A Comprehensive Review on Current Advances in Peptide Drug Development and Design


Abstract
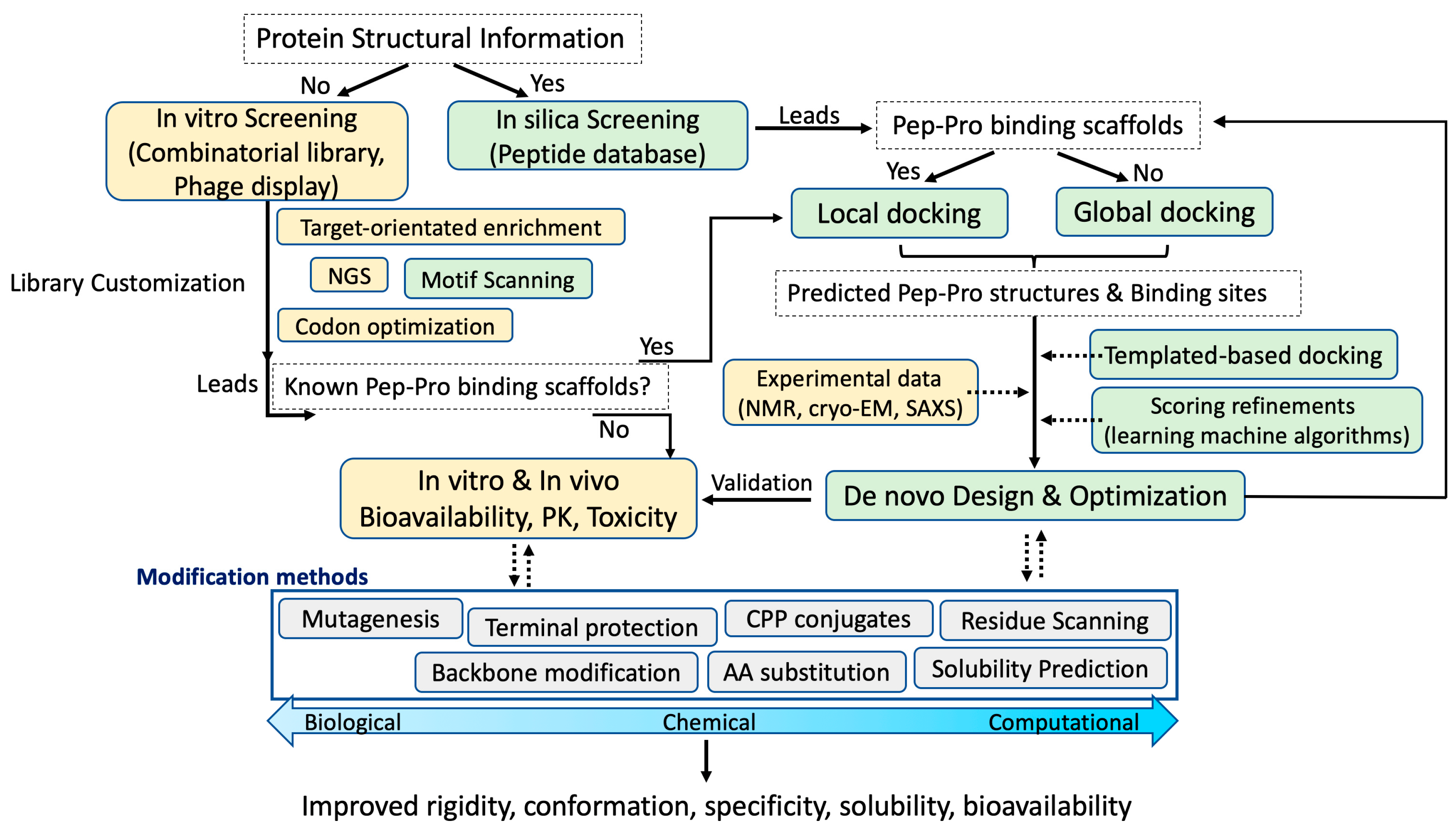
:1. Introduction
2. Key Aspects in Bioactive Peptide Drug Development
2.1. Historic Overview
2.2. Overcoming Intrinsic Drawbacks of Peptide Drugs
2.2.1. Termini Protection
2.2.2. Non-Chemical Methods: Identifying Critical Residues
2.2.3. Synthetic Amino Acid Substitution and Backbone Modification
2.2.4. Computational Methods for Improving Aqueous Solubility and Membrane Permeability
2.2.5. Membrane Protein-Facilitated Intracellular Peptide Uptake
2.3. High-Throughput Screening (HTS) for New Peptide Leads
3. Peptides and Protein–Protein Interactions
3.1. Promising Developments for Interfering Peptides
3.2. Experimental and Computational Methods for Determining PPI
3.2.1. Computational Docking Strategies
3.2.2. Sequence- or Structural-based Predictions
4. Innovations and Computational Methods for Peptide—Protein Interactions
4.1. Selection of Initial Peptide Scaffolds
4.2. Docking Peptide–Protein Interactions
4.2.1. Local and Global Docking Methods
4.2.2. Global Docking Methods
4.2.3. Template-Based Docking Method
5. Concluding Remarks
Funding
Conflicts of Interest
References
- Press, O.W.; Eary, J.F.; Appelbaum, F.R.; Martin, P.J.; Nelp, W.B.; Glenn, S.; Fisher, D.R.; Porter, B.; Matthews, D.C.; Gooley, T.; et al. Phase II trial of 131I-B1 (anti-CD20) antibody therapy with autologous stem cell transplantation for relapsed B cell lymphomas. Lancet 1995, 346, 336–340. [Google Scholar] [CrossRef]
- Goldenberg, D.M.; Deland, F.; Kim, E.; Bennett, S.; Primus, F.J.; van Nagell, J.R., Jr.; Estes, N.; DeSimone, P.; Rayburn, P. Use of radiolabeled antibodies to carcinoembryonic antigen for the detection and localization of diverse cancers by external photoscanning. N. Engl. J. Med. 1978, 298, 1384–1386. [Google Scholar] [CrossRef] [PubMed]
- Krenning, E.P.; Bakker, W.H.; Breeman, W.A.; Koper, J.W.; Kooij, P.P.; Ausema, L.; Lameris, J.S.; Reubi, J.C.; Lamberts, S.W. Localisation of endocrine-related tumours with radioiodinated analogue of somatostatin. Lancet 1989, 1, 242–244. [Google Scholar] [CrossRef]
- Banting, F.G.; Best, C.H.; Collip, J.B.; Campbell, W.R.; Fletcher, A.A. Pancreatic Extracts in the Treatment of Diabetes Mellitus. Can. Med. Assoc. J. 1922, 12, 141–146. [Google Scholar]
- Birk, A.V.; Liu, S.; Soong, Y.; Mills, W.; Singh, P.; Warren, J.D.; Seshan, S.V.; Pardee, J.D.; Szeto, H.H. The mitochondrial-targeted compound SS-31 re-energizes ischemic mitochondria by interacting with cardiolipin. J. Am. Soc. Nephrol. 2013, 24, 1250–1261. [Google Scholar] [CrossRef]
- Chang, Y.S.; Graves, B.; Guerlavais, V.; Tovar, C.; Packman, K.; To, K.H.; Olson, K.A.; Kesavan, K.; Gangurde, P.; Mukherjee, A.; et al. Stapled alpha-helical peptide drug development: A potent dual inhibitor of MDM2 and MDMX for p53-dependent cancer therapy. Proc. Natl. Acad. Sci. USA 2013, 110, 3445–3454. [Google Scholar] [CrossRef]
- Yan, Z.; Wang, J. Specificity quantification of biomolecular recognition and its implication for drug discovery. Sci. Rep. 2012, 2, 309. [Google Scholar] [CrossRef] [PubMed]
- Thomas, D. A Big Year for Novel Drugs Approvals; Biotechnology Innovation Organization: Washington, DC, USA, 2013. [Google Scholar]
- Wells, J.A.; McClendon, C.L. Reaching for high-hanging fruit in drug discovery at protein-protein interfaces. Nature 2007, 450, 1001–1009. [Google Scholar] [CrossRef] [PubMed]
- Hewitt, W.M.; Leung, S.S.; Pye, C.R.; Ponkey, A.R.; Bednarek, M.; Jacobson, M.P.; Lokey, R.S. Cell-permeable cyclic peptides from synthetic libraries inspired by natural products. J. Am. Chem. Soc. 2015, 137, 715–721. [Google Scholar] [CrossRef] [PubMed]
- Driggers, E.M.; Hale, S.P.; Lee, J.; Terrett, N.K. The exploration of macrocycles for drug discovery-an underexploited structural class. Nat. Rev. Drug. Discov. 2008, 7, 608–624. [Google Scholar] [CrossRef] [PubMed]
- Matsson, P.; Doak, B.C.; Over, B.; Kihlberg, J. Cell permeability beyond the rule of 5. Adv. Drug. Deliv. Rev. 2016, 101, 42–61. [Google Scholar] [CrossRef]
- Bliss, M. Banting’s, Best’s, and Collip’s accounts of the discovery of insulin. Bull. Hist. Med. 1982, 56, 554–568. [Google Scholar]
- Research, T.M. Global Industry Analysis, Size, Share, Growth, Trends and Forecast. Pept. Mark. 2016, 2016–2024. [Google Scholar]
- Fosgerau, K.; Hoffmann, T. Peptide therapeutics: Current status and future directions. Drug Discov. Today 2015, 20, 122–128. [Google Scholar] [CrossRef]
- Puente, X.S.; Gutierrez-Fernandez, A.; Ordonez, G.R.; Hillier, L.W.; Lopez-Otin, C. Comparative genomic analysis of human and chimpanzee proteases. Genomics 2005, 86, 638–647. [Google Scholar] [CrossRef] [PubMed]
- Werle, M.; Bernkop-Schnurch, A. Strategies to improve plasma half life time of peptide and protein drugs. Amino Acids 2006, 30, 351–367. [Google Scholar] [CrossRef]
- Jambunathan, K.; Galande, A.K. Design of a serum stability tag for bioactive peptides. Protein Pept. Lett. 2014, 21, 32–38. [Google Scholar] [CrossRef]
- Di, L. Strategic approaches to optimizing peptide ADME properties. AAPS. J. 2015, 17, 134–143. [Google Scholar] [CrossRef] [PubMed]
- Weiss, G.A.; Watanabe, C.K.; Zhong, A.; Goddard, A.; Sidhu, S.S. Rapid mapping of protein functional epitopes by combinatorial alanine scanning. Proc. Natl. Acad. Sci. USA 2000, 97, 8950–8954. [Google Scholar] [CrossRef]
- Morrison, K.L.; Weiss, G.A. Combinatorial alanine-scanning. Curr. Opin. Chem. Biol. 2001, 5, 302–307. [Google Scholar] [CrossRef]
- Eustache, S.; Leprince, J.; Tuffery, P. Progress with peptide scanning to study structure-activity relationships: The implications for drug discovery. Expert. Opin. Drug Discov. 2016, 11, 771–784. [Google Scholar] [CrossRef]
- Weinstock, M.T.; Francis, J.N.; Redman, J.S.; Kay, M.S. Protease-resistant peptide design-empowering nature’s fragile warriors against HIV. Biopolymers 2012, 98, 431–442. [Google Scholar] [CrossRef] [PubMed]
- Wisniewski, K.; Galyean, R.; Tariga, H.; Alagarsamy, S.; Croston, G.; Heitzmann, J.; Kohan, A.; Wisniewska, H.; Laporte, R.; Riviere, P.J.; et al. New, potent, selective, and short-acting peptidic V1a receptor agonists. J. Med. Chem. 2011, 54, 4388–4398. [Google Scholar] [CrossRef]
- Frey, V.; Viaud, J.; Subra, G.; Cauquil, N.; Guichou, J.F.; Casara, P.; Grassy, G.; Chavanieu, A. Structure-activity relationships of Bak derived peptides: Affinity and specificity modulations by amino acid replacement. Eur. J. Med. Chem. 2008, 43, 966–972. [Google Scholar] [CrossRef] [PubMed]
- Finan, B.; Yang, B.; Ottaway, N.; Smiley, D.L.; Ma, T.; Clemmensen, C.; Chabenne, J.; Zhang, L.; Habegger, K.M.; Fischer, K.; et al. A rationally designed monomeric peptide triagonist corrects obesity and diabetes in rodents. Nat. Med. 2015, 21, 27–36. [Google Scholar] [CrossRef]
- Werner, H.M.; Cabalteja, C.C.; Horne, W.S. Peptide Backbone Composition and Protease Susceptibility: Impact of Modification Type.; Position, and Tandem Substitution. ChemBioChem 2016, 17, 712–718. [Google Scholar] [CrossRef] [PubMed]
- Liskamp, R.M.; Rijkers, D.T.; Kruijtzer, J.A.; Kemmink, J. Peptides and proteins as a continuing exciting source of inspiration for peptidomimetics. ChemBioChem 2011, 12, 1626–1653. [Google Scholar] [CrossRef] [PubMed]
- Cabrele, C.; Martinek, T.A.; Reiser, O.; Berlicki, L. Peptides containing beta-amino acid patterns: Challenges and successes in medicinal chemistry. J. Med. Chem. 2014, 57, 9718–9739. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, J.; Rechenmacher, F.; Kessler, H. N-methylation of peptides and proteins: An. important element for modulating biological functions. Angew. Chem. Int. Ed. Engl. 2013, 52, 254–269. [Google Scholar] [CrossRef] [PubMed]
- Perez, J.J. Designing Peptidomimetics. Curr. Top Med. Chem. 2018, 18, 566–590. [Google Scholar] [CrossRef]
- Chingle, R.; Proulx, C.; Lubell, W.D. Azapeptide Synthesis Methods for Expanding Side-Chain Diversity for Biomedical Applications. ACC Chem. Res. 2017, 50, 1541–1556. [Google Scholar] [CrossRef] [PubMed]
- Pelay-Gimeno, M.; Glas, A.; Koch, O.; Grossmann, T.N. Structure-Based Design of Inhibitors of Protein-Protein Interactions: Mimicking Peptide Binding Epitopes. Angew. Chem. Int. Ed. Engl. 2015, 54, 8896–8927. [Google Scholar] [CrossRef] [PubMed]
- Kluskens, L.D.; Nelemans, S.A.; Rink, R.; de Vries, L.; Meter-Arkema, A.; Wang, Y.; Walther, T.; Kuipers, A.; Moll, G.N.; Haas, M. Angiotensin-(1-7) with thioether bridge: An. angiotensin-converting enzyme-resistant, potent angiotensin-(1-7) analog. J. Pharm. Exp. 2009, 328, 849–854. [Google Scholar] [CrossRef]
- Decoene, K.W.; Vannecke, W.; Passioura, T.; Suga, H.; Madder, A. Pyrrole-Mediated Peptide Cyclization Identified through Genetically Reprogrammed Peptide Synthesis. Biomedicines 2018, 6, 99. [Google Scholar] [CrossRef]
- Wu, S.J.; Luo, J.; O’Neil, K.T.; Kang, J.; Lacy, E.R.; Canziani, G.; Baker, A.; Huang, M.; Tang, Q.M.; Raju, T.S.; et al. Structure-based engineering of a monoclonal antibody for improved solubility. Protein Eng. Des. Sel. 2010, 23, 643–651. [Google Scholar] [CrossRef]
- Mant, C.T.; Kovacs, J.M.; Kim, H.M.; Pollock, D.D.; Hodges, R.S. Intrinsic amino acid side-chain hydrophilicity/hydrophobicity coefficients determined by reversed-phase high-performance liquid chromatography of model peptides: Comparison with other hydrophilicity/hydrophobicity scales. Biopolymers 2009, 92, 573–595. [Google Scholar] [CrossRef]
- Cortes, C.; Vapnik, V. Support-Vector Networks. Mach. Learn. 1995, 20, 273–297. [Google Scholar] [CrossRef]
- Agostini, F.; Cirillo, D.; Livi, C.M.; Ponti, R.D.; Tartaglia, G.G. cc SOL omics: A webserver for solubility prediction of endogenous and heterologous expression in Escherichia coli. Bioinformatics 2014, 30, 2975–2977. [Google Scholar] [CrossRef]
- Smialowski, P.; Doose, G.; Torkler, P.; Kaufmann, S.; Frishman, D. PROSO II-a new method for protein solubility prediction. FEBS J. 2012, 279, 2192–2200. [Google Scholar] [CrossRef]
- Derakhshankhah, H.; Jafari, S. Cell penetrating peptides: A concise review with emphasis on biomedical applications. Biomed. Pharm. 2018, 108, 1090–1096. [Google Scholar] [CrossRef]
- Copolovici, D.M.; Langel, K.; Eriste, E.; Langel, U. Cell-penetrating peptides: Design, synthesis, and applications. ACS Nano. 2014, 8, 1972–1994. [Google Scholar] [CrossRef]
- Mahlapuu, M.; Hakansson, J.; Ringstad, L.; Bjorn, C. Antimicrobial Peptides: An. Emerging Category of Therapeutic Agents. Front Cell Infect. Microbiol. 2016, 6, 194. [Google Scholar] [CrossRef]
- Qian, Z.; LaRochelle, J.R.; Jiang, B.; Lian, W.; Hard, R.L.; Selner, N.G.; Luechapanichkul, R.; Barrios, A.M.; Pei, D. Early endosomal escape of a cyclic cell-penetrating peptide allows effective cytosolic cargo delivery. Biochemistry 2014, 53, 4034–4046. [Google Scholar] [CrossRef] [PubMed]
- Qian, Z.; Martyna, A.; Hard, R.L.; Wang, J.; Appiah-Kubi, G.; Coss, C.; Phelps, M.A.; Rossman, J.S.; Pei, D. Discovery and Mechanism of Highly Efficient Cyclic Cell-Penetrating Peptides. Biochemistry 2016, 55, 2601–2612. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Xing, P.; Su, R.; Shi, G.; Ma, Z.S.; Zou, Q. CPPred-RF: A Sequence-based Predictor for Identifying Cell-Penetrating Peptides and Their Uptake Efficiency. J. Proteome. Res. 2017, 16, 2044–2053. [Google Scholar] [CrossRef] [PubMed]
- Holton, T.A.; Pollastri, G.; Shields, D.C.; Mooney, C. CPPpred: Prediction of cell penetrating peptides. Bioinformatics 2013, 29, 3094–3096. [Google Scholar] [CrossRef] [PubMed]
- Pandey, P.; Patel, V.; George, N.V.; Mallajosyula, S.S. KELM-CPPpred: Kernel Extreme Learning Machine Based Prediction Model. for Cell-Penetrating Peptides. J. Proteome. Res. 2018, 17, 3214–3222. [Google Scholar] [CrossRef]
- Eiriksdottir, E.; Konate, K.; Langel, U.; Divita, G.; Deshayes, S. Secondary structure of cell-penetrating peptides controls membrane interaction and insertion. Biochim. Biophys. Acta 2010, 1798, 1119–1128. [Google Scholar] [CrossRef]
- Gautam, A.; Chaudhary, K.; Kumar, R.; Sharma, A.; Kapoor, P.; Tyagi, A. Raghava GPS, consortium Osdd. In silico approaches for designing highly effective cell penetrating peptides. J. Transl. Med. 2013, 11, 74. [Google Scholar] [CrossRef]
- Agrawal, P.; Bhalla, S.; Usmani, S.S.; Singh, S.; Chaudhary, K.; Raghava, G.P.; Gautam, A. CPPsite 2.0: A repository of experimentally validated cell-penetrating peptides. Nucleic Acids Res. 2016, 44, 1098–1103. [Google Scholar] [CrossRef]
- Lian, W.; Jiang, B.; Qian, Z.; Pei, D. Cell-permeable bicyclic peptide inhibitors against intracellular proteins. J. Am. Chem. Soc. 2014, 136, 9830–9833. [Google Scholar] [CrossRef]
- Trinh, T.B.; Upadhyaya, P.; Qian, Z.; Pei, D. Discovery of a Direct Ras Inhibitor by Screening a Combinatorial Library of Cell-Permeable Bicyclic Peptides. ACS Comb. Sci. 2016, 18, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Carter, E.; Lau, C.Y.; Tosh, D.; Ward, S.G.; Mrsny, R.J. Cell penetrating peptides fail to induce an innate immune response in epithelial cells in vitro: Implications for continued therapeutic use. Eur. J. Pharm. Biopharm. 2013, 85, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.P. Filamentous fusion phage: Novel expression vectors that display cloned antigens on the virion surface. Science 1985, 228, 1315–1317. [Google Scholar] [CrossRef]
- McCafferty, J.; Griffiths, A.D.; Winter, G.; Chiswell, D.J. Phage antibodies: Filamentous phage displaying antibody variable domains. Nature 1990, 348, 552–554. [Google Scholar] [CrossRef] [PubMed]
- Omidfar, K.; Daneshpour, M. Advances in phage display technology for drug discovery. Expert. Opin. Drug Discov. 2015, 10, 651–669. [Google Scholar] [CrossRef]
- Matochko, W.L.; Derda, R. Next-generation sequencing of phage-displayed peptide libraries. Methods Mol. Biol. 2015, 1248, 249–266. [Google Scholar] [PubMed]
- Ng, S.; Derda, R. Phage-displayed macrocyclic glycopeptide libraries. Org. Biomol. Chem. 2016, 14, 5539–5545. [Google Scholar] [CrossRef] [PubMed]
- Heinis, C.; Winter, G. Encoded libraries of chemically modified peptides. Curr. Opin. Chem. Biol. 2015, 26, 89–98. [Google Scholar] [CrossRef]
- Rolland, T.; Tasan, M.; Charloteaux, B.; Pevzner, S.J.; Zhong, Q.; Sahni, N.; Yi, S.; Lemmens, I.; Fontanillo, C.; Mosca, R.; et al. A proteome-scale map of the human interactome network. Cell 2014, 159, 1212–1226. [Google Scholar] [CrossRef]
- Cunningham, A.D.; Qvit, N.; Mochly-Rosen, D. Peptides and peptidomimetics as regulators of protein-protein interactions. Curr. Opin. Struct. Biol. 2017, 44, 59–66. [Google Scholar] [CrossRef]
- Petta, I.; Lievens, S.; Libert, C.; Tavernier, J.; de Bosscher, K. Modulation of Protein-Protein Interactions for the Development of Novel Therapeutics. Mol. Ther. 2016, 24, 707–718. [Google Scholar] [CrossRef]
- Warso, M.A.; Richards, J.M.; Mehta, D.; Christov, K.; Schaeffer, C.; Bressler, L.R.; Yamada, T.; Majumdar, D.; Kennedy, S.A.; Beattie, C.W.; et al. A first-in-class, first-in-human, phase I trial of p28, a non-HDM2-mediated peptide inhibitor of p53 ubiquitination in patients with advanced solid tumours. Br. J. Cancer 2013, 108, 1061–1070. [Google Scholar] [CrossRef]
- Tabernero, J.; Dirix, L.; Schoffski, P.; Cervantes, A.; Capdevila, J.; Baselga, J.; Beijsterveldt, L.V.; Winkler, H.; Kraljevic, S.; Zhuang, S.H. Phase I pharmacokinetic (PK) and pharmacodynamic (PD) study of HDM-2 antagonist JNJ-26854165 in patients with advanced refractory solid tumors. J. Clin. Oncol. 2009, 27, 3514. [Google Scholar]
- Wong, D.; Kandagatla, P.; Korz, W.; Chinni, S.R. Targeting CXCR4 with CTCE-9908 inhibits prostate tumor metastasis. BMC Urol. 2014, 14, 12. [Google Scholar] [CrossRef]
- Huang, E.H.; Singh, B.; Cristofanilli, M.; Gelovani, J.; Wei, C.; Vincent, L.; Cook, K.R.; Lucci, A. A CXCR4 antagonist CTCE-9908 inhibits primary tumor growth and metastasis of breast cancer. J. Surg. Res. 2009, 155, 231–236. [Google Scholar] [CrossRef] [PubMed]
- Chiquet, C.; Aptel, F.; Creuzot-Garcher, C.; Berrod, J.P.; Kodjikian, L.; Massin, P.; Deloche, C.; Perino, J.; Kirwan, B.A.; de Brouwer, S.; et al. Postoperative Ocular Inflammation: A Single Subconjunctival Injection of XG-102 Compared to Dexamethasone Drops in a Randomized Trial. Am. J. Ophthalmol. 2017, 174, 76–84. [Google Scholar] [CrossRef] [PubMed]
- Borsello, T.; Clarke, P.G.; Hirt, L.; Vercelli, A.; Repici, M.; Schorderet, D.F.; Bogousslavsky, J.; Bonny, C. A peptide inhibitor of c-Jun N-terminal kinase protects against excitotoxicity and cerebral ischemia. Nat. Med. 2003, 9, 1180–1186. [Google Scholar] [CrossRef] [PubMed]
- Lau, Y.H.; de Andrade, P.; Wu, Y.; Spring, D.R. Peptide stapling techniques based on different macrocyclisation chemistries. Chem. Soc. Rev. 2015, 44, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Mine, Y.; Munir, H.; Nakanishi, Y.; Sugiyama, D. Biomimetic Peptides for the Treatment of Cancer. Anticancer Res. 2016, 36, 3565–3570. [Google Scholar] [PubMed]
- Ellert-Miklaszewska, A.; Poleszak, K.; Kaminska, B. Short peptides interfering with signaling pathways as new therapeutic tools for cancer treatment. Future Med. Chem. 2017, 9, 199–221. [Google Scholar] [CrossRef]
- Sliwoski, G.; Kothiwale, S.; Meiler, J.; Lowe, E.W., Jr. Computational methods in drug discovery. Pharm. Rev. 2014, 66, 334–395. [Google Scholar] [CrossRef] [PubMed]
- De Vries, S.J.; Schindler, C.E.; de Beauchene, I.C.; Zacharias, M. A web interface for easy flexible protein-protein docking with ATTRACT. Biophys. J. 2015, 108, 462–465. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.M.; Serohijos, A.W.R.; Murphy, S.; Lucarelli, D.; Lofranco, L.L.; Feldman, A.; Shakhnovich, E.I. Minimalistic predictor of protein binding energy: Contribution of solvation factor to protein binding. Biophys. J. 2015, 108, 795–798. [Google Scholar] [CrossRef] [PubMed]
- Yugandhar, K.; Gromiha, M.M. Protein-protein binding affinity prediction from amino acid sequence. Bioinformatics 2014, 30, 3583–3589. [Google Scholar] [CrossRef] [PubMed]
- Yugandhar, K.; Gromiha, M.M. Feature selection and classification of protein-protein complexes based on their binding affinities using machine learning approaches. Proteins 2014, 82, 2088–2096. [Google Scholar] [CrossRef] [PubMed]
- Lensink, M.F.; Velankar, S.; Wodak, S.J. Modeling protein-protein and protein-peptide complexes: CAPRI 6th edition. Proteins 2017, 85, 359–377. [Google Scholar] [CrossRef] [PubMed]
- Van Zundert, G.C.P.; Rodrigues, J.; Trellet, M.; Schmitz, C.; Kastritis, P.L.; Karaca, E.; Melquiond, A.S.J.; van Dijk, M.; de Vries, S.J.; Bonvin, A. The HADDOCK2.2 Web Server: User-Friendly Integrative Modeling of Biomolecular Complexes. J. Mol. Biol. 2016, 428, 720–725. [Google Scholar] [CrossRef]
- Lensink, M.F.; Velankar, S.; Kryshtafovych, A.; Huang, S.Y.; Schneidman-Duhovny, D.; Sali, A.; Segura, J.; Fernandez-Fuentes, N.; Viswanath, S.; Elber, R.; et al. Prediction of homoprotein and heteroprotein complexes by protein docking and template-based modeling: A CASP-CAPRI experiment. Proteins 2016, 84, 323–348. [Google Scholar] [CrossRef] [PubMed]
- Comeau, S.R.; Gatchell, D.W.; Vajda, S.; Camacho, C.J. ClusPro: An automated docking and discrimination method for the prediction of protein complexes. Bioinformatics 2004, 20, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Vanhee, P.; Reumers, J.; Stricher, F.; Baeten, L.; Serrano, L.; Schymkowitz, J.; Rousseau, F. PepX: A structural database of non-redundant protein-peptide complexes. Nucleic Acids Res. 2010, 38, 545–551. [Google Scholar] [CrossRef]
- London, N.; Movshovitz-Attias, D.; Schueler-Furman, O. The structural basis of peptide-protein binding strategies. Structure 2010, 18, 188–199. [Google Scholar] [CrossRef]
- Clackson, T.; Wells, J.A. A hot spot of binding energy in a hormone-receptor interface. Science 1995, 267, 383–386. [Google Scholar] [CrossRef]
- Buonfiglio, R.; Recanatini, M.; Masetti, M. Protein Flexibility in Drug Discovery: From Theory to Computation. ChemMedChem 2015, 10, 1141–1148. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, S.; Leiss, M.; Moser, M.; Ohashi, T.; Kitao, T.; Heckmann, D.; Pfeifer, A.; Kessler, H.; Takagi, J.; Erickson, H.P.; et al. The RGD motif in fibronectin is essential for development but dispensable for fibril assembly. J. Cell Biol. 2007, 178, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Mohapatra, S.; Saha, A.; Mondal, P.; Jana, B.; Ghosh, S.; Biswas, A.; Ghosh, S. Synergistic Anticancer Effect of Peptide-Docetaxel Nanoassembly Targeted to Tubulin: Toward Development of Dual Warhead Containing Nanomedicine. Adv. Healthc. Mater. 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Inaba, H.; Matsuura, K. Peptide Nanomaterials Designed from Natural Supramolecular Systems. Chem. Rec. 2018. [Google Scholar] [CrossRef]
- Inaba, H.; Yamamoto, T.; Kabir, A.M.R.; Kakugo, A.; Sada, K.; Matsuura, K. Molecular Encapsulation Inside Microtubules Based on Tau-Derived Peptides. Chem. Eur. J. 2018, 24, 14958–14967. [Google Scholar] [CrossRef]
- Jana, B.; Mondal, P.; Saha, A.; Adak, A.; Das, G.; Mohapatra, S.; Kurkute, P.; Ghosh, S. Designed Tetrapeptide Interacts with Tubulin and Microtubule. Langmuir 2018, 34, 1123–1132. [Google Scholar] [CrossRef] [PubMed]
- Antes, I. DynaDock: A new molecular dynamics-based algorithm for protein-peptide docking including receptor flexibility. Proteins 2010, 78, 1084–1104. [Google Scholar] [CrossRef] [PubMed]
- Raveh, B.; London, N.; Schueler-Furman, O. Sub-angstrom modeling of complexes between flexible peptides and globular proteins. Proteins 2010, 78, 2029–2040. [Google Scholar] [CrossRef]
- London, N.; Raveh, B.; Cohen, E.; Fathi, G.; Schueler-Furman, O. Rosetta FlexPepDock web server--high resolution modeling of peptide-protein interactions. Nucleic Acids Res. 2011, 39, 249–253. [Google Scholar] [CrossRef] [PubMed]
- Donsky, E.; Wolfson, H.J. PepCrawler: A fast RRT-based algorithm for high-resolution refinement and binding affinity estimation of peptide inhibitors. Bioinformatics 2011, 27, 2836–2842. [Google Scholar] [CrossRef]
- Raveh, B.; London, N.; Zimmerman, L.; Schueler-Furman, O. Rosetta FlexPepDock ab-initio: Simultaneous folding, docking and refinement of peptides onto their receptors. PLoS ONE 2011, 6, e18934. [Google Scholar] [CrossRef]
- Trellet, M.; Melquiond, A.S.; Bonvin, A.M. A unified conformational selection and induced fit approach to protein-peptide docking. PLoS ONE 2013, 8, e58769. [Google Scholar] [CrossRef] [PubMed]
- Verdonk, M.L.; Cole, J.C.; Hartshorn, M.J.; Murray, C.W.; Taylor, R.D. Improved protein-ligand docking using GOLD. Proteins 2003, 52, 609–623. [Google Scholar] [CrossRef]
- Rentzsch, R.; Renard, B.Y. Docking small peptides remains a great challenge: An assessment using AutoDock Vina. Brief. Bioinform. 2015, 16, 1045–1056. [Google Scholar] [CrossRef]
- Jain, A.N. Surflex: Fully automatic flexible molecular docking using a molecular similarity-based search engine. J. Med. Chem. 2003, 46, 499–511. [Google Scholar] [CrossRef]
- Hauser, A.S.; Windshugel, B. LEADS-PEP: A Benchmark Data Set for Assessment of Peptide Docking Performance. J. Chem. Inf. Model 2016, 56, 188–200. [Google Scholar] [CrossRef] [PubMed]
- Trabuco, L.G.; Lise, S.; Petsalaki, E.; Russell, R.B. PepSite: Prediction of peptide-binding sites from protein surfaces. Nucleic Acids Res. 2012, 40, 423–427. [Google Scholar] [CrossRef]
- Petsalaki, E.; Stark, A.; Garcia-Urdiales, E.; Russell, R.B. Accurate prediction of peptide binding sites on protein surfaces. PLoS Comput. Biol. 2009, 5, e1000335. [Google Scholar] [CrossRef]
- Porter, K.A.; Xia, B.; Beglov, D.; Bohnuud, T.; Alam, N.; Schueler-Furman, O.; Kozakov, D. ClusPro PeptiDock: Efficient global docking of peptide recognition motifs using FFT. Bioinformatics 2017, 33, 3299–3301. [Google Scholar] [CrossRef]
- De Vries, S.J.; Rey, J.; Schindler, C.E.M.; Zacharias, M.; Tuffery, P. The pepATTRACT web server for blind, large-scale peptide-protein docking. Nucleic Acids Res. 2017, 45, 361–364. [Google Scholar] [CrossRef]
- Lavi, A.; Ngan, C.H.; Movshovitz-Attias, D.; Bohnuud, T.; Yueh, C.; Beglov, D.; Schueler-Furman, O.; Kozakov, D. Detection of peptide-binding sites on protein surfaces: The first step toward the modeling and targeting of peptide-mediated interactions. Proteins 2013, 81, 2096–2105. [Google Scholar] [CrossRef]
- Kurcinski, M.; Jamroz, M.; Blaszczyk, M.; Kolinski, A.; Kmiecik, S. CABS-dock web server for the flexible docking of peptides to proteins without prior knowledge of the binding site. Nucleic Acids Res. 2015, 43, 419–424. [Google Scholar] [CrossRef]
- Ben-Shimon, A.; Niv, M.Y. AnchorDock: Blind. and Flexible Anchor-Driven Peptide Docking. Structure 2015, 23, 929–940. [Google Scholar] [CrossRef]
- Zhou, P.; Jin, B.; Li, H.; Huang, S.Y. HPEPDOCK: A web server for blind peptide-protein docking based on a hierarchical algorithm. Nucleic Acids Res. 2018, 46, 443–450. [Google Scholar] [CrossRef]
- Lee, H.; Heo, L.; Lee, M.S.; Seok, C. GalaxyPepDock: A protein-peptide docking tool based on interaction similarity and energy optimization. Nucleic Acids Res. 2015, 43, 431–435. [Google Scholar] [CrossRef]
- Taherzadeh, G.; Zhou, Y.; Liew, A.W.; Yang, Y. Structure-based prediction of protein- peptide binding regions using Random Forest. Bioinformatics 2018, 34, 477–484. [Google Scholar] [CrossRef]
- Iqbal, S.; Hoque, M.T. PBRpredict-Suite: A suite of models to predict peptide-recognition domain residues from protein sequence. Bioinformatics 2018, 34, 3289–3299. [Google Scholar] [CrossRef]
- Obarska-Kosinska, A.; Iacoangeli, A.; Lepore, R.; Tramontano, A. PepComposer: Computational design of peptides binding to a given protein surface. Nucleic Acids Res. 2016, 44, 522–528. [Google Scholar] [CrossRef]
- Wang, S.H.; Lee, A.C.; Chen, I.J.; Chang, N.C.; Wu, H.C.; Yu, H.M.; Chang, Y.J.; Lee, T.W.; Yu, J.C.; Yu, A.L.; et al. Structure-based optimization of GRP78-binding peptides that enhances efficacy in cancer imaging and therapy. Biomaterials 2016, 94, 31–44. [Google Scholar] [CrossRef]
- Yu, J.; Andreani, J.; Ochsenbein, F.; Guerois, R. Lessons from (co-)evolution in the docking of proteins and peptides for CAPRI Rounds 28–35. Proteins: Struct. Funct. Bioinform. 2017, 85, 378–390. [Google Scholar] [CrossRef]


{kind=link}
| Method | Learning Machine Model | Input Length (aa) | Input Format | Multiple Entry | Database | Web Server | Refs |
|---|---|---|---|---|---|---|---|
| Peptide Solubility | |||||||
| ccSOL omics | Super vector machine (SVM) | – | FASTA | Yes (up to 104) | Target Track (non-redundant) (http://sbkb.org/tt/) | http://s.tartaglialab.com/static_files/shared/tutorial_ccsol_omics.html | [39] |
| PROSO II | Super vector machine (SVM) | 21 to 2000 | FASTA | Yes (up to 50) | Target Track (http://sbkb.org/tt/) | http://mbiljj45.bio.med.uni-muenchen.de:8888/prosoII/prosoII.seam | [40] |
| Cell-Penetrating Peptides | |||||||
| CPPpred | Artificial neural networks (ANN) | 5 to 30 | FASTA | Yes | CPPsite | http://bioware.ucd.ie/cpppred | [47] |
| CPPpred-RF | Random forest (RF) | – | FASTA | Yes | CPP924 and CPPsite3 | http://server.malab.cn/CPPred-RF | [46] |
| KELM-CPPpred | Kernel extreme learning model (KELM) | 5 to 30 | FASTA | Yes | Curated 408 CPP/non-CPP | http://sairam.people.iitgn.ac.in/KELM-CPPpred.html | [48] |
| CellPPD | Super vector machine (SVM) | – | FASTA | Yes | CPPsite1,2,3 | http://crdd.osdd.net/raghava/cellppd/multi_pep.php | [50] |
| CPPsite 2.0 | – | – | FASTA | Yes | 1855 uniquely curated | http://crdd.osdd.net/raghava/cppsite/ | [51] |
| Methods | Key Features | Model Quality # | Web Server | Refs |
|---|---|---|---|---|
| Local Docking | ||||
| DynaDock |
| Near-native | Not available to public | [92] |
| Rosetta FlexPepDock |
| Sub-angstrom * | http://flexpepdock.furmanlab.cs.huji.ac.il or http://www.rosettacommons.org/software | [93] |
| PepCrawler |
| Near-native * | http://bioinfo3d.cs.tau.ac.il/PepCrawler | [95] |
| Rosetta FlexPepDock ab initio |
| Near-native to Sub-angstrom § | http://www.rosettacommons.org/software | [96] |
| HADDOCK peptide docking |
| Near-native * | http://haddock.science.uu.nl/services/HADDOCK2.2/ | [79,97] |
| PepSite 2.0 |
| Medium † | http://pepsite2.russelllab.org | [102] |
| Global Docking | ||||
| ClusPro PeptiDock |
| Near-native to Sub-angstrom § | https://peptidock.cluspro.org/ | [81,104] |
| pepATTRACT |
| Near-native to Sub-angstrom § | http://bioserv.rpbs.univ-paris-diderot.fr/services/pepATTRACT/ | [105] |
| HPEPDOCK |
| Near-native to Sub-angstrom § | http://huanglab.phys.hust.edu.cn/hpepdock/ | [109] |
| Template-based | ||||
| GalaxyPepDock |
| Medium (ligand); Near-native (interface) | http://galaxy.seoklab.org/pepdock | [110] |
| SPRINT-Str |
| N/A | http://sparks-lab.org/server/SPRINT-Str | [111] |
| PBRpredict-Suite |
| N/A | http://cs.uno.edu/~tamjid/Software/PBRpredict/pbrpredict-suite.zip | [112] |
| PepComposer |
| Near-native | https://cassandra.med.uniroma1.it/pepcomposer/webserver/pepcomposer.php | [113] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, A.C.-L.; Harris, J.L.; Khanna, K.K.; Hong, J.-H. A Comprehensive Review on Current Advances in Peptide Drug Development and Design. Int. J. Mol. Sci. 2019, 20, 2383. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20102383
Lee AC-L, Harris JL, Khanna KK, Hong J-H. A Comprehensive Review on Current Advances in Peptide Drug Development and Design. International Journal of Molecular Sciences. 2019; 20(10):2383. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20102383
Chicago/Turabian StyleLee, Andy Chi-Lung, Janelle Louise Harris, Kum Kum Khanna, and Ji-Hong Hong. 2019. "A Comprehensive Review on Current Advances in Peptide Drug Development and Design" International Journal of Molecular Sciences 20, no. 10: 2383. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20102383