Binding of Organometallic Ruthenium Anticancer Complexes to DNA: Thermodynamic Base and Sequence Selectivity


Abstract
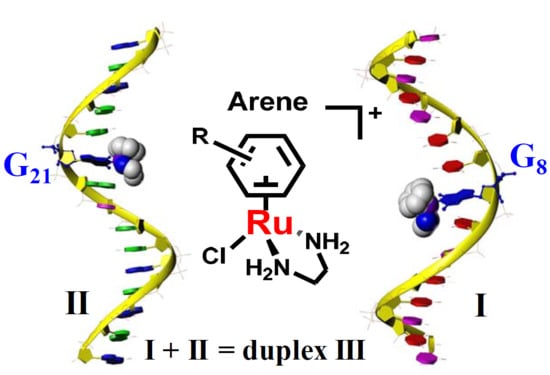
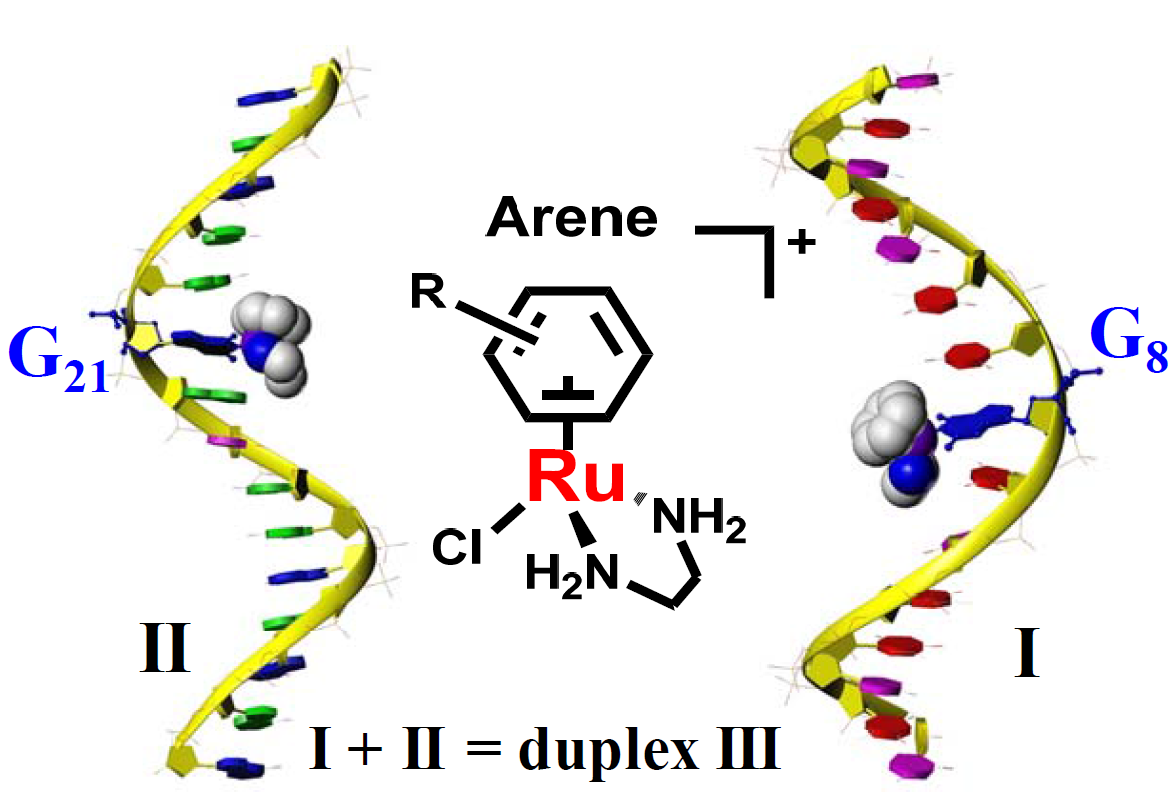
:
1. Introduction
2. Results
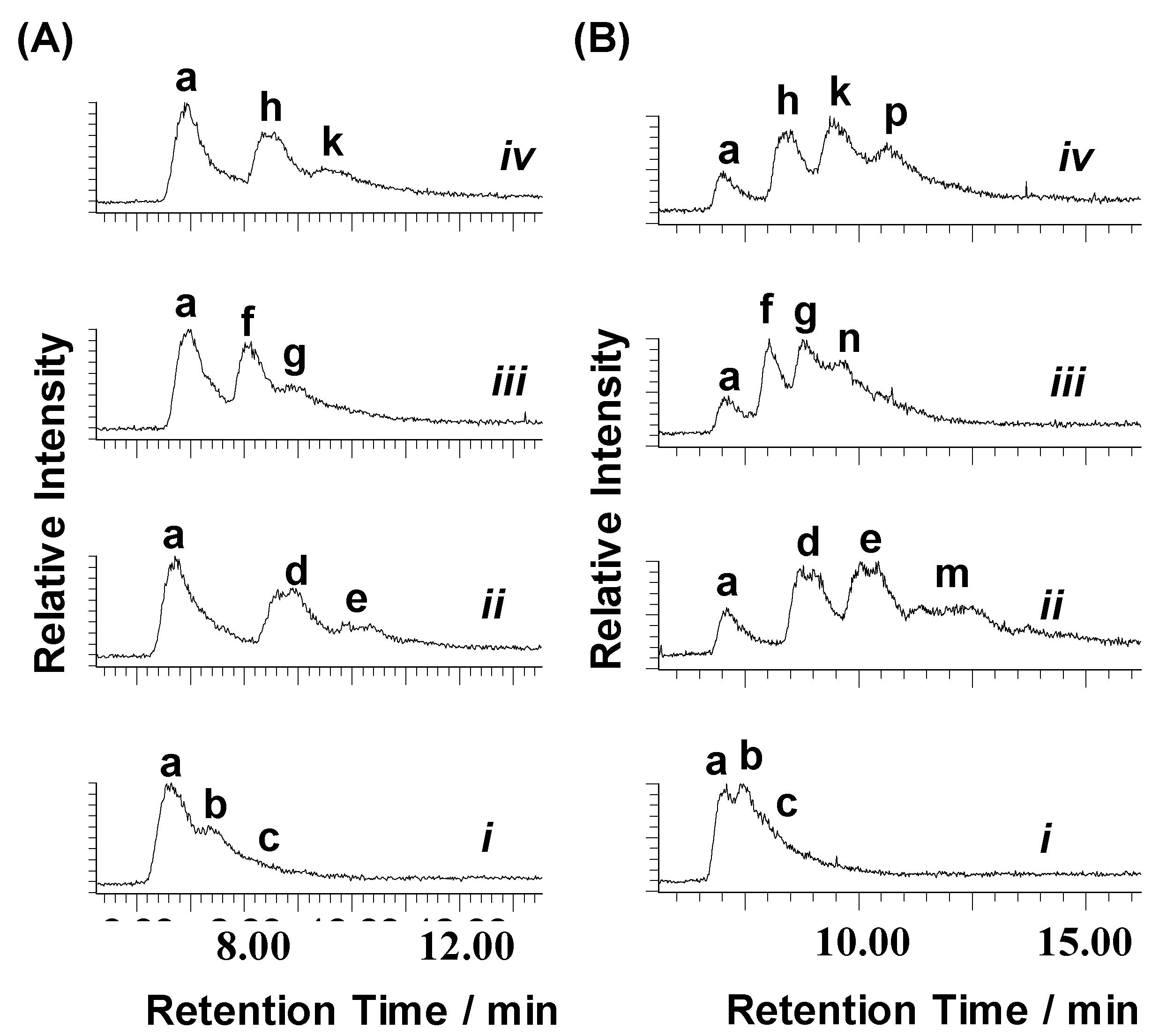
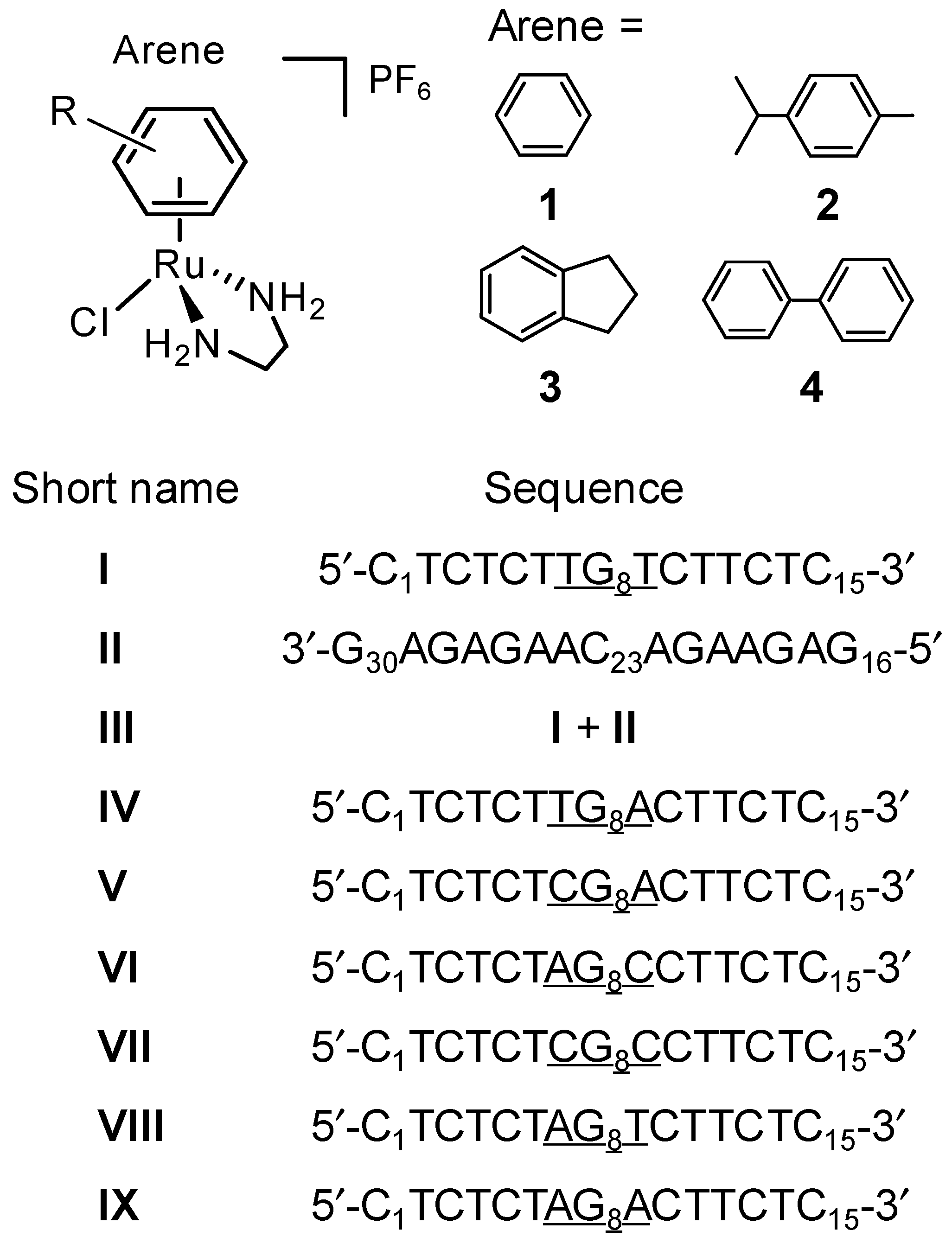
2.1. Reactions of Organometallic Ruthenium(II) Complexes with One-G-centered Single-Stranded ODNs
2.2. Reactions of Organometallic Ruthenium(II) Complexes with Single-Strand II
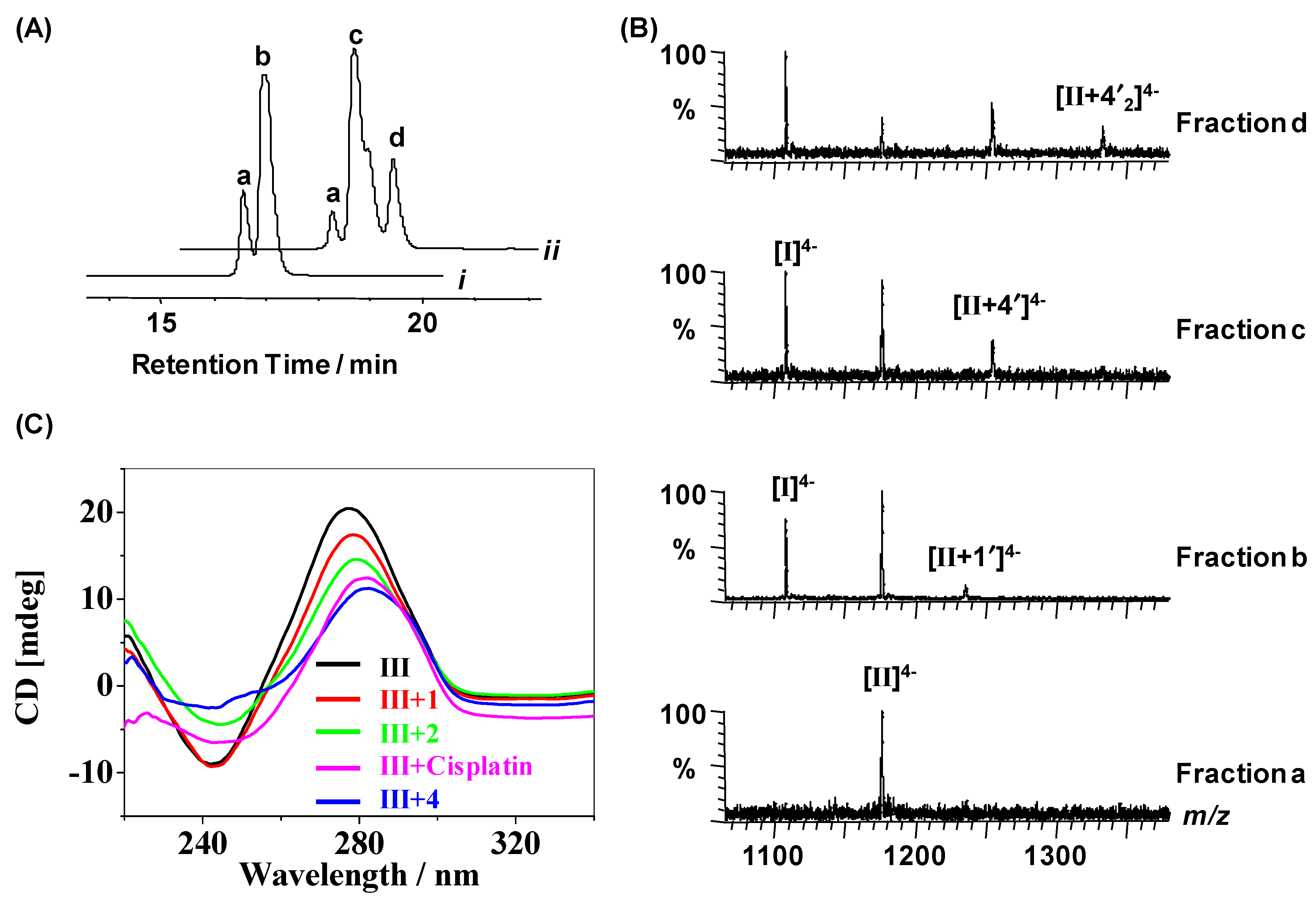
2.3. Reactions of Organometallic Ruthenium(II) Complexes with Duplex III
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Sample Preparation
4.3. High Performance Liquid Chromatography (HPLC)
4.4. Electrospray Ionization Mass Spectroscopy (ESI-MS)
4.5. Circular Dichroism (CD) Spectroscopy
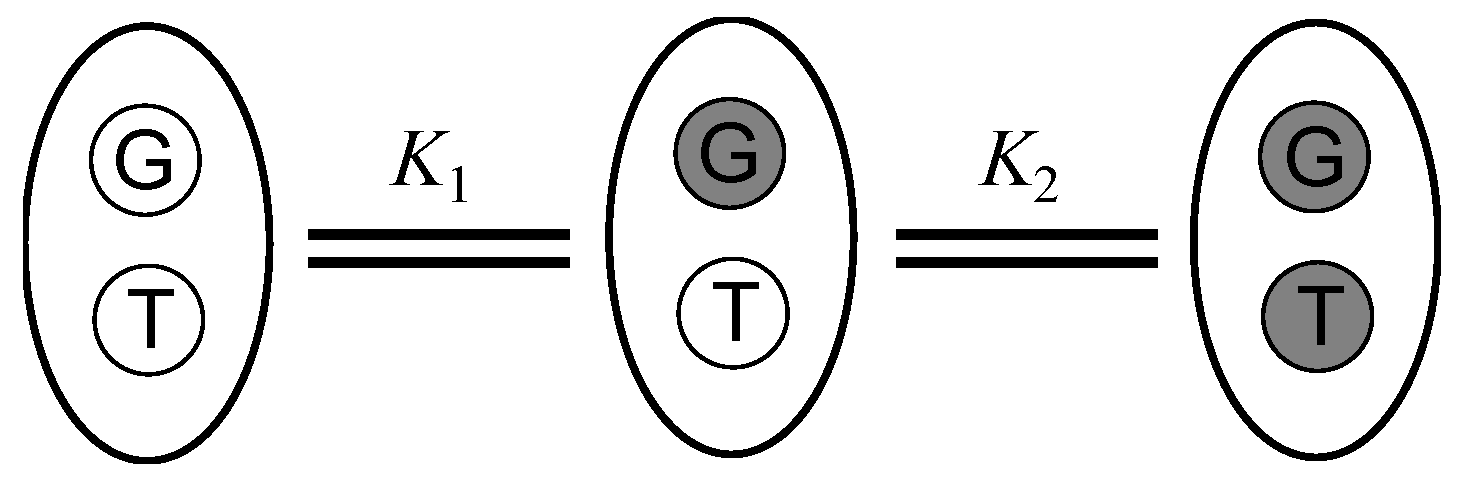
4.6. Determination of Equilibrium Binding Constants of Organometallic Ruthenium Complexes to ODNs
5. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Aird, R.E.; Cummings, J.; Ritchie, A.A.; Muir, M.; Morris, R.E.; Chen, H.; Sadler, P.J.; Jodrell, D.I. In vitro and in vivo activity and cross resistance profiles of novel ruthenium(II) organometallic arene complexes in human ovarian cancer. Br. J. Cancer 2002, 86, 1652–1657. [Google Scholar] [CrossRef] [PubMed]
- Morris, R.E.; Aird, R.E.; Murdoch, P.D.; Chen, H.M.; Cummings, J.; Hughes, N.D.; Parsons, S.; Parkin, A.; Boyd, G.; Jodrell, D.I.; et al. Inhibition of cancer cell growth by ruthenium(II) arene complexes. J. Med. Chem. 2001, 44, 3616–3621. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.K.; Melchart, M.; Habtemariam, A.; Sadler, P.J. Organometallic chemistry, biology and medicine: Ruthenium arene anticancer complexes. Chem. Commun. 2005, 4764–4776. [Google Scholar] [CrossRef] [PubMed]
- Sava, G.; Bergamo, A.; Dyson, P.J. Metal-based antitumour drugs in the post-genomic era: What comes next? Dalton Trans. 2011, 40, 9069–9075. [Google Scholar] [CrossRef] [PubMed]
- Hartinger, C.G.; Dyson, P.J. Bioorganometallic chemistry-from teaching paradigms to medicinal applications. Chem. Soc. Rev. 2009, 38, 391–401. [Google Scholar] [CrossRef] [PubMed]
- Meggers, E. Targeting proteins with metal complexes. Chem. Commun. 2009, 1001–1010. [Google Scholar] [CrossRef] [PubMed]
- Hambley, T.W. Developing new metal-based therapeutics: Challenges and opportunities. Dalton Trans. 2007, 4929–4937. [Google Scholar] [CrossRef] [PubMed]
- Mjos, K.D.; Orvig, C. Metallodrugs in Medicinal Inorganic Chemistry. Chem. Rev. 2014, 114, 4540–4563. [Google Scholar] [CrossRef] [PubMed]
- Suss-Fink, G. Arene ruthenium complexes as anticancer agents. Dalton Trans. 2010, 39, 1673–1688. [Google Scholar] [CrossRef] [PubMed]
- Bennett, M.A.; Byrnes, M.J.; Kovacik, I. The fragment bis(acetylacetonato)ruthenium: A meeting-point of coordination and organometallic chemistry. J. Organomet. Chem. 2004, 689, 4463–4474. [Google Scholar] [CrossRef]
- Jamieson, E.R.; Lippard, S.J. Structure, recognition, and processing of cisplatin-DNA adducts. Chem. Rev. 1999, 99, 2467–2498. [Google Scholar] [CrossRef] [PubMed]
- Sava, G.; Bergamo, A. Ruthenium-based compounds and tumour growth control. Int. J. Oncol. 2000, 17, 353–365. [Google Scholar] [CrossRef] [PubMed]
- Galanski, M.; Arion, V.B.; Jakupec, M.A.; Keppler, B.K. Recent developments in the field of tumor-inhibiting metal complexes. Curr. Pharm. Des. 2003, 9, 2078–2089. [Google Scholar] [CrossRef] [PubMed]
- Rademaker-Lakhai, J.M.; van den Bongard, D.; Pluim, D.; Beijnen, J.H.; Schellens, J.H.M. A phase I and pharmacological study with imidazolium-trans-DMSO-imidazole-tetrachlororuthenate, a novel ruthenium anticancer agent. Clin. Cancer Res. 2004, 10, 3717–3727. [Google Scholar] [CrossRef] [PubMed]
- Jakupec, M.A.; Galanski, M.; Arion, V.B.; Hartinger, C.G.; Keppler, B.K. Antitumour metal compounds: More than theme and variations. Dalton Trans. 2008, 183–194. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.M.; Parkinson, J.A.; Morris, R.E.; Sadler, P.J. Highly selective binding of organometallic ruthenium ethylenediamine complexes to nucleic acids: Novel recognition mechanisms. J. Am. Chem. Soc. 2003, 125, 173–186. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.M.; Parkinson, J.A.; Parsons, S.; Coxall, R.A.; Gould, R.O.; Sadler, P.J. Organometallic ruthenium(II) diamine anticancer complexes: Arene-nucleobase stacking and stereospecific hydrogen-bonding in guanine adducts. J. Am. Chem. Soc. 2002, 124, 3064–3082. [Google Scholar] [CrossRef] [PubMed]
- Novakova, O.; Kasparkova, J.; Bursova, V.; Hofr, C.; Vojtiskova, M.; Chen, H.M.; Sadler, P.J.; Brabec, V. Conformation of DNA modified by monofunctional Ru(II) arene complexes: Recognition by DNA binding proteins and repair. Relationship to cytotoxicity. Chem. Biol. 2005, 12, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.; Luo, Q.; Hu, W.B.; Li, X.C.; Wang, F.Y.; Xiong, S.X.; Sadler, P.J. Mechanism of interstrand migration of organoruthenium anticancer complexes within a DNA duplex. Metallomics 2012, 4, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.K.; Berners-Price, S.J.; Wang, F.Y.; Parkinson, J.A.; Xu, J.J.; Bella, J.; Sadler, P.J. Diversity in guanine-selective DNA binding modes for an organometallic ruthenium arene complex. Angew. Chem. Int. Ed. 2006, 45, 8153–8156. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.K.; Wang, F.Y.; Parkinson, J.A.; Bella, J.; Sadler, P.J. Ruthenation of duplex and single-stranded d(CGGCCG) by organometallic anticancer complexes. Chem. Eur. J. 2006, 12, 6151–6165. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.Y.; Bella, J.; Parkinson, J.A.; Sadler, P.J. Competitive reactions of a ruthenium arene anticancer complex with histidine, cytochrome c and an oligonucleotide. J. Biol. Inorg. Chem. 2005, 10, 147–155. [Google Scholar] [CrossRef] [PubMed]
- Novakova, O.; Chen, H.M.; Vrana, O.; Rodger, A.; Sadler, P.J.; Brabec, V. DNA interactions of monofunctional organometallic ruthenium(II) antitumor complexes in cell-free media. Biochemistry 2003, 42, 11544–11554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, K.; Hu, W.B.; Luo, Q.; Li, X.C.; Xiong, S.X.; Sadler, P.J.; Wang, F.Y. Competitive Binding Sites of a Ruthenium Arene Anticancer Complex on Oligonucleotides Studied by Mass Spectrometry: Ladder-Sequencing versus Top-Down. J. Am. Soc. Mass Spectrom. 2013, 24, 410–420. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.; Liu, S.Y.; Luo, Q.; Hu, W.B.; Li, X.C.; Wang, F.Y.; Zheng, R.H.; Cui, J.; Sadler, P.J.; Xiang, J.F.; et al. Thymines in Single-Stranded Oligonucleotides and G-Quadruplex DNA Are Competitive with Guanines for Binding to an Organoruthenium Anticancer Complex. Inorg. Chem. 2013, 52, 11332–11342. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Chen, H.M.; Parsons, S.; Oswald, L.D.H.; Davidson, J.E.; Sadler, P.J. Kinetics of aquation and anation of Ruthenium(II) arene anticancer complexes, acidity and X-ray structures of aqua adducts. Chem. Eur. J. 2003, 9, 5810–5820. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.Y.; Habtemariam, A.; van der Geer, E.P.L.; Fernandez, R.; Melchart, M.; Deeth, R.J.; Aird, R.; Guichard, S.; Fabbiani, F.P.A.; Lozano-Casal, P.; et al. Controlling ligand substitution reactions of organometallic complexes: Tuning cancer cell cytotoxicity. Proc. Natl. Acad. Sci. USA 2005, 102, 18269–18274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Z.J.; Sadler, P.J. Medicinal inorganic chemistry. Adv. Inorg. Chem. 2000, 49, 183–306. [Google Scholar]
- Dijt, F.J.; Chottard, J.C.; Girault, J.P.; Reedijk, J. Formation and Structure of Reaction-Products of Cis-Ptcl2(Nh3)2 with D(Apg) and or D(Gpa) in Dinucleotide, Trinucleotide and Penta-Nucleotide—Preference for Gpa Chelation over Apg Chelation. Eur. J. Biochem. 1989, 179, 333–344. [Google Scholar] [CrossRef]
- Davies, M.S.; Berners-Price, S.J.; Hambley, T.W. Rates of platination of AG and GA containing double-stranded oligonucleotides: Insights into why cisplatin binds to GG and AG but not GA sequences in DNA. J. Am. Chem. Soc. 1998, 120, 11380–11390. [Google Scholar] [CrossRef]
- Legendre, F.; Kozelka, J.; Chottard, J.C. GG versus AG platination: A kinetic study on hairpin-stabilized duplex oligonucleotides. Inorg. Chem. 1998, 37, 3964–3967. [Google Scholar] [CrossRef] [PubMed]
- Reeder, F.; Guo, Z.J.; Murdoch, P.D.; Corazza, A.; Hambley, T.W.; Berners-Price, S.J.; Chottard, J.C.; Sadler, P.J. Platination of a GG site on single-stranded and double-stranded forms of a 14-base oligonucleotide with diaqua cisplatin followed by NMR and HPLC—Influence of the platinum ligands and base sequence on 5′-G versus 3′-G platination selectivity. Eur. J. Biochem. 1997, 249, 370–382. [Google Scholar] [CrossRef] [PubMed]
- Reeder, F.; Gonnet, F.; Kozelka, J.; Chottard, J.C. Reactions of the double-stranded oligonucleotide d(TTGGCCAA)2 with cis-[Pt(NH3)2(H2O)(2)]2+ and [Pt(NH3)3(H2O)]2+. Chem.-A Eur. J. 1996, 2, 1068–1076. [Google Scholar] [CrossRef]
- Garbett, N.C.; Ragazzon, P.A.; Chaires, J.B. Circular dichroism to determine binding mode and affinity of ligand-DNA interactions. Nat. Protoc. 2007, 2, 3166–3172. [Google Scholar] [CrossRef] [PubMed]
- Boger, D.L.; Fink, B.E.; Brunette, S.R.; Tse, W.C.; Hedrick, M.P. A simple, high-resolution method for establishing DNA binding affinity and sequence selectivity. J. Am. Chem. Soc. 2001, 123, 5878–5891. [Google Scholar] [CrossRef] [PubMed]
- Neidle, S. Recent developments in triple-helix regulation of gene expression. Anticancer Drug Des. 1997, 12, 433–442. [Google Scholar] [PubMed]
- Choo, Y.; Sanchezgarcia, I.; Klug, A. In-Vivo Repression by a Site-Specific DNA-Binding Protein Designed against an Oncogenic Sequence. Nature 1994, 372, 642–645. [Google Scholar] [CrossRef] [PubMed]
- Mrksich, M.; Parks, M.E.; Dervan, P.B. Hairpin Peptide Motif—A New Class of Oligopeptides for Sequence-Specific Recognition in the Minor-Groove of Double-Helical DNA. J. Am. Chem. Soc. 1994, 116, 7983–7988. [Google Scholar] [CrossRef]
- Werstuck, G.; Green, M.R. Controlling gene expression in living cells through small molecule-RNA interactions. Science 1998, 282, 296–298. [Google Scholar] [CrossRef] [PubMed]
- Kimura, E.; Kitamura, H.; Ohtani, K.; Koike, T. Elaboration of selective and efficient recognition of thymine base in dinucleotides (TpT, ApT, CpT, and GpT), single-stranded d(GTGACGCC), and double-stranded d(CGCTAGCC)2 by Zn2+-acridinylcyclen (acridinylcyclen = (9-acridinyl)methyl-1,4,7,10-tetraazacyclododecane). J. Am. Chem. Soc. 2000, 122, 4668–4677. [Google Scholar]
- Shionoya, M.; Kimura, E.; Shiro, M. A New Ternary Zinc(II) Complex with [12]Anen(4) (=1,4,7,10-Tetraazacyclododecane) and Azt (=3′-Azido-3′-Deoxythymidine)—Highly Selective Recognition of Thymidine and Its Related Nucleosides by a Zinc(II) Macrocyclic Tetraamine Complex with Novel Complementary Associations. J. Am. Chem. Soc. 1993, 115, 6730–6737. [Google Scholar]
- Chen, H.M.; Parkinson, J.A.; Novakova, O.; Bella, J.; Wang, F.Y.; Dawson, A.; Gould, R.; Parsons, S.; Brabec, V.; Sadler, P.J. Induced-fit recognition of DNA by organometallic complexes with dynamic stereogenic centers. Proc. Natl. Acad. Sci. USA 2003, 100, 14623–14628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.K.; Parkinson, J.A.; Bella, J.; Wang, F.Y.; Sadler, P.J. Penetrative DNA intercalation and G-base selectivity of an organometallic tetrahydroanthracene RuII anticancer complex. Chem. Sci. 2010, 1, 258–270. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.K.; Sadler, P.J. Metal Complexes as DNA Intercalators. Acc. Chem. Res. 2011, 44, 349–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, J.S.; Jenkins, T.C.; Chaires, J.B. Energetics of DNA intercalation reactions. Biochemistry 2000, 39, 8439–8447. [Google Scholar] [CrossRef] [PubMed]
- Erkkila, K.E.; Odom, D.T.; Barton, J.K. Recognition and reaction of metallointercalators with DNA. Chem. Rev. 1999, 99, 2777–2795. [Google Scholar] [CrossRef] [PubMed]
- Shionoya, M.; Ikeda, T.; Kimura, E.; Shiro, M. Novel Multipoint Molecular Recognition of Nucleobases by a New Zinc(II) Complex of Acridine-Pendant Cyclen (Cyclen=1,4,7,10-Tetraazacyclododecane). J. Am. Chem. Soc. 1994, 116, 3848–3859. [Google Scholar] [CrossRef]
- Bierbach, U.; Farrell, N. Modulation of nucleotide binding of trans-platinum(II) complexes by planar ligands. A combined proton NMR and molecular mechanics study. Inorg. Chem. 1997, 36, 3657–3665. [Google Scholar] [CrossRef] [PubMed]
- Bjorndal, M.T.; Fygenson, D.K. DNA melting in the presence of fluorescent intercalating oxazole yellow dyes measured with a gel-based assay. Biopolymers 2002, 65, 40–44. [Google Scholar] [CrossRef] [PubMed]
- Maeda, Y.; Nunomura, K.; Ohtsubo, E. Differential scanning calorimetrics study of the effect of intercalators and other kinds of DNA-binding drugs on the stepwise melting of plasmid DNA. J. Mol. Biol. 1990, 215, 321–329. [Google Scholar] [CrossRef]
- Rodger, A.; Norden, B. Circular Dichroism and Linear Dichroism; Oxford University Press: Oxford, UK; New York, NY, USA; Tokyo, Japan, 1997. [Google Scholar]
- Gelasco, A.; Lippard, S.J. NMR solution structure of a DNA dodecamer duplex containing a cis-diammineplatinum(II) d(GpG) intrastrand cross-link, the major adduct of the anticancer drug cisplatin. Biochemistry 1998, 37, 9230–9239. [Google Scholar] [CrossRef] [PubMed]
- Keenea, F.R.; Smith, J.A.; Collins, J.G. Metal complexes as structure-selective binding agents for nucleic acids. Coord. Chem. Rev. 2009, 253, 2021–2035. [Google Scholar] [CrossRef]
- Elmroth, S.K.C.; Lippard, S.J. Surface and Electrostatic Contributions to DNA-Promoted Reactions of Platinum(II) Complexes with Short Oligonucleotides—A Kinetic-Study. Inorg. Chem. 1995, 34, 5234–5243. [Google Scholar] [CrossRef]
- Elmroth, S.K.C.; Lippard, S.J. Platinum binding to d(GpG) target sequences and phosphorothioate linkages in DNA occurs more rapidly with increasing oligonucleotide length. J. Am. Chem. Soc. 1994, 116, 3633–3634. [Google Scholar] [CrossRef]
- Pizarro, A.M.; Sadler, P.J. Unusual DNA binding modes for metal anticancer complexes. Biochimie 2009, 91, 1198–1211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saenger, W. Principles of Nucleic Acid Structure. In Springer Advanced Texts in Chemistry; Cantor, C.R., Ed.; Springer: New York, NY, USA, 1984; pp. 201–219. [Google Scholar]
- Lippert, B. Cisplatin—Chemistry and Biochemistry of a Leading Anticancer Drug; Wiley-VCH: Weinheim, Germany, 1999. [Google Scholar]
- Fichtinger-Schepman, A.M.J.; Vanderveer, J.L.; Denhartog, J.H.J.; Lohman, P.H.M.; Reedijk, J. Adducts of the Antitumor Drug Cis-Diamminedichloroplatinum(II) with DNA—Formation, Identification, and Quantitation. Biochemistry 1985, 24, 707–713. [Google Scholar] [CrossRef] [PubMed]
- Kozelka, J. Molecular origin of the sequence-dependent kinetics of reactions between cisplatin derivatives and DNA. Inorg. Chim. Acta 2009, 362, 651–668. [Google Scholar] [CrossRef]
- Monjardet-Bas, V.; Elizondo-Riojas, M.A.; Chottard, J.C.; Kozelka, J. A combined effect of molecular electrostatic potential and N7 accessibility explains sequence-dependent binding of cis-[Pt(NH3)2(H2O)2]2+ to DNA duplexes. Angew. Chem. Int. Ed. 2002, 41, 2998–3001. [Google Scholar] [CrossRef]
- Benasutti, M.; Ejadi, S.; Whitlow, M.D.; Loechler, E.L. Mapping the binding site of aflatoxin B1 in DNA: Systematic analysis of the reactivity of aflatoxin B1 with guanines in different DNA sequences. Biochemistry 1988, 27, 472–481. [Google Scholar] [CrossRef] [PubMed]
- Mattes, W.B.; Hartley, J.A.; Kohn, K.W. DNA sequence selectivity of guanine–N7 alkylation by nitrogen mustards. Nucleic Acids Res. 1986, 14, 2971–2987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zelonka, R.A.; Baird, M.C. Benzene Complexes of Ruthenium(II). Can. J. Chem. 1972, 50, 3063–3072. [Google Scholar] [CrossRef] [Green Version]
- Bennett, M.A.; Smith, A.K. Arene Ruthenium(II) Complexes Formed by Dehydrogenation of Cyclohexadienes with Ruthenium(III) Trichloride. J. Chem. Soc. Dalton Trans. 1974, 233–241. [Google Scholar] [CrossRef]
- Liu, S.Y.; Wu, K.; Zheng, W.; Zhao, Y.; Luo, Q.; Xiong, S.X.; Wang, F.Y. Identification and discrimination of binding sites of an organoruthenium anticancer complex to single-stranded oligonucleotides by mass spectrometry. Analyst 2014, 139, 4491–4496. [Google Scholar] [CrossRef] [PubMed]
- Klotz, I.M. Ligand Receptor Energetics: A Guide for the Perplexed; John Wiley & Sons, Inc.: New York, NY, USA, 1997. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
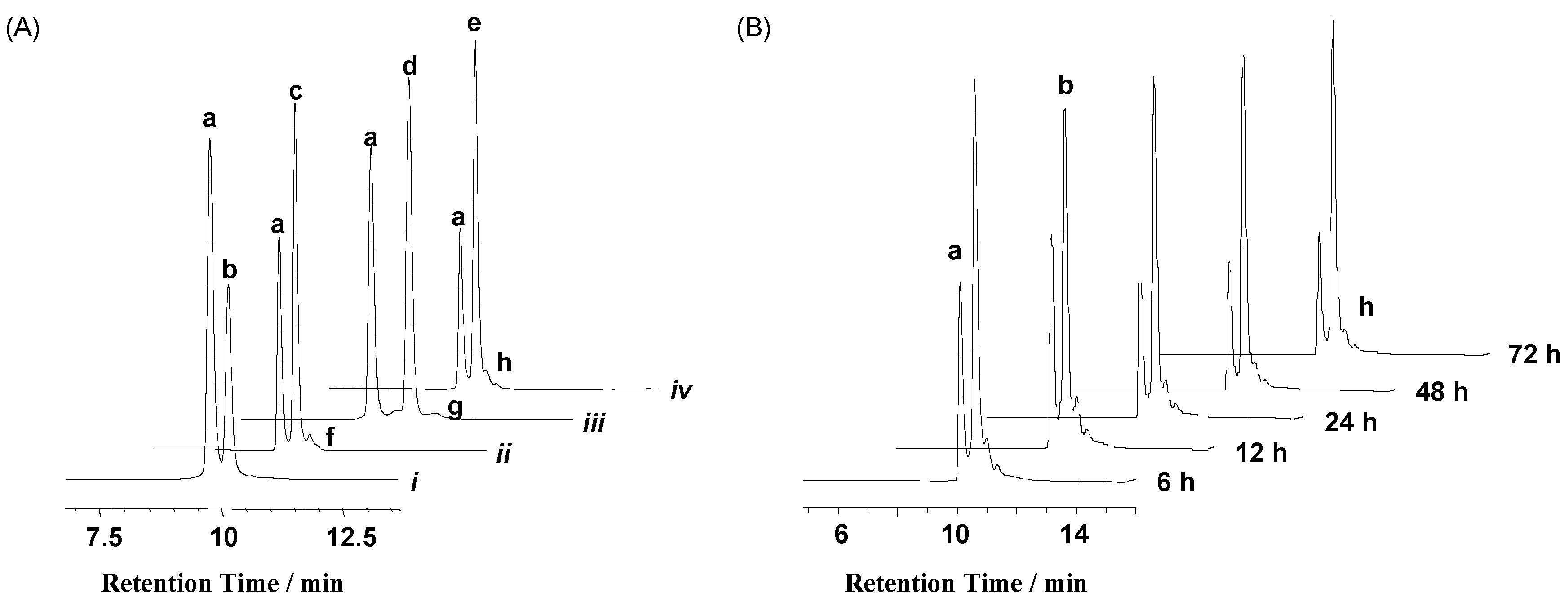
| Ru Complex | m/z: Observed (Calculated) | ||
|---|---|---|---|
| [I]3− | [I + {Ru} 1]3− | [I + {Ru}2]3− | |
| 1 | a: 1477.22 (1477.24) | b: 1556.56 (1556.58) | — 2 |
| 2 | a: 1477.22 (1477.24) | c: 1575.25 (1575.27) | f: 1672.96 (1672.95) |
| 3 | a: 1477.22 (1477.24) | d: 1569.91 (1569.92) | g: 1662.65 (1662.60) |
| 4 | a: 1477.22 (1477.24) | e: 1581.89 (1581.92) | h: 1686.66 (1686.60) |
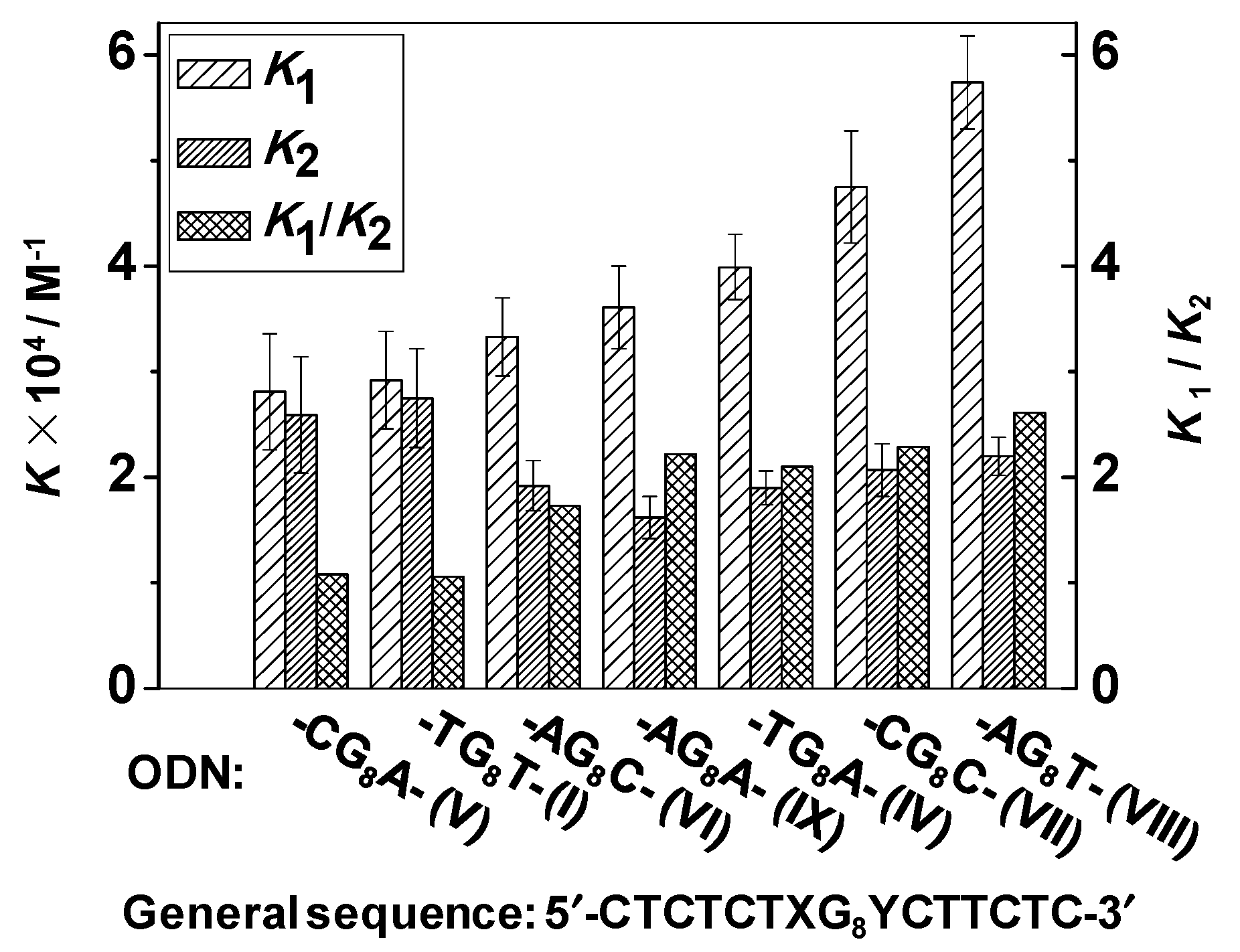
| Ru Complex | K1 (104 M−1) | K2 (104 M−1) | K1/K2 |
|---|---|---|---|
| 1 | 0.79 ± 0.04 | 0.18 ± 0.03 | 4.39 |
| 2 | 3.94 ± 0.24 | 0.51 ± 0.05 | 7.72 |
| 3 | 2.52 ± 0.13 | 0.43 ± 0.04 | 5.86 |
| 4 | 2.92 ± 0.46 | 2.75 ± 0.47 | 1.06 |
| Ru Complex | m/z: Observed (Calculated) | |||
|---|---|---|---|---|
| [II + {Ru} 1]3− | [II + {Ru}2]3− | [II + {Ru}3]3− | [II + {Ru}4]3− | |
| 1 | b: 1647.31 (1647.28) | c: 1726.69 (1726.62) | c: 1805.81 (1805.62) | — 2 |
| 2 | d: 1666.32 (1666.30) | e: 1764.34 (1764.33) | m: 1862.10 (1862.02) | — 2 |
| 3 | f: 1660.96 (1660.96) | g: 1753.37 (1753.30) | n: 1846.01 (1845.99) | n: 1938.42 (1938.34) |
| 4 | h: 1672.96 (1672.96) | k: 1777.33 (1777.30) | p: 1882.04 (1881.99) | p: 1986.32 (1986.33) |
| Ru Complex | Observed (Calculated) m/z | Observed Ions |
|---|---|---|
| 1 | 1107.89 (1107.91) | [I]4− |
| 1167.44 (1167.43) | [I-1′ 1]4− | |
| 1175.92 (1175.96) | [II]4− | |
| 1235.44 (1235.46) | [II-1′]4− | |
| 1294.69 (1294.71) | [II-1′2]4− | |
| 1342.93 (1342.96) | [II-1′3]4− | |
| 4 | 1186.40 (1186.44) | [I-4′1]4− |
| 1264.67 (1264.70) | [I-4′2]4− | |
| 1343.18 (1343.21) | [I-4′3]4− | |
| 1477.57 (1477.57) | [I]3− | |
| 1582.23 (1582.25) | [I-4′]3− | |
| 1686.69 (1686.60) | [I-4′2]3− | |
| 1791.38 (1791.28) | [I-4′3]3− | |
| 1254.19 (1254.22) | [II-4′]4− | |
| 1332.76 (1332.73) | [II-4′2]4− | |
| 1411.23 (1411.24) | [II-4′3]4− | |
| 1489.46 (1489.50) | [II-4′4]4− | |
| 1567.73 (1567.76) | [II-4′5]4− | |
| 1646.30 (1646.27) | [II-4′6]4− | |
| 1568.36 (1568.28) | [II]3− | |
| 1672.66 (1672.63) | [II-4′]3− | |
| 1777.35 (1777.30) | [II-4′2]3− | |
| 1882.07(1881.99) | [II-4′3]3− | |
| 1986.45(1986.33) | [II-4′4]3− | |
| 2090.80(2090.68) | [II-4′5]3− |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, S.; Liang, A.; Wu, K.; Zeng, W.; Luo, Q.; Wang, F. Binding of Organometallic Ruthenium Anticancer Complexes to DNA: Thermodynamic Base and Sequence Selectivity. Int. J. Mol. Sci. 2018, 19, 2137. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19072137
Liu S, Liang A, Wu K, Zeng W, Luo Q, Wang F. Binding of Organometallic Ruthenium Anticancer Complexes to DNA: Thermodynamic Base and Sequence Selectivity. International Journal of Molecular Sciences. 2018; 19(7):2137. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19072137
Chicago/Turabian StyleLiu, Suyan, Aihua Liang, Kui Wu, Wenjuan Zeng, Qun Luo, and Fuyi Wang. 2018. "Binding of Organometallic Ruthenium Anticancer Complexes to DNA: Thermodynamic Base and Sequence Selectivity" International Journal of Molecular Sciences 19, no. 7: 2137. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19072137