Hypoxia-Induced Epithelial-To-Mesenchymal Transition Mediates Fibroblast Abnormalities via ERK Activation in Cutaneous Wound Healing

 , ,
, ,
Abstract
:
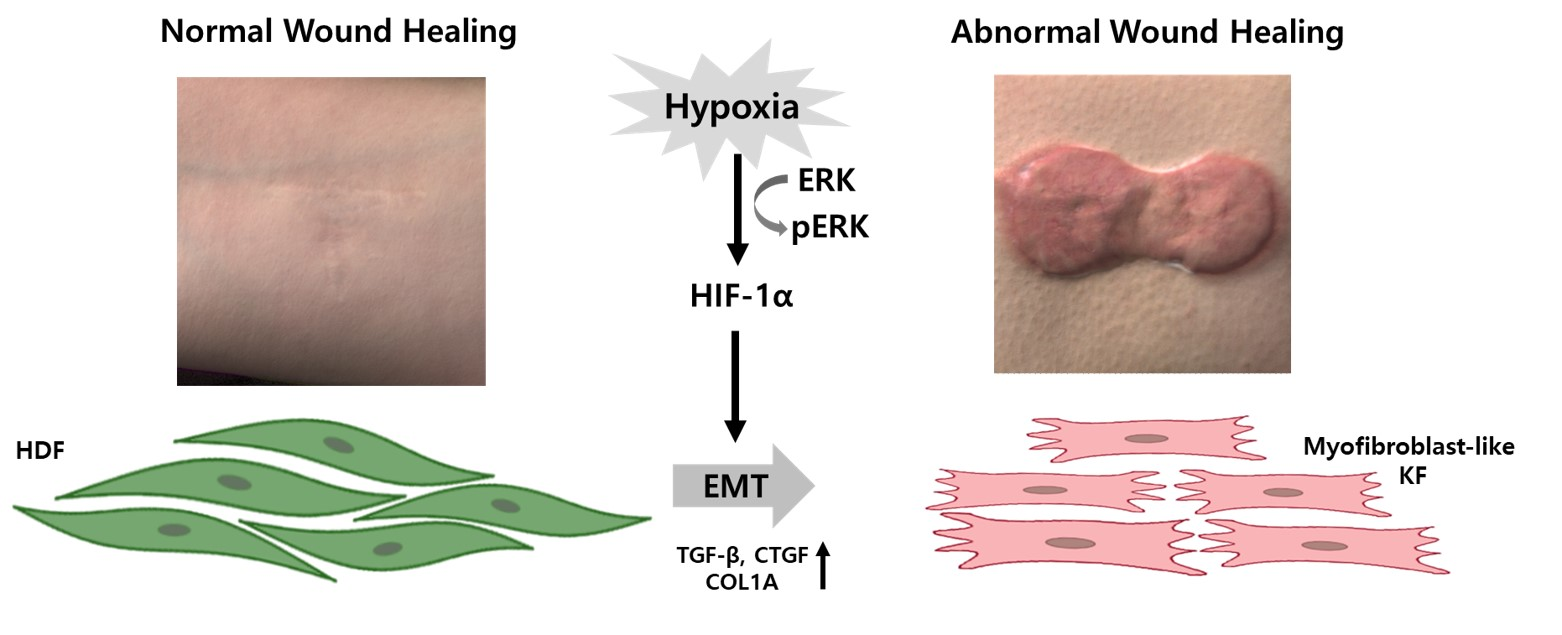
1. Introduction
2. Results
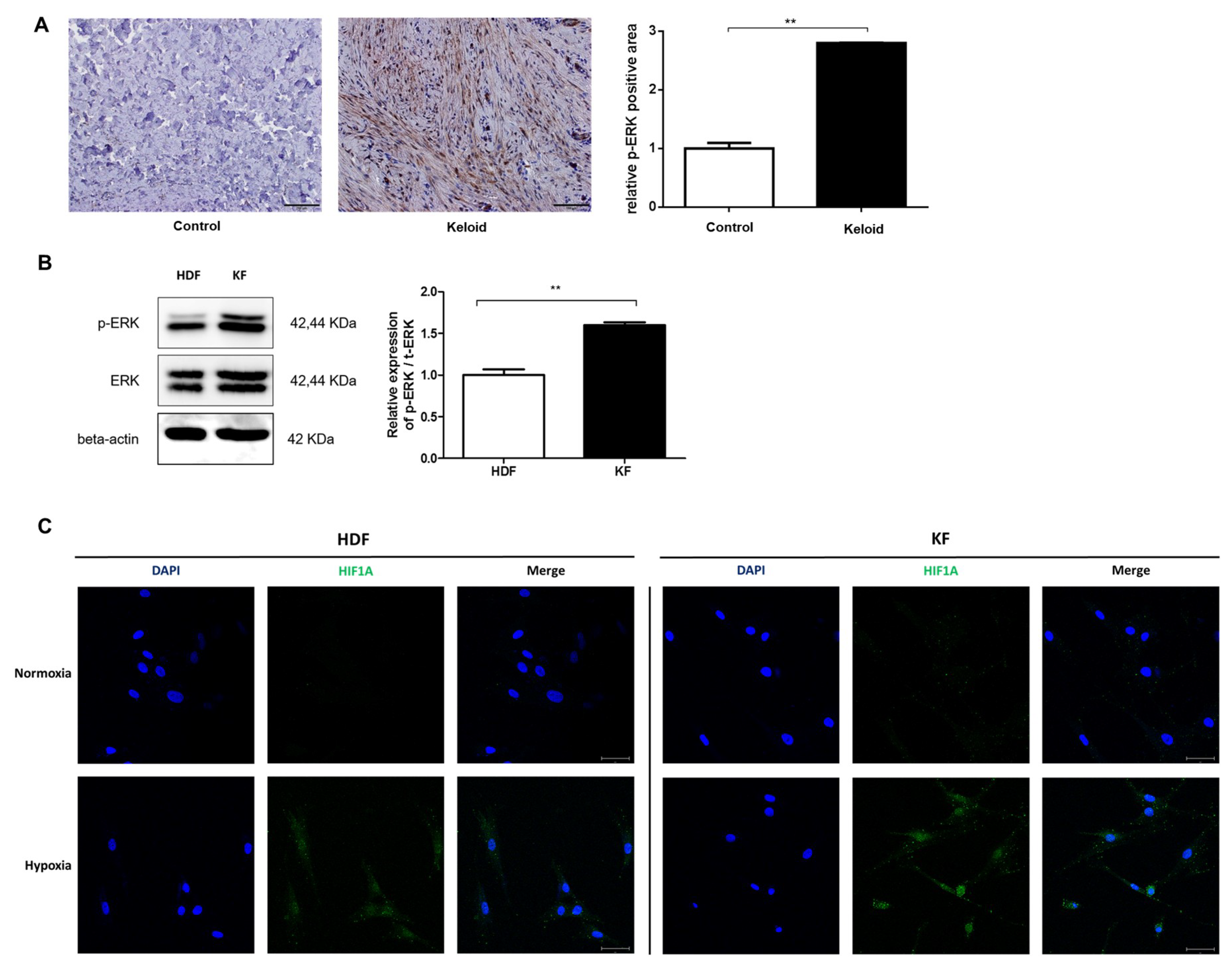
2.1. p-ERK and HIF-1α Levels Are Elevated in Keloid Tissue and Fibroblasts
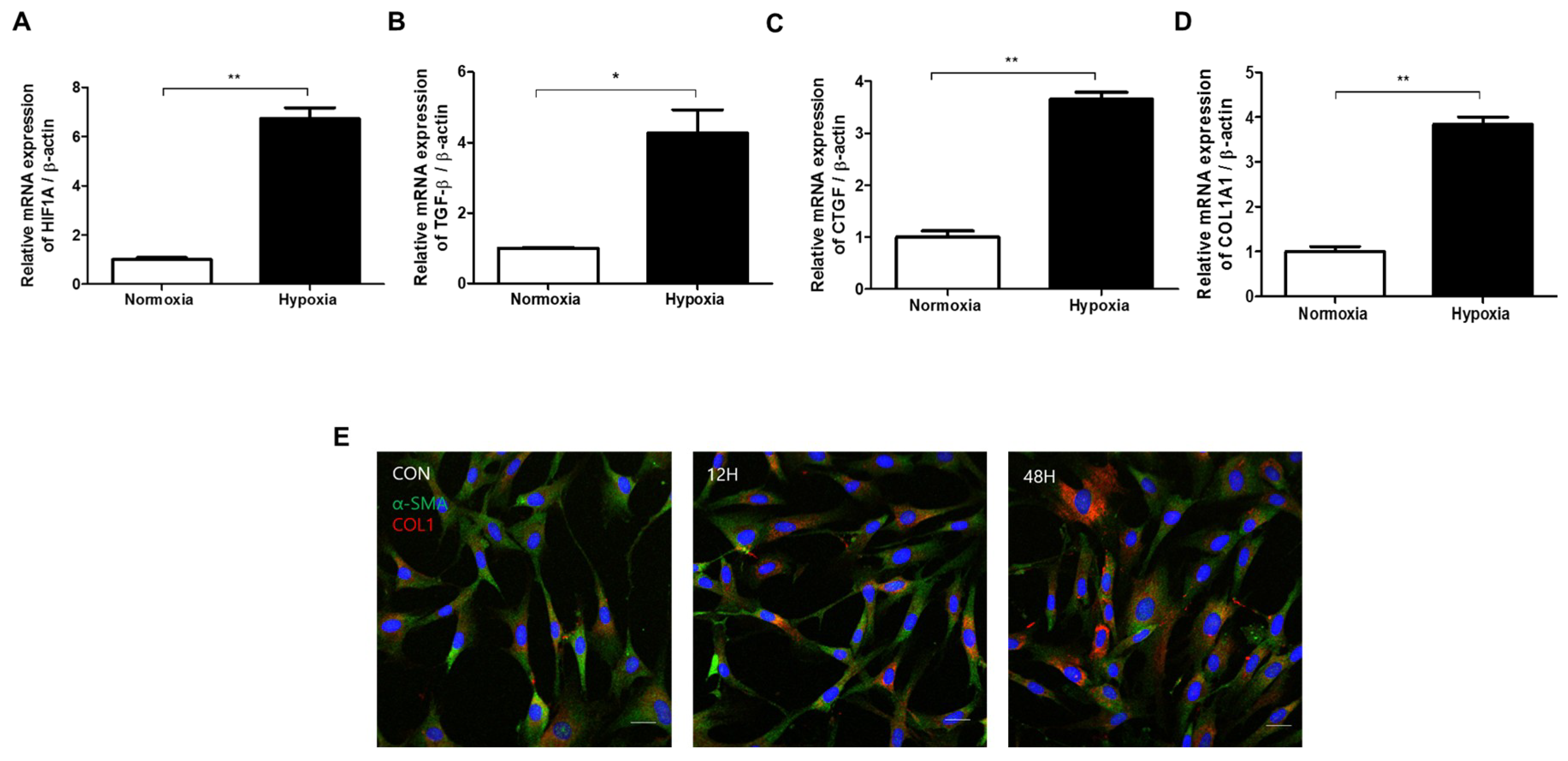
2.2. Hypoxia Activates TGF-β Signaling and Induces EMT in HDFs
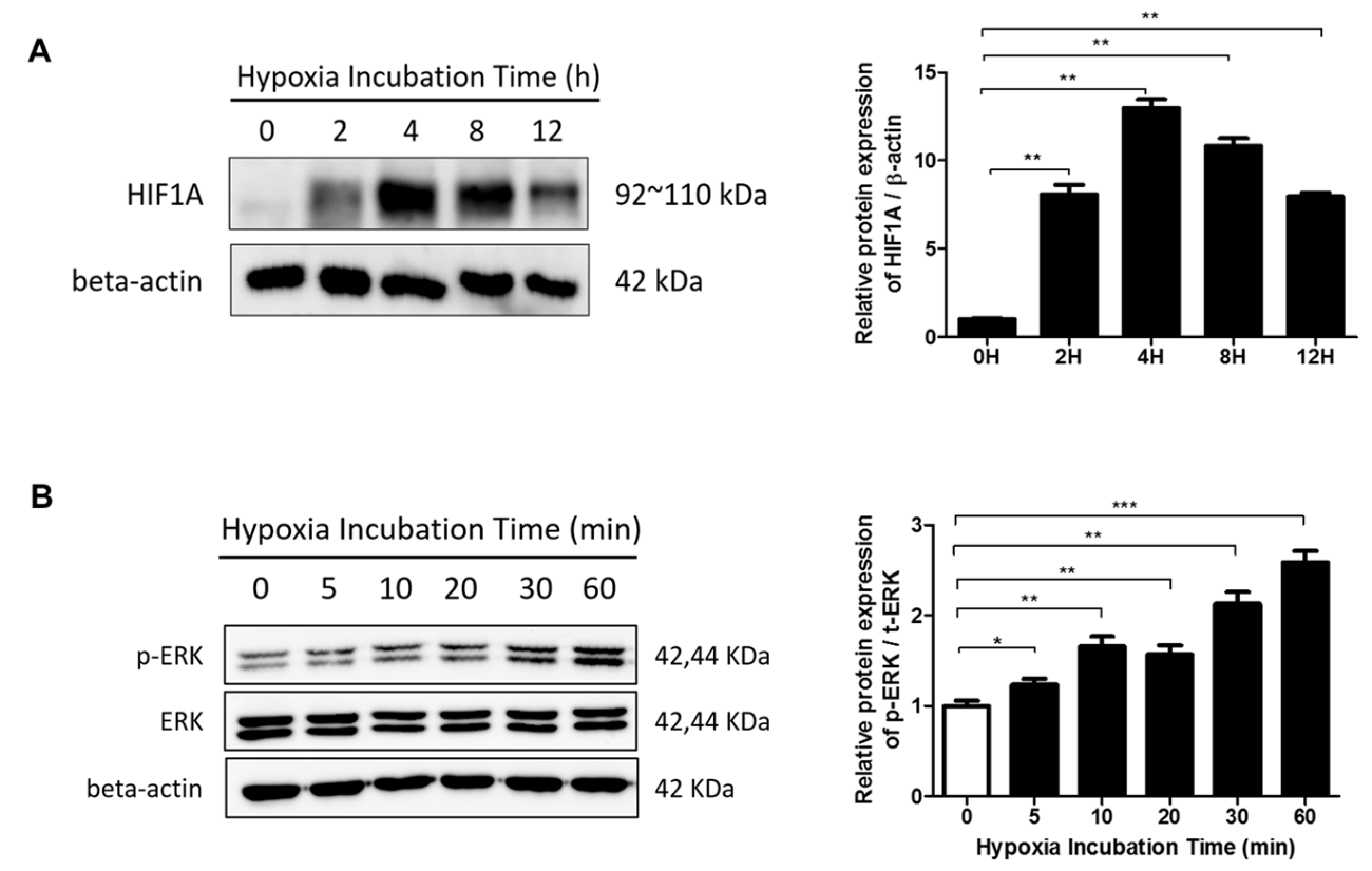
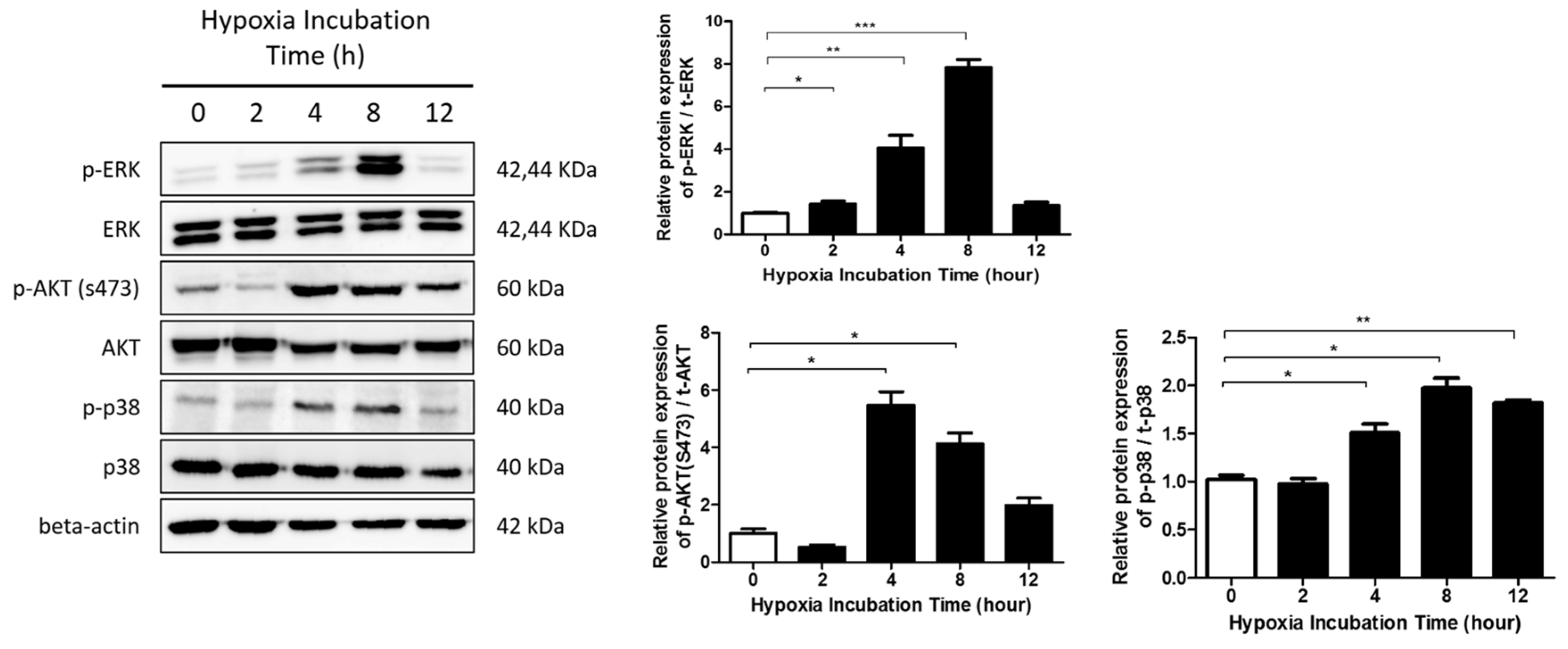
2.3. The ERK/MAPK Pathway Is Involved in Hypoxia-Induced EMT
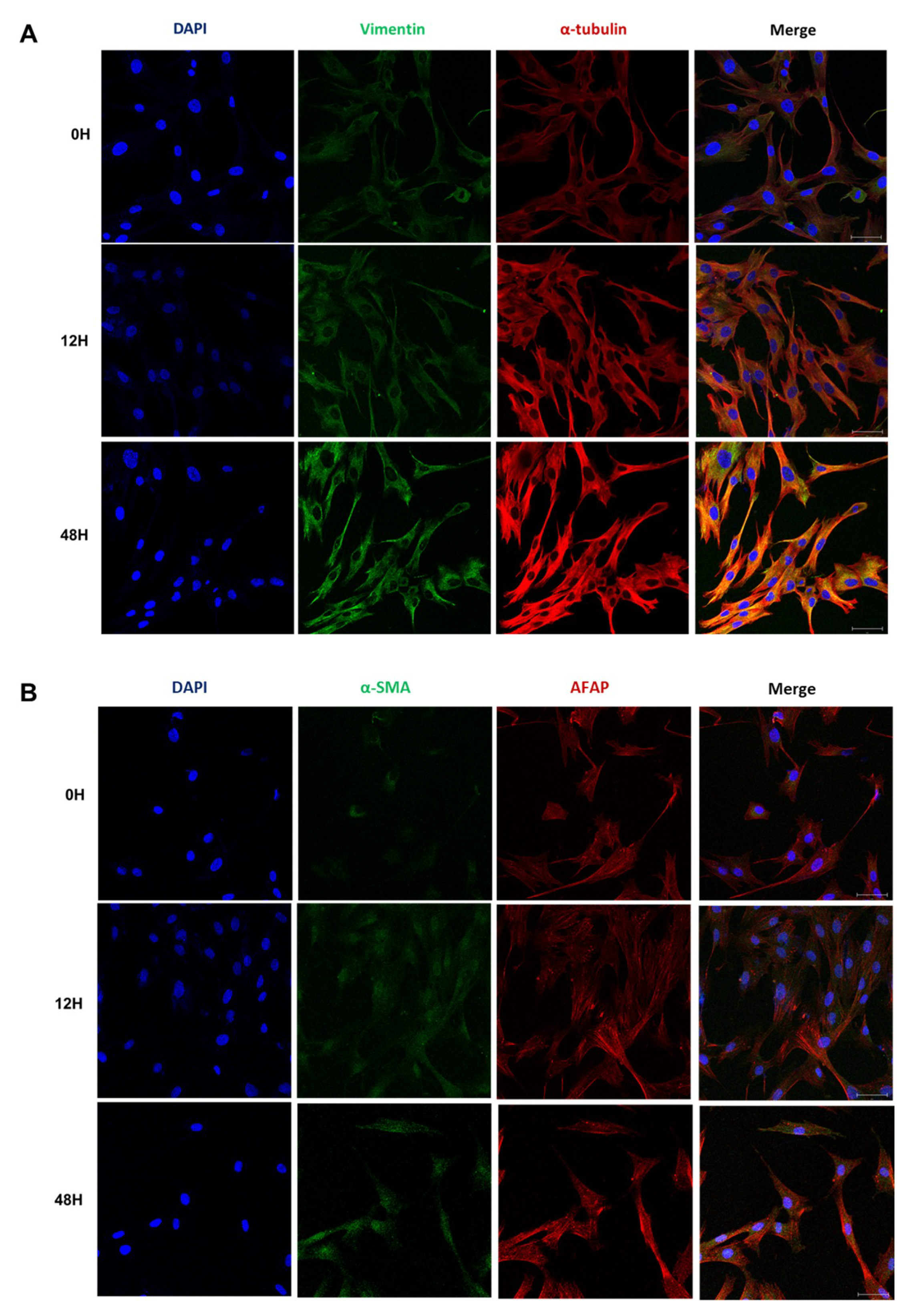
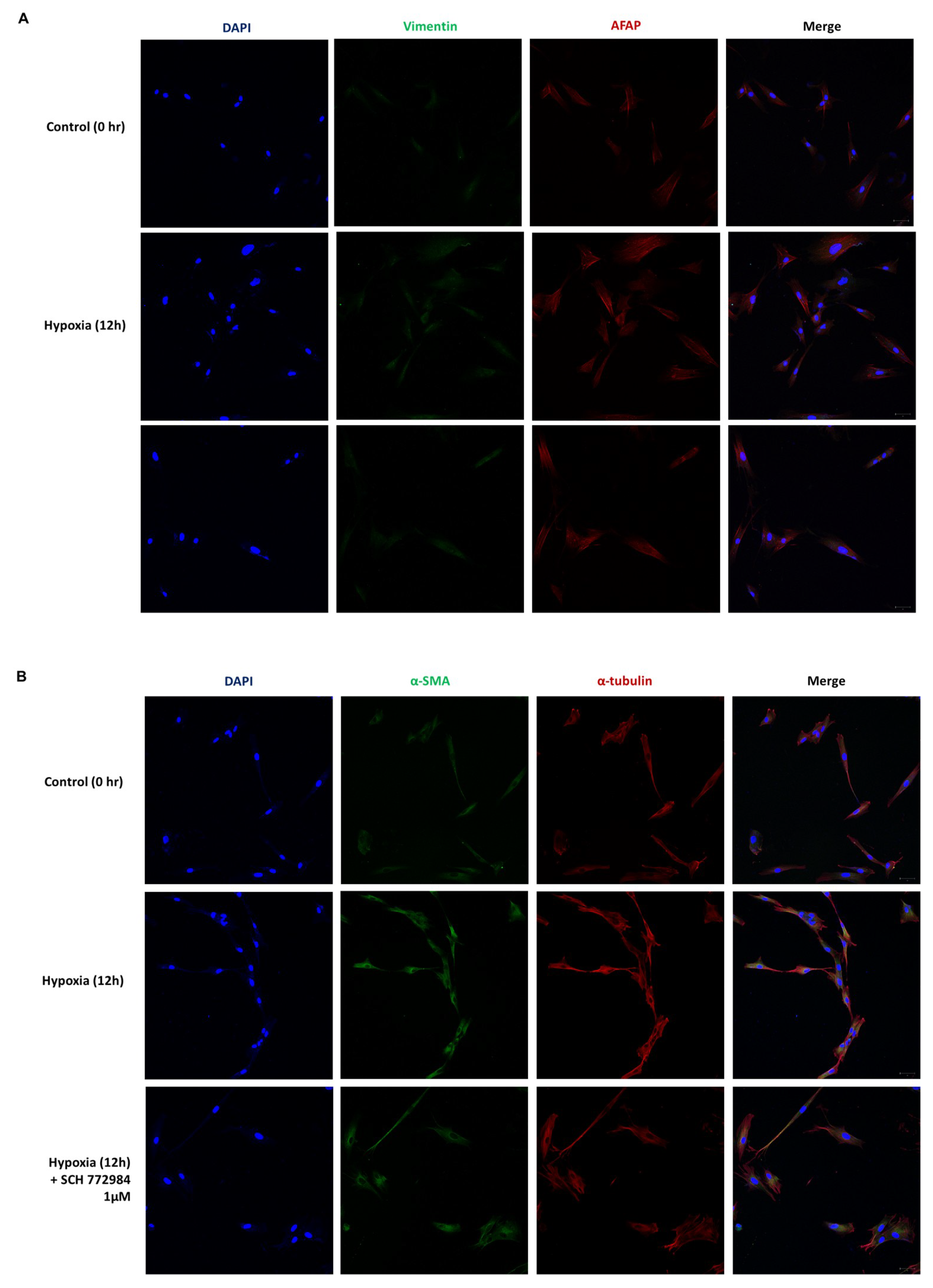
2.4. Phenotypic EMT Markers Are Expressed in HDFs under Hypoxia
2.5. Hypoxia Induces a Phenotypic Switch of Fibroblasts to Myofibroblasts
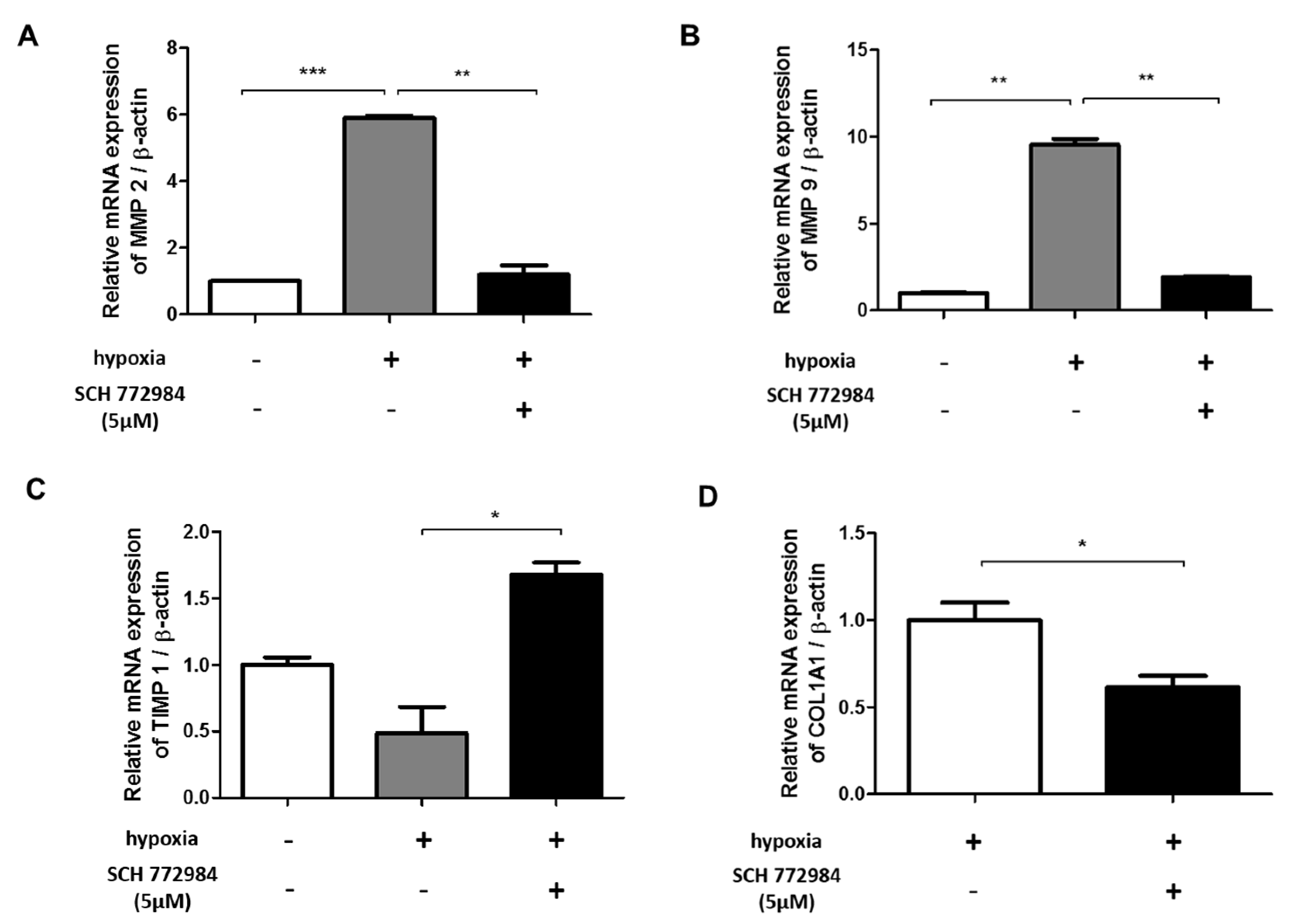
2.6. ERK Inhibition Reduces Hypoxia-Induced ECM Deposition
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Keloid-Derived Fibroblasts
4.2. Real-Time PCR Analysis
4.3. Western Blotting Analysis
4.4. Cell Viability Analysis
4.5. Immunohistochemistry
4.6. Antibodies and Reagents
4.7. Confocal Imaging
4.8. Statistical Analysis
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AFAP | Actin filament associated protein 1 |
| CTGF | Connective tissue growth factor |
| ECM | Extracellular matrix |
| EMT | Epithelial-mesenchymal transition |
| ERK | Extracellular signal-regulated kinase |
| HDF | Human dermal fibroblast |
| HIF-1α | Hypoxia-inducible factor-1α |
| KF | Keloid fibroblast |
| MAPK | Mitogen-activated protein kinase |
| MMP | Matrix metalloproteinase |
| PBS | Phosphate-buffered saline |
| PI3K | Phosphatidylinositol-4,5-bisphosphate 3-kinase |
| TGF-β | Transforming growth factor-β |
| TIMP | Tissue inhibitor of metalloproteinases |
References
- Gauglitz, G.G.; Korting, H.C.; Pavicic, T.; Ruzicka, T.; Jeschke, M.G. Hypertrophic Scarring and Keloids: Pathomechanisms and Current and Emerging Treatment Strategies. Mol. Med. 2011, 17, 113–125. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Akaishi, S.; Hyakusoku, H.; Ogawa, R. Are keloid and hypertrophic scar different forms of the same disorder? A fibroproliferative skin disorder hypothesis based on keloid findings. Int. Wound J. 2014, 11, 517–522. [Google Scholar] [CrossRef]
- Huang, C.; Akaishi, S.; Ogawa, R. Mechanosignaling pathways in cutaneous scarring. Arch. Dermatol. Res. 2012, 304, 589–597. [Google Scholar] [CrossRef] [PubMed]
- Song, N.; Wu, X.; Gao, Z.; Zhou, G.; Zhang, W.J.; Liu, W. Enhanced expression of membrane transporter and drug resistance in keloid fibroblasts. Hum. Pathol. 2012, 43, 2024–2032. [Google Scholar] [CrossRef]
- Qu, M.; Song, N.; Chai, G.; Wu, X.; Liu, W. Pathological niche environment transforms dermal stem cells to keloid stem cells: A hypothesis of keloid formation and development. Med. Hypotheses 2013, 81, 807–812. [Google Scholar] [CrossRef]
- Lee, W.J.; Park, J.H.; Shin, J.U.; Noh, H.; Lew, D.H.; Yang, W.I.; Yun, C.O.; Lee, K.H.; Lee, J.H. Endothelial-to-mesenchymal transition induced by Wnt 3a in keloid pathogenesis. Wound Repair Regen. 2015, 23, 435–442. [Google Scholar] [CrossRef] [PubMed]
- Vincent, A.S.; Phan, T.T.; Mukhopadhyay, A.; Lim, H.Y.; Halliwell, B.; Wong, K.P. Human skin keloid fibroblasts display bioenergetics of cancer cells. J. Investig. Dermatol. 2008, 128, 702–709. [Google Scholar] [CrossRef]
- Lee, H.J.; Jang, Y.J. Recent Understandings of Biology, Prophylaxis and Treatment Strategies for Hypertrophic Scars and Keloids. Int. J. Mol. Sci. 2018, 19, 711. [Google Scholar] [CrossRef]
- Yan, L.; Cao, R.; Wang, L.Z.; Liu, Y.B.; Pan, B.; Yin, Y.H.; Lv, X.Y.; Zhuang, Q.; Sun, X.J.; Xiao, R. Epithelial-mesenchymal transition in keloid tissues and TGF-beta 1-induced hair follicle outer root sheath keratinocytes. Wound Repair Regen. 2015, 23, 601–610. [Google Scholar] [CrossRef]
- Jumper, N.; Hodgkinson, T.; Paus, R.; Bayat, A. Site-specific gene expression profiling as a novel strategy for unravelling keloid disease pathobiology. PLoS ONE 2017, 12, e0172955. [Google Scholar] [CrossRef]
- Higgins, D.F.; Kimura, K.; Iwano, M.; Haase, V.H. Hypoxia-inducible factor signaling in the development of tissue fibrosis. Cell Cycle 2008, 7, 1128–1132. [Google Scholar] [CrossRef]
- Sun, S.R.; Ning, X.X.; Zhai, Y.; Du, R.; Lu, Y.Y.; He, L.J.; Li, R.; Wu, W.N.; Sun, W.J.; Wang, H.M. Egr-1 Mediates Chronic Hypoxia-Induced Renal Interstitial Fibrosis via the PKC/ERK Pathway. Am. J. Nephrol. 2014, 39, 436–448. [Google Scholar] [CrossRef]
- Zhang, Q.Z.; Wu, Y.D.; Chau, C.H.; Ann, D.K.; Bertolami, C.N.; Le, A.D. Crosstalk of hypoxia-mediated signaling pathways in upregulating plasminogen activator inhibitor-1 expression in keloid fibroblasts. J. Cell Physiol. 2004, 199, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.Y.; Chen, J.; Xu, B.; Long, X.; Qin, H.; Zhao, R.C.; Wang, X.J. Keloid-derived keratinocytes acquire a fibroblast-like appearance and an enhanced invasive capacity in a hypoxic microenvironment in vitro. Int. J. Mol. Med. 2015, 35, 1246–1256. [Google Scholar] [CrossRef]
- Chang, L.F.; Karin, M. Mammalian MAP kinase signalling cascades. Nature 2001, 410, 37–40. [Google Scholar] [CrossRef] [PubMed]
- Leivonen, S.K.; Hakkinen, L.; Liu, D.; Kahari, V.M. Smad3 and ERK1/2 coordinately mediate transforming growth factor-beta-induced expression of connective tissue growth factor in human fibroblasts. J. Investig. Dermatol. 2005, 125, A66. [Google Scholar]
- Hu, Y.B.; Peng, J.W.; Feng, D.Y.; Chu, L.; Li, X.A.; Jin, Z.Y.; Lin, Z.; Zeng, Q.F. Role of extracellular signal-regulated kinase, p38 kinase, and activator protein-1 in transforming growth factor-beta 1-induced alpha smooth muscle actin expression in human fetal lung fibroblasts in vitro. Lung 2006, 184, 33–42. [Google Scholar] [CrossRef]
- Das, M.; Bouchey, D.M.; Moore, M.J.; Hopkins, D.C.; Nemenoff, R.A.; Stenmark, K.R. Hypoxia-induced proliferative response of vascular adventitial fibroblasts is dependent on G protein-mediated activation of mitogen-activated protein kinases. J. Biol. Chem. 2001, 276, 15631–15640. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.L.; Zhang, H.B.; Sun, L.; Gao, Y.Q.; Jin, H.F.; Liang, S.H.; Wang, Y.X.; Dong, M.Q.; Shi, Y.Q.; Li, Z.C.; et al. ERK/MAPK activation involves hypoxia-induced MGr1-Ag/37LRP expression and contributes to apoptosis resistance in gastric cancer. Int. J. Cancer 2010, 127, 820–829. [Google Scholar] [CrossRef]
- Hong, K.H.; Yoo, S.A.; Kang, S.S.; Choi, J.J.; Kim, W.U.; Cho, C.S. Hypoxia induces expression of connective tissue growth factor in scleroderma skin fibroblasts. Clin. Exp. Immunol. 2006, 146, 362–370. [Google Scholar] [CrossRef]
- Minet, E.; Arnould, T.; Michel, G.; Roland, I.; Mottet, D.; Raes, M.; Remacle, J.; Michiels, C. ERK activation upon hypoxia: Involvement in HIF-1 activation. FEBS Lett. 2000, 468, 53–58. [Google Scholar] [CrossRef]
- Lundgren, K.; Nordenskjold, B.; Landberg, G. Hypoxia, Snail and incomplete epithelial-mesenchymal transition in breast cancer. Br. J. Cancer 2009, 101, 1769–1781. [Google Scholar] [CrossRef]
- Tomasek, J.J.; Gabbiani, G.; Hinz, B.; Chaponnier, C.; Brown, R.A. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat. Rev. Mol. Cell Biol. 2002, 3, 349–363. [Google Scholar] [CrossRef]
- Hinz, B.; Celetta, G.; Tomasek, J.J.; Gabbiani, G.; Chaponnier, C. Alpha-smooth muscle actin expression upregulates fibroblast contractile activity. Mol. Biol. Cell 2001, 12, 2730–2741. [Google Scholar] [CrossRef]
- Shankar, J.; Nabi, I.R. Actin Cytoskeleton Regulation of Epithelial Mesenchymal Transition in Metastatic Cancer Cells. PLoS ONE 2015, 10, e0119954. [Google Scholar] [CrossRef]
- O’Toole, E.A.; Van Koningsveld, R.; Chen, M.; Woodley, D.T. Hypoxia induces epidermal keratinocyte matrix metalloproteinase-9 secretion via the protein kinase C pathway. J. Cell Physiol. 2008, 214, 47–55. [Google Scholar] [CrossRef]
- Kuwahara, H.; Tosa, M.; Egawa, S.; Murakami, M.; Mohammad, G.; Ogawa, R. Examination of Epithelial Mesenchymal Transition in Keloid Tissues and Possibility of Keloid Therapy Target. Prs-Glob. Open 2016, 4, e1138. [Google Scholar] [CrossRef]
- Liu, M.N.; Ning, X.X.; Li, R.; Yang, Z.; Yang, X.X.; Sun, S.R.; Qian, Q. Signalling pathways involved in hypoxia-induced renal fibrosis. J. Cell Mol. Med. 2017, 21, 1248–1259. [Google Scholar] [CrossRef]
- Leask, A. MEK/ERK inhibitors: Proof-of-concept studies in lung fibrosis. J. Cell Commun. Signal. 2012, 6, 59–60. [Google Scholar] [CrossRef]
- Samatar, A.A.; Poulikakos, P.I. Targeting RAS-ERK signalling in cancer: Promises and challenges. Nat. Rev. Drug Discov. 2014, 13, 928. [Google Scholar] [CrossRef]
- Kim, J.; Park, J.C.; Lee, M.H.; Yang, C.E.; Lee, J.H.; Lee, W.J. High-Mobility Group Box 1 Mediates Fibroblast Activity via RAGE-MAPK and NF-B Signaling in Keloid Scar Formation. Int. J. Mol. Sci 2018, 19, 76. [Google Scholar] [CrossRef]
- Yun, I.S.; Lee, M.H.; Rah, D.K.; Lew, D.H.; Park, J.C.; Lee, W.J. Heat Shock Protein 90 Inhibitor (17-AAG) Induces Apoptosis and Decreases Cell Migration/Motility of Keloid Fibroblasts. Plast. Reconstr. Surg. 2015, 136, 44e–53e. [Google Scholar] [CrossRef]
- Pakyari, M.; Farrokhi, A.; Maharlooei, M.K.; Ghahary, A. Critical Role of Transforming Growth Factor Beta in Different Phases of Wound Healing. Adv. Wound Care 2013, 2, 215–224. [Google Scholar] [CrossRef] [Green Version]
- Higgins, D.F.; Biju, M.P.; Akai, Y.; Wutz, A.; Johnson, R.S.; Haase, V.H. Hypoxic induction of Ctgf is directly mediated by Hif-1. Am. J. Physiol. Ren. 2004, 287, F1223–F1232. [Google Scholar] [CrossRef]
- Cheng, Y.; Lin, C.H.; Chen, J.Y.; Li, C.H.; Liu, Y.T.; Chen, B.C. Induction of Connective Tissue Growth Factor Expression by Hypoxia in Human Lung Fibroblasts via the MEKK1/MEK1/ERK1/GLI-1/GLI-2 and AP-1 Pathways. PLoS ONE 2016, 11, e0160593. [Google Scholar] [CrossRef]
- Kakudo, N.; Morimoto, N.; Ogawa, T.; Taketani, S.; Kusumoto, K. Hypoxia Enhances Proliferation of Human Adipose-Derived Stem Cells via HIF-1a Activation. PLoS ONE 2015, 10, e0139890. [Google Scholar] [CrossRef]
- Fotia, C.; Massa, A.; Boriani, F.; Baldini, N.; Granchi, D. Hypoxia enhances proliferation and stemness of human adipose-derived mesenchymal stem cells. Cytotechnology 2015, 67, 1073–1084. [Google Scholar] [CrossRef]
- Kietzmann, T.; Mennerich, D.; Dimova, E. Hypoxia-Inducible Factors (HIFs) and Phosphorylation: Impact on Stability, Localization, and Transactivity. Front. Cell Dev. Biol. 2016, 4, 11. [Google Scholar] [CrossRef]
- Sumbayev, V.V.; Yasinska, I.M. Regulation of MAP kinase-dependent apoptotic pathway: Implication of reactive oxygen and nitrogen species. Arch. Biochem. Biophys. 2005, 436, 406–412. [Google Scholar] [CrossRef]
- Wang, B.; Jiang, H.Y.; Ma, N.; Wang, Y.J. Phosphorylated-p38 mitogen-activated protein kinase expression is associated with clinical factors in invasive breast cancer. SpringerPlus 2016, 5, 934. [Google Scholar] [CrossRef]
- Lue, H.Q.; Kapurniotu, A.; Fingerle-Rowson, G.; Roger, T.; Leng, L.; Thiele, M.; Calandra, T.; Bucala, R.; Bernhagen, J. Rapid and transient activation of the ERK MAPK signalling pathway by macrophage migration inhibitory factor (MIF) and dependence on JAB1/CSN5 and Src kinase activity. Cell Signal. 2006, 18, 688–703. [Google Scholar] [CrossRef]
- Principe, D.R.; Diaz, A.M.; Torres, C.; Mangan, R.J.; DeCant, B.; McKinney, R.; Tsao, M.S.; Lowy, A.; Munshi, H.G.; Jung, B.; et al. TGFbeta engages MEK/ERK to differentially regulate benign and malignant pancreas cell function. Oncogene 2017, 36, 4336–4348. [Google Scholar] [CrossRef]
- Lu, X.; Law, B.K.; Chytil, A.M.; Brown, K.A.; Aakre, M.E.; Moses, H.L. Activation of the Erk pathway is required for TGF-beta 1-induced EMT in vitro. Neoplasia 2004, 6, 603–610. [Google Scholar] [CrossRef]
- Mottet, D.; Dumont, V.; Deccache, Y.; Demazy, C.; Ninane, N.; Raes, M.; Michiels, C. Regulation of hypoxia-inducible factor-1 alpha protein level during hypoxic conditions by the phosphatidylinositol 3-kinase/Akt/glycogen synthase kinase 3 beta pathway in HepG2 cells. J. Biol. Chem. 2003, 278, 31277–31285. [Google Scholar] [CrossRef]
- Jun, E.K.; Zhang, Q.; Yoon, B.S.; Moon, J.H.; Lee, G.; Park, G.; Kang, P.J.; Lee, J.H.; Kim, A.; You, S. Hypoxic Conditioned Medium from Human Amniotic Fluid-Derived Mesenchymal Stem Cells Accelerates Skin Wound Healing through TGF-beta/SMAD2 and PI3K/Akt Pathways. Int. J. Mol. Sci. 2014, 15, 605–628. [Google Scholar] [CrossRef]
- Wang, W.; Qu, M.; Xu, L.; Wu, X.; Gao, Z.; Gu, T.; Zhang, W.; Ding, X.; Liu, W.; Chen, Y.L. Sorafenib exerts an anti-keloid activity by antagonizing TGF-beta/Smad and MAPK/ERK signaling pathways. J. Mol. Med. 2016, 94, 1181–1194. [Google Scholar] [CrossRef]
- Soini, T.; Eloranta, K.; Pihlajoki, M.; Kyronlahti, A.; Akinrinade, O.; Andersson, N.; Lohi, J.; Pakarinen, M.P.; Wilson, D.B.; Heikinheimo, M. Transcription factor GATA4 associates with mesenchymal-like gene expression in human hepatoblastoma cells. Tumour Biol. 2018, 40. [Google Scholar] [CrossRef] [Green Version]
- Xiong, Y.; Liu, Y.; Xiong, W.Q.; Zhang, L.; Liu, H.W.; Du, Y.; Li, N. Hypoxia-inducible factor 1 alpha-induced epithelial-mesenchymal transition of endometrial epithelial cells may contribute to the development of endometriosis. Hum. Reprod. 2016, 31, 1327–1338. [Google Scholar] [CrossRef]
- Rao, Q.; Chen, Y.; Yeh, C.R.; Ding, J.; Li, L.; Chang, C.S.; Yeh, S.Y. Recruited mast cells in the tumor microenvironment enhance bladder cancer metastasis via modulation of ER beta/CCL2/CCR2 EMT/MMP9 signals. Oncotarget 2016, 7, 7842–7855. [Google Scholar] [CrossRef]
- Chen, S.; Liu, J.; Yang, M.; Lai, W.; Ye, L.; Chen, J.; Hou, X.; Ding, H.; Zhang, W.; Wu, Y.; et al. Fn14, a Downstream Target of the TGF-beta Signaling Pathway, Regulates Fibroblast Activation. PLoS ONE 2015, 10, e0143802. [Google Scholar] [CrossRef]
- Yalu, R.; Oyesiji, A.E.; Eisenberg, I.; Imbar, T.; Meidan, R. HIF1A-dependent increase in endothelin 2 levels in granulosa cells: Role of hypoxia, LH/cAMP, and reactive oxygen species. Reproduction 2015, 149, 11–20. [Google Scholar] [CrossRef]
- Wang, H.H.; Zhong, Q.; Yang, T.S.; Qi, Y.; Fu, M.C.; Yang, X.; Qiao, L.; Ling, Q.; Liu, S.F.; Zhao, Y.M. Comparative characterization of SHED and DPSCs during extended cultivation in vitro. Mol. Med. Rep. 2018, 17, 6551–6559. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target Gene | Primer Sequences (5′–3′) or Assay ID | Reference |
|---|---|---|
| E-cadherin | Forward: CACCACGGGCTTGGATTTTG Reverse: TGGGGGCTTCATTCACATCC | [47] |
| N-cadherin | Forward: TCAGGCGTCTGTAGAGGCTT Reverse: ATGCACATCCTTCGATAAGACTG | [48] |
| Vimentin | Forward: GACGCCATCAACACCGAGTT Reverse: CTTTGTCGTTGGTTAGCTGGT | [49] |
| TGF-beta | Forward: ACCCACAACGAAATCTATGACA Reverse: GCTGAGGTATCGCCAGGAAT | [50] |
| HIF1A | Forward: ACTCATCCATGTGACCACG Reverse: TAGTTCTCCCCCGGCTAG | [51] |
| COL1A1 | Forward: AAGGTGTTGTGCGATGACG Reverse: TGGTCGGTGGGTGACTCTG | [50] |
| Beta-actin | Forward: CTACCTCATGAAGATCCTCACCGA Reverse: TTCTCCTTAATGTCACGCACGATT | [52] |
| CTGF | Hs00170014_m1 | |
| MMP9 | Hs00957562_m1 | |
| MMP2 | Hs01548727_m1 | |
| TIMP2 | HS00234278_m1 | |
| TIMP1 | Hs01092512_g1 | |
| COL1A1 | Hs00164004_m1 | |
| Beta-actin | Hs01060665_g1 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, J.; Kim, B.; Kim, S.M.; Yang, C.E.; Song, S.Y.; Lee, W.J.; Lee, J.H. Hypoxia-Induced Epithelial-To-Mesenchymal Transition Mediates Fibroblast Abnormalities via ERK Activation in Cutaneous Wound Healing. Int. J. Mol. Sci. 2019, 20, 2546. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20102546
Kim J, Kim B, Kim SM, Yang CE, Song SY, Lee WJ, Lee JH. Hypoxia-Induced Epithelial-To-Mesenchymal Transition Mediates Fibroblast Abnormalities via ERK Activation in Cutaneous Wound Healing. International Journal of Molecular Sciences. 2019; 20(10):2546. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20102546
Chicago/Turabian StyleKim, Jihee, Bomi Kim, Soo Min Kim, Chae Eun Yang, Seung Yong Song, Won Jai Lee, and Ju Hee Lee. 2019. "Hypoxia-Induced Epithelial-To-Mesenchymal Transition Mediates Fibroblast Abnormalities via ERK Activation in Cutaneous Wound Healing" International Journal of Molecular Sciences 20, no. 10: 2546. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20102546