance in obese individuals. Gastroenterology 2013, 145, 366–374. [Google Scholar] [CrossRef] [PubMed]
- Westlake, S.L.; Colebatch, A.N.; Baird, J.; Kiely, P.; Quinn, M.; Choy, E.; Ostor, A.J.; Edwards, C.J. The effect of methotrexate on cardiovascular disease in patients with rheumatoid arthritis: A systematic literature review. Rheumatology 2010, 49, 295–307. [Google Scholar] [CrossRef] [PubMed]
- Vlachopoulos, C.; Gravos, A.; Georgiopoulos, G.; Terentes-Printzios, D.; Ioakeimidis, N.; Vassilopoulos, D.; Stamatelopoulos, K.; Tousoulis, D. The effect of TNF—A antagonists on aortic stiffness and wave reflections: A meta-analysis. Clin. Rheumatol. 2018, 37, 515–526. [Google Scholar] [CrossRef] [PubMed]
- Protogerou, A.D.; Zampeli, E.; Fragiadaki, K.; Stamatelopoulos, K.; Papamichael, C.; Sfikakis, P.P. A pilot study of endothelial dysfunction and aortic stiffness after interleukin-6 receptor inhibition in rheumatoid arthritis. Atherosclerosis 2011, 219, 734–736. [Google Scholar] [CrossRef] [PubMed]
- Tam, L.-S.; Kitas, G.D.; Gonzalez-Gay, M.A. Can suppression of inflammation by anti-TNF prevent progression of subclinical atherosclerosis in inflammatory arthritis? Rheumatology 2014, 53, 1108–1119. [Google Scholar] [CrossRef] [PubMed]
- Low, A.S.; Symmons, D.P.; Lunt, M.; Mercer, L.K.; Gale, C.P.; Watson, K.D.; Dixon, W.G.; Hyrich, K.L. British Society for Rheumatology Biologics Register for Rheumatoid Arthritis (BSRBR-RA) and the BSRBR Control Centre Consortium, Relationship between exposure to tumour necrosis factor inhibitor therapy and incidence and severity of myocardial infarction in patients with rheumatoid arthritis. Ann. Rheum. Dis. 2017, 76, 654–660. [Google Scholar] [CrossRef] [PubMed]
- Roubille, C.; Richer, V.; Starnino, T.; McCourt, C.; McFarlane, A.; Fleming, P.; Siu, S.; Kraft, J.; Lynde, C.; Pope, J.; Gulliver, W. The effects of tumour necrosis factor inhibitors, methotrexate, non-steroidal anti-inflammatory drugs and corticosteroids on cardiovascular events in rheumatoid arthritis, psoriasis and psoriatic arthritis: A systematic review and meta-analysis. Ann. Rheum. Dis. 2015, 74, 480–489. [Google Scholar] [CrossRef] [PubMed]
- Ketelhuth, D.F.J.; Hansson, G.K. Modulation of autoimmunity and atherosclerosis—common targets and promising translational approaches against disease. Circ. J. 2015, 79, 924–933. [Google Scholar] [CrossRef] [PubMed]
- Koga, M.; Kai, H.; Yasukawa, H.; Yamamoto, T.; Kawai, Y.; Kato, S.; Kusaba, K.; Kai, M.; Egashira, K.; Kataoka, Y.; Imaizumi, T. Inhibition of Progression and Stabilization of Plaques by Postnatal Interferon-Function Blocking in ApoE-Knockout Mice. Circ. Res. 2007, 101, 348–356. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef] [PubMed]
- Dessein, P.H.; Tsang, L.; Woodiwiss, A.J.; Norton, G.R.; Solomon, A. Circulating Concentrations of the Novel Adipokine Chemerin Are Associated with Cardiovascular Disease Risk in Rheumatoid Arthritis. J. Rheumatol. 2014, 41, 1746–1754. [Google Scholar] [CrossRef] [PubMed]
- Makrilakis, K.; Fragiadaki, K.; Sfikakis, P.P.; Kitas, G.D. Chemerin and Cardiovascular Risk in Rheumatoid Arthritis after Interleukin 6 Receptor Blockade. J. Rheumatol. 2015, 42, 349. [Google Scholar] [CrossRef] [PubMed]
- Thomas, G.; Mancini, J.; Jourde-Chiche, N.; Sarlon, G.; Amoura, Z.; Harlé, J.R.; Jougla, E.; Chiche, L. Mortality Associated With Systemic Lupus Erythematosus in France Assessed by Multiple-Cause-of-Death Analysis. Arthritis Rheumatol. 2014, 66, 2503–2511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elfving, P.; Puolakka, K.; Kautiainen, H.; Virta, L.J.; Pohjolainen, T.; Kaipiainen-Seppänen, O. Mortality and causes of death among incident cases of systemic lupus erythematosus in Finland 2000–2008. Lupus 2014, 23, 1430–1434. [Google Scholar] [CrossRef] [PubMed]
- Wong, K.; Gladman, D.D.; Husted, J.; Long, J.A.; Farewell, V.T.; Long, J.A. Mortality studies in psoriatic arthritis: Results from a single outpatient clinic. I. Causes and risk of death. Arthritis Rheum. 1997, 40, 1868–1872. [Google Scholar] [CrossRef] [PubMed]
- Ali, Y.; Tom, B.D.; Schentag, C.T.; Farewell, V.T.; Gladman, D.D. Improved survival in psoriatic arthritis with calendar time. Arthritis Rheumatol. 2007, 56, 2708–2714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathieu, S.; Pereira, B.; Soubrier, M. Cardiovascular events in ankylosing spondylitis: An updated meta-analysis. Semin. Arthritis Rheum. 2015, 44, 551–555. [Google Scholar] [CrossRef] [PubMed]
- Wainstein, M.V.; Mossmann, M.; Araujo, G.N.; Gonçalves, S.C.; Gravina, G.L.; Sangalli, M.; Veadrigo, F.; Matte, R.; Reich, R.; Costa, F.G.; et al. Elevated serum interleukin-6 is predictive of coronary artery disease in intermediate risk overweight patients referred for coronary angiography. Diabetol. Metab. Syndr. 2017, 9, 67. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.H.; Glei, D.; Weinstein, M.; Wu, S.I.; Chien, K.L. Additive value of interleukin-6 and C-reactive protein in risk prediction for all-cause and cardiovascular mortality among a representative adult cohort in Taiwan. J. Formos. Med. Assoc. 2017, 116, 982–992. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| RA | SLE | AS | PsA | ||
|---|---|---|---|---|---|
| CVD risk or mortality | Comparative risk to DM [8,9,29] 1.5-fold risk compared to the general population [1] More than 1.5 risk of fatal CV event [31] | 2- to 3-fold mortality (up to 16-fold) [12,13] | ↑ vs. controls [32,33,34] | Prevalence resembles that of RA [35] ↑ mortality [32] | |
| CV events | CHD | Similar risk as DM [10] 1.6–2.1 rate ratio for MI [17,30,31] | 2- to 3-fold risk (up to 52- fold in young SLE women) [12,13] | 1.4 relative risk of MI [18,36] | 1.4 relative risk of MI [17] |
| Stroke | 1.9 rate ratio [31] | 2-fold risk [12,13] | 1.3-1.4 relative risk [18,36] | Similar or slightly increased prevalence [37] | |
| Subclinical CVD | cIMT | ↑ vs. controls [38] | ↑ vs. controls [14,16] | ↑ vs. controls [23] | ↑ vs. controls [39] |
| PWV | ↑ vs. controls [40] | ↑ vs. controls [41,42,43] | ↑ vs. controls [44,45] | ↑ vs. controls [46,47] | |
| FMD | ↓ vs. controls [48,49] | ↓ vs. controls [16] | ↓ vs. controls [45] | ↓ vs. controls [39] | |
| Aix | ↑ vs. controls [40] | ↑ vs. controls [50,51] | Similar to controls [23] | -- | |
| Plaques | ↑ carotid vs. controls [38] femoral analogous to DM [52] | 2-fold risk [14,15,16], comparable to RA and DM [15] | Similar to controls [23] | 3-fold risk vs. controls [39] | |
| Classical Risk factors |
|
|
|
| |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arida, A.; Protogerou, A.D.; Kitas, G.D.; Sfikakis, P.P. Systemic Inflammatory Response and Atherosclerosis: The Paradigm of Chronic Inflammatory Rheumatic Diseases. Int. J. Mol. Sci. 2018, 19, 1890. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19071890
Arida A, Protogerou AD, Kitas GD, Sfikakis PP. Systemic Inflammatory Response and Atherosclerosis: The Paradigm of Chronic Inflammatory Rheumatic Diseases. International Journal of Molecular Sciences. 2018; 19(7):1890. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19071890
Chicago/Turabian StyleArida, Aikaterini, Athanasios D. Protogerou, George D. Kitas, and Petros P. Sfikakis. 2018. "Systemic Inflammatory Response and Atherosclerosis: The Paradigm of Chronic Inflammatory Rheumatic Diseases" International Journal of Molecular Sciences 19, no. 7: 1890. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19071890