New Therapeutic Implications of Endothelial Nitric Oxide Synthase (eNOS) Function/Dysfunction in Cardiovascular Disease

 ,
, 
 and
and 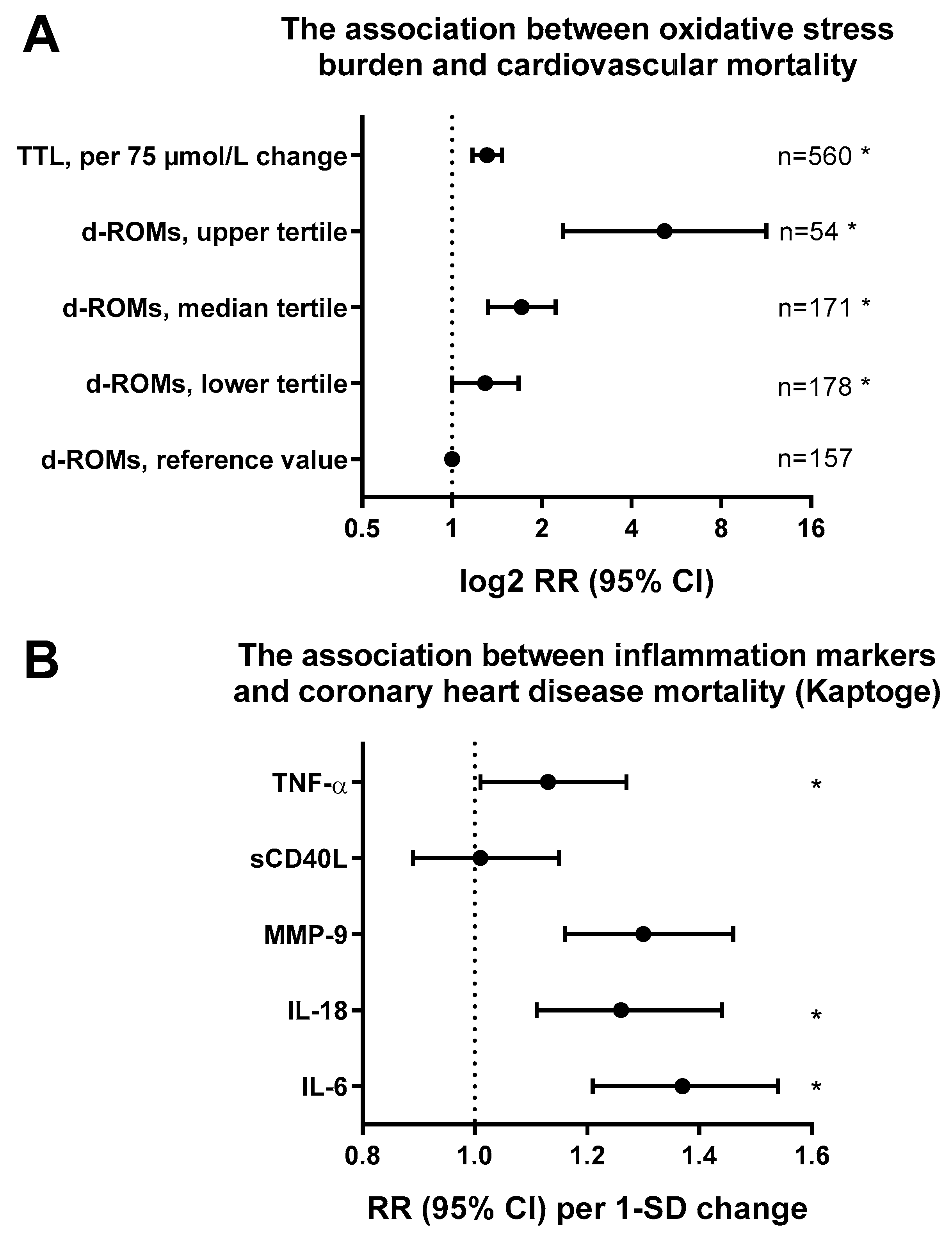{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. eNOS Impairment, Vascular Dysfunction and Cardiovascular Disease: Socioeconomic Impact and Molecular Triggers
1.1. Global Burden of Disease Study and Cardiovascular Risk Factors
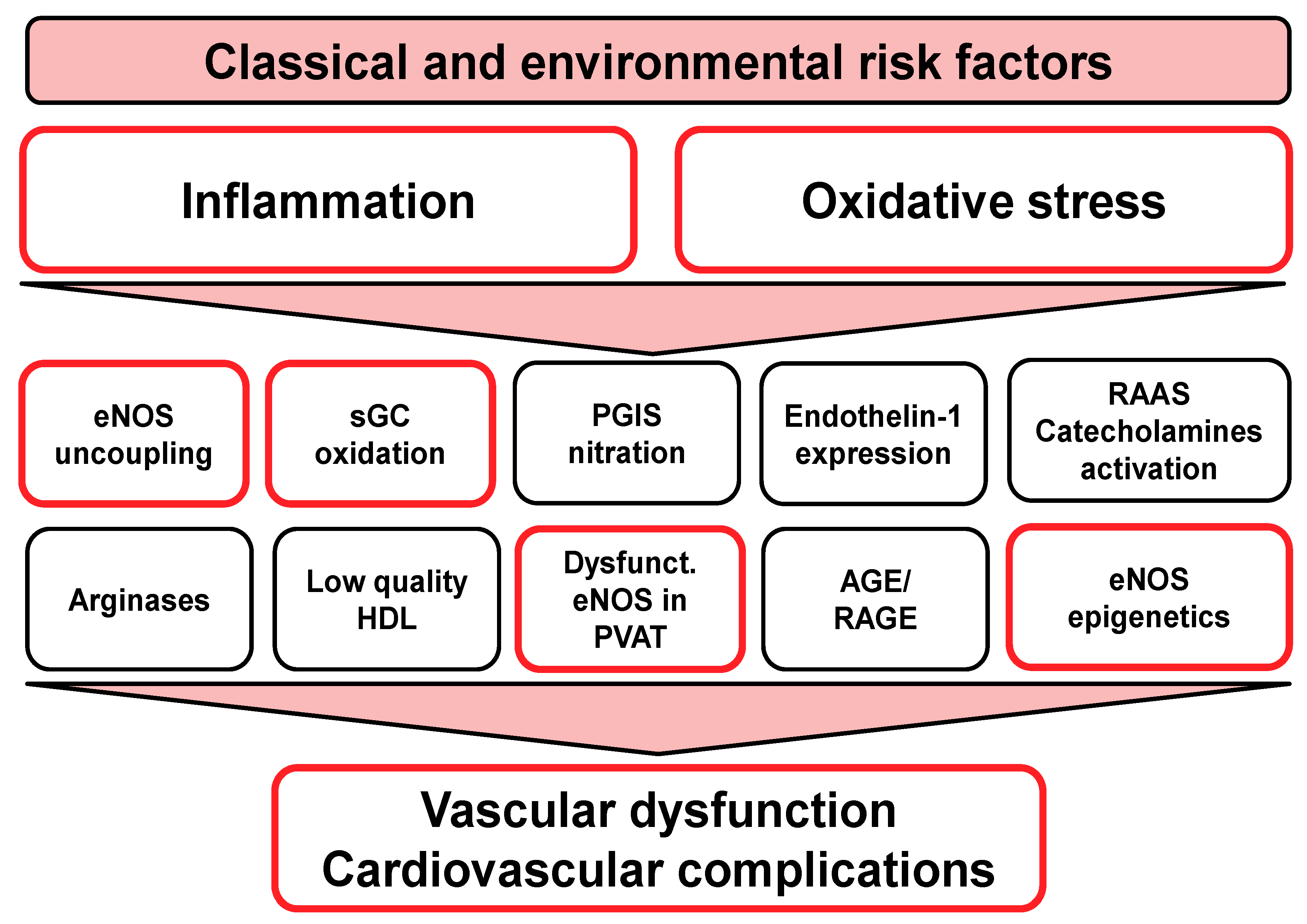
1.2. Oxidative Stress as a Unifying Molecular Trigger of Endothelial Dysfunction, Atherosclerosis, and Cardiovascular Disease
1.3. Role of Inflammation for Endothelial Dysfunction and Cardiovascular Disease
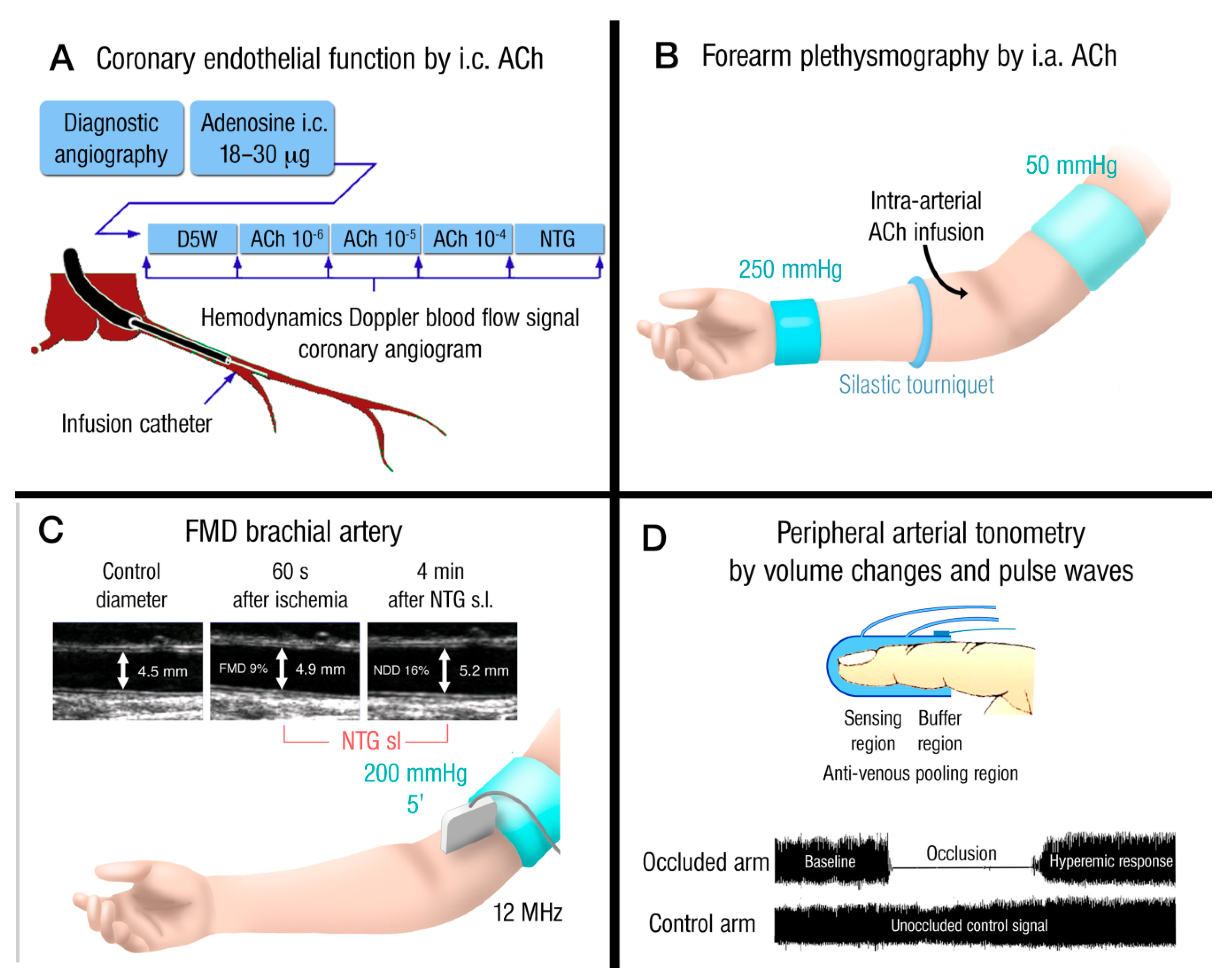
1.4. Prognostic Value of Endothelial Dysfunction and Measurement in Human Subjects
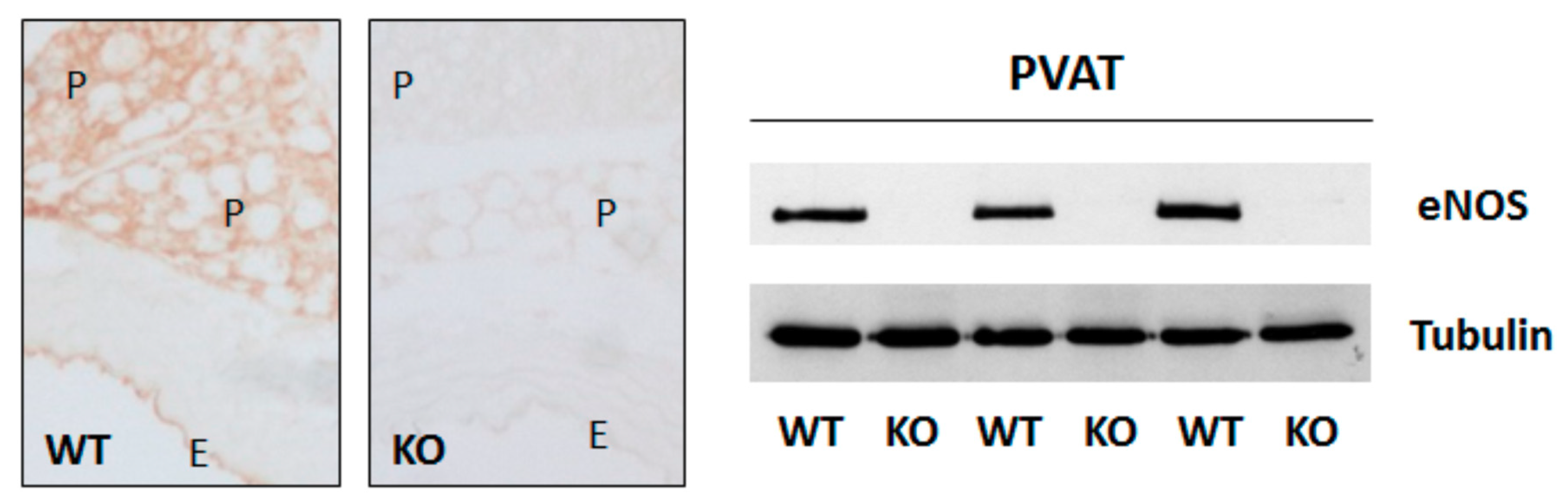
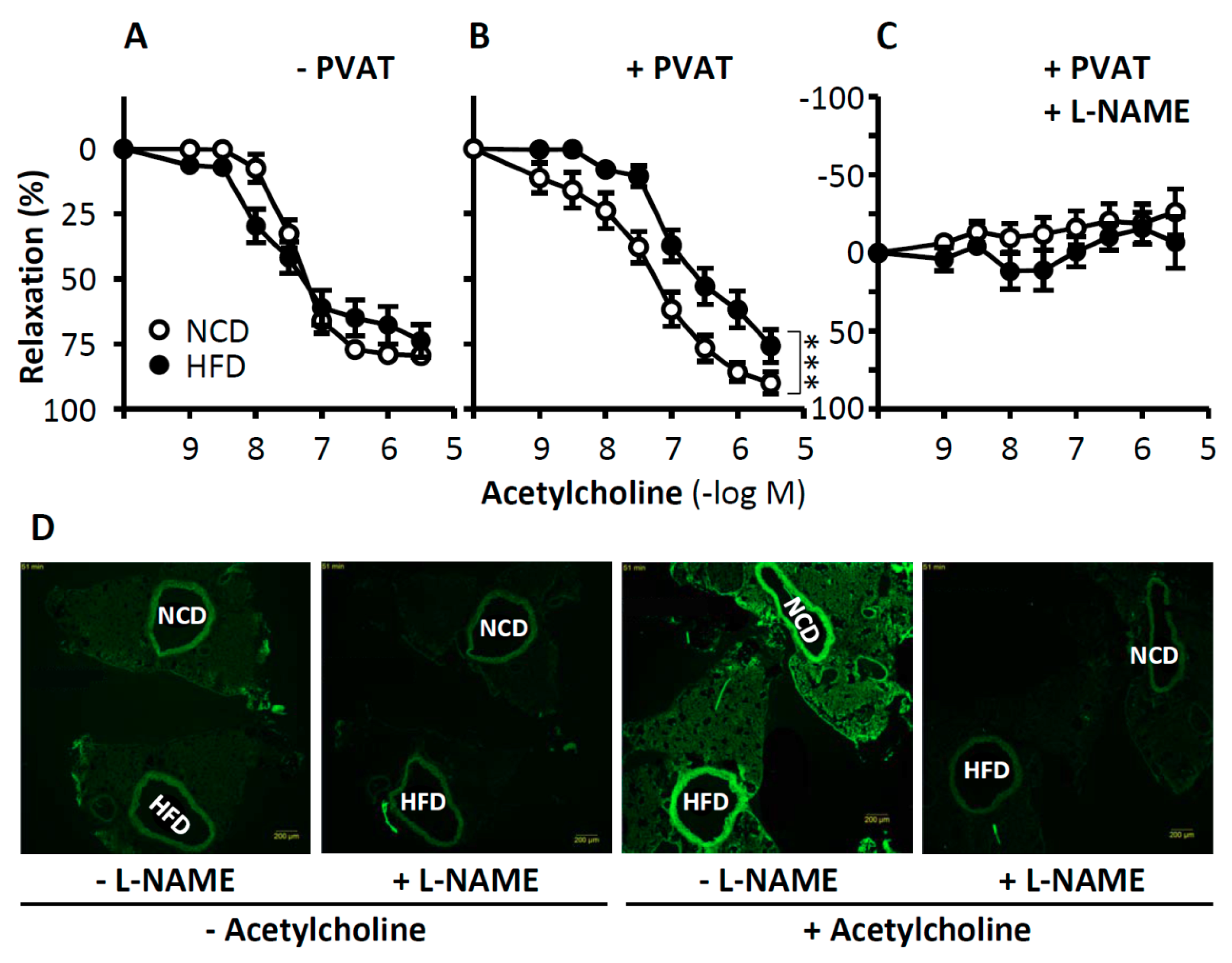
1.5. Role of eNOS in Perivascular Adipose Tissue for Vascular Function
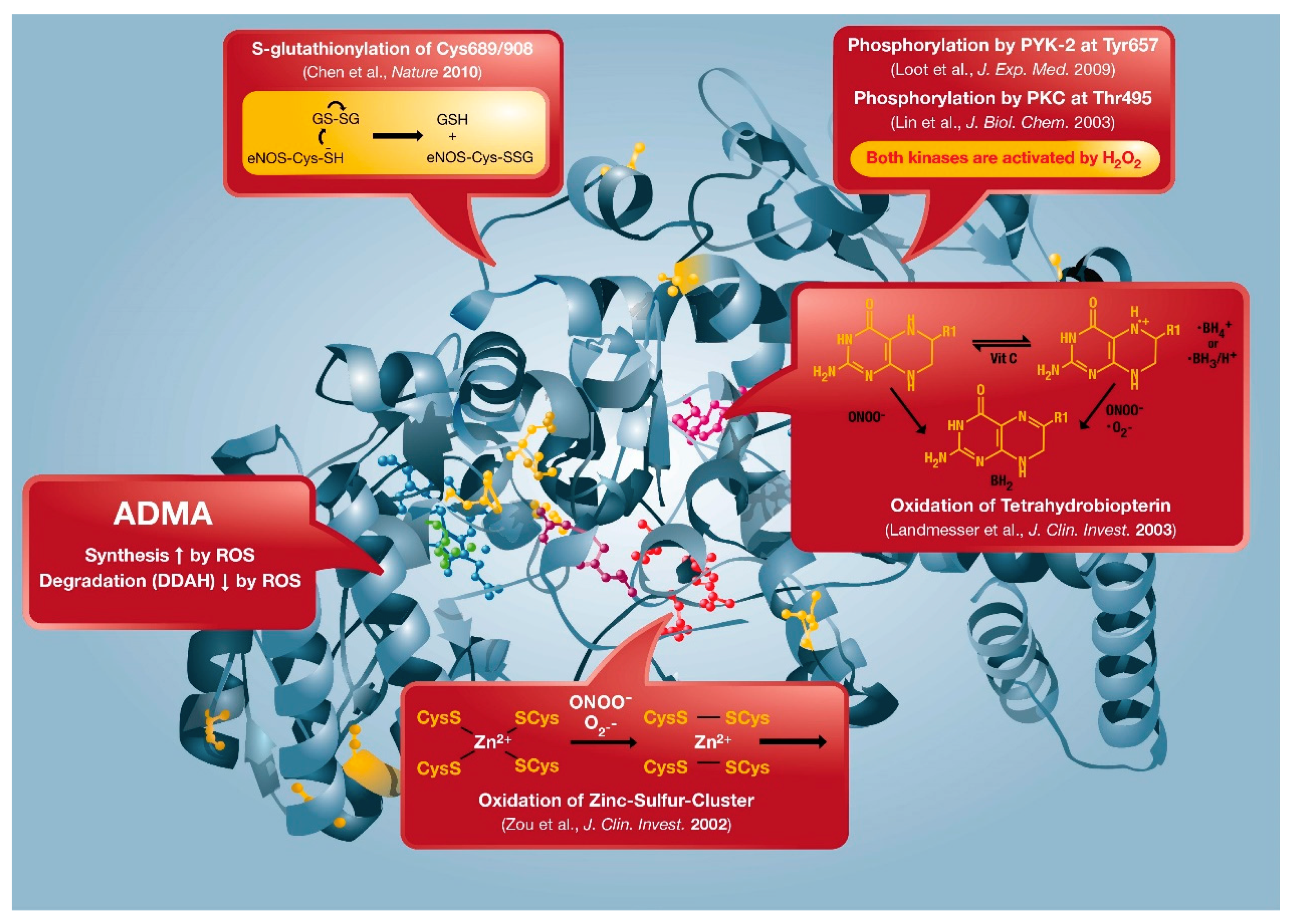
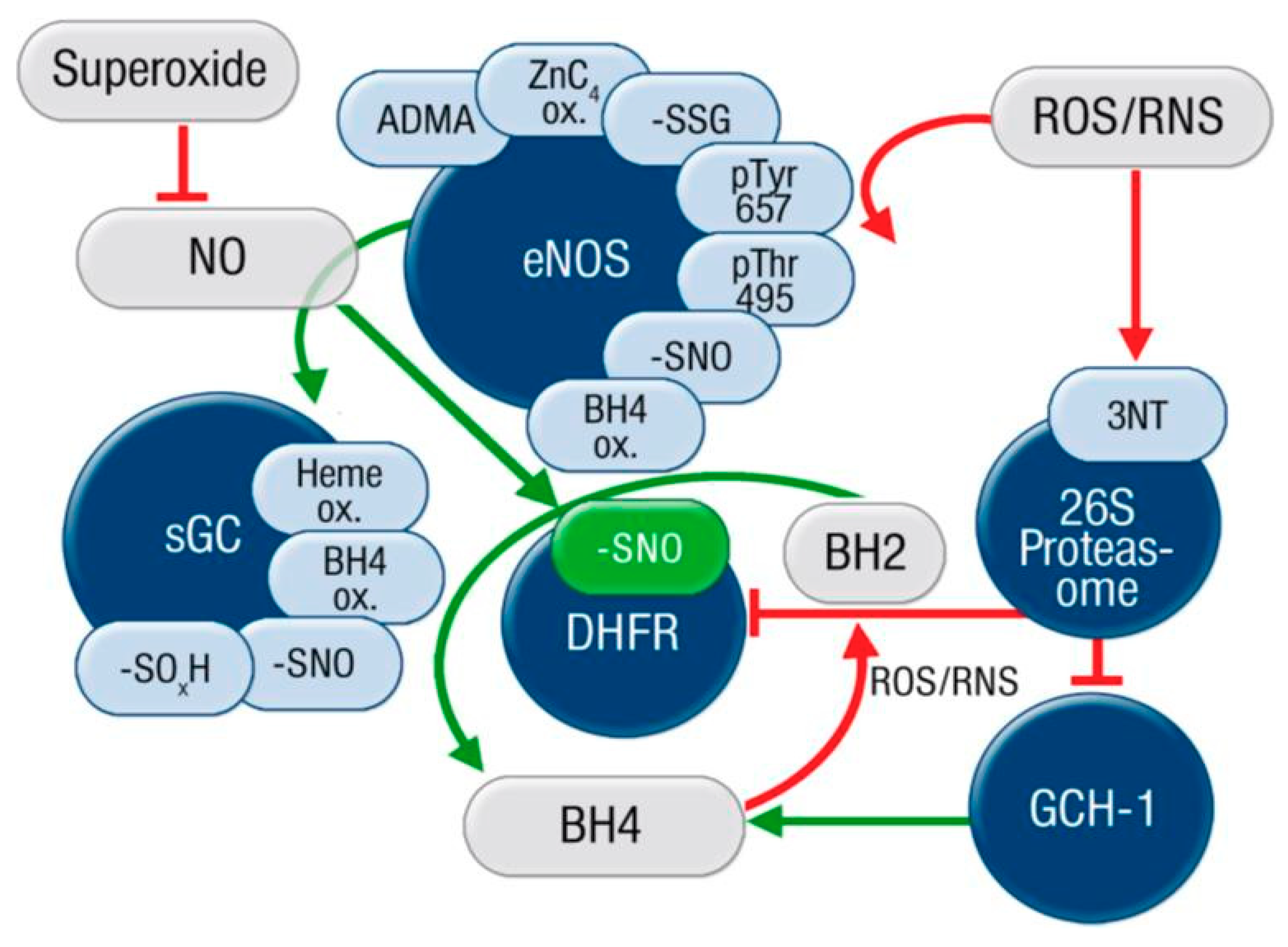
2. “Redox Switches” in Endothelial Nitric Oxide Synthase (eNOS) and Associated Pathways for Therapeutic Targeting
2.1. Oxidative Depletion of Tetrahydrobiopterin
2.2. Oxidative Disruption of the Zinc-Sulfur-Complex (ZnCys4) in the Binding Region of the eNOS Dimer
2.3. S-glutathionylation of the eNOS Reductase Domain
2.4. Phosphorylation at Thr495 and Tyr657
2.5. ADMA Formation and Degradation by DDAH
2.6. L-Arginine Deficiency
3. Other State-of-the-Art and Future Therapeutic Strategies for Targeting eNOS Dysfunction/Uncoupling
3.1. Established Cardiovascular Drugs: Statins, ACE-Inhibitors and AT1-Receptor Blockers
3.2. Therapeutic Targeting of Cascades up- and Down-Stream of the eNOS/NO/sGC/cGMP Axis
3.3. Therapeutic Targeting of eNOS Enzyme
3.4. Epigenetic Regulation of eNOS Expression
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Munzel, T.; Gori, T.; Keaney, J.F., Jr.; Maack, C.; Daiber, A. Pathophysiological role of oxidative stress in systolic and diastolic heart failure and its therapeutic implications. Eur. Heart J. 2015, 36, 2555–2564. [Google Scholar] [CrossRef] [Green Version]
- Lim, S.S.; Vos, T.; Flaxman, A.D.; Danaei, G.; Shibuya, K.; Adair-Rohani, H.; Amann, M.; Anderson, H.R.; Andrews, K.G.; Aryee, M.; et al. A comparative risk assessment of burden of disease and injury attributable to 67 risk factors and risk factor clusters in 21 regions, 1990–2010: A systematic analysis for the global burden of disease study 2010. Lancet 2012, 380, 2224–2260. [Google Scholar] [CrossRef]
- Murray, C.J.; Ezzati, M.; Flaxman, A.D.; Lim, S.; Lozano, R.; Michaud, C.; Naghavi, M.; Salomon, J.A.; Shibuya, K.; Vos, T.; et al. GBD 2010: Design, definitions, and metrics. Lancet 2012, 380, 2063–2066. [Google Scholar] [CrossRef]
- Cohen, A.J.; Brauer, M.; Burnett, R.; Anderson, H.R.; Frostad, J.; Estep, K.; Balakrishnan, K.; Brunekreef, B.; Dandona, L.; Dandona, R.; et al. Estimates and 25-year trends of the global burden of disease attributable to ambient air pollution: An analysis of data from the global burden of diseases study 2015. Lancet 2017, 389, 1907–1918. [Google Scholar] [CrossRef]
- Munzel, T.; Gori, T.; Al-Kindi, S.; Deanfield, J.; Lelieveld, J.; Daiber, A.; Rajagopalan, S. Effects of gaseous and solid constituents of air pollution on endothelial function. Eur. Heart J. 2018, 39, 3543–3550. [Google Scholar] [CrossRef]
- Munzel, T.; Schmidt, F.P.; Steven, S.; Herzog, J.; Daiber, A.; Sorensen, M. Environmental noise and the cardiovascular system. J. Am. Coll. Cardiol. 2018, 71, 688–697. [Google Scholar] [CrossRef]
- Yusuf, S.; Hawken, S.; Ounpuu, S.; Dans, T.; Avezum, A.; Lanas, F.; McQueen, M.; Budaj, A.; Pais, P.; Varigos, J.; et al. Effect of potentially modifiable risk factors associated with myocardial infarction in 52 countries (the INTERHEART study): Case-control study. Lancet 2004, 364, 937–952. [Google Scholar] [CrossRef]
- Rosengren, A.; Hawken, S.; Ounpuu, S.; Sliwa, K.; Zubaid, M.; Almahmeed, W.A.; Blackett, K.N.; Sitthi-amorn, C.; Sato, H.; Yusuf, S.; et al. Association of psychosocial risk factors with risk of acute myocardial infarction in 11119 cases and 13648 controls from 52 countries (the INTERHEART study): Case-control study. Lancet 2004, 364, 953–962. [Google Scholar] [CrossRef]
- Siegrist, J.; Sies, H. Disturbed redox homeostasis in oxidative distress: A molecular link from chronic psychosocial work stress to coronary heart disease? Circ. Res. 2017, 121, 103–105. [Google Scholar] [CrossRef]
- Schiavone, S.; Jaquet, V.; Trabace, L.; Krause, K.H. Severe life stress and oxidative stress in the brain: From animal models to human pathology. Antioxid. Redox Signal. 2013, 18, 1475–1490. [Google Scholar] [CrossRef]
- Vita, J.A.; Treasure, C.B.; Nabel, E.G.; McLenachan, J.M.; Fish, R.D.; Yeung, A.C.; Vekshtein, V.I.; Selwyn, A.P.; Ganz, P. Coronary vasomotor response to acetylcholine relates to risk factors for coronary artery disease. Circulation 1990, 81, 491–497. [Google Scholar] [CrossRef]
- Panza, J.A.; Quyyumi, A.A.; Brush, J.E., Jr.; Epstein, S.E. Abnormal endothelium-dependent vascular relaxation in patients with essential hypertension. N. Engl. J. Med. 1990, 323, 22–27. [Google Scholar] [CrossRef]
- Daiber, A.; Steven, S.; Weber, A.; Shuvaev, V.V.; Muzykantov, V.R.; Laher, I.; Li, H.; Lamas, S.; Munzel, T. Targeting vascular (endothelial) dysfunction. Br. J. Pharmacol. 2017, 174, 1591–1619. [Google Scholar] [CrossRef]
- Munzel, T.; Daiber, A. Environmental stressors and their impact on health and disease with focus on oxidative stress. Antioxid. Redox Signal. 2018, 28, 735–740. [Google Scholar] [CrossRef]
- Griendling, K.K.; FitzGerald, G.A. Oxidative stress and cardiovascular injury: Part I: Basic mechanisms and in vivo monitoring of ROS. Circulation 2003, 108, 1912–1916. [Google Scholar] [CrossRef]
- Griendling, K.K.; FitzGerald, G.A. Oxidative stress and cardiovascular injury: Part II: Animal and human studies. Circulation 2003, 108, 2034–2040. [Google Scholar] [CrossRef]
- Sies, H. Oxidative stress: A concept in redox biology and medicine. Redox Biol. 2015, 4, 180–183. [Google Scholar] [CrossRef]
- Ohara, Y.; Peterson, T.E.; Harrison, D.G. Hypercholesterolemia increases endothelial superoxide anion production. J. Clin. Investig. 1993, 91, 2546–2551. [Google Scholar] [CrossRef]
- Harrison, D.G.; Ohara, Y. Physiologic consequences of increased vascular oxidant stresses in hypercholesterolemia and atherosclerosis: Implications for impaired vasomotion. Am. J. Cardiol. 1995, 75, 75B–81B. [Google Scholar] [CrossRef]
- Schulz, E.; Wenzel, P.; Munzel, T.; Daiber, A. Mitochondrial redox signaling: Interaction of mitochondrial reactive oxygen species with other sources of oxidative stress. Antioxid. Redox Signal. 2014, 20, 308–324. [Google Scholar] [CrossRef]
- Forstermann, U.; Munzel, T. Endothelial nitric oxide synthase in vascular disease: From marvel to menace. Circulation 2006, 113, 1708–1714. [Google Scholar] [CrossRef]
- Munzel, T.; Daiber, A.; Ullrich, V.; Mulsch, A. Vascular consequences of endothelial nitric oxide synthase uncoupling for the activity and expression of the soluble guanylyl cyclase and the cGMP-dependent protein kinase. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1551–1557. [Google Scholar] [CrossRef]
- Daiber, A.; Oelze, M.; Daub, S.; Steven, S.; Schuff, A.; Kroller-Schon, S.; Hausding, M.; Wenzel, P.; Schulz, E.; Gori, T.; et al. Vascular redox signaling, redox switches in endothelial nitric oxide synthase and endothelial dysfunction. In Systems Biology of Free Radicals and Antioxidants; Laher, I., Ed.; Springer: Berlin/Heidelberg, Germany, 2014; pp. 1177–1211. [Google Scholar]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef]
- Daiber, A. Redox signaling (cross-talk) from and to mitochondria involves mitochondrial pores and reactive oxygen species. Biochim. Biophys. Acta 2010, 1797, 897–906. [Google Scholar] [CrossRef] [Green Version]
- Heitzer, T.; Schlinzig, T.; Krohn, K.; Meinertz, T.; Munzel, T. Endothelial dysfunction, oxidative stress, and risk of cardiovascular events in patients with coronary artery disease. Circulation 2001, 104, 2673–2678. [Google Scholar] [CrossRef]
- Schachinger, V.; Britten, M.B.; Zeiher, A.M. Prognostic impact of coronary vasodilator dysfunction on adverse long-term outcome of coronary heart disease. Circulation 2000, 101, 1899–1906. [Google Scholar] [CrossRef]
- Nakazono, K.; Watanabe, N.; Matsuno, K.; Sasaki, J.; Sato, T.; Inoue, M. Does superoxide underlie the pathogenesis of hypertension? Proc. Natl. Acad. Sci. USA 1991, 88, 10045–10048. [Google Scholar] [CrossRef] [Green Version]
- Rajagopalan, S.; Kurz, S.; Munzel, T.; Tarpey, M.; Freeman, B.A.; Griendling, K.K.; Harrison, D.G. Angiotensin II-mediated hypertension in the rat increases vascular superoxide production via membrane NADH/NADPH oxidase activation. Contribution to alterations of vasomotor tone. J. Clin. Investig. 1996, 97, 1916–1923. [Google Scholar] [CrossRef]
- Mollnau, H.; Wendt, M.; Szocs, K.; Lassegue, B.; Schulz, E.; Oelze, M.; Li, H.; Bodenschatz, M.; August, M.; Kleschyov, A.L.; et al. Effects of angiotensin ii infusion on the expression and function of NAD(P)H oxidase and components of nitric oxide/cGMP signaling. Circ. Res. 2002, 90, e58–e65. [Google Scholar] [CrossRef]
- Landmesser, U.; Cai, H.; Dikalov, S.; McCann, L.; Hwang, J.; Jo, H.; Holland, S.M.; Harrison, D.G. Role of p47(phox) in vascular oxidative stress and hypertension caused by angiotensin II. Hypertension 2002, 40, 511–515. [Google Scholar] [CrossRef]
- Kuzkaya, N.; Weissmann, N.; Harrison, D.G.; Dikalov, S. Interactions of peroxynitrite, tetrahydrobiopterin, ascorbic acid, and thiols: Implications for uncoupling endothelial nitric-oxide synthase. J. Biol. Chem. 2003, 278, 22546–22554. [Google Scholar] [CrossRef] [PubMed]
- Landmesser, U.; Dikalov, S.; Price, S.R.; McCann, L.; Fukai, T.; Holland, S.M.; Mitch, W.E.; Harrison, D.G. Oxidation of tetrahydrobiopterin leads to uncoupling of endothelial cell nitric oxide synthase in hypertension. J. Clin. Investig. 2003, 111, 1201–1209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fratta Pasini, A.; Albiero, A.; Stranieri, C.; Cominacini, M.; Pasini, A.; Mozzini, C.; Vallerio, P.; Cominacini, L.; Garbin, U. Serum oxidative stress-induced repression of Nrf2 and GSH depletion: A mechanism potentially involved in endothelial dysfunction of young smokers. PLoS ONE 2012, 7, e30291. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, M.I.; Saglam, M.; Caglar, K.; Cakir, E.; Sonmez, A.; Ozgurtas, T.; Aydin, A.; Eyileten, T.; Ozcan, O.; Acikel, C.; et al. The determinants of endothelial dysfunction in CKD: Oxidative stress and asymmetric dimethylarginine. Am. J. Kidney Dis. 2006, 47, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Jurado-Gamez, B.; Fernandez-Marin, M.C.; Gomez-Chaparro, J.L.; Munoz-Cabrera, L.; Lopez-Barea, J.; Perez-Jimenez, F.; Lopez-Miranda, J. Relationship of oxidative stress and endothelial dysfunction in sleep apnoea. Eur. Respir. J. 2011, 37, 873–879. [Google Scholar] [CrossRef] [PubMed]
- Blankenberg, S.; Rupprecht, H.J.; Bickel, C.; Torzewski, M.; Hafner, G.; Tiret, L.; Smieja, M.; Cambien, F.; Meyer, J.; Lackner, K.J. Glutathione peroxidase 1 activity and cardiovascular events in patients with coronary artery disease. N. Engl. J. Med. 2003, 349, 1605–1613. [Google Scholar] [CrossRef] [PubMed]
- Schottker, B.; Brenner, H.; Jansen, E.H.; Gardiner, J.; Peasey, A.; Kubinova, R.; Pajak, A.; Topor-Madry, R.; Tamosiunas, A.; Saum, K.U.; et al. Evidence for the free radical/oxidative stress theory of ageing from the CHANCES consortium: A meta-analysis of individual participant data. BMC Med. 2015, 13, 300. [Google Scholar] [CrossRef]
- Khaw, K.T.; Bingham, S.; Welch, A.; Luben, R.; Wareham, N.; Oakes, S.; Day, N. Relation between plasma ascorbic acid and mortality in men and women in EPIC-Norfolk prospective study: A prospective population study. European prospective investigation into cancer and nutrition. Lancet 2001, 357, 657–663. [Google Scholar] [CrossRef]
- Munzel, T.; Gori, T.; Bruno, R.M.; Taddei, S. Is oxidative stress a therapeutic target in cardiovascular disease? Eur. Heart J. 2010, 31, 2741–2748. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, H.H.; Stocker, R.; Vollbracht, C.; Paulsen, G.; Riley, D.; Daiber, A.; Cuadrado, A. Antioxidants in translational medicine. Antioxid. Redox Signal. 2015, 23, 1130–1143. [Google Scholar] [CrossRef]
- Shuaib, A.; Lees, K.R.; Lyden, P.; Grotta, J.; Davalos, A.; Davis, S.M.; Diener, H.C.; Ashwood, T.; Wasiewski, W.W.; Emeribe, U.; et al. NXY-059 for the treatment of acute ischemic stroke. N. Engl. J. Med. 2007, 357, 562–571. [Google Scholar] [CrossRef] [PubMed]
- Karbach, S.; Wenzel, P.; Waisman, A.; Munzel, T.; Daiber, A. eNOS uncoupling in cardiovascular diseases—The role of oxidative stress and inflammation. Curr. Pharm. Des. 2014, 20, 3579–3594. [Google Scholar] [CrossRef] [PubMed]
- Harrison, D.G.; Guzik, T.J.; Lob, H.E.; Madhur, M.S.; Marvar, P.J.; Thabet, S.R.; Vinh, A.; Weyand, C.M. Inflammation, immunity, and hypertension. Hypertension 2011, 57, 132–140. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, P.; Kossmann, S.; Munzel, T.; Daiber, A. Redox regulation of cardiovascular inflammation—Immunomodulatory function of mitochondrial and Nox-derived reactive oxygen and nitrogen species. Free Radic. Biol. Med. 2017, 109, 48–60. [Google Scholar] [CrossRef] [PubMed]
- Kaptoge, S.; Seshasai, S.R.; Gao, P.; Freitag, D.F.; Butterworth, A.S.; Borglykke, A.; Di Angelantonio, E.; Gudnason, V.; Rumley, A.; Lowe, G.D.; et al. Inflammatory cytokines and risk of coronary heart disease: New prospective study and updated meta-analysis. Eur. Heart J. 2014, 35, 578–589. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef] [PubMed]
- Cave, A.C.; Brewer, A.C.; Narayanapanicker, A.; Ray, R.; Grieve, D.J.; Walker, S.; Shah, A.M. NADPH oxidases in cardiovascular health and disease. Antioxid. Redox Signal. 2006, 8, 691–728. [Google Scholar] [CrossRef]
- Guzik, T.J.; Hoch, N.E.; Brown, K.A.; McCann, L.A.; Rahman, A.; Dikalov, S.; Goronzy, J.; Weyand, C.; Harrison, D.G. Role of the T cell in the genesis of angiotensin II induced hypertension and vascular dysfunction. J. Exp. Med. 2007, 204, 2449–2460. [Google Scholar] [CrossRef] [Green Version]
- Wenzel, P.; Knorr, M.; Kossmann, S.; Stratmann, J.; Hausding, M.; Schuhmacher, S.; Karbach, S.H.; Schwenk, M.; Yogev, N.; Schulz, E.; et al. Lysozyme M-positive monocytes mediate angiotensin II-induced arterial hypertension and vascular dysfunction. Circulation 2011, 124, 1370–1381. [Google Scholar] [CrossRef]
- Bulua, A.C.; Simon, A.; Maddipati, R.; Pelletier, M.; Park, H.; Kim, K.Y.; Sack, M.N.; Kastner, D.L.; Siegel, R.M. Mitochondrial reactive oxygen species promote production of proinflammatory cytokines and are elevated in TNFR1-associated periodic syndrome (TRAPS). J. Exp. Med. 2011, 208, 519–533. [Google Scholar] [CrossRef] [Green Version]
- West, A.P.; Brodsky, I.E.; Rahner, C.; Woo, D.K.; Erdjument-Bromage, H.; Tempst, P.; Walsh, M.C.; Choi, Y.; Shadel, G.S.; Ghosh, S. TLR signalling augments macrophage bactericidal activity through mitochondrial ROS. Nature 2011, 472, 476–480. [Google Scholar] [CrossRef]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Mittal, M.; Siddiqui, M.R.; Tran, K.; Reddy, S.P.; Malik, A.B. Reactive oxygen species in inflammation and tissue injury. Antioxid. Redox Signal. 2014, 20, 1126–1167. [Google Scholar] [CrossRef] [PubMed]
- El Assar, M.; Angulo, J.; Rodriguez-Manas, L. Oxidative stress and vascular inflammation in aging. Free Radic. Biol. Med. 2013, 65, 380–401. [Google Scholar] [CrossRef] [PubMed]
- Mikhed, Y.; Daiber, A.; Steven, S. Mitochondrial oxidative stress, mitochondrial DNA damage and their role in age-related vascular dysfunction. Int. J. Mol. Sci. 2015, 16, 15918–15953. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Zheng, X.; Mahdi, A.; Zhou, Z.; Tratsiakovich, Y.; Jiao, T.; Kiss, A.; Kovamees, O.; Alvarsson, M.; Catrina, S.B.; et al. Red blood cells in type 2 diabetes impair cardiac post-ischemic recovery through an arginase-dependent modulation of nitric oxide synthase and reactive oxygen species. JACC Basic Transl. Sci. 2018, 3, 450–463. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Mahdi, A.; Tratsiakovich, Y.; Zahoran, S.; Kovamees, O.; Nordin, F.; Uribe Gonzalez, A.E.; Alvarsson, M.; Ostenson, C.G.; Andersson, D.C.; et al. Erythrocytes from patients with type 2 diabetes induce endothelial dysfunction via arginase I. J. Am. Coll. Cardiol. 2018, 72, 769–780. [Google Scholar] [CrossRef] [PubMed]
- Soltesz, P.; Kerekes, G.; Der, H.; Szucs, G.; Szanto, S.; Kiss, E.; Bodolay, E.; Zeher, M.; Timar, O.; Szodoray, P.; et al. Comparative assessment of vascular function in autoimmune rheumatic diseases: Considerations of prevention and treatment. Autoimmun. Rev. 2011, 10, 416–425. [Google Scholar] [CrossRef] [PubMed]
- Murdaca, G.; Colombo, B.M.; Cagnati, P.; Gulli, R.; Spano, F.; Puppo, F. Endothelial dysfunction in rheumatic autoimmune diseases. Atherosclerosis 2012, 224, 309–317. [Google Scholar] [CrossRef]
- Vena, G.A.; Vestita, M.; Cassano, N. Psoriasis and cardiovascular disease. Dermatol. Ther. 2010, 23, 144–151. [Google Scholar] [CrossRef] [PubMed]
- Hak, A.E.; Karlson, E.W.; Feskanich, D.; Stampfer, M.J.; Costenbader, K.H. Systemic lupus erythematosus and the risk of cardiovascular disease: Results from the nurses’ health study. Arthritis Care Res. 2009, 61, 1396–1402. [Google Scholar] [CrossRef] [PubMed]
- Mehta, N.N.; Azfar, R.S.; Shin, D.B.; Neimann, A.L.; Troxel, A.B.; Gelfand, J.M. Patients with severe psoriasis are at increased risk of cardiovascular mortality: Cohort study using the general practice research database. Eur. Heart J. 2010, 31, 1000–1006. [Google Scholar] [CrossRef] [PubMed]
- Peters, M.J.; Symmons, D.P.; McCarey, D.; Dijkmans, B.A.; Nicola, P.; Kvien, T.K.; McInnes, I.B.; Haentzschel, H.; Gonzalez-Gay, M.A.; Provan, S.; et al. EULAR evidence-based recommendations for cardiovascular risk management in patients with rheumatoid arthritis and other forms of inflammatory arthritis. Ann. Rheum. Dis. 2010, 69, 325–331. [Google Scholar] [CrossRef] [PubMed]
- Herrera, J.; Ferrebuz, A.; MacGregor, E.G.; Rodriguez-Iturbe, B. Mycophenolate mofetil treatment improves hypertension in patients with psoriasis and rheumatoid arthritis. J. Am. Soc. Nephrol. 2006, 17, S218–S225. [Google Scholar] [CrossRef] [PubMed]
- Sodergren, A.; Karp, K.; Boman, K.; Eriksson, C.; Lundstrom, E.; Smedby, T.; Soderlund, L.; Rantapaa-Dahlqvist, S.; Wallberg-Jonsson, S. Atherosclerosis in early rheumatoid arthritis: Very early endothelial activation and rapid progression of intima media thickness. Arthritis Res. Ther. 2010, 12, R158. [Google Scholar] [CrossRef] [PubMed]
- Balci, D.D.; Balci, A.; Karazincir, S.; Ucar, E.; Iyigun, U.; Yalcin, F.; Seyfeli, E.; Inandi, T.; Egilmez, E. Increased carotid artery intima-media thickness and impaired endothelial function in psoriasis. J. Eur. Acad. Dermatol. Venereol. 2009, 23, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Cesare, A.; Di Meglio, P.; Nestle, F.O. The IL-23/Th17 axis in the immunopathogenesis of psoriasis. J. Investig. Dermatol. 2009, 129, 1339–1350. [Google Scholar] [CrossRef]
- Leonardi, C.; Matheson, R.; Zachariae, C.; Cameron, G.; Li, L.; Edson-Heredia, E.; Braun, D.; Banerjee, S. Anti-interleukin-17 monoclonal antibody ixekizumab in chronic plaque psoriasis. N. Engl. J. Med. 2012, 366, 1190–1199. [Google Scholar] [CrossRef]
- Papp, K.A.; Leonardi, C.; Menter, A.; Ortonne, J.P.; Krueger, J.G.; Kricorian, G.; Aras, G.; Li, J.; Russell, C.B.; Thompson, E.H.; et al. Brodalumab, an anti-interleukin-17-receptor antibody for psoriasis. N. Engl. J. Med. 2012, 366, 1181–1189. [Google Scholar] [CrossRef]
- Crispin, J.C.; Tsokos, G.C. IL-17 in systemic lupus erythematosus. J. Biomed. Biotechnol. 2010, 2010, 943254. [Google Scholar] [CrossRef]
- Choy, E. Understanding the dynamics: Pathways involved in the pathogenesis of rheumatoid arthritis. Rheumatology 2012, 51 (Suppl. 5), v3–v11. [Google Scholar] [CrossRef]
- Pasceri, V.; Yeh, E.T. A tale of two diseases: Atherosclerosis and rheumatoid arthritis. Circulation 1999, 100, 2124–2126. [Google Scholar] [CrossRef] [PubMed]
- Perticone, F.; Ceravolo, R.; Pujia, A.; Ventura, G.; Iacopino, S.; Scozzafava, A.; Ferraro, A.; Chello, M.; Mastroroberto, P.; Verdecchia, P.; et al. Prognostic significance of endothelial dysfunction in hypertensive patients. Circulation 2001, 104, 191–196. [Google Scholar] [CrossRef] [PubMed]
- Calver, A.; Collier, J.; Vallance, P. Inhibition and stimulation of nitric oxide synthesis in the human forearm arterial bed of patients with insulin-dependent diabetes. J. Clin. Investig. 1992, 90, 2548–2554. [Google Scholar] [CrossRef] [PubMed]
- Celermajer, D.S.; Sorensen, K.E.; Georgakopoulos, D.; Bull, C.; Thomas, O.; Robinson, J.; Deanfield, J.E. Cigarette smoking is associated with dose-related and potentially reversible impairment of endothelium-dependent dilation in healthy young adults. Circulation 1993, 88, 2149–2155. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, M.A.; Selwyn, A.P. Endothelial function, inflammation, and prognosis in cardiovascular disease. Am. J. Med. 2003, 115 (Suppl. 1), 99–106. [Google Scholar] [CrossRef]
- Pauriah, M.; Khan, F.; Lim, T.K.; Elder, D.H.; Godfrey, V.; Kennedy, G.; Belch, J.J.; Booth, N.A.; Struthers, A.D.; Lang, C.C. B-type natriuretic peptide is an independent predictor of endothelial function in man. Clin. Sci. 2012, 123, 307–312. [Google Scholar] [CrossRef] [Green Version]
- Okui, H.; Hamasaki, S.; Ishida, S.; Kataoka, T.; Orihara, K.; Fukudome, T.; Ogawa, M.; Oketani, N.; Saihara, K.; Shinsato, T.; et al. Adiponectin is a better predictor of endothelial function of the coronary artery than HOMA-R, body mass index, immunoreactive insulin, or triglycerides. Int. J. Cardiol. 2008, 126, 53–61. [Google Scholar] [CrossRef]
- Schmidt, F.P.; Basner, M.; Kroger, G.; Weck, S.; Schnorbus, B.; Muttray, A.; Sariyar, M.; Binder, H.; Gori, T.; Warnholtz, A.; et al. Effect of nighttime aircraft noise exposure on endothelial function and stress hormone release in healthy adults. Eur. Heart J. 2013, 34, 3508–3514. [Google Scholar] [CrossRef]
- Schmidt, F.; Kolle, K.; Kreuder, K.; Schnorbus, B.; Wild, P.; Hechtner, M.; Binder, H.; Gori, T.; Munzel, T. Nighttime aircraft noise impairs endothelial function and increases blood pressure in patients with or at high risk for coronary artery disease. Clin. Res. Cardiol. 2015, 104, 23–30. [Google Scholar] [CrossRef]
- Mills, N.L.; Tornqvist, H.; Robinson, S.D.; Gonzalez, M.; Darnley, K.; MacNee, W.; Boon, N.A.; Donaldson, K.; Blomberg, A.; Sandstrom, T.; et al. Diesel exhaust inhalation causes vascular dysfunction and impaired endogenous fibrinolysis. Circulation 2005, 112, 3930–3936. [Google Scholar] [CrossRef] [PubMed]
- Brook, R.D.; Urch, B.; Dvonch, J.T.; Bard, R.L.; Speck, M.; Keeler, G.; Morishita, M.; Marsik, F.J.; Kamal, A.S.; Kaciroti, N.; et al. Insights into the mechanisms and mediators of the effects of air pollution exposure on blood pressure and vascular function in healthy humans. Hypertension 2009, 54, 659–667. [Google Scholar] [CrossRef] [PubMed]
- Ghiadoni, L.; Donald, A.E.; Cropley, M.; Mullen, M.J.; Oakley, G.; Taylor, M.; O’Connor, G.; Betteridge, J.; Klein, N.; Steptoe, A.; et al. Mental stress induces transient endothelial dysfunction in humans. Circulation 2000, 102, 2473–2478. [Google Scholar] [CrossRef] [PubMed]
- Broadley, A.J.; Korszun, A.; Abdelaal, E.; Moskvina, V.; Jones, C.J.; Nash, G.B.; Ray, C.; Deanfield, J.; Frenneaux, M.P. Inhibition of cortisol production with metyrapone prevents mental stress-induced endothelial dysfunction and baroreflex impairment. J. Am. Coll. Cardiol. 2005, 46, 344–350. [Google Scholar] [CrossRef]
- Munzel, T.; Sinning, C.; Post, F.; Warnholtz, A.; Schulz, E. Pathophysiology, diagnosis and prognostic implications of endothelial dysfunction. Ann. Med. 2008, 40, 180–196. [Google Scholar] [CrossRef]
- Heitzer, T.; Ylä-Herttuala, S.; Luoma, J.; Kurz, S.; Munzel, T.; Just, H.; Olschewski, M.; Drexler, H. Cigarette smoking potentiates endothelial dysfunction of forearm resistance vessels in patients with hypercholesterolemia: Role of oxidized LDL. Circulation 1996, 93, 1346–1353. [Google Scholar] [CrossRef]
- Schnabel, R.B.; Wild, P.S.; Schulz, A.; Zeller, T.; Sinning, C.R.; Wilde, S.; Kunde, J.; Lubos, E.; Lackner, K.J.; Warnholtz, A.; et al. Multiple endothelial biomarkers and noninvasive vascular function in the general population: The Gutenberg Health Study. Hypertension 2012, 60, 288–295. [Google Scholar] [CrossRef]
- Garcia, M.M.; Lima, P.R.; Correia, L.C. Prognostic value of endothelial function in patients with atherosclerosis: Systematic review. Arq. Bras. Cardiol. 2012, 99, 857–865. [Google Scholar] [CrossRef]
- Suessenbacher, A.; Dorler, J.; Wunder, J.; Hohenwarter, F.; Alber, H.F.; Pachinger, O.; Frick, M. Comparison of brachial artery wall thickness versus endothelial function to predict late cardiovascular events in patients undergoing elective coronary angiography. Am. J. Cardiol. 2013, 111, 671–675. [Google Scholar] [CrossRef]
- Roos, W.P.; Kaina, B. DNA damage-induced cell death: From specific DNA lesions to the DNA damage response and apoptosis. Cancer Lett. 2013, 332, 237–248. [Google Scholar] [CrossRef]
- Schnabel, R.B.; Schulz, A.; Wild, P.S.; Sinning, C.R.; Wilde, S.; Eleftheriadis, M.; Herkenhoff, S.; Zeller, T.; Lubos, E.; Lackner, K.J.; et al. Noninvasive vascular function measurement in the community: Cross-sectional relations and comparison of methods. Circ. Cardiovasc. Imaging 2011, 4, 371–380. [Google Scholar] [CrossRef]
- Mitchell, G.F. Arterial stiffness: Insights from Framingham and Iceland. Curr. Opin. Nephrol. Hypertens. 2015, 24, 1–7. [Google Scholar] [CrossRef]
- Ashfaq, S.; Abramson, J.L.; Jones, D.P.; Rhodes, S.D.; Weintraub, W.S.; Hooper, W.C.; Vaccarino, V.; Harrison, D.G.; Quyyumi, A.A. The relationship between plasma levels of oxidized and reduced thiols and early atherosclerosis in healthy adults. J. Am. Coll. Cardiol. 2006, 47, 1005–1011. [Google Scholar] [CrossRef]
- Soltis, E.E.; Cassis, L.A. Influence of perivascular adipose tissue on rat aortic smooth muscle responsiveness. Clin. Exp. Hypertens. Part A Theory Pract. 1991, 13, 277–296. [Google Scholar] [CrossRef]
- Xia, N.; Li, H. The role of perivascular adipose tissue in obesity-induced vascular dysfunction. Br. J. Pharmacol. 2017, 174, 3425–3442. [Google Scholar] [CrossRef]
- Dashwood, M.R.; Dooley, A.; Shi-Wen, X.; Abraham, D.J.; Souza, D.S. Does periadventitial fat-derived nitric oxide play a role in improved saphenous vein graft patency in patients undergoing coronary artery bypass surgery? J. Vasc. Res. 2007, 44, 175–181. [Google Scholar] [CrossRef]
- Xia, N.; Horke, S.; Habermeier, A.; Closs, E.I.; Reifenberg, G.; Gericke, A.; Mikhed, Y.; Munzel, T.; Daiber, A.; Forstermann, U.; et al. Uncoupling of endothelial nitric oxide synthase in perivascular adipose tissue of diet-induced obese mice. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Gil-Ortega, M.; Stucchi, P.; Guzman-Ruiz, R.; Cano, V.; Arribas, S.; Gonzalez, M.C.; Ruiz-Gayo, M.; Fernandez-Alfonso, M.S.; Somoza, B. Adaptative nitric oxide overproduction in perivascular adipose tissue during early diet-induced obesity. Endocrinology 2010, 151, 3299–3306. [Google Scholar] [CrossRef]
- Victorio, J.A.; Fontes, M.T.; Rossoni, L.V.; Davel, A.P. Different anti-contractile function and nitric oxide production of thoracic and abdominal perivascular adipose tissues. Front. Physiol. 2016, 7, 295. [Google Scholar] [CrossRef] [PubMed]
- Virdis, A.; Duranti, E.; Rossi, C.; Dell’Agnello, U.; Santini, E.; Anselmino, M.; Chiarugi, M.; Taddei, S.; Solini, A. Tumour necrosis factor-alpha participates on the endothelin-1/nitric oxide imbalance in small arteries from obese patients: Role of perivascular adipose tissue. Eur. Heart J. 2015, 36, 784–794. [Google Scholar] [CrossRef] [PubMed]
- Aghamohammadzadeh, R.; Unwin, R.D.; Greenstein, A.S.; Heagerty, A.M. Effects of obesity on perivascular adipose tissue vasorelaxant function: Nitric oxide, inflammation and elevated systemic blood pressure. J. Vasc. Res. 2015, 52, 299–305. [Google Scholar] [CrossRef]
- Bussey, C.E.; Withers, S.B.; Aldous, R.G.; Edwards, G.; Heagerty, A.M. Obesity-related perivascular adipose tissue damage is reversed by sustained weight loss in the rat. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 1377–1385. [Google Scholar] [CrossRef]
- Li, H.; Forstermann, U. Nitric oxide in the pathogenesis of vascular disease. J. Pathol. 2000, 190, 244–254. [Google Scholar] [CrossRef] [Green Version]
- Withers, S.B.; Simpson, L.; Fattah, S.; Werner, M.E.; Heagerty, A.M. cGMP-dependent protein kinase (PKG) mediates the anticontractile capacity of perivascular adipose tissue. Cardiovasc. Res. 2014, 101, 130–137. [Google Scholar] [CrossRef]
- Weston, A.H.; Egner, I.; Dong, Y.; Porter, E.L.; Heagerty, A.M.; Edwards, G. Stimulated release of a hyperpolarizing factor (ADHF) from mesenteric artery perivascular adipose tissue: Involvement of myocyte BKCa channels and adiponectin. Br. J. Pharmacol. 2013, 169, 1500–1509. [Google Scholar] [CrossRef]
- Xia, N.; Weisenburger, S.; Koch, E.; Burkart, M.; Reifenberg, G.; Forstermann, U.; Li, H. Restoration of perivascular adipose tissue function in diet-induced obese mice without changing bodyweight. Br. J. Pharmacol. 2017, 174, 3443–3453. [Google Scholar] [CrossRef] [Green Version]
- Xia, N.; Forstermann, U.; Li, H. Effects of resveratrol on eNOS in the endothelium and the perivascular adipose tissue. Ann. N. Y. Acad. Sci. 2017, 1403, 132–141. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Li, J.; Xiao, N.; Wang, M.; Kou, J.; Qi, L.; Huang, F.; Liu, B.; Liu, K. Pharmacological activation of AMPK ameliorates perivascular adipose/endothelial dysfunction in a manner interdependent on AMPK and SIRT1. Pharmacol. Res. 2014, 89, 19–28. [Google Scholar] [CrossRef]
- Chen, Y.; Xu, X.; Zhang, Y.; Liu, K.; Huang, F.; Liu, B.; Kou, J. Diosgenin regulates adipokine expression in perivascular adipose tissue and ameliorates endothelial dysfunction via regulation of AMPK. J. Steroid Biochem. Mol. Biol. 2016, 155, 155–165. [Google Scholar] [CrossRef]
- Daiber, A.; Di Lisa, F.; Oelze, M.; Kroller-Schon, S.; Steven, S.; Schulz, E.; Munzel, T. Crosstalk of mitochondria with NADPH oxidase via reactive oxygen and nitrogen species signalling and its role for vascular function. Br. J. Pharmacol. 2017, 174, 1670–1689. [Google Scholar] [CrossRef]
- Gryglewski, R.J.; Palmer, R.M.; Moncada, S. Superoxide anion is involved in the breakdown of endothelium-derived vascular relaxing factor. Nature 1986, 320, 454–456. [Google Scholar] [CrossRef] [PubMed]
- Munzel, T.; Daiber, A.; Gori, T. Nitrate therapy: New aspects concerning molecular action and tolerance. Circulation 2011, 123, 2132–2144. [Google Scholar] [CrossRef] [PubMed]
- Vasquez-Vivar, J.; Kalyanaraman, B.; Martasek, P.; Hogg, N.; Masters, B.S.; Karoui, H.; Tordo, P.; Pritchard, K.A., Jr. Superoxide generation by endothelial nitric oxide synthase: The influence of cofactors. Proc. Natl. Acad. Sci. USA 1998, 95, 9220–9225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guzik, T.J.; Mussa, S.; Gastaldi, D.; Sadowski, J.; Ratnatunga, C.; Pillai, R.; Channon, K.M. Mechanisms of increased vascular superoxide production in human diabetes mellitus: Role of NAD(P)H oxidase and endothelial nitric oxide synthase. Circulation 2002, 105, 1656–1662. [Google Scholar] [CrossRef] [PubMed]
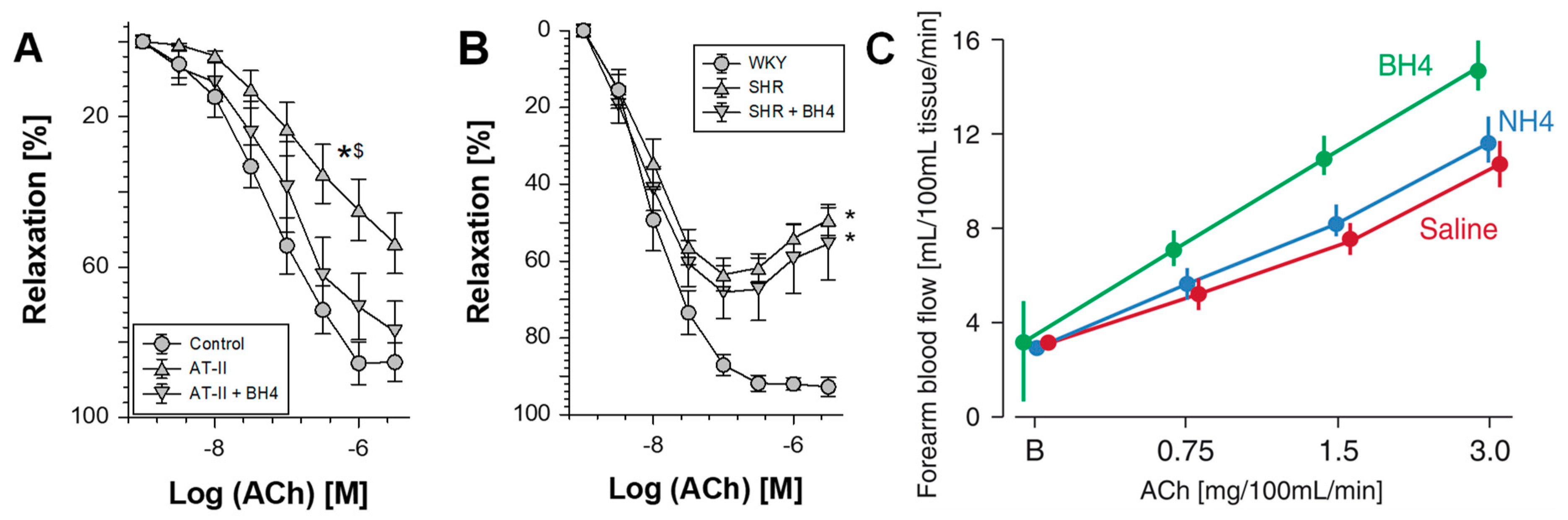
- Alp, N.J.; Mussa, S.; Khoo, J.; Cai, S.; Guzik, T.; Jefferson, A.; Goh, N.; Rockett, K.A.; Channon, K.M. Tetrahydrobiopterin-dependent preservation of nitric oxide-mediated endothelial function in diabetes by targeted transgenic GTP-cyclohydrolase I overexpression. J. Clin. Investig. 2003, 112, 725–735. [Google Scholar] [CrossRef]
- Bendall, J.K.; Alp, N.J.; Warrick, N.; Cai, S.; Adlam, D.; Rockett, K.; Yokoyama, M.; Kawashima, S.; Channon, K.M. Stoichiometric relationships between endothelial tetrahydrobiopterin, endothelial no synthase (eNOS) activity, and eNOS coupling in vivo: Insights from transgenic mice with endothelial-targeted GTP cyclohydrolase 1 and eNOS overexpression. Circ. Res. 2005, 97, 864–871. [Google Scholar] [CrossRef]
- Laursen, J.B.; Somers, M.; Kurz, S.; McCann, L.; Warnholtz, A.; Freeman, B.A.; Tarpey, M.; Fukai, T.; Harrison, D.G. Endothelial regulation of vasomotion in apoE-deficient mice: Implications for interactions between peroxynitrite and tetrahydrobiopterin. Circulation 2001, 103, 1282–1288. [Google Scholar] [CrossRef]
- Schulz, E.; Jansen, T.; Wenzel, P.; Daiber, A.; Munzel, T. Nitric oxide, tetrahydrobiopterin, oxidative stress, and endothelial dysfunction in hypertension. Antioxid. Redox Signal. 2008, 10, 1115–1126. [Google Scholar] [CrossRef]
- Harrison, D.G.; Chen, W.; Dikalov, S.; Li, L. Regulation of endothelial cell tetrahydrobiopterin pathophysiological and therapeutic implications. Adv. Pharmacol. 2010, 60, 107–132. [Google Scholar]
- Bendall, J.K.; Douglas, G.; McNeill, E.; Channon, K.M.; Crabtree, M.J. Tetrahydrobiopterin in cardiovascular health and disease. Antioxid. Redox Signal. 2014, 20, 3040–3077. [Google Scholar] [CrossRef]
- Vasquez-Vivar, J. Tetrahydrobiopterin, superoxide, and vascular dysfunction. Free Radic. Biol. Med. 2009, 47, 1108–1119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alp, N.J.; Channon, K.M. Regulation of endothelial nitric oxide synthase by tetrahydrobiopterin in vascular disease. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Wang, S.; Wu, Y.; Song, P.; Zou, M.H. Tyrosine nitration of PA700 activates the 26S proteasome to induce endothelial dysfunction in mice with angiotensin II-induced hypertension. Hypertension 2009, 54, 625–632. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Wu, Y.; Song, P.; Zhang, M.; Wang, S.; Zou, M.H. Proteasome-dependent degradation of guanosine 5′-triphosphate cyclohydrolase I causes tetrahydrobiopterin deficiency in diabetes mellitus. Circulation 2007, 116, 944–953. [Google Scholar] [CrossRef] [PubMed]
- Whitsett, J.; Picklo, M.J., Sr.; Vasquez-Vivar, J. 4-hydroxy-2-nonenal increases superoxide anion radical in endothelial cells via stimulated GTP cyclohydrolase proteasomal degradation. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 2340–2347. [Google Scholar] [CrossRef] [PubMed]
- Munzel, T.; Daiber, A. Does endothelial tetrahydrobiopterin control the endothelial no synthase coupling state in arterial resistance arteries? Br. J. Pharmacol. 2017, 174, 2422–2424. [Google Scholar] [CrossRef] [PubMed]
- Chuaiphichai, S.; Crabtree, M.J.; McNeill, E.; Hale, A.B.; Trelfa, L.; Channon, K.M.; Douglas, G. A key role for tetrahydrobiopterin-dependent endothelial NOS regulation in resistance arteries: Studies in endothelial cell tetrahydrobiopterin-deficient mice. Br. J. Pharmacol. 2017, 174, 657–671. [Google Scholar] [CrossRef] [PubMed]
- Douglas, G.; Hale, A.B.; Patel, J.; Chuaiphichai, S.; Al Haj Zen, A.; Rashbrook, V.S.; Trelfa, L.; Crabtree, M.J.; McNeill, E.; Channon, K.M. Roles for endothelial cell and macrophage Gch1 and tetrahydrobiopterin in atherosclerosis progression. Cardiovasc. Res. 2018, 114, 1385–1399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munzel, T.; Daiber, A. Role of endothelial and macrophage tetrahydrobiopterin in development and progression of atherosclerosis: BH4 puzzle solved? Cardiovasc. Res. 2018, 114, 1310–1312. [Google Scholar] [CrossRef] [PubMed]
- Kossmann, S.; Hu, H.; Steven, S.; Schonfelder, T.; Fraccarollo, D.; Mikhed, Y.; Brahler, M.; Knorr, M.; Brandt, M.; Karbach, S.H.; et al. Inflammatory monocytes determine endothelial nitric-oxide synthase uncoupling and nitro-oxidative stress induced by angiotensin II. J. Biol. Chem. 2014, 289, 27540–27550. [Google Scholar] [CrossRef] [PubMed]
- Chalupsky, K.; Cai, H. Endothelial dihydrofolate reductase: Critical for nitric oxide bioavailability and role in angiotensin II uncoupling of endothelial nitric oxide synthase. Proc. Natl. Acad. Sci. USA 2005, 102, 9056–9061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ionova, I.A.; Vasquez-Vivar, J.; Whitsett, J.; Herrnreiter, A.; Medhora, M.; Cooley, B.C.; Pieper, G.M. Deficient BH4 production via de novo and salvage pathways regulates no responses to cytokines in adult cardiac myocytes. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H2178–H2187. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Lu, Q.; Ding, Y.; Wang, Q.; Xiao, L.; Song, P.; Zou, M.H. Endothelial nitric oxide synthase-derived nitric oxide prevents dihydrofolate reductase degradation via promoting S-nitrosylation. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 2366–2373. [Google Scholar] [CrossRef] [PubMed]
- Munzel, T.; Daiber, A. Redox regulation of dihydrofolate reductase: Friend or troublemaker? Arterioscler. Thromb. Vasc. Biol. 2015, 35, 2261–2262. [Google Scholar] [CrossRef] [PubMed]
- Zou, M.H.; Shi, C.; Cohen, R.A. Oxidation of the zinc-thiolate complex and uncoupling of endothelial nitric oxide synthase by peroxynitrite. J. Clin. Investig. 2002, 109, 817–826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hein, T.W.; Singh, U.; Vasquez-Vivar, J.; Devaraj, S.; Kuo, L.; Jialal, I. Human C-reactive protein induces endothelial dysfunction and uncoupling of eNOS in vivo. Atherosclerosis 2009, 206, 61–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.H.; Bugaj, L.J.; Oh, Y.J.; Bivalacqua, T.J.; Ryoo, S.; Soucy, K.G.; Santhanam, L.; Webb, A.; Camara, A.; Sikka, G.; et al. Arginase inhibition restores NOS coupling and reverses endothelial dysfunction and vascular stiffness in old rats. J. Appl. Physiol. 2009, 107, 1249–1257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemarie, C.A.; Shbat, L.; Marchesi, C.; Angulo, O.J.; Deschenes, M.E.; Blostein, M.D.; Paradis, P.; Schiffrin, E.L. Mthfr deficiency induces endothelial progenitor cell senescence via uncoupling of eNOS and downregulation of SIRT1. Am. J. Physiol. Heart Circ. Physiol. 2011, 300, H745–H753. [Google Scholar] [CrossRef] [PubMed]
- Tokutomi, Y.; Kataoka, K.; Yamamoto, E.; Nakamura, T.; Fukuda, M.; Nako, H.; Toyama, K.; Dong, Y.F.; Ahmed, K.A.; Sawa, T.; et al. Vascular responses to 8-nitro-cyclic GMP in non-diabetic and diabetic mice. Br. J. Pharmacol. 2011, 162, 1884–1893. [Google Scholar] [CrossRef] [PubMed]
- Lobysheva, I.; Rath, G.; Sekkali, B.; Bouzin, C.; Feron, O.; Gallez, B.; Dessy, C.; Balligand, J.L. Moderate caveolin-1 downregulation prevents NADPH oxidase-dependent endothelial nitric oxide synthase uncoupling by angiotensin II in endothelial cells. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 2098–2105. [Google Scholar] [CrossRef] [PubMed]
- Serizawa, K.; Yogo, K.; Aizawa, K.; Tashiro, Y.; Ishizuka, N. Nicorandil prevents endothelial dysfunction due to antioxidative effects via normalisation of NADPH oxidase and nitric oxide synthase in streptozotocin diabetic rats. Cardiovasc. Diabetol. 2011, 10, 105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faria, A.M.; Papadimitriou, A.; Silva, K.C.; Lopes de Faria, J.M.; Lopes de Faria, J.B. Uncoupling endothelial nitric oxide synthase is ameliorated by green tea in experimental diabetes by re-establishing tetrahydrobiopterin levels. Diabetes 2012, 61, 1838–1847. [Google Scholar] [CrossRef] [PubMed]
- Fike, C.D.; Dikalova, A.; Kaplowitz, M.R.; Cunningham, G.; Summar, M.; Aschner, J.L. Rescue treatment with L-citrulline inhibits hypoxia-induced pulmonary hypertension in newborn pigs. Am. J. Respir. Cell Mol. Biol. 2015, 53, 255–264. [Google Scholar] [CrossRef] [PubMed]
- Davel, A.P.; Brum, P.C.; Rossoni, L.V. Isoproterenol induces vascular oxidative stress and endothelial dysfunction via a Giα-coupled β2-adrenoceptor signaling pathway. PLoS ONE 2014, 9, e91877. [Google Scholar] [CrossRef] [PubMed]
- Solini, A.; Rossi, C.; Duranti, E.; Taddei, S.; Natali, A.; Virdis, A. Saxagliptin prevents vascular remodeling and oxidative stress in db/db mice. Role of endothelial nitric oxide synthase uncoupling and cyclooxygenase. Vasc. Pharmacol. 2016, 76, 62–71. [Google Scholar] [CrossRef] [PubMed]
- Musicki, B.; Burnett, A.L. Constitutive NOS uncoupling and NADPH oxidase upregulation in the penis of type 2 diabetic men with erectile dysfunction. Andrology 2017, 5, 294–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Octavia, Y.; Kararigas, G.; de Boer, M.; Chrifi, I.; Kietadisorn, R.; Swinnen, M.; Duimel, H.; Verheyen, F.K.; Brandt, M.M.; Fliegner, D.; et al. Folic acid reduces doxorubicin-induced cardiomyopathy by modulating endothelial nitric oxide synthase. J. Cell. Mol. Med. 2017, 21, 3277–3287. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; Zhang, X.; Wang, B.; Wu, Q.; Li, B.; Li, A.; Zhang, H.; Xiu, R. Functional status of microvascular vasomotion is impaired in spontaneously hypertensive rat. Sci. Rep. 2017, 7, 17080. [Google Scholar] [CrossRef]
- Couto, G.K.; Paula, S.M.; Gomes-Santos, I.L.; Negrao, C.E.; Rossoni, L.V. Exercise training induces eNOS coupling and restores relaxation in coronary arteries of heart failure rats. Am. J. Physiol. Heart Circ. Physiol. 2018, 314, H878–H887. [Google Scholar] [CrossRef]
- Santana, M.N.S.; Souza, D.S.; Miguel-Dos-Santos, R.; Rabelo, T.K.; Vasconcelos, C.M.L.; Navia-Pelaez, J.M.; Jesus, I.C.G.; Silva-Neto, J.A.D.; Lauton-Santos, S.; Capettini, L.; et al. Resistance exercise mediates remote ischemic preconditioning by limiting cardiac eNOS uncoupling. J. Mol. Cell. Cardiol. 2018, 125, 61–72. [Google Scholar] [CrossRef]
- Suvorava, T.; Nagy, N.; Pick, S.; Lieven, O.; Ruther, U.; Dao, V.T.; Fischer, J.W.; Weber, M.; Kojda, G. Impact of eNOS-dependent oxidative stress on endothelial function and neointima formation. Antioxid. Redox Signal. 2015, 23, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Suvorava, T.; Pick, S.; Kojda, G. Selective impairment of blood pressure reduction by endothelial nitric oxide synthase dimer destabilization in mice. J. Hypertens. 2017, 35, 76–88. [Google Scholar] [CrossRef] [PubMed]
- Benson, M.A.; Batchelor, H.; Chuaiphichai, S.; Bailey, J.; Zhu, H.; Stuehr, D.J.; Bhattacharya, S.; Channon, K.M.; Crabtree, M.J. A pivotal role for tryptophan 447 in enzymatic coupling of human endothelial nitric oxide synthase (eNOS): Effects on tetrahydrobiopterin-dependent catalysis and eNOS dimerization. J. Biol. Chem. 2013, 288, 29836–29845. [Google Scholar] [CrossRef] [PubMed]
- Daiber, A.; Frein, D.; Namgaladze, D.; Ullrich, V. Oxidation and nitrosation in the nitrogen monoxide/superoxide system. J. Biol. Chem. 2002, 277, 11882–11888. [Google Scholar] [CrossRef] [PubMed]
- Campos-Mota, G.P.; Navia-Pelaez, J.M.; Araujo-Souza, J.C.; Stergiopulos, N.; Capettini, L.S.A. Role of ERK1/2 activation and nNOS uncoupling on endothelial dysfunction induced by lysophosphatidylcholine. Atherosclerosis 2017, 258, 108–118. [Google Scholar] [CrossRef]
- Chen, C.A.; Wang, T.Y.; Varadharaj, S.; Reyes, L.A.; Hemann, C.; Talukder, M.A.; Chen, Y.R.; Druhan, L.J.; Zweier, J.L. S-glutathionylation uncouples eNOS and regulates its cellular and vascular function. Nature 2010, 468, 1115–1118. [Google Scholar] [CrossRef]
- Zweier, J.L.; Chen, C.A.; Druhan, L.J. S-glutathionylation reshapes our understanding of endothelial nitric oxide synthase uncoupling and nitric oxide/reactive oxygen species-mediated signaling. Antioxid. Redox Signal. 2011, 14, 1769–1775. [Google Scholar] [CrossRef]
- Oelze, M.; Knorr, M.; Kroller-Schon, S.; Kossmann, S.; Gottschlich, A.; Rummler, R.; Schuff, A.; Daub, S.; Doppler, C.; Kleinert, H.; et al. Chronic therapy with isosorbide-5-mononitrate causes endothelial dysfunction, oxidative stress, and a marked increase in vascular endothelin-1 expression. Eur. Heart J. 2013, 34, 3206–3216. [Google Scholar] [CrossRef]
- Kroller-Schon, S.; Steven, S.; Kossmann, S.; Scholz, A.; Daub, S.; Oelze, M.; Xia, N.; Hausding, M.; Mikhed, Y.; Zinssius, E.; et al. Molecular mechanisms of the crosstalk between mitochondria and NADPH oxidase through reactive oxygen species-studies in white blood cells and in animal models. Antioxid. Redox Signal. 2014, 20, 247–266. [Google Scholar] [CrossRef]
- Oelze, M.; Kroller-Schon, S.; Steven, S.; Lubos, E.; Doppler, C.; Hausding, M.; Tobias, S.; Brochhausen, C.; Li, H.; Torzewski, M.; et al. Glutathione peroxidase-1 deficiency potentiates dysregulatory modifications of endothelial nitric oxide synthase and vascular dysfunction in aging. Hypertension 2014, 63, 390–396. [Google Scholar] [CrossRef]
- Speer, T.; Owala, F.O.; Holy, E.W.; Zewinger, S.; Frenzel, F.L.; Stahli, B.E.; Razavi, M.; Triem, S.; Cvija, H.; Rohrer, L.; et al. Carbamylated low-density lipoprotein induces endothelial dysfunction. Eur. Heart J. 2014, 35, 3021–3032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, F.; Szczepaniak, W.S.; Shiva, S.; Liu, H.; Wang, Y.; Wang, L.; Wang, Y.; Kelley, E.E.; Chen, A.F.; Gladwin, M.T.; et al. Nox2-dependent glutathionylation of endothelial NOS leads to uncoupled superoxide production and endothelial barrier dysfunction in acute lung injury. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2014, 307, L987–L997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Deel, E.D.; Octavia, Y.; de Boer, M.; Juni, R.P.; Tempel, D.; van Haperen, R.; de Crom, R.; Moens, A.L.; Merkus, D.; Duncker, D.J. Normal and high eNOS levels are detrimental in both mild and severe cardiac pressure-overload. J. Mol. Cell. Cardiol. 2015, 88, 145–154. [Google Scholar] [CrossRef] [PubMed]
- Musicki, B.; Hannan, J.L.; Lagoda, G.; Bivalacqua, T.J.; Burnett, A.L. Mechanistic link between erectile dysfunction and systemic endothelial dysfunction in type 2 diabetic rats. Andrology 2016, 4, 977–983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kroller-Schon, S.; Daiber, A.; Steven, S.; Oelze, M.; Frenis, K.; Kalinovic, S.; Heimann, A.; Schmidt, F.P.; Pinto, A.; Kvandova, M.; et al. Crucial role for nox2 and sleep deprivation in aircraft noise-induced vascular and cerebral oxidative stress, inflammation, and gene regulation. Eur. Heart J. 2018, 39, 3528–3539. [Google Scholar] [CrossRef] [PubMed]
- Munzel, T.; Daiber, A.; Steven, S.; Tran, L.P.; Ullmann, E.; Kossmann, S.; Schmidt, F.P.; Oelze, M.; Xia, N.; Li, H.; et al. Effects of noise on vascular function, oxidative stress, and inflammation: Mechanistic insight from studies in mice. Eur. Heart J. 2017, 38, 2838–2849. [Google Scholar] [CrossRef] [PubMed]
- Van Deel, E.D.; Octavia, Y.; de Waard, M.C.; de Boer, M.; Duncker, D.J. Exercise training has contrasting effects in myocardial infarction and pressure overload due to divergent endothelial nitric oxide synthase regulation. Int. J. Mol. Sci. 2018, 19, 1968. [Google Scholar] [CrossRef]
- Du, Y.; Navab, M.; Shen, M.; Hill, J.; Pakbin, P.; Sioutas, C.; Hsiai, T.K.; Li, R. Ambient ultrafine particles reduce endothelial nitric oxide production via S-glutathionylation of eNOS. Biochem. Biophys. Res. Commun. 2013, 436, 462–466. [Google Scholar] [CrossRef] [Green Version]
- De Pascali, F.; Hemann, C.; Samons, K.; Chen, C.A.; Zweier, J.L. Hypoxia and reoxygenation induce endothelial nitric oxide synthase uncoupling in endothelial cells through tetrahydrobiopterin depletion and S-glutathionylation. Biochemistry 2014, 53, 3679–3688. [Google Scholar] [CrossRef]
- Chen, C.A.; De Pascali, F.; Basye, A.; Hemann, C.; Zweier, J.L. Redox modulation of endothelial nitric oxide synthase by glutaredoxin-1 through reversible oxidative post-translational modification. Biochemistry 2013, 52, 6712–6723. [Google Scholar] [CrossRef]
- Shang, Q.; Bao, L.; Guo, H.; Hao, F.; Luo, Q.; Chen, J.; Guo, C. Contribution of glutaredoxin-1 to S-glutathionylation of endothelial nitric oxide synthase for mesenteric nitric oxide generation in experimental necrotizing enterocolitis. Transl. Res. 2017, 188, 92–105. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Li, X.; Shang, Q.; Gao, Z.; Hao, F.; Guo, H.; Guo, C. Fecal microbiota transplantation (FMT) could reverse the severity of experimental necrotizing enterocolitis (NEC) via oxidative stress modulation. Free Radic. Biol. Med. 2017, 108, 32–43. [Google Scholar] [CrossRef] [PubMed]
- Espinosa-Diez, C.; Miguel, V.; Vallejo, S.; Sanchez, F.J.; Sandoval, E.; Blanco, E.; Cannata, P.; Peiro, C.; Sanchez-Ferrer, C.F.; Lamas, S. Role of glutathione biosynthesis in endothelial dysfunction and fibrosis. Redox Biol. 2018, 14, 88–99. [Google Scholar] [CrossRef] [PubMed]
- Crabtree, M.J.; Brixey, R.; Batchelor, H.; Hale, A.B.; Channon, K.M. Integrated redox sensor and effector functions for tetrahydrobiopterin- and glutathionylation-dependent endothelial nitric-oxide synthase uncoupling. J. Biol. Chem. 2013, 288, 561–569. [Google Scholar] [CrossRef] [PubMed]
- Idigo, W.O.; Reilly, S.; Zhang, M.H.; Zhang, Y.H.; Jayaram, R.; Carnicer, R.; Crabtree, M.J.; Balligand, J.L.; Casadei, B. Regulation of endothelial nitric-oxide synthase (NOS) S-glutathionylation by neuronal NOS: Evidence of a functional interaction between myocardial constitutive NOS isoforms. J. Biol. Chem. 2012, 287, 43665–43673. [Google Scholar] [CrossRef] [PubMed]
- Dimmeler, S.; Fleming, I.; Fisslthaler, B.; Hermann, C.; Busse, R.; Zeiher, A.M. Activation of nitric oxide synthase in endothelial cells by AKT-dependent phosphorylation. Nature 1999, 399, 601–605. [Google Scholar] [CrossRef] [PubMed]
- Loot, A.E.; Schreiber, J.G.; Fisslthaler, B.; Fleming, I. Angiotensin ii impairs endothelial function via tyrosine phosphorylation of the endothelial nitric oxide synthase. J. Exp. Med. 2009, 206, 2889–2896. [Google Scholar] [CrossRef]
- Fleming, I.; Fisslthaler, B.; Dimmeler, S.; Kemp, B.E.; Busse, R. Phosphorylation of Thr(495) regulates Ca2+/calmodulin-dependent endothelial nitric oxide synthase activity. Circ. Res. 2001, 88, e68–e75. [Google Scholar] [CrossRef]
- Lin, M.I.; Fulton, D.; Babbitt, R.; Fleming, I.; Busse, R.; Pritchard, K.A., Jr.; Sessa, W.C. Phosphorylation of threonine 497 in endothelial nitric-oxide synthase coordinates the coupling of L-arginine metabolism to efficient nitric oxide production. J. Biol. Chem. 2003, 278, 44719–44726. [Google Scholar] [CrossRef]
- Matsubara, M.; Hayashi, N.; Jing, T.; Titani, K. Regulation of endothelial nitric oxide synthase by protein kinase C. J. Biochem. 2003, 133, 773–781. [Google Scholar] [CrossRef]
- Rathore, R.; Zheng, Y.M.; Niu, C.F.; Liu, Q.H.; Korde, A.; Ho, Y.S.; Wang, Y.X. Hypoxia activates NADPH oxidase to increase [ROS]i and [Ca2+]i through the mitochondrial ROS-PKCε signaling axis in pulmonary artery smooth muscle cells. Free Radic. Biol. Med. 2008, 45, 1223–1231. [Google Scholar] [CrossRef] [PubMed]
- Lin, D.; Takemoto, D.J. Oxidative activation of protein kinase Cγ through the C1 domain. Effects on gap junctions. J. Biol. Chem. 2005, 280, 13682–13693. [Google Scholar] [CrossRef] [PubMed]
- Signorello, M.G.; Segantin, A.; Passalacqua, M.; Leoncini, G. Homocysteine decreases platelet NO level via protein kinase C activation. Nitric Oxide 2009, 20, 104–113. [Google Scholar] [CrossRef] [PubMed]
- Singh, U.; Devaraj, S.; Vasquez-Vivar, J.; Jialal, I. C-reactive protein decreases endothelial nitric oxide synthase activity via uncoupling. J. Mol. Cell. Cardiol. 2007, 43, 780–791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lenasi, H.; Kohlstedt, K.; Fichtlscherer, B.; Mulsch, A.; Busse, R.; Fleming, I. Amlodipine activates the endothelial nitric oxide synthase by altering phosphorylation on Ser1177 and Thr495. Cardiovasc. Res. 2003, 59, 844–853. [Google Scholar] [CrossRef]
- Barauna, V.G.; Mantuan, P.R.; Magalhaes, F.C.; Campos, L.C.; Krieger, J.E. AT1 receptor blocker potentiates shear-stress induced nitric oxide production via modulation of eNOS phosphorylation of residues Thr495 and Ser1177. Biochem. Biophys. Res. Commun. 2013, 441, 713–719. [Google Scholar] [CrossRef] [PubMed]
- Knorr, M.; Hausding, M.; Kroller-Schuhmacher, S.; Steven, S.; Oelze, M.; Heeren, T.; Scholz, A.; Gori, T.; Wenzel, P.; Schulz, E.; et al. Nitroglycerin-induced endothelial dysfunction and tolerance involve adverse phosphorylation and S-glutathionylation of endothelial nitric oxide synthase: Beneficial effects of therapy with the AT1 receptor blocker telmisartan. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 2223–2231. [Google Scholar] [CrossRef]
- Almansob, M.A.; Xu, B.; Zhou, L.; Hu, X.X.; Chen, W.; Chang, F.J.; Ci, H.B.; Yao, J.P.; Xu, Y.Q.; Yao, F.J.; et al. Simvastatin reduces myocardial injury undergoing noncoronary artery cardiac surgery: A randomized controlled trial. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2304–2313. [Google Scholar] [CrossRef]
- Yang, L.; Jia, Z.; Yang, L.; Zhu, M.; Zhang, J.; Liu, J.; Wu, P.; Tian, W.; Li, J.; Qi, Z.; et al. Exercise protects against chronic beta-adrenergic remodeling of the heart by activation of endothelial nitric oxide synthase. PLoS ONE 2014, 9, e96892. [Google Scholar]
- Calvert, J.W.; Condit, M.E.; Aragon, J.P.; Nicholson, C.K.; Moody, B.F.; Hood, R.L.; Sindler, A.L.; Gundewar, S.; Seals, D.R.; Barouch, L.A.; et al. Exercise protects against myocardial ischemia-reperfusion injury via stimulation of β3-adrenergic receptors and increased nitric oxide signaling: Role of nitrite and nitrosothiols. Circ. Res. 2011, 108, 1448–1458. [Google Scholar] [CrossRef]
- Guterbaum, T.J.; Braunstein, T.H.; Fossum, A.; Holstein-Rathlou, N.H.; Torp-Pedersen, C.T.; Dominguez, H. Endothelial nitric oxide synthase phosphorylation at threonine 495 and mitochondrial reactive oxygen species formation in response to a high H2O2 concentration. J. Vasc. Res. 2013, 50, 410–420. [Google Scholar] [CrossRef] [PubMed]
- Carlstrom, M.; Liu, M.; Yang, T.; Zollbrecht, C.; Huang, L.; Peleli, M.; Borniquel, S.; Kishikawa, H.; Hezel, M.; Persson, A.E.; et al. Cross-talk between nitrate-nitrite-NO and NO synthase pathways in control of vascular NO homeostasis. Antioxid. Redox Signal. 2015, 23, 295–306. [Google Scholar] [CrossRef]
- Boger, R.H. Association of asymmetric dimethylarginine and endothelial dysfunction. Clin. Chem. Lab. Med. 2003, 41, 1467–1472. [Google Scholar] [CrossRef] [PubMed]
- Sydow, K.; Munzel, T. ADMA and oxidative stress. Atheroscler. Suppl. 2003, 4, 41–51. [Google Scholar] [CrossRef]
- Schnabel, R.; Blankenberg, S.; Lubos, E.; Lackner, K.J.; Rupprecht, H.J.; Espinola-Klein, C.; Jachmann, N.; Post, F.; Peetz, D.; Bickel, C.; et al. Asymmetric dimethylarginine and the risk of cardiovascular events and death in patients with coronary artery disease: Results from The AtheroGene Study. Circ. Res. 2005, 97, e53–e59. [Google Scholar] [CrossRef]
- Boger, R.H. When the endothelium cannot say ‘NO’ anymore. ADMA, an endogenous inhibitor of NO synthase, promotes cardiovascular disease. Eur. Heart J. 2003, 24, 1901–1902. [Google Scholar] [CrossRef] [PubMed]
- Tousoulis, D.; Papageorgiou, N.; Androulakis, E.; Siasos, G.; Latsios, G.; Tentolouris, K.; Stefanadis, C. Diabetes mellitus-associated vascular impairment: Novel circulating biomarkers and therapeutic approaches. J. Am. Coll. Cardiol. 2013, 62, 667–676. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Ma, P.; Xiong, A.; Xu, Y.; Wang, Y.; Xu, Q. Protective effects of low-dose rosuvastatin on isoproterenol-induced chronic heart failure in rats by regulation of DDAH-ADMA-NO pathway. Cardiovasc. Ther. 2017, 35, e12241. [Google Scholar] [CrossRef]
- Hsu, C.P.; Zhao, J.F.; Lin, S.J.; Shyue, S.K.; Guo, B.C.; Lu, T.M.; Lee, T.S. Asymmetric dimethylarginine limits the efficacy of simvastatin activating endothelial nitric oxide synthase. J. Am. Heart Assoc. 2016, 5, e003327. [Google Scholar] [CrossRef]
- Lin, Y.; Feng, M.; Lu, C.W.; Lei, Y.P.; He, Z.M.; Xiong, Y. Preservation of vascular DDAH activity contributes to the protection of captopril against endothelial dysfunction in hyperlipidemic rabbits. Eur. J. Pharmacol. 2017, 798, 43–48. [Google Scholar] [CrossRef]
- Bode-Boger, S.M.; Scalera, F.; Ignarro, L.J. The L-arginine paradox: Importance of the L-arginine/asymmetrical dimethylarginine ratio. Pharmacol. Ther. 2007, 114, 295–306. [Google Scholar] [CrossRef]
- Lass, A.; Suessenbacher, A.; Wolkart, G.; Mayer, B.; Brunner, F. Functional and analytical evidence for scavenging of oxygen radicals by L-arginine. Mol. Pharmacol. 2002, 61, 1081–1088. [Google Scholar] [CrossRef]
- Closs, E.I.; Ostad, M.A.; Simon, A.; Warnholtz, A.; Jabs, A.; Habermeier, A.; Daiber, A.; Forstermann, U.; Munzel, T. Impairment of the extrusion transporter for asymmetric dimethyl-L-arginine: A novel mechanism underlying vasospastic angina. Biochem. Biophys. Res. Commun. 2012, 423, 218–223. [Google Scholar] [CrossRef] [PubMed]
- Watson, C.P.; Pazarentzos, E.; Fidanboylu, M.; Padilla, B.; Brown, R.; Thomas, S.A. The transporter and permeability interactions of asymmetric dimethylarginine (ADMA) and L-arginine with the human blood-brain barrier in vitro. Brain Res. 2016, 1648, 232–242. [Google Scholar] [CrossRef]
- Speranza, L.; Franceschelli, S.; D’Orazio, N.; Gaeta, R.; Bucciarelli, T.; Felaco, M.; Grilli, A.; Riccioni, G. The biological effect of pharmacological treatment on dimethylaminohydrolases (DDAH-1) and cationic amino acid transporter-1 (CAT-1) expression in patients with acute congestive heart failure. Microvasc. Res. 2011, 82, 391–396. [Google Scholar] [CrossRef]
- Yang, Z.; Ming, X.F. Arginase: The emerging therapeutic target for vascular oxidative stress and inflammation. Front. Immunol. 2013, 4, 149. [Google Scholar] [CrossRef] [PubMed]
- Pitt, B.; Stier, C.T., Jr.; Rajagopalan, S. Mineralocorticoid receptor blockade: New insights into the mechanism of action in patients with cardiovascular disease. J. Renin Angiotensin Aldosterone Syst. 2003, 4, 164–168. [Google Scholar] [CrossRef] [PubMed]
- Mehler, P.S.; Coll, J.R.; Estacio, R.; Esler, A.; Schrier, R.W.; Hiatt, W.R. Intensive blood pressure control reduces the risk of cardiovascular events in patients with peripheral arterial disease and type 2 diabetes. Circulation 2003, 107, 753–756. [Google Scholar] [CrossRef] [PubMed]
- Kintscher, U.; Bramlage, P.; Paar, W.D.; Thoenes, M.; Unger, T. Irbesartan for the treatment of hypertension in patients with the metabolic syndrome: A sub analysis of the Treat to Target post authorization survey. Prospective observational, two armed study in 14,200 patients. Cardiovasc. Diabetol. 2007, 6, 12. [Google Scholar] [CrossRef] [Green Version]
- Parving, H.H.; Brenner, B.M.; McMurray, J.J.; de Zeeuw, D.; Haffner, S.M.; Solomon, S.D.; Chaturvedi, N.; Ghadanfar, M.; Weissbach, N.; Xiang, Z.; et al. Aliskiren trial in type 2 diabetes using cardio-renal endpoints (ALTITUDE): Rationale and study design. Nephrol. Dial. Transplant. 2009, 24, 1663–1671. [Google Scholar] [CrossRef]
- Koh, K.K.; Lim, S.; Choi, H.; Lee, Y.; Han, S.H.; Lee, K.; Oh, P.C.; Sakuma, I.; Shin, E.K.; Quon, M.J. Combination pravastatin and valsartan treatment has additive beneficial effects to simultaneously improve both metabolic and cardiovascular phenotypes beyond that of monotherapy with either drug in patients with primary hypercholesterolemia. Diabetes 2013, 62, 3547–3552. [Google Scholar] [CrossRef] [PubMed]
- Patel, T.N.; Shishehbor, M.H.; Bhatt, D.L. A review of high-dose statin therapy: Targeting cholesterol and inflammation in atherosclerosis. Eur. Heart J. 2007, 28, 664–672. [Google Scholar] [CrossRef] [PubMed]
- Goodfriend, T.L.; Elliott, M.E.; Catt, K.J. Angiotensin receptors and their antagonists. N. Engl. J. Med. 1996, 334, 1649–1654. [Google Scholar] [CrossRef] [PubMed]
- Steven, S.; Munzel, T.; Daiber, A. Exploiting the pleiotropic antioxidant effects of established drugs in cardiovascular disease. Int. J. Mol. Sci. 2015, 16, 18185–18223. [Google Scholar] [CrossRef] [PubMed]
- Hibbert, B.; Simard, T.; Ramirez, F.D.; Pourdjabbar, A.; Raizman, J.E.; Maze, R.; Wilson, K.R.; Hawken, S.; O’Brien, E.R. The effect of statins on circulating endothelial progenitor cells in humans: A systematic review. J. Cardiovasc. Pharmacol. 2013, 62, 491–496. [Google Scholar] [CrossRef] [PubMed]
- Broeders, M.A.; Doevendans, P.A.; Bekkers, B.C.; Bronsaer, R.; van Gorsel, E.; Heemskerk, J.W.; Egbrink, M.G.; van Breda, E.; Reneman, R.S.; van Der Zee, R. Nebivolol: A third-generation beta-blocker that augments vascular nitric oxide release: Endothelial β2-adrenergic receptor-mediated nitric oxide production. Circulation 2000, 102, 677–684. [Google Scholar] [CrossRef] [PubMed]
- Tzemos, N.; Lim, P.O.; MacDonald, T.M. Nebivolol reverses endothelial dysfunction in essential hypertension: A randomized, double-blind, crossover study. Circulation 2001, 104, 511–514. [Google Scholar] [CrossRef]
- Oelze, M.; Daiber, A.; Brandes, R.P.; Hortmann, M.; Wenzel, P.; Hink, U.; Schulz, E.; Mollnau, H.; von Sandersleben, A.; Kleschyov, A.L.; et al. Nebivolol inhibits superoxide formation by NADPH oxidase and endothelial dysfunction in angiotensin II-treated rats. Hypertension 2006, 48, 677–684. [Google Scholar] [CrossRef]
- Sorrentino, S.A.; Doerries, C.; Manes, C.; Speer, T.; Dessy, C.; Lobysheva, I.; Mohmand, W.; Akbar, R.; Bahlmann, F.; Besler, C.; et al. Nebivolol exerts beneficial effects on endothelial function, early endothelial progenitor cells, myocardial neovascularization, and left ventricular dysfunction early after myocardial infarction beyond conventional beta1-blockade. J. Am. Coll. Cardiol. 2011, 57, 601–611. [Google Scholar] [CrossRef]
- Magee, L.A.; Abalos, E.; von Dadelszen, P.; Sibai, B.; Easterling, T.; Walkinshaw, S.; Group, C.S. How to manage hypertension in pregnancy effectively. Br. J. Clin. Pharmacol. 2011, 72, 394–401. [Google Scholar] [CrossRef] [Green Version]
- Taylor, A.L.; Lindenfeld, J.; Ziesche, S.; Walsh, M.N.; Mitchell, J.E.; Adams, K.; Tam, S.W.; Ofili, E.; Sabolinski, M.L.; Worcel, M.; et al. Outcomes by gender in the African-American heart failure trial. J. Am. Coll. Cardiol. 2006, 48, 2263–2267. [Google Scholar] [CrossRef] [PubMed]
- Taylor, A.L.; Ziesche, S.; Yancy, C.; Carson, P.; D’Agostino, R., Jr.; Ferdinand, K.; Taylor, M.; Adams, K.; Sabolinski, M.; Worcel, M.; et al. Combination of isosorbide dinitrate and hydralazine in blacks with heart failure. N. Engl. J. Med. 2004, 351, 2049–2057. [Google Scholar] [CrossRef] [PubMed]
- Munzel, T.; Meinertz, T.; Tebbe, U.; Schneider, H.T.; Stalleicken, D.; Wargenau, M.; Gori, T.; Klingmann, I.; Investigators, C.S. Efficacy of the long-acting nitro vasodilator pentaerithrityl tetranitrate in patients with chronic stable angina pectoris receiving anti-anginal background therapy with beta-blockers: A 12-week, randomized, double-blind, placebo-controlled trial. Eur. Heart J. 2014, 35, 895–903. [Google Scholar] [CrossRef] [PubMed]
- Oberle, S.; Abate, A.; Grosser, N.; Vreman, H.J.; Dennery, P.A.; Schneider, H.T.; Stalleicken, D.; Schroder, H. Heme oxygenase-1 induction may explain the antioxidant profile of pentaerythrityl trinitrate. Biochem. Biophys. Res. Commun. 2002, 290, 1539–1544. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, P.; Oelze, M.; Coldewey, M.; Hortmann, M.; Seeling, A.; Hink, U.; Mollnau, H.; Stalleicken, D.; Weiner, H.; Lehmann, J.; et al. Heme oxygenase-1: A novel key player in the development of tolerance in response to organic nitrates. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 1729–1735. [Google Scholar] [CrossRef]
- Schuhmacher, S.; Oelze, M.; Bollmann, F.; Kleinert, H.; Otto, C.; Heeren, T.; Steven, S.; Hausding, M.; Knorr, M.; Pautz, A.; et al. Vascular dysfunction in experimental diabetes is improved by pentaerithrityl tetranitrate but not isosorbide-5-mononitrate therapy. Diabetes 2011, 60, 2608–2616. [Google Scholar] [CrossRef] [PubMed]
- Boswell-Smith, V.; Spina, D.; Page, C.P. Phosphodiesterase inhibitors. Br. J. Pharmacol. 2006, 147 (Suppl. 1), S252–S257. [Google Scholar] [CrossRef]
- Evgenov, O.V.; Pacher, P.; Schmidt, P.M.; Hasko, G.; Schmidt, H.H.; Stasch, J.P. NO-independent stimulators and activators of soluble guanylate cyclase: Discovery and therapeutic potential. Nat. Rev. Drug Discov. 2006, 5, 755–768. [Google Scholar] [CrossRef]
- Schuhmacher, S.; Wenzel, P.; Schulz, E.; Oelze, M.; Mang, C.; Kamuf, J.; Gori, T.; Jansen, T.; Knorr, M.; Karbach, S.; et al. Pentaerythritol tetranitrate improves angiotensin II-induced vascular dysfunction via induction of heme oxygenase-1. Hypertension 2010, 55, 897–904. [Google Scholar] [CrossRef]
- Wenzel, P.; Schulz, E.; Oelze, M.; Muller, J.; Schuhmacher, S.; Alhamdani, M.S.; Debrezion, J.; Hortmann, M.; Reifenberg, K.; Fleming, I.; et al. AT1-receptor blockade by telmisartan upregulates GTP-cyclohydrolase I and protects eNOS in diabetic rats. Free Radic. Biol. Med. 2008, 45, 619–626. [Google Scholar] [CrossRef]
- Wenzel, P.; Daiber, A.; Oelze, M.; Brandt, M.; Closs, E.; Xu, J.; Thum, T.; Bauersachs, J.; Ertl, G.; Zou, M.H.; et al. Mechanisms underlying recoupling of eNOS by HMG-CoA reductase inhibition in a rat model of streptozotocin-induced diabetes mellitus. Atherosclerosis 2008, 198, 65–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heitzer, T.; Brockhoff, C.; Mayer, B.; Warnholtz, A.; Mollnau, H.; Henne, S.; Meinertz, T.; Munzel, T. Tetrahydrobiopterin improves endothelium-dependent vasodilation in chronic smokers: Evidence for a dysfunctional nitric oxide synthase. Circ. Res. 2000, 86, E36–E41. [Google Scholar] [CrossRef] [PubMed]
- Heitzer, T.; Krohn, K.; Albers, S.; Meinertz, T. Tetrahydrobiopterin improves endothelium-dependent vasodilation by increasing nitric oxide activity in patients with type ii diabetes mellitus. Diabetologia 2000, 43, 1435–1438. [Google Scholar] [CrossRef] [PubMed]
- Antoniades, C.; Shirodaria, C.; Warrick, N.; Cai, S.; de Bono, J.; Lee, J.; Leeson, P.; Neubauer, S.; Ratnatunga, C.; Pillai, R.; et al. 5-methyltetrahydrofolate rapidly improves endothelial function and decreases superoxide production in human vessels: Effects on vascular tetrahydrobiopterin availability and endothelial nitric oxide synthase coupling. Circulation 2006, 114, 1193–1201. [Google Scholar] [CrossRef] [PubMed]
- Gori, T.; Burstein, J.M.; Ahmed, S.; Miner, S.E.; Al-Hesayen, A.; Kelly, S.; Parker, J.D. Folic acid prevents nitroglycerin-induced nitric oxide synthase dysfunction and nitrate tolerance: A human in vivo study. Circulation 2001, 104, 1119–1123. [Google Scholar] [CrossRef] [PubMed]
- De Maria, R.; Campolo, J.; Frontali, M.; Taroni, F.; Federico, A.; Inzitari, D.; Tavani, A.; Romano, S.; Puca, E.; Orzi, F.; et al. Effects of sapropterin on endothelium-dependent vasodilation in patients with CADASIL: A randomized controlled trial. Stroke 2014, 45, 2959–2966. [Google Scholar] [CrossRef] [PubMed]
- Reverter, E.; Mesonero, F.; Seijo, S.; Martinez, J.; Abraldes, J.G.; Penas, B.; Berzigotti, A.; Deulofeu, R.; Bosch, J.; Albillos, A.; et al. Effects of sapropterin on portal and systemic hemodynamics in patients with cirrhosis and portal hypertension: A bicentric double-blind placebo-controlled study. Am. J. Gastroenterol. 2015, 110, 985–992. [Google Scholar] [CrossRef] [PubMed]
- Jeong, J.H.; Lee, N.; Tucker, M.A.; Rodriguez-Miguelez, P.; Looney, J.; Thomas, J.; Derella, C.C.; El-Marakby, A.A.; Musall, J.B.; Sullivan, J.C.; et al. Tetrahydrobiopterin improves endothelial function in patients with cystic fibrosis. J. Appl. Physiol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Machin, D.R.; Clifton, H.L.; Richardson, R.S.; Wray, D.W.; Donato, A.J.; Frech, T.M. Acute oral tetrahydrobiopterin administration ameliorates endothelial dysfunction in systemic sclerosis. Clin. Exp. Rheumatol. 2017, 35 (Suppl. 106), 167–172. [Google Scholar]
- Maki-Petaja, K.M.; Day, L.; Cheriyan, J.; Hall, F.C.; Ostor, A.J.; Shenker, N.; Wilkinson, I.B. Tetrahydrobiopterin supplementation improves endothelial function but does not alter aortic stiffness in patients with rheumatoid arthritis. J. Am. Heart Assoc. 2016, 5, e002762. [Google Scholar] [CrossRef] [PubMed]
- McCarty, M.F. Supplementation with phycocyanobilin, citrulline, taurine, and supranutritional doses of folic acid and biotin-potential for preventing or slowing the progression of diabetic complications. Healthcare 2017, 5, 15. [Google Scholar] [CrossRef] [PubMed]
- Fraccarollo, D.; Widder, J.D.; Galuppo, P.; Thum, T.; Tsikas, D.; Hoffmann, M.; Ruetten, H.; Ertl, G.; Bauersachs, J. Improvement in left ventricular remodeling by the endothelial nitric oxide synthase enhancer AVE9488 after experimental myocardial infarction. Circulation 2008, 118, 818–827. [Google Scholar] [CrossRef] [PubMed]
- Westermann, D.; Riad, A.; Richter, U.; Jager, S.; Savvatis, K.; Schuchardt, M.; Bergmann, N.; Tolle, M.; Nagorsen, D.; Gotthardt, M.; et al. Enhancement of the endothelial no synthase attenuates experimental diastolic heart failure. Basic Res. Cardiol. 2009, 104, 499–509. [Google Scholar] [CrossRef] [PubMed]
- Crabtree, M.J.; Tatham, A.L.; Al-Wakeel, Y.; Warrick, N.; Hale, A.B.; Cai, S.; Channon, K.M.; Alp, N.J. Quantitative regulation of intracellular endothelial nitric-oxide synthase (eNOS) coupling by both tetrahydrobiopterin-eNOS stoichiometry and biopterin redox status: Insights from cells with TET-regulated GTP cyclohydrolase I expression. J. Biol. Chem. 2009, 284, 1136–1144. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Witte, K.; August, M.; Brausch, I.; Godtel-Armbrust, U.; Habermeier, A.; Closs, E.I.; Oelze, M.; Munzel, T.; Forstermann, U. Reversal of endothelial nitric oxide synthase uncoupling and up-regulation of endothelial nitric oxide synthase expression lowers blood pressure in hypertensive rats. J. Am. Coll. Cardiol. 2006, 47, 2536–2544. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, K.; Heeschen, C.; Aicher, A.; Ziebart, T.; Honold, J.; Urbich, C.; Rossig, L.; Koehl, U.; Koyanagi, M.; Mohamed, A.; et al. Ex vivo pretreatment of bone marrow mononuclear cells with endothelial NO synthase enhancer AVE9488 enhances their functional activity for cell therapy. Proc. Natl. Acad. Sci. USA 2006, 103, 14537–14541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frantz, S.; Adamek, A.; Fraccarollo, D.; Tillmanns, J.; Widder, J.D.; Dienesch, C.; Schafer, A.; Podolskaya, A.; Held, M.; Ruetten, H.; et al. The eNOS enhancer AVE 9488: A novel cardioprotectant against ischemia reperfusion injury. Basic Res. Cardiol. 2009, 104, 773–779. [Google Scholar] [CrossRef]
- Riad, A.; Westermann, D.; Van Linthout, S.; Mohr, Z.; Uyulmaz, S.; Becher, P.M.; Rutten, H.; Wohlfart, P.; Peters, H.; Schultheiss, H.P.; et al. Enhancement of endothelial nitric oxide synthase production reverses vascular dysfunction and inflammation in the hindlimbs of a rat model of diabetes. Diabetologia 2008, 51, 2325–2332. [Google Scholar] [CrossRef]
- Yang, Q.; Xue, H.M.; Wong, W.T.; Tian, X.Y.; Huang, Y.; Tsui, S.K.; Ng, P.K.; Wohlfart, P.; Li, H.; Xia, N.; et al. AVE3085, an enhancer of endothelial nitric oxide synthase, restores endothelial function and reduces blood pressure in spontaneously hypertensive rats. Br. J. Pharmacol. 2011, 163, 1078–1085. [Google Scholar] [CrossRef] [Green Version]
- Cheang, W.S.; Wong, W.T.; Tian, X.Y.; Yang, Q.; Lee, H.K.; He, G.W.; Yao, X.; Huang, Y. Endothelial nitric oxide synthase enhancer reduces oxidative stress and restores endothelial function in db/db mice. Cardiovasc. Res. 2011, 92, 267–275. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Chen, C.; Feng, C.; Tang, A.; Ma, Y.; He, X.; Li, Y.; He, J.; Dong, Y. AVE 3085, a novel endothelial nitric oxide synthase enhancer, attenuates cardiac remodeling in mice through the Smad signaling pathway. Arch. Biochem. Biophys. 2015, 570, 8–13. [Google Scholar] [CrossRef] [PubMed]
- Hou, H.T.; Wang, J.; Zhang, X.; Wang, Z.Q.; Chen, T.N.; Zhang, J.L.; Yang, Q.; He, G.W. Endothelial nitric oxide synthase enhancer AVE3085 reverses endothelial dysfunction induced by homocysteine in human internal mammary arteries. Nitric Oxide 2018, 81, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Schafer, A.; Fraccarollo, D.; Widder, J.; Eigenthaler, M.; Ertl, G.; Bauersachs, J. Inhibition of platelet activation in rats with severe congestive heart failure by a novel endothelial nitric oxide synthase transcription enhancer. Eur. J. Heart Fail. 2009, 11, 336–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallerath, T.; Li, H.; Godtel-Ambrust, U.; Schwarz, P.M.; Forstermann, U. A blend of polyphenolic compounds explains the stimulatory effect of red wine on human endothelial no synthase. Nitric Oxide 2005, 12, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Xia, N.; Forstermann, U.; Li, H. Resveratrol and endothelial nitric oxide. Molecules 2014, 19, 16102–16121. [Google Scholar] [CrossRef] [PubMed]
- Matouk, C.C.; Marsden, P.A. Epigenetic regulation of vascular endothelial gene expression. Circ. Res. 2008, 102, 873–887. [Google Scholar] [CrossRef]
- Chan, Y.; Fish, J.E.; D’Abreo, C.; Lin, S.; Robb, G.B.; Teichert, A.M.; Karantzoulis-Fegaras, F.; Keightley, A.; Steer, B.M.; Marsden, P.A. The cell-specific expression of endothelial nitric-oxide synthase: A role for DNA methylation. J. Biol. Chem. 2004, 279, 35087–35100. [Google Scholar] [CrossRef]
- Fish, J.E.; Matouk, C.C.; Rachlis, A.; Lin, S.; Tai, S.C.; D’Abreo, C.; Marsden, P.A. The expression of endothelial nitric-oxide synthase is controlled by a cell-specific histone code. J. Biol. Chem. 2005, 280, 24824–24838. [Google Scholar] [CrossRef]
- Gan, Y.; Shen, Y.H.; Wang, J.; Wang, X.; Utama, B.; Wang, J.; Wang, X.L. Role of histone deacetylation in cell-specific expression of endothelial nitric-oxide synthase. J. Biol. Chem. 2005, 280, 16467–16475. [Google Scholar] [CrossRef]
- Gluckman, P.D.; Hanson, M.A.; Buklijas, T.; Low, F.M.; Beedle, A.S. Epigenetic mechanisms that underpin metabolic and cardiovascular diseases. Nat. Rev. Endocrinol. 2009, 5, 401–408. [Google Scholar] [CrossRef]
- Krause, B.J.; Costello, P.M.; Munoz-Urrutia, E.; Lillycrop, K.A.; Hanson, M.A.; Casanello, P. Role of DNA methyltransferase 1 on the altered eNOS expression in human umbilical endothelium from intrauterine growth restricted fetuses. Epigenetics 2013, 8, 944–952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herrera, E.A.; Cifuentes-Zuniga, F.; Figueroa, E.; Villanueva, C.; Hernandez, C.; Alegria, R.; Arroyo-Jousse, V.; Penaloza, E.; Farias, M.; Uauy, R.; et al. N-acetylcysteine, a glutathione precursor, reverts vascular dysfunction and endothelial epigenetic programming in intrauterine growth restricted guinea pigs. J. Physiol. 2017, 595, 1077–1092. [Google Scholar] [CrossRef] [PubMed]
- Grandvuillemin, I.; Buffat, C.; Boubred, F.; Lamy, E.; Fromonot, J.; Charpiot, P.; Simoncini, S.; Sabatier, F.; Dignat-George, F.; Peyter, A.C.; et al. Arginase upregulation and eNOS uncoupling contribute to impaired endothelium-dependent vasodilation in a rat model of intrauterine growth restriction. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2018, 315, R509–R520. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Forstermann, U. Uncoupling of endothelial NO synthase in atherosclerosis and vascular disease. Curr. Opin. Pharmacol. 2013, 13, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Siuda, D.; Xia, N.; Reifenberg, G.; Daiber, A.; Munzel, T.; Forstermann, U.; Li, H. Maternal treatment of spontaneously hypertensive rats with pentaerythritol tetranitrate reduces blood pressure in female offspring. Hypertension 2015, 65, 232–237. [Google Scholar] [CrossRef] [PubMed]








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Daiber, A.; Xia, N.; Steven, S.; Oelze, M.; Hanf, A.; Kröller-Schön, S.; Münzel, T.; Li, H. New Therapeutic Implications of Endothelial Nitric Oxide Synthase (eNOS) Function/Dysfunction in Cardiovascular Disease. Int. J. Mol. Sci. 2019, 20, 187. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20010187
Daiber A, Xia N, Steven S, Oelze M, Hanf A, Kröller-Schön S, Münzel T, Li H. New Therapeutic Implications of Endothelial Nitric Oxide Synthase (eNOS) Function/Dysfunction in Cardiovascular Disease. International Journal of Molecular Sciences. 2019; 20(1):187. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20010187
Chicago/Turabian StyleDaiber, Andreas, Ning Xia, Sebastian Steven, Matthias Oelze, Alina Hanf, Swenja Kröller-Schön, Thomas Münzel, and Huige Li. 2019. "New Therapeutic Implications of Endothelial Nitric Oxide Synthase (eNOS) Function/Dysfunction in Cardiovascular Disease" International Journal of Molecular Sciences 20, no. 1: 187. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20010187