Senescent Microvesicles: A Novel Advance in Molecular Mechanisms of Atherosclerotic Calcification

 , , and
, , and
Abstract
:1. Introduction
2. Atherosclerosis
3. Senescence
3.1. SASP
3.1.1. Replicative Senescence
3.1.2. Aging
3.2. SIPS
3.2.1. Hyperlipidaemia
3.2.2. Diabetes Mellitus
3.2.3. Hypertension
3.2.4. Smoking
3.2.5. Uremic Toxins
4. Microvesicles in Atherosclerosis
5. Microvesicles in Atherosclerotic Calcification
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AP | Atherosclerotic plaque |
| CVDs | Cardiovascular diseases |
| VC | Vascular calcification |
| EVs | Extracellular vesicles |
| MVs | Microvesicles |
| miRNAs | MicroRNAs |
| ECs | Endothelial cells |
| HT | Hypertension |
| ROS | Reactive oxygen species |
| HBP | High blood pressure |
| RNS | Reactive nitrogen species |
| VCAM-1 | Vascular cell adhesion molecule-1 |
| ICAM-1 | Intercellular adhesion molecule-1 |
| MMPs | Metalloproteinases |
| NO | Nitric oxide |
| nLDL | Native low-density lipoproteins |
| oxLDL | Oxidized low-density lipoproteins |
| CKDs | Chronic kidney diseases |
| SASP | Senescence-associated secretory phenotype |
| SIPS | Stress-induced premature senescence |
| OIS | Oncogene-induced senescence |
| pRb | Retinoblastoma protein |
| SCAPs | Senescent cell anti-apoptotic pathways |
| SAMD | Senescence-associated mitochondrial dysfunction |
| SA-β-gal | Senescence-associated-β-galactosidase |
| BrU | 5-bromodeoxyuridine |
| 3H-dT | 3H-Thymidine |
| PCNA | Proliferating cell nuclear antigen |
| SAHFs | Senescence-associated heterochromatin foci |
| DAPI | 4′,6-diamidino-2-phenylindole |
| SDF | Senescence-associated DNA damage foci |
| γ-H2AX | phosphorylated histone H2AX |
| 53BP1 | p53-binding protein-1 |
| X-gal substrate | 5-bromo-4-chloro-3-indolyl-d-galactoside |
| C12FDG | 5-Dodecanoylaminofluorescein di-β-d-galactopyranoside |
| Hsp70 | Heat shock protein 70 |
| MFRTA | Mitochondrial free radical theory of aging |
| CHD | Coronary heart disease |
| NEFA | Non-esterified free fatty acid |
| AGEs | Advanced glycosylation end products |
| HT | Hypertension |
| EUTOX | European Uremic Toxin Work Group |
| NF-κB | Nuclear factor-κB |
| BMP2 | Bone morphogenetic protein 2 |
| CEVs | Calcifying extracellular vesicles |
| vWF | von Willebrand factor |
| CD40L | Cluster of differentiation 40 ligand |
| Gla | γ-carboxyglutamic acid |
| NADPH | Nicotinamide adenine dinucleotide phosphate |
| SOD2 | Superoxide dismutase-2 |
| TSG101 | Tumor susceptibility gene 101 protein |
| S100A9 | S100 calcium-binding protein A9 |
| EMVs | Endothelial microvesicles |
| MMVs | Macrophages microvesicles |
| ALP | Alkaline phosphatase |
References
- Marinou, K.; Christodoulides, C.; Antoniades, C.; Koutsilieris, M. Wnt signalling in cardiovascular physiology. Trends Endocrinol. Metab. 2012, 23, 628–636. [Google Scholar] [CrossRef] [PubMed]
- Tsaousi, A.; Mill, C.; George, S.J. The wnt pathways in vascular disease: Lessons from vascular development. Curr. Opin. Lipidol. 2011, 22, 350–357. [Google Scholar] [CrossRef] [PubMed]
- Tsai, J.S.; Chen, C.Y.; Chen, Y.L.; Chuang, L.M. Rosiglitazone inhibits monocyte/macrophage adhesion through de novo adiponectin production in human monocytes. J. Cell. Biochem. 2010, 110, 1410–1419. [Google Scholar] [CrossRef] [PubMed]
- Wadley, A.J.; Veldhuijzen van Zanten, J.J.; Aldred, S. The interactions of oxidative stress and inflammation with vascular dysfunction in ageing: The vascular health triad. Age 2013, 35, 705–718. [Google Scholar] [CrossRef] [PubMed]
- Uryga, A.K.; Bennett, M.R. Ageing induced vascular smooth muscle cell senescence in atherosclerosis. J. Physiol. 2016, 594, 2115–2124. [Google Scholar] [CrossRef] [PubMed]
- Thompson, B.; Towler, D.A. Arterial calcification and bone physiology: Role of the bone-vascular axis. Nat. Rev. Endocrinol. 2012, 8, 529–543. [Google Scholar] [CrossRef] [PubMed]
- Boulanger, C.M.; Loyer, X.; Rautou, P.E.; Amabile, N. Extracellular vesicles in coronary artery disease. Nat. Rev. Cardiol. 2017, 14, 259–272. [Google Scholar] [CrossRef] [PubMed]
- Colombo, M.; Raposo, G.; Théry, C. Biogenesis, secretion and intercellular interactions of exosomes and other extracellular vesicles. Annu. Rev. Cell Dev. Biol. 2014, 30, 255–289. [Google Scholar] [CrossRef] [PubMed]
- Yáñez-Mó, M.; Siljander, P.R.; Andreu, Z.; Zavec, A.B.; Borràs, F.E.; Buzas, E.I.; Buzas, K.; Casal, E.; Cappello, F.; Carvalho, J.; et al. Biological properties of extracellular vesicles and their physiological functions. J. Extracell. Vesicles 2015, 4, 27066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandler, W.L. Measurement of microvesicle levels in human blood using flow cytometry. Cytom. B Clin. Cytom. 2016, 90, 326–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Craige, S.M. Reactive oxygen species in endothelial function—From disease to adaptation. Circ. J. 2015, 79, 1145–1155. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, B.D.; Paine, M.S.; Brooks, A.M.; McCubrey, J.A.; Renegar, R.H.; Wang, R.; Terrian, D.M. Senescence-associated exosome release from human prostate cancer cells. Cancer Res. 2008, 68, 7864–7871. [Google Scholar] [CrossRef] [PubMed]
- Takasugi, M. Emerging roles of extracellular vesicles in cellular senescence and aging. Aging Cell 2018, 17, e12734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, A.; Okada, R.; Nagao, K.; Kawamata, Y.; Hanyu, A.; Yoshimoto, S.; Takasugi, M.; Watanabe, S.; Kanemaki, M.T.; Obuse, C.; et al. Exosomes maintain cellular homeostasis by excreting harmful dna from cells. Nat. Commun. 2017, 8, 15287. [Google Scholar] [CrossRef] [PubMed]
- Anderson, H.C.; Mulhall, D.; Garimella, R. Role of extracellular membrane vesicles in the pathogenesis of various diseases, including cancer, renal diseases, atherosclerosis and arthritis. Lab. Investig. 2010, 90, 1549–1557. [Google Scholar] [CrossRef] [PubMed]
- Gustafson, D.; Veitch, S.; Fish, J.E. Extracellular vesicles as protagonists of diabetic cardiovascular pathology. Front. Cardiovasc. Med. 2017, 4, 71. [Google Scholar] [CrossRef] [PubMed]
- Fleury, A.; Martinez, M.C.; Le Lay, S. Extracellular vesicles as therapeutic tools in cardiovascular diseases. Front. Immunol. 2014, 5, 370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, M.L.; Williams, K.J. Microvesicles: Potential markers and mediators of endothelial dysfunction. Curr. Opin. Endocrinol. Diabetes Obes. 2012, 19, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Badimon, L.; Suades, R.; Arderiu, G.; Peña, E.; Chiva-Blanch, G.; Padró, T. Microvesicles in atherosclerosis and angiogenesis: From bench to bedside and reverse. Front. Cardiovasc. Med. 2017, 4, 77. [Google Scholar] [CrossRef] [PubMed]
- Van der Vorst, E.P.C.; de Jong, R.J.; Donners, M.M.P.C. Message in a microbottle: Modulation of vascular inflammation and atherosclerosis by extracellular vesicles. Front. Cardiovasc. Med. 2018, 5, 2. [Google Scholar] [CrossRef] [PubMed]
- Chistiakov, D.A.; Orekhov, A.N.; Bobryshev, Y.V. Extracellular vesicles and atherosclerotic disease. Cell. Mol. Life Sci. 2015, 72, 2697–2708. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Wu, C.; Xiao, J.; Li, D.; Sun, Z.; Li, M. Endothelial extracellular vesicles modulate the macrophage phenotype: Potential implications in atherosclerosis. Scand. J. Immunol. 2018, 87, e12648. [Google Scholar] [CrossRef] [PubMed]
- Soriano, S.; Carmona, A.; Triviño, F.; Rodriguez, M.; Alvarez-Benito, M.; Martín-Malo, A.; Alvarez-Lara, M.A.; Ramírez, R.; Aljama, P.; Carracedo, J. Endothelial damage and vascular calcification in patients with chronic kidney disease. Am. J. Physiol. Renal Physiol. 2014, 307, F1302–F1311. [Google Scholar] [CrossRef] [PubMed]
- New, S.E.; Aikawa, E. Role of extracellular vesicles in de novo mineralization: An additional novel mechanism of cardiovascular calcification. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1753–1758. [Google Scholar] [CrossRef] [PubMed]
- New, S.E.; Goettsch, C.; Aikawa, M.; Marchini, J.F.; Shibasaki, M.; Yabusaki, K.; Libby, P.; Shanahan, C.M.; Croce, K.; Aikawa, E. Macrophage-derived matrix vesicles: An alternative novel mechanism for microcalcification in atherosclerotic plaques. Circ. Res. 2013, 113, 72–77. [Google Scholar] [CrossRef] [PubMed]
- Kapustin, A.N.; Chatrou, M.L.; Drozdov, I.; Zheng, Y.; Davidson, S.M.; Soong, D.; Furmanik, M.; Sanchis, P.; De Rosales, R.T.; Alvarez-Hernandez, D.; et al. Vascular smooth muscle cell calcification is mediated by regulated exosome secretion. Circ. Res. 2015, 116, 1312–1323. [Google Scholar] [CrossRef] [PubMed]
- Krohn, J.B.; Hutcheson, J.D.; Martínez-Martínez, E.; Aikawa, E. Extracellular vesicles in cardiovascular calcification: Expanding current paradigms. J. Physiol. 2016, 594, 2895–2903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krohn, J.B.; Hutcheson, J.D.; Martínez-Martínez, E.; Irvin, W.S.; Bouten, C.V.; Bertazzo, S.; Bendeck, M.P.; Aikawa, E. Discoidin domain receptor-1 regulates calcific extracellular vesicle release in vascular smooth muscle cell fibrocalcific response via transforming growth factor-β signalling. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 525–533. [Google Scholar] [CrossRef] [PubMed]
- Blankenberg, S.; Barbaux, S.; Tiret, L. Adhesion molecules and atherosclerosis. Atherosclerosis 2003, 170, 191–203. [Google Scholar] [CrossRef]
- Rodier, F.; Campisi, J. Four faces of cellular senescence. J. Cell Biol. 2011, 192, 547–556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coppé, J.P.; Patil, C.K.; Rodier, F.; Sun, Y.; Muñoz, D.P.; Goldstein, J.; Nelson, P.S.; Desprez, P.Y.; Campisi, J. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic ras and the p53 tumor suppressor. PLoS Biol. 2008, 6, 2853–2868. [Google Scholar] [CrossRef] [PubMed]
- Hubackova, S.; Krejcikova, K.; Bartek, J.; Hodny, Z. Il1- and tgfβ-nox4 signalling, oxidative stress and dna damage response are shared features of replicative, oncogene-induced and drug-induced paracrine ‘bystander senescence’. Aging 2012, 4, 932–951. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.Y.; Awad, E.M.; Oszwald, A.; Mayr, M.; Yin, X.; Waltenberger, B.; Stuppner, H.; Lipovac, M.; Uhrin, P.; Breuss, J.M. Premature senescence of endothelial cells upon chronic exposure to tnfα can be prevented by n-acetyl cysteine and plumericin. Sci. Rep. 2017, 7, 39501. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T.; Iguchi, A. Possibility of the regression of atherosclerosis through the prevention of endothelial senescence by the regulation of nitric oxide and free radical scavengers. Geriatr. Gerontol. Int. 2010, 10, 115–130. [Google Scholar] [CrossRef] [PubMed]
- Minamino, T.; Miyauchi, H.; Yoshida, T.; Ishida, Y.; Yoshida, H.; Komuro, I. Endothelial cell senescence in human atherosclerosis: Role of telomere in endothelial dysfunction. Circulation 2002, 105, 1541–1544. [Google Scholar] [CrossRef] [PubMed]
- Donato, A.J.; Morgan, R.G.; Walker, A.E.; Lesniewski, L.A. Cellular and molecular biology of aging endothelial cells. J. Mol. Cell. Cardiol. 2015, 89, 122–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davignon, J.; Ganz, P. Role of endothelial dysfunction in atherosclerosis. Circulation 2004, 109, III27–III32. [Google Scholar] [CrossRef] [PubMed]
- Christ, A.; Bekkering, S.; Latz, E.; Riksen, N.P. Long-term activation of the innate immune system in atherosclerosis. Semin. Immunol. 2016, 28, 384–393. [Google Scholar] [CrossRef] [PubMed]
- Foncea, R.; Carvajal, C.; Almarza, C.; Leighton, F. Endothelial cell oxidative stress and signal transduction. Biol. Res. 2000, 33, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Lusis, A.J. Atherosclerosis. Nature 2000, 407, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Beckman, J.A.; Creager, M.A.; Libby, P. Diabetes and atherosclerosis: Epidemiology, pathophysiology and management. JAMA 2002, 287, 2570–2581. [Google Scholar] [CrossRef] [PubMed]
- Jeffrey, J.; Siracuse, E.L.C. The Pathogenesis of Diabetic Atherosclerosis. In Diabetes and Peripheral Vascular Disease; Humana Press: Totowa, NJ, USA, 2012; pp. 13–26. [Google Scholar]
- Shrikhande, G.V.; Scali, S.T.; da Silva, C.G.; Damrauer, S.M.; Csizmadia, E.; Putheti, P.; Matthey, M.; Arjoon, R.; Patel, R.; Siracuse, J.J.; et al. O-glycosylation regulates ubiquitination and degradation of the anti-inflammatory protein a20 to accelerate atherosclerosis in diabetic apoe-null mice. PLoS ONE 2010, 5, e14240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vengrenyuk, Y.; Carlier, S.; Xanthos, S.; Cardoso, L.; Ganatos, P.; Virmani, R.; Einav, S.; Gilchrist, L.; Weinbaum, S. A hypothesis for vulnerable plaque rupture due to stress-induced debonding around cellular microcalcifications in thin fibrous caps. Proc. Natl. Acad. Sci. USA 2006, 103, 14678–14683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Howson, J.M.M.; Zhao, W.; Barnes, D.R.; Ho, W.K.; Young, R.; Paul, D.S.; Waite, L.L.; Freitag, D.F.; Fauman, E.B.; Salfati, E.L.; et al. Fifteen new risk loci for coronary artery disease highlight arterial-wall-specific mechanisms. Nat. Genet. 2017, 49, 1113–1119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klarin, D.; Zhu, Q.M.; Emdin, C.A.; Chaffin, M.; Horner, S.; McMillan, B.J.; Leed, A.; Weale, M.E.; Spencer, C.C.A.; Aguet, F.; et al. Genetic analysis in uk biobank links insulin resistance and transendothelial migration pathways to coronary artery disease. Nat. Genet. 2017, 49, 1392–1397. [Google Scholar] [CrossRef] [PubMed]
- Albanese, I.; Khan, K.; Barratt, B.; Al-Kindi, H.; Schwertani, A. Atherosclerotic calcification: Wnt is the hint. J. Am. Heart Assoc. 2018, 7, e007356. [Google Scholar] [CrossRef] [PubMed]
- Tesauro, M.; Mauriello, A.; Rovella, V.; Annicchiarico-Petruzzelli, M.; Cardillo, C.; Melino, G.; Di Daniele, N. Arterial ageing: From endothelial dysfunction to vascular calcification. J. Intern. Med. 2017, 281, 471–482. [Google Scholar] [CrossRef] [PubMed]
- Alique, M.; Ruíz-Torres, M.P.; Bodega, G.; Noci, M.V.; Troyano, N.; Bohórquez, L.; Luna, C.; Luque, R.; Carmona, A.; Carracedo, J.; et al. Microvesicles from the plasma of elderly subjects and from senescent endothelial cells promote vascular calcification. Aging 2017, 9, 778–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polfus, L.M.; Smith, J.A.; Shimmin, L.C.; Bielak, L.F.; Morrison, A.C.; Kardia, S.L.; Peyser, P.A.; Hixson, J.E. Genome-wide association study of gene by smoking interactions in coronary artery calcification. PLoS ONE 2013, 8, e74642. [Google Scholar] [CrossRef] [PubMed]
- Van Setten, J.; Isgum, I.; Smolonska, J.; Ripke, S.; de Jong, P.A.; Oudkerk, M.; de Koning, H.; Lammers, J.W.; Zanen, P.; Groen, H.J.; et al. Genome-wide association study of coronary and aortic calcification implicates risk loci for coronary artery disease and myocardial infarction. Atherosclerosis 2013, 228, 400–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Donnell, C.J.; Kavousi, M.; Smith, A.V.; Kardia, S.L.; Feitosa, M.F.; Hwang, S.J.; Sun, Y.V.; Province, M.A.; Aspelund, T.; Dehghan, A.; et al. Genome-wide association study for coronary artery calcification with follow-up in myocardial infarction. Circulation 2011, 124, 2855–2864. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, C.J.; Cupples, L.A.; D’Agostino, R.B.; Fox, C.S.; Hoffmann, U.; Hwang, S.J.; Ingellson, E.; Liu, C.; Murabito, J.M.; Polak, J.F.; et al. Genome-wide association study for subclinical atherosclerosis in major arterial territories in the nhlbi’s framingham heart study. BMC Med. Genet. 2007, 8 (Suppl. 1), S4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wojczynski, M.K.; Li, M.; Bielak, L.F.; Kerr, K.F.; Reiner, A.P.; Wong, N.D.; Yanek, L.R.; Qu, L.; White, C.C.; Lange, L.A.; et al. Genetics of coronary artery calcification among african americans, a meta-analysis. BMC Med. Genet. 2013, 14, 75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Divers, J.; Palmer, N.D.; Langefeld, C.D.; Brown, W.M.; Lu, L.; Hicks, P.J.; Smith, S.C.; Xu, J.; Terry, J.G.; Register, T.C.; et al. Genome-wide association study of coronary artery calcified atherosclerotic plaque in african americans with type 2 diabetes. BMC Genet. 2017, 18, 105. [Google Scholar] [CrossRef] [PubMed]
- Martin, G.M. The biology of aging: 1985–2010 and beyond. FASEB J. 2011, 25, 3756–3762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kadota, T.; Yoshioka, Y.; Fujita, Y.; Kuwano, K.; Ochiya, T. Extracellular vesicles in lung cancer-from bench to bedside. Semin. Cell Dev. Biol. 2017, 67, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.J.; Cai, G.Y.; Chen, X.M. Cellular senescence, senescence-associated secretory phenotype and chronic kidney disease. Oncotarget 2017, 8, 64520–64533. [Google Scholar] [CrossRef] [PubMed]
- St Sauver, J.L.; Boyd, C.M.; Grossardt, B.R.; Bobo, W.V.; Finney Rutten, L.J.; Roger, V.L.; Ebbert, J.O.; Therneau, T.M.; Yawn, B.P.; Rocca, W.A. Risk of developing multimorbidity across all ages in an historical cohort study: Differences by sex and ethnicity. BMJ Open 2015, 5, e006413. [Google Scholar] [CrossRef] [PubMed]
- Kirkland, J.L.; Tchkonia, T. Cellular senescence: A translational perspective. EBioMedicine 2017, 21, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Van Deursen, J.M. The role of senescent cells in ageing. Nature 2014, 509, 439–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blagosklonny, M.V. Geroconversion: Irreversible step to cellular senescence. Cell Cycle 2014, 13, 3628–3635. [Google Scholar] [CrossRef] [PubMed]
- Shay, J.W.; Wright, W.E. Hayflick, his limit and cellular ageing. Nat. Rev. Mol. Cell Biol. 2000, 1, 72–76. [Google Scholar] [CrossRef] [PubMed]
- Goligorsky, M.S.; Chen, J.; Patschan, S. Stress-induced premature senescence of endothelial cells: A perilous state between recovery and point of no return. Curr. Opin. Hematol. 2009, 16, 215–219. [Google Scholar] [CrossRef] [PubMed]
- Campisi, J. Aging, cellular senescence and cancer. Annu. Rev. Physiol. 2013, 75, 685–705. [Google Scholar] [CrossRef] [PubMed]
- Seoane, M.; Costoya, J.A.; Arce, V.M. Uncoupling oncogene-induced senescence (OIS) and dna damage response (DDR) triggered by dna hyper-replication: Lessons from primary mouse embryo astrocytes (MEA). Sci. Rep. 2017, 7, 12991. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.C.; Bennett, M. Aging and atherosclerosis: Mechanisms, functional consequences and potential therapeutics for cellular senescence. Circ. Res. 2012, 111, 245–259. [Google Scholar] [CrossRef] [PubMed]
- Head, T.; Daunert, S.; Goldschmidt-Clermont, P.J. The aging risk and atherosclerosis: A fresh look at arterial homeostasis. Front. Genet. 2017, 8, 216. [Google Scholar] [CrossRef] [PubMed]
- Urbanelli, L.; Buratta, S.; Sagini, K.; Tancini, B.; Emiliani, C. Extracellular vesicles as new players in cellular senescence. Int. J. Mol. Sci. 2016, 17, 1408. [Google Scholar] [CrossRef] [PubMed]
- Carmona, A.; Guerrero, F.; Buendia, P.; Obrero, T.; Aljama, P.; Carracedo, J. Microvesicles derived from indoxyl sulphate treated endothelial cells induce endothelial progenitor cells dysfunction. Front. Physiol. 2017, 8, 666. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Tchkonia, T.; Pirtskhalava, T.; Gower, A.C.; Ding, H.; Giorgadze, N.; Palmer, A.K.; Ikeno, Y.; Hubbard, G.B.; Lenburg, M.; et al. The achilles’ heel of senescent cells: From transcriptome to senolytic drugs. Aging Cell 2015, 14, 644–658. [Google Scholar] [CrossRef] [PubMed]
- Baar, M.P.; Brandt, R.M.C.; Putavet, D.A.; Klein, J.D.D.; Derks, K.W.J.; Bourgeois, B.R.M.; Stryeck, S.; Rijksen, Y.; van Willigenburg, H.; Feijtel, D.A.; et al. Targeted apoptosis of senescent cells restores tissue homeostasis in response to chemotoxicity and aging. Cell 2017, 169, 132–147. [Google Scholar] [CrossRef] [PubMed]
- Burton, D.G.A.; Stolzing, A. Cellular senescence: Immunosurveillance and future immunotherapy. Ageing Res. Rev. 2018, 43, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Campisi, J.; d’Adda di Fagagna, F. Cellular senescence: When bad things happen to good cells. Nat. Rev. Mol. Cell Biol. 2007, 8, 729–740. [Google Scholar] [CrossRef] [PubMed]
- Burton, D.G.; Sheerin, A.N.; Ostler, E.L.; Smith, K.; Giles, P.J.; Lowe, J.; Rhys-Williams, W.; Kipling, D.G.; Faragher, R.G. Cyclin d1 overexpression permits the reproducible detection of senescent human vascular smooth muscle cells. Ann. N. Y. Acad. Sci. 2007, 1119, 20–31. [Google Scholar] [CrossRef] [PubMed]
- Freund, A.; Laberge, R.M.; Demaria, M.; Campisi, J. Lamin b1 loss is a senescence-associated biomarker. Mol. Biol. Cell 2012, 23, 2066–2075. [Google Scholar] [CrossRef] [PubMed]
- Narita, M.; Nũnez, S.; Heard, E.; Lin, A.W.; Hearn, S.A.; Spector, D.L.; Hannon, G.J.; Lowe, S.W. Rb-mediated heterochromatin formation and silencing of e2f target genes during cellular senescence. Cell. 2003, 113, 703–716. [Google Scholar] [CrossRef]
- Lee, B.Y.; Han, J.A.; Im, J.S.; Morrone, A.; Johung, K.; Goodwin, E.C.; Kleijer, W.J.; DiMaio, D.; Hwang, E.S. Senescence-associated beta-galactosidase is lysosomal beta-galactosidase. Aging Cell 2006, 5, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Debacq-Chainiaux, F.; Erusalimsky, J.D.; Campisi, J.; Toussaint, O. Protocols to detect senescence-associated beta-galactosidase (sa-betagal) activity, a biomarker of senescent cells in culture and in vivo. Nat. Protoc. 2009, 4, 1798–1806. [Google Scholar] [CrossRef] [PubMed]
- Krishnamurthy, J.; Torrice, C.; Ramsey, M.R.; Kovalev, G.I.; Al-Regaiey, K.; Su, L.; Sharpless, N.E. Ink4a/arf expression is a biomarker of aging. J. Clin. Investig. 2004, 114, 1299–1307. [Google Scholar] [CrossRef] [PubMed]
- Beauséjour, C.M.; Krtolica, A.; Galimi, F.; Narita, M.; Lowe, S.W.; Yaswen, P.; Campisi, J. Reversal of human cellular senescence: Roles of the p53 and p16 pathways. EMBO J. 2003, 22, 4212–4222. [Google Scholar] [CrossRef] [PubMed]
- Itahana, K.; Zou, Y.; Itahana, Y.; Martinez, J.L.; Beausejour, C.; Jacobs, J.J.; Van Lohuizen, M.; Band, V.; Campisi, J.; Dimri, G.P. Control of the replicative life span of human fibroblasts by p16 and the polycomb protein bmi-1. Mol. Cell. Biol. 2003, 23, 389–401. [Google Scholar] [CrossRef] [PubMed]
- Campisi, J. Senescent cells, tumor suppression and organismal aging: Good citizens, bad neighbors. Cell 2005, 120, 513–522. [Google Scholar] [CrossRef] [PubMed]
- Correia-Melo, C.; Hewitt, G.; Passos, J.F. Telomeres, oxidative stress and inflammatory factors: Partners in cellular senescence? Longev. Healthspan 2014, 3, 1. [Google Scholar] [CrossRef] [PubMed]
- Hayflick, L.; Moorhead, P.S. The serial cultivation of human diploid cell strains. Exp. Cell Res. 1961, 25, 585–621. [Google Scholar] [CrossRef]
- Jaskelioff, M.; Muller, F.L.; Paik, J.H.; Thomas, E.; Jiang, S.; Adams, A.C.; Sahin, E.; Kost-Alimova, M.; Protopopov, A.; Cadiñanos, J.; et al. Telomerase reactivation reverses tissue degeneration in aged telomerase-deficient mice. Nature 2011, 469, 102–106. [Google Scholar] [CrossRef] [PubMed]
- Kong, Y.; Trabucco, S.E.; Zhang, H. Oxidative stress, mitochondrial dysfunction and the mitochondria theory of aging. Interdiscip. Top. Gerontol. 2014, 39, 86–107. [Google Scholar] [PubMed]
- Grootaert, M.O.J.; Moulis, M.; Roth, L.; Martinet, W.; Vindis, C.; Bennett, M.R.; De Meyer, G.R.Y. Vascular smooth muscle cell death, autophagy and senescence in atherosclerosis. Cardiovasc. Res. 2018, 114, 622–634. [Google Scholar] [CrossRef] [PubMed]
- McHugh, D.; Gil, J. Senescence and aging: Causes, consequences and therapeutic avenues. J. Cell Biol. 2018, 217, 65–77. [Google Scholar] [CrossRef] [PubMed]
- De la Fuente, M.; Miquel, J. An update of the oxidation-inflammation theory of aging: The involvement of the immune system in oxi-inflamm-aging. Curr. Pharm. Des. 2009, 15, 3003–3026. [Google Scholar] [CrossRef] [PubMed]
- Martínez de Toda, I.; De la Fuente, M. The role of hsp70 in oxi-inflamm-aging and its use as a potential biomarker of lifespan. Biogerontology 2015, 16, 709–721. [Google Scholar] [CrossRef] [PubMed]
- Sagini, K.; Costanzi, E.; Emiliani, C.; Buratta, S.; Urbanelli, L. Extracellular vesicles as conveyors of membrane-derived bioactive lipids in immune system. Int. J. Mol. Sci. 2018, 19, 1227. [Google Scholar] [CrossRef] [PubMed]
- Cui, H.; Kong, Y.; Zhang, H. Oxidative stress, mitochondrial dysfunction and aging. J. Signal Transduct. 2012, 2012, 646354. [Google Scholar] [CrossRef] [PubMed]
- Theurey, P.; Pizzo, P. The aging mitochondria. Genes 2018, 9, 22. [Google Scholar] [CrossRef] [PubMed]
- Fan, Q.; Chen, L.; Cheng, S.; Li, F.; Lau, W.B.; Wang, l.F.; Liu, J.H. Aging aggravates nitrate-mediated ros/rns changes. Oxid. Med. Cell. Longev. 2014, 2014, 376515. [Google Scholar] [CrossRef] [PubMed]
- Bratic, A.; Larsson, N.G. The role of mitochondria in aging. J. Clin. Investig. 2013, 123, 951–957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, M.D.; Cooke, M.S. Oxidative damage to dna in non-malignant disease: Biomarker or biohazard? In Genome and Disease; Karger Publishers: Basel, Switzerland, 2006; Volume 1, pp. 53–66. [Google Scholar]
- Ademowo, O.S.; Dias, H.K.I.; Burton, D.G.A.; Griffiths, H.R. Lipid (per) oxidation in mitochondria: An emerging target in the ageing process? Biogerontology 2017, 18, 859–879. [Google Scholar] [CrossRef] [PubMed]
- Childs, B.G.; Durik, M.; Baker, D.J.; van Deursen, J.M. Cellular senescence in aging and age-related disease: From mechanisms to therapy. Nat. Med. 2015, 21, 1424–1435. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Espín, D.; Serrano, M. Cellular senescence: From physiology to pathology. Nat. Rev. Mol. Cell Biol. 2014, 15, 482–496. [Google Scholar] [CrossRef] [PubMed]
- Dzau, V.J. Atherosclerosis and hypertension: Mechanisms and interrelationships. J. Cardiovasc. Pharmacol. 1990, 15 (Suppl. 5), S59–S64. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.T.; Park, H.; Yoon, H.J.; Yang, S.Y. Long-term treatment of native ldl induces senescence of cultured human endothelial cells. Oxid. Med. Cell. Longev. 2017, 2017, 6487825. [Google Scholar] [PubMed]
- Ling, W.; Lougheed, M.; Suzuki, H.; Buchan, A.; Kodama, T.; Steinbrecher, U.P. Oxidized or acetylated low density lipoproteins are rapidly cleared by the liver in mice with disruption of the scavenger receptor class a type I/II gene. J. Clin. Investig. 1997, 100, 244–252. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Liu, J. Association between circulating oxidized low-density lipoprotein and atherosclerotic cardiovascular disease. Chronic Dis. Transl. Med. 2017, 3, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Trpkovic, A.; Resanovic, I.; Stanimirovic, J.; Radak, D.; Mousa, S.A.; Cenic-Milosevic, D.; Jevremovic, D.; Isenovic, E.R. Oxidized low-density lipoprotein as a biomarker of cardiovascular diseases. Crit. Rev. Clin. Lab. Sci. 2015, 52, 70–85. [Google Scholar] [CrossRef] [PubMed]
- Kugiyama, K.; Kerns, S.A.; Morrisett, J.D.; Roberts, R.; Henry, P.D. Impairment of endothelium-dependent arterial relaxation by lysolecithin in modified low-density lipoproteins. Nature 1990, 344, 160–162. [Google Scholar] [CrossRef] [PubMed]
- Khan, B.V.; Parthasarathy, S.S.; Alexander, R.W.; Medford, R.M. Modified low density lipoprotein and its constituents augment cytokine-activated vascular cell adhesion molecule-1 gene expression in human vascular endothelial cells. J. Clin. Investig. 1995, 95, 1262–1270. [Google Scholar] [CrossRef] [PubMed]
- Galle, J.; Bengen, J.; Schollmeyer, P.; Wanner, C. Impairment of endothelium-dependent dilation in rabbit renal arteries by oxidized lipoprotein(a). Role of oxygen-derived radicals. Circulation 1995, 92, 1582–1589. [Google Scholar] [CrossRef] [PubMed]
- Dimmeler, S.; Haendeler, J.; Galle, J.; Zeiher, A.M. Oxidized low-density lipoprotein induces apoptosis of human endothelial cells by activation of cpp32-like proteases. A mechanistic clue to the ‘response to injury’ hypothesis. Circulation 1997, 95, 1760–1763. [Google Scholar] [CrossRef] [PubMed]
- Imanishi, T.; Hano, T.; Sawamura, T.; Nishio, I. Oxidized low-density lipoprotein induces endothelial progenitor cell senescence, leading to cellular dysfunction. Clin. Exp. Pharmacol. Physiol. 2004, 31, 407–413. [Google Scholar] [CrossRef] [PubMed]
- Bonello-Palot, N.; Simoncini, S.; Robert, S.; Bourgeois, P.; Sabatier, F.; Levy, N.; Dignat-George, F.; Badens, C. Prelamin a accumulation in endothelial cells induces premature senescence and functional impairment. Atherosclerosis 2014, 237, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Yentrapalli, R.; Azimzadeh, O.; Kraemer, A.; Malinowsky, K.; Sarioglu, H.; Becker, K.F.; Atkinson, M.J.; Moertl, S.; Tapio, S. Quantitative and integrated proteome and microrna analysis of endothelial replicative senescence. J. Proteom. 2015, 126, 12–23. [Google Scholar] [CrossRef] [PubMed]
- Turner, R. Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). Uk prospective diabetes study (ukpds) group. Lancet 1998, 352, 837–853. [Google Scholar]
- Nathan, D.M.; Cleary, P.A.; Backlund, J.Y.; Genuth, S.M.; Lachin, J.M.; Orchard, T.J.; Raskin, P.; Zinman, B.; Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications (DCCT/EDIC) Study Research Group. Intensive diabetes treatment and cardiovascular disease in patients with type 1 diabetes. N. Engl. J. Med. 2005, 353, 2643–2653. [Google Scholar] [PubMed]
- Zhao, Y.; Banerjee, S.; Dey, N.; LeJeune, W.S.; Sarkar, P.S.; Brobey, R.; Rosenblatt, K.P.; Tilton, R.G.; Choudhary, S. Klotho depletion contributes to increased inflammation in kidney of the db/db mouse model of diabetes via rela (serine)536 phosphorylation. Diabetes 2011, 60, 1907–1916. [Google Scholar] [CrossRef] [PubMed]
- Alique, M.; Luna, C.; Carracedo, J.; Ramírez, R. Ldl biochemical modifications: A link between atherosclerosis and aging. Food Nutr. Res. 2015, 5, 29240. [Google Scholar] [CrossRef] [PubMed]
- Martínez, M.C.; Andriantsitohaina, R. Extracellular vesicles in metabolic syndrome. Circ. Res. 2017, 120, 1674–1686. [Google Scholar] [CrossRef] [PubMed]
- Pardo, F.; Villalobos-Labra, R.; Sobrevia, B.; Toledo, F.; Sobrevia, L. Extracellular vesicles in obesity and diabetes mellitus. Mol. Asp. Med. 2018, 60, 81–91. [Google Scholar] [CrossRef] [PubMed]
- Lakhter, A.J.; Sims, E.K. Minireview: Emerging roles for extracellular vesicles in diabetes and related metabolic disorders. Mol. Endocrinol. 2015, 29, 1535–1548. [Google Scholar] [CrossRef] [PubMed]
- Gong, M.; Hubner, N. Molecular genetics of human hypertension. Clin. Sci. 2006, 110, 315–326. [Google Scholar] [CrossRef] [PubMed]
- Hollander, W. Role of hypertension in atherosclerosis and cardiovascular disease. Am. J. Cardiol. 1976, 38, 786–800. [Google Scholar] [CrossRef]
- Baradaran, A.; Nasri, H.; Rafieian-Kopaei, M. Oxidative stress and hypertension: Possibility of hypertension therapy with antioxidants. J. Res. Med. Sci. 2014, 19, 358–367. [Google Scholar] [PubMed]
- Grossman, E. Does increased oxidative stress cause hypertension? Diabetes Care 2008, 31 (Suppl. 2), S185–S189. [Google Scholar] [CrossRef] [PubMed]
- Ceriello, A. Possible role of oxidative stress in the pathogenesis of hypertension. Diabetes Care 2008, 31 (Suppl. 2), S181–S184. [Google Scholar] [CrossRef] [PubMed]
- Preston, R.A.; Jy, W.; Jimenez, J.J.; Mauro, L.M.; Horstman, L.L.; Valle, M.; Aime, G.; Ahn, Y.S. Effects of severe hypertension on endothelial and platelet microparticles. Hypertension 2003, 41, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Rosińska, J.; Łukasik, M.; Kozubski, W. The impact of vascular disease treatment on platelet-derived microvesicles. Cardiovasc. Drugs Ther. 2017, 31, 627–644. [Google Scholar] [CrossRef] [PubMed]
- Ambrose, J.A.; Barua, R.S. The pathophysiology of cigarette smoking and cardiovascular disease: An update. J. Am. Coll. Cardiol. 2004, 43, 1731–1737. [Google Scholar] [CrossRef] [PubMed]
- Mercado, C.; Jaimes, E.A. Cigarette smoking as a risk factor for atherosclerosis and renal disease: Novel pathogenic insights. Curr. Hypertens. Rep. 2007, 9, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Messner, B.; Bernhard, D. Smoking and cardiovascular disease: Mechanisms of endothelial dysfunction and early atherogenesis. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 509–515. [Google Scholar] [CrossRef] [PubMed]
- Csordas, A.; Bernhard, D. The biology behind the atherothrombotic effects of cigarette smoke. Nat. Rev. Cardiol. 2013, 10, 219–230. [Google Scholar] [CrossRef] [PubMed]
- Fujita, Y.; Araya, J.; Ito, S.; Kobayashi, K.; Kosaka, N.; Yoshioka, Y.; Kadota, T.; Hara, H.; Kuwano, K.; Ochiya, T. Suppression of autophagy by extracellular vesicles promotes myofibroblast differentiation in copd pathogenesis. J. Extracell. Vesicles 2015, 4, 28388. [Google Scholar] [CrossRef] [PubMed]
- Benedikter, B.J.; Volgers, C.; Haenen, G.R.; Savelkoul, P.H.; Wouters, E.F.; Rohde, G.G.; Weseler, A.R.; Stassen, F.R. Cigarette smoke extract induces the release of extracellular vesicles by airway epithelial cells via cellular carbonyl stress. Eur. Respir. J. 2015, 46, PA5113. [Google Scholar]
- Bæk, R.; Varming, K.; Jørgensen, M.M. Does smoking, age or gender affect the protein phenotype of extracellular vesicles in plasma? Transfus. Apher. Sci. 2016, 55, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Foley, R.N.; Parfrey, P.S.; Sarnak, M.J. Epidemiology of cardiovascular disease in chronic renal disease. J. Am. Soc. Nephrol. 1998, 9, S162–S163. [Google Scholar] [CrossRef]
- Stack, A.G.; Bloembergen, W.E. Prevalence and clinical correlates of coronary artery disease among new dialysis patients in the united states: A cross-sectional study. J. Am. Soc. Nephrol. 2001, 12, 1516–1523. [Google Scholar] [PubMed]
- Go, A.S.; Chertow, G.M.; Fan, D.; McCulloch, C.E.; Hsu, C.Y. Chronic kidney disease and the risks of death, cardiovascular events and hospitalization. N. Engl. J. Med. 2004, 351, 1296–1305. [Google Scholar] [CrossRef] [PubMed]
- Levey, A.S.; Rocco, M.V.; Anderson, S.; Andreoli, S.P.; Bailie, G.R.; Bakris, G.L.; Callahan, M.B.; Greene, J.H.; Johnson, C.A.; Lash, J.P. K/doqi clinical practice guidelines on hypertension and antihypertensive agents in chronic kidney disease. Am. J. Kidney Dis. 2004, 43, S12–S90. [Google Scholar]
- Ronco, C.; Haapio, M.; House, A.A.; Anavekar, N.; Bellomo, R. Cardiorenal syndrome. J. Am. Coll. Cardiol. 2008, 52, 1527–1539. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Yoshida, M. Protein-bound uremic toxins: New culprits of cardiovascular events in chronic kidney disease patients. Toxins 2014, 6, 665–678. [Google Scholar] [CrossRef] [PubMed]
- Childs, B.G.; Baker, D.J.; Wijshake, T.; Conover, C.A.; Campisi, J.; van Deursen, J.M. Senescent intimal foam cells are deleterious at all stages of atherosclerosis. Science 2016, 354, 472–477. [Google Scholar] [CrossRef] [PubMed]
- Duranton, F.; Cohen, G.; De Smet, R.; Rodriguez, M.; Jankowski, J.; Vanholder, R.; Argiles, A.; European Uremic Toxin Work Group. Normal and pathologic concentrations of uremic toxins. J. Am. Soc. Nephrol. 2012, 23, 1258–1270. [Google Scholar] [CrossRef] [PubMed]
- Lekawanvijit, S.; Kompa, A.R.; Wang, B.H.; Kelly, D.J.; Krum, H. Cardiorenal syndrome: The emerging role of protein-bound uremic toxins. Circ. Res. 2012, 111, 1470–1483. [Google Scholar] [CrossRef] [PubMed]
- Tumur, Z.; Shimizu, H.; Enomoto, A.; Miyazaki, H.; Niwa, T. Indoxyl sulphate upregulates expression of icam-1 and mcp-1 by oxidative stress-induced nf-kappab activation. Am. J. Nephrol. 2010, 31, 435–441. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Osaka, M.; Higuchi, Y.; Nishijima, F.; Ishii, H.; Yoshida, M. Indoxyl sulphate induces leukocyte-endothelial interactions through up-regulation of e-selectin. J. Biol. Chem. 2010, 285, 38869–38875. [Google Scholar] [CrossRef] [PubMed]
- Adelibieke, Y.; Shimizu, H.; Muteliefu, G.; Bolati, D.; Niwa, T. Indoxyl sulphate induces endothelial cell senescence by increasing reactive oxygen species production and p53 activity. J. Ren. Nutr. 2012, 22, 86–89. [Google Scholar] [CrossRef] [PubMed]
- Ryu, J.H.; Kim, S.J. Clopidogrel effectively suppresses endothelial microparticle generation induced by indoxyl sulphate via inhibition of the p38 mitogen-activated protein kinase pathway. Blood Purif. 2011, 32, 186–194. [Google Scholar] [CrossRef] [PubMed]
- Meijers, B.K.; Van Kerckhoven, S.; Verbeke, K.; Dehaen, W.; Vanrenterghem, Y.; Hoylaerts, M.F.; Evenepoel, P. The uremic retention solute p-cresyl sulphate and markers of endothelial damage. Am. J. Kidney Dis. 2009, 54, 891–901. [Google Scholar] [CrossRef] [PubMed]
- Adijiang, A.; Goto, S.; Uramoto, S.; Nishijima, F.; Niwa, T. Indoxyl sulphate promotes aortic calcification with expression of osteoblast-specific proteins in hypertensive rats. Nephrol. Dial. Transpl. 2008, 23, 1892–1901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanimura, A.; McGregor, D.H.; Anderson, H.C. Matrix vesicles in atherosclerotic calcification. Proc. Soc. Exp. Biol. Med. 1983, 172, 173–177. [Google Scholar] [CrossRef] [PubMed]
- Baron, M.; Boulanger, C.M.; Staels, B.; Tailleux, A. Cell-derived microparticles in atherosclerosis: Biomarkers and targets for pharmacological modulation? J. Cell. Mol. Med. 2012, 16, 1365–1376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, M.; Loyer, X.; Boulanger, C.M. Extracellular vesicles as new pharmacological targets to treat atherosclerosis. Eur. J. Pharmacol. 2015, 763, 90–103. [Google Scholar] [CrossRef] [PubMed]
- Rautou, P.E.; Vion, A.C.; Amabile, N.; Chironi, G.; Simon, A.; Tedgui, A.; Boulanger, C.M. Microparticles, vascular function and atherothrombosis. Circ. Res. 2011, 109, 593–606. [Google Scholar] [CrossRef] [PubMed]
- Leroyer, A.S.; Rautou, P.E.; Silvestre, J.S.; Castier, Y.; Lesèche, G.; Devue, C.; Duriez, M.; Brandes, R.P.; Lutgens, E.; Tedgui, A.; et al. Cd40 ligand+ microparticles from human atherosclerotic plaques stimulate endothelial proliferation and angiogenesis a potential mechanism for intraplaque neovascularization. J. Am. Coll. Cardiol. 2008, 52, 1302–1311. [Google Scholar] [CrossRef] [PubMed]
- Leroyer, A.S.; Tedgui, A.; Boulanger, C.M. Role of microparticles in atherothrombosis. J. Intern. Med. 2008, 263, 528–537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milasan, A.; Tessandier, N.; Tan, S.; Brisson, A.; Boilard, E.; Martel, C. Extracellular vesicles are present in mouse lymph and their level differs in atherosclerosis. J. Extracell. Vesicles 2016, 5, 31427. [Google Scholar] [CrossRef] [PubMed]
- Perrotta, I.; Aquila, S. Exosomes in human atherosclerosis: An ultrastructural analysis study. Ultrastruct. Pathol. 2016, 40, 101–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goetzl, E.J.; Schwartz, J.B.; Mustapic, M.; Lobach, I.V.; Daneman, R.; Abner, E.L.; Jicha, G.A. Altered cargo proteins of human plasma endothelial cell-derived exosomes in atherosclerotic cerebrovascular disease. FASEB J. 2017, 31, 3689–3694. [Google Scholar] [CrossRef] [PubMed]
- Buzas, E.I.; György, B.; Nagy, G.; Falus, A.; Gay, S. Emerging role of extracellular vesicles in inflammatory diseases. Nat. Rev. Rheumatol. 2014, 10, 356–364. [Google Scholar] [CrossRef] [PubMed]
- Pan, Q.; Liu, H.; Zheng, C.; Zhao, Y.; Liao, X.; Wang, Y.; Chen, Y.; Zhao, B.; Lazartigues, E.; Yang, Y.; et al. Microvesicles derived from inflammation-challenged endothelial cells modulate vascular smooth muscle cell functions. Front. Physiol. 2016, 7, 692. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Li, G.; Liu, M.L. Microvesicles as emerging biomarkers and therapeutic targets in cardiometabolic diseases. Genom. Proteom. Bioinform. 2018, 16, 50–62. [Google Scholar] [CrossRef] [PubMed]
- Hulsmans, M.; Holvoet, P. Microrna-containing microvesicles regulating inflammation in association with atherosclerotic disease. Cardiovasc. Res. 2013, 100, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; DiDonato, J.A.; Buffa, J.; Comhair, S.A.; Aronica, M.A.; Dweik, R.A.; Lee, N.A.; Lee, J.J.; Thomassen, M.J.; Kavuru, M.; et al. Eosinophil peroxidase catalyzed protein carbamylation participates in asthma. J. Biol. Chem. 2016, 291, 22118–22135. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.B.; Thuy Ly, T.B.; Bernhardt, I. Microvesicles released from human red blood cells: Properties and potential applications. In Novel Implications of Exosomes in Diagnosis and Treatment of Cancer and Infectious Diseases; Intechopen: Rijeka, Croatia, 2018; pp. 1–26. [Google Scholar]
- Cai, J.; Guan, W.; Tan, X.; Chen, C.; Li, L.; Wang, N.; Zou, X.; Zhou, F.; Wang, J.; Pei, F.; et al. Sry gene transferred by extracellular vesicles accelerates atherosclerosis by promotion of leucocyte adherence to endothelial cells. Clin. Sci. 2015, 129, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Jansen, F.; Li, Q.; Pfeifer, A.; Werner, N. Endothelial- and immune cell-derived extracellular vesicles in the regulation of cardiovascular health and disease. JACC Basic Transl. Sci. 2017, 2, 790–807. [Google Scholar] [CrossRef]
- Canault, M.; Leroyer, A.S.; Peiretti, F.; Lesèche, G.; Tedgui, A.; Bonardo, B.; Alessi, M.C.; Boulanger, C.M.; Nalbone, G. Microparticles of human atherosclerotic plaques enhance the shedding of the tumor necrosis factor-alpha converting enzyme/adam17 substrates, tumor necrosis factor and tumor necrosis factor receptor-1. Am. J. Pathol. 2007, 171, 1713–1723. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Li, S. Tctap a-040 endothelial microparticles-mediated transfer of microrna-19b promotes atherosclerosis via activating perivascular adipose tissue inflammation in apoe−/− mice. J. Am. Coll. Cardiol. 2018, 71, S23. [Google Scholar] [CrossRef]
- Bakhshian Nik, A.; Hutcheson, J.D.; Aikawa, E. Extracellular vesicles as mediators of cardiovascular calcification. Front. Cardiovasc. Med. 2017, 4, 78. [Google Scholar] [CrossRef] [PubMed]
- Hutcheson, J.D.; Goettsch, C.; Pham, T.; Iwashita, M.; Aikawa, M.; Singh, S.A.; Aikawa, E. Enrichment of calcifying extracellular vesicles using density-based ultracentrifugation protocol. J. Extracell. Vesicles 2014, 3, 25129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bei, Y.; Das, S.; Rodosthenous, R.S.; Holvoet, P.; Vanhaverbeke, M.; Monteiro, M.C.; Monteiro, V.V.S.; Radosinska, J.; Bartekova, M.; Jansen, F.; et al. Extracellular vesicles in cardiovascular theranostics. Theranostics 2017, 7, 4168–4182. [Google Scholar] [CrossRef] [PubMed]
- Leopold, J.A. Vascular calcification: Mechanisms of vascular smooth muscle cell calcification. Trends Cardiovasc. Med. 2015, 25, 267–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, N.X.; O’Neill, K.D.; Moe, S.M. Matrix vesicles induce calcification of recipient vascular smooth muscle cells through multiple signalling pathways. Kidney Int. 2018, 93, 343–354. [Google Scholar] [CrossRef] [PubMed]
- Aikawa, E. Extracellular vesicles in cardiovascular disease: Focus on vascular calcification. J. Physiol. 2016, 594, 2877–2880. [Google Scholar] [CrossRef] [PubMed]
- Kapustin, A.N.; Davies, J.D.; Reynolds, J.L.; McNair, R.; Jones, G.T.; Sidibe, A.; Schurgers, L.J.; Skepper, J.N.; Proudfoot, D.; Mayr, M.; et al. Calcium regulates key components of vascular smooth muscle cell-derived matrix vesicles to enhance mineralization. Circ. Res. 2011, 109, e1–e12. [Google Scholar] [CrossRef] [PubMed]
- Abbas, M.; Jesel, L.; Auger, C.; Amoura, L.; Messas, N.; Manin, G.; Rumig, C.; León-González, A.J.; Ribeiro, T.P.; Silva, G.C.; et al. Endothelial microparticles from acute coronary syndrome patients induce premature coronary artery endothelial cell aging and thrombogenicity: Role of the ang II/at1 receptor/nadph oxidase-mediated activation of mapks and pi3-kinase pathways. Circulation 2017, 135, 280–296. [Google Scholar] [CrossRef] [PubMed]
- Tsubouchi, K.; Araya, J.; Minagawa, S.; Hara, H.; Ichikawa, A.; Saito, N.; Kadota, T.; Sato, N.; Yoshida, M.; Kurita, Y.; et al. Azithromycin attenuates myofibroblast differentiation and lung fibrosis development through proteasomal degradation of nox4. Autophagy 2017, 13, 1420–1434. [Google Scholar] [CrossRef] [PubMed]

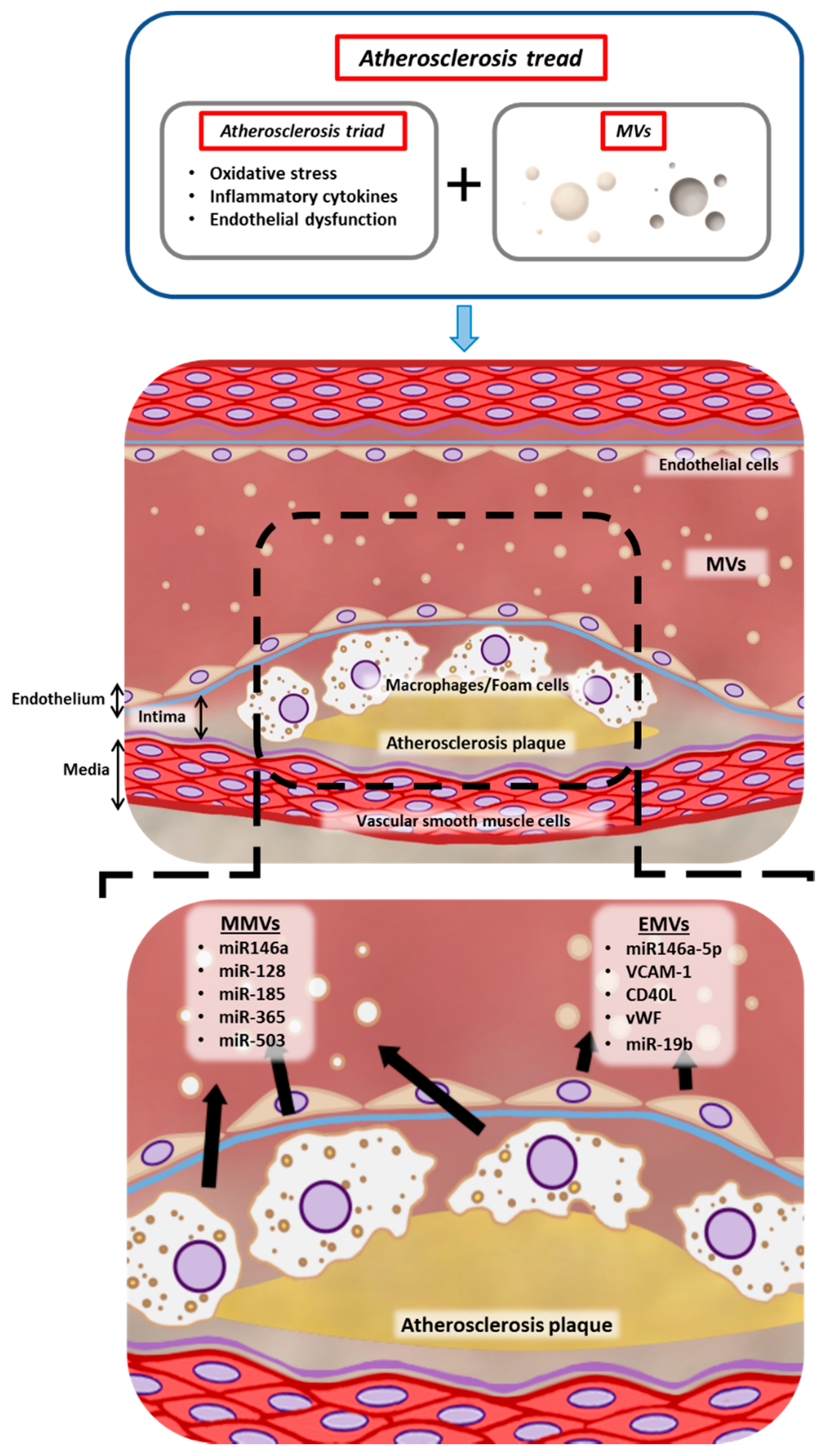


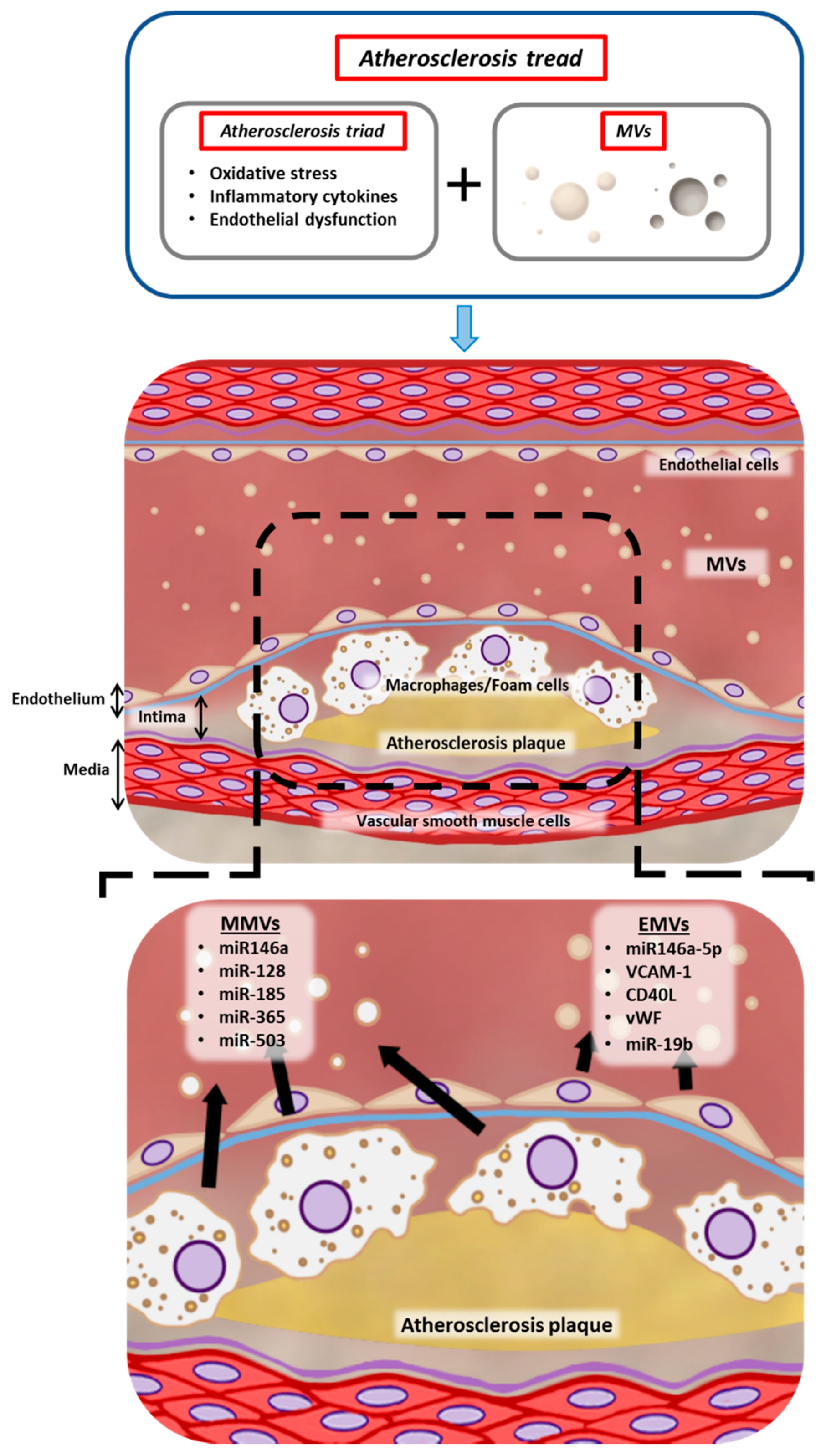
{kind=link}
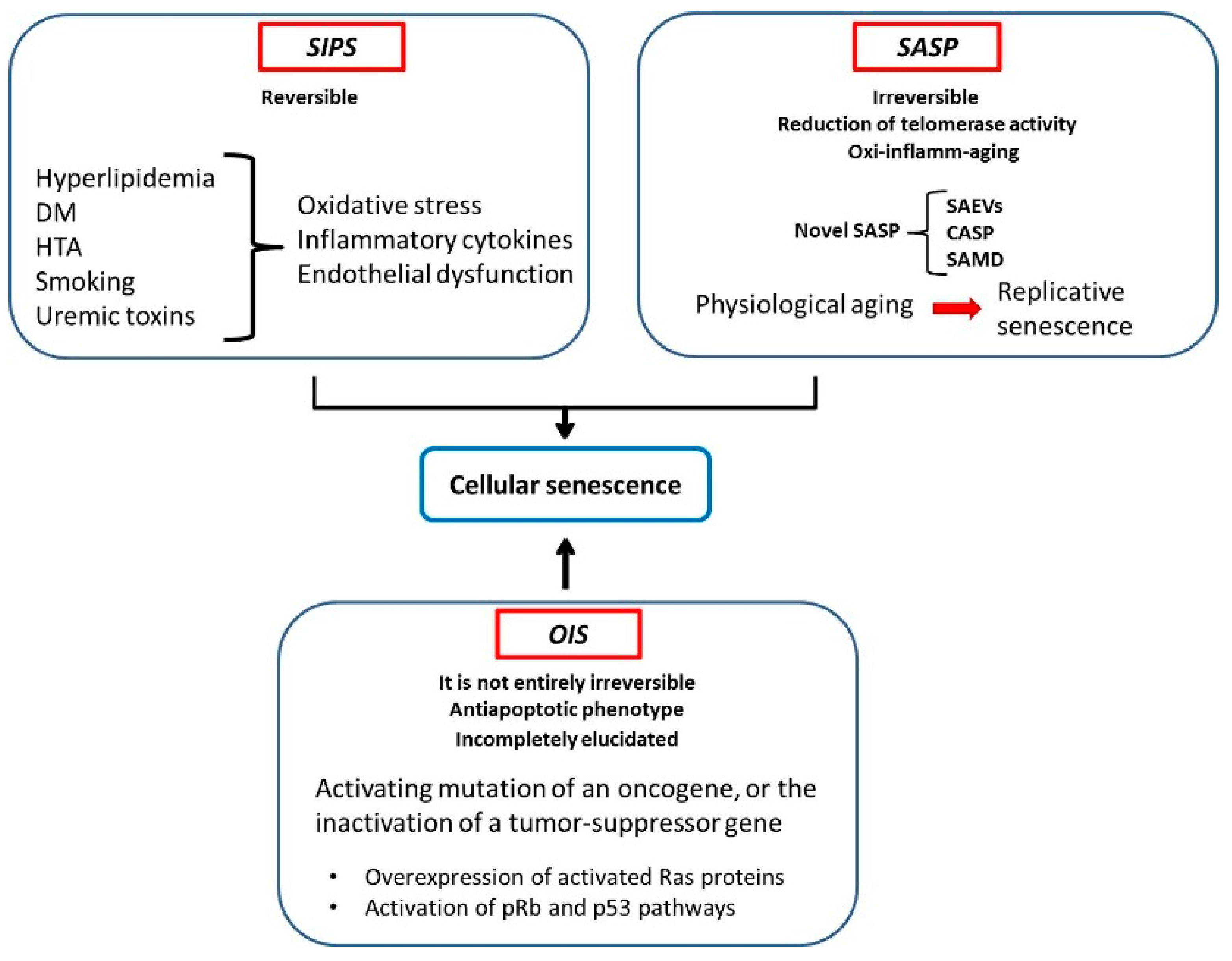
{kind=link}
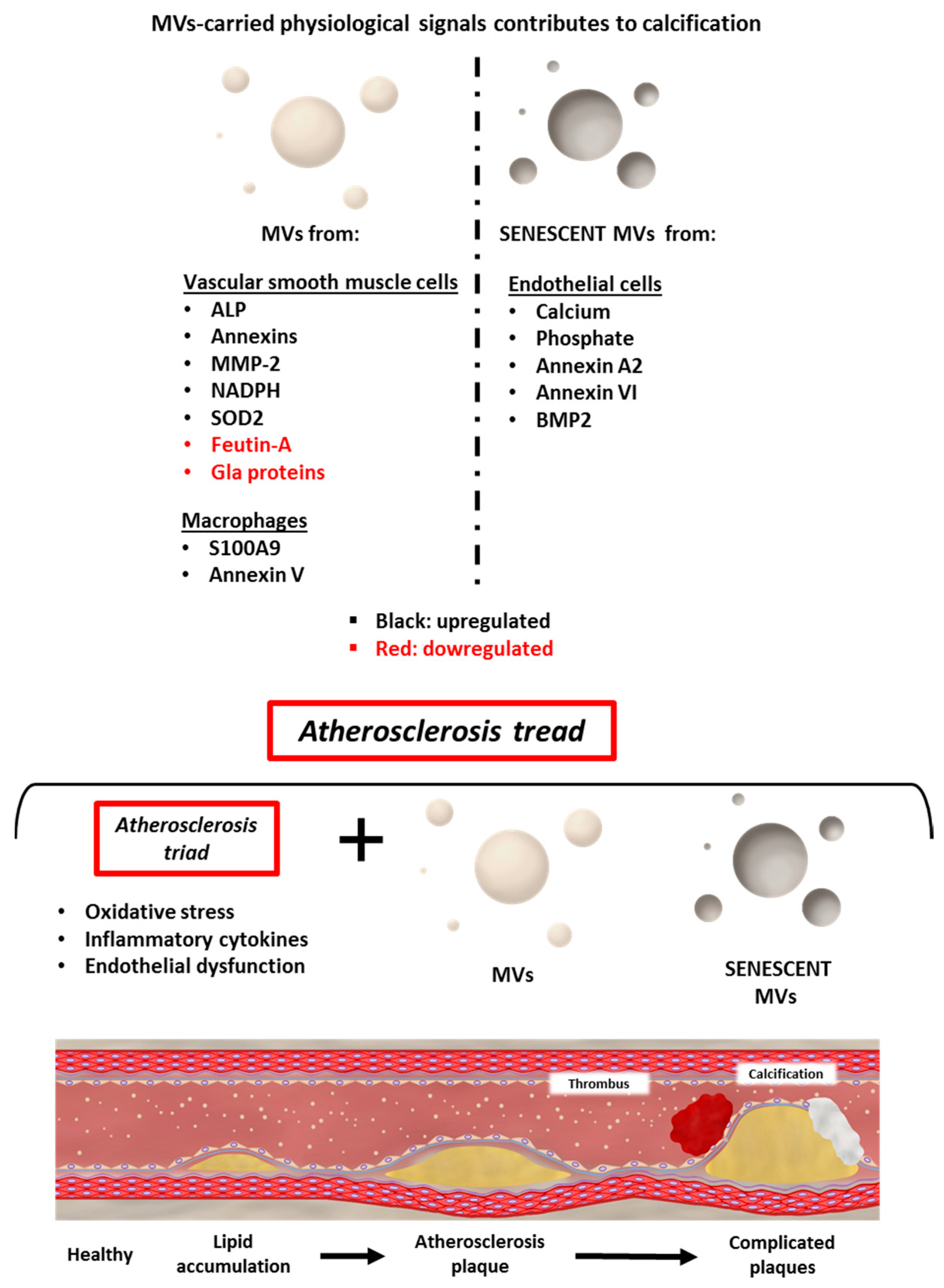{kind=link}
| Process | Characteristics | Markers | Regulation | References |
|---|---|---|---|---|
| DNA regulation | DNA replication | BrdU | ↓ | [74] |
| 3H-dT | ||||
| PCNA | ||||
| Ki-67 | ||||
| SAHFs | DAPI (DNA dye) | ↑ | [74,77] | |
| SDF | γ-H2AX | ↑ | [74] | |
| 53BP1 | ||||
| Lysosomal βgalactosidase activity | SA-β-gal activity | X-gal substrate | ↑ | [78,79] |
| C12FDG | ||||
| Cycle control | Cell cycle arrest proteins | p16 | ↑ | [80,81,82] |
| p21 | [65,83] | |||
| p53 | ||||
| Cyclin D1 | [75] | |||
| Lamin B1 | ↓ | [76] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alique, M.; Ramírez-Carracedo, R.; Bodega, G.; Carracedo, J.; Ramírez, R. Senescent Microvesicles: A Novel Advance in Molecular Mechanisms of Atherosclerotic Calcification. Int. J. Mol. Sci. 2018, 19, 2003. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19072003
Alique M, Ramírez-Carracedo R, Bodega G, Carracedo J, Ramírez R. Senescent Microvesicles: A Novel Advance in Molecular Mechanisms of Atherosclerotic Calcification. International Journal of Molecular Sciences. 2018; 19(7):2003. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19072003
Chicago/Turabian StyleAlique, Matilde, Rafael Ramírez-Carracedo, Guillermo Bodega, Julia Carracedo, and Rafael Ramírez. 2018. "Senescent Microvesicles: A Novel Advance in Molecular Mechanisms of Atherosclerotic Calcification" International Journal of Molecular Sciences 19, no. 7: 2003. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19072003