Prostate Cancer and Bone Metastases: The Underlying Mechanisms

 , ,
, ,  , and
, and 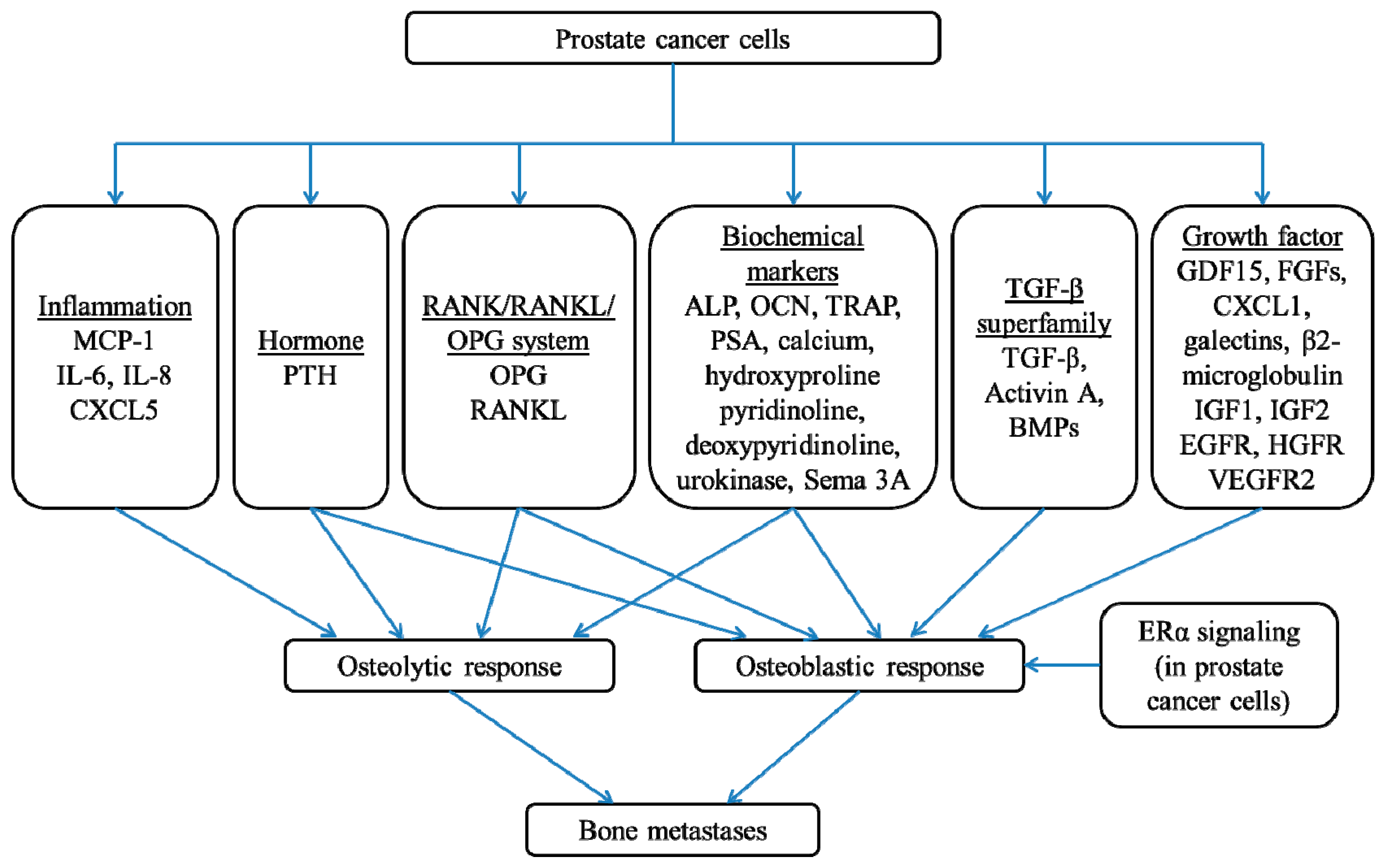{kind=link}
Abstract
:1. Introduction
2. The Underlying Molecular Mechanisms in Prostate Cancer Bone Metastases
2.1. The Role of Parathyroid Hormone (PTH)
2.2. The Role of the RANK/RANKL/OPG System
2.3. The Role of the TGF-β Signaling Axis
2.4. The Role of Bone Morphogenetic Protein (BMP)
2.5. The Role of Other Growth Factors
2.6. The Role of Inflammation
2.7. The Role of Biochemical Markers
2.8. The Role of Estrogen
3. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ALP | Alkaline phosphatase |
| AR | Androgen receptor |
| BMC | Bone mineral content |
| BMD | Bone mineral density |
| BMP | Bone morphogenetic protein |
| CTP | Pyridinoline cross-linked carboxyterminal telopeptide |
| CXCL1 | C-X-C motif chemokine ligand 1 |
| CXCL5 | C-X-C motif chemokine ligand 5 |
| EGFR | Epidermal growth factor receptor |
| ERα | Estrogen receptor-alpha |
| ERBB2 | Erb-B2 receptor tyrosine kinase 2 |
| ET-1 | Endothelin-1 |
| ETAR | Endothelin-A receptor |
| FDA | Food and Drug Administration |
| FGF | Fibroblast growth factor |
| GDF15 | Growth differentiation factor 15 |
| HGF | Hepatocyte growth factor |
| HGFR | Hepatocyte growth factor receptor |
| IGF | Insulin-like growth factor |
| IL-8 | Interleukin-8 |
| MAPK | Mitogen-activated protein kinase |
| MCP-1 | Monocyte chemotactic protein-1 |
| M-CSF | Macrophage-colony stimulating factors |
| NF-κB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| OCN | Osteocalcin |
| OHS | Osteoblast-derived sarcoma cells |
| OPG | Osteoprotegerin |
| PSA | Prostate-specific antigen |
| PTH | Parathyroid hormone |
| PTHrP | Parathyroid hormone-related peptide |
| RANK | Receptor activator of nuclear factor-kappa B |
| RANKL | Receptor activator of nuclear factor-kappa B ligand |
| Runx2 | Runt-related transcription factor 2 |
| SCID | Severe combined immunodeficient |
| Sema 3A | Semaphorin 3A |
| STAT3 | Signal transducer and activator of transcription 3 |
| TGF-β | Transforming growth factor-beta |
| TRAP | Tartrate-resistant acid phosphatase |
| VEGF-A | Vascular endothelial growth factor-A |
| VEGFR2 | Vascular endothelial growth factor receptor 2 |
| Wnt | Wingless |
References
- Baade, P.D.; Youlden, D.R.; Cramb, S.M.; Dunn, J.; Gardiner, R.A. Epidemiology of prostate cancer in the Asia-Pacific region. Prostate Int. 2013, 1, 47–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bubendorf, L.; Schöpfer, A.; Wagner, U.; Sauter, G.; Moch, H.; Willi, N.; Gasser, T.C.; Mihatsch, M.J. Metastatic patterns of prostate cancer: An autopsy study of 1,589 patients. Hum. Pathol. 2000, 31, 578–583. [Google Scholar] [CrossRef]
- Paget, S. The distribution of secondary growths in cancer of the breast. Lancet 1889, 133, 571–573. [Google Scholar] [CrossRef]
- Bagi, C. Skeletal implications of prostate cancer. J. Musculoskelet. Neuronal. Interact 2003, 3, 112–117. [Google Scholar] [PubMed]
- Kingsley, L.A.; Fournier, P.G.; Chirgwin, J.M.; Guise, T.A. Molecular biology of bone metastasis. Mol Cancer Ther. 2007, 6, 2609–2617. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.J.; Pollock, C.B.; Kelly, K. Mechanisms of cancer metastasis to the bone. Cell Res. 2005, 15, 57. [Google Scholar] [CrossRef] [PubMed]
- Westendorf, J.J.; Kahler, R.A.; Schroeder, T.M. Wnt signaling in osteoblasts and bone diseases. Gene 2004, 341, 19–39. [Google Scholar] [CrossRef] [PubMed]
- Roodman, G.D. Mechanisms of bone metastasis. N. Engl. J. Med. 2004, 350, 1655–1664. [Google Scholar] [CrossRef]
- Abou-Samra, A.B.; Juppner, H.; Force, T.; Freeman, M.W.; Kong, X.F.; Schipani, E.; Urena, P.; Richards, J.; Bonventre, J.V.; Potts, J.T., Jr.; et al. Expression cloning of a common receptor for parathyroid hormone and parathyroid hormone-related peptide from rat osteoblast-like cells: A single receptor stimulates intracellular accumulation of both cAMP and inositol trisphosphates and increases intracellular free calcium. Proc. Natl. Acad. Sci. USA 1992, 89, 2732–2736. [Google Scholar] [PubMed]
- Hall, C.L.; Kang, S.; MacDougald, O.A.; Keller, E.T. Role of Wnts in prostate cancer bone metastases. J. Cell Biochem. 2006, 97, 661–672. [Google Scholar] [CrossRef]
- Ellis, W.J.; Vessella, R.L.; Buhler, K.R.; Bladou, F.; True, L.D.; Bigler, S.A.; Curtis, D.; Lange, P.H. Characterization of a novel androgen-sensitive, prostate-specific antigen-producing prostatic carcinoma xenograft: LuCaP 23. Clin. Cancer Res. 1996, 2, 1039–1048. [Google Scholar]
- Lim, D.J.; Liu, X.L.; Sutkowski, D.M.; Braun, E.J.; Lee, C.; Kozlowski, J.M. Growth of an androgen-sensitive human prostate cancer cell line, LNCaP, in nude mice. Prostate 1993, 22, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Liu, A.Y.; Brubaker, K.D.; Goo, Y.A.; Quinn, J.E.; Kral, S.; Sorensen, C.M.; Vessella, R.L.; Belldegrun, A.S.; Hood, L.E. Lineage relationship between LNCaP and LNCaP-derived prostate cancer cell lines. Prostate 2004, 60, 98–108. [Google Scholar] [CrossRef] [PubMed]
- Li, T.-H.; Zhao, H.; Peng, Y.; Beliakoff, J.; Brooks, J.D.; Sun, Z. A promoting role of androgen receptor in androgen-sensitive and -insensitive prostate cancer cells. Nucleic Acids Res. 2007, 35, 2767–2776. [Google Scholar] [CrossRef] [PubMed]
- Al Nakouzi, N.; Bawa, O.; Le Pape, A.; Lerondel, S.; Gaudin, C.; Opolon, P.; Gonin, P.; Fizazi, K.; Chauchereau, A. The IGR-CaP1 xenograft model recapitulates mixed osteolytic/blastic bone lesions observed in metastatic prostate cancer. Neoplasia 2012, 14, 376–387. [Google Scholar] [CrossRef]
- Huang, J.C.; Sakata, T.; Pfleger, L.L.; Bencsik, M.; Halloran, B.P.; Bikle, D.D.; Nissenson, R.A. PTH differentially regulates expression of RANKL and OPG. J. Bone Miner Res. 2004, 19, 235–244. [Google Scholar] [CrossRef]
- Kandil, E.; Noureldine, S.; Khalek, M.A.; Daroca, P.; Friedlander, P. Ectopic secretion of parathyroid hormone in a neuroendocrine tumor: A case report and review of the literature. Int. J. Clin. Exp. Med. 2011, 4, 234–240. [Google Scholar]
- Demura, M.; Yoneda, T.; Wang, F.; Zen, Y.; Karashima, S.; Zhu, A.; Cheng, Y.; Yamagishi, M.; Takeda, Y. Ectopic production of parathyroid hormone in a patient with sporadic medullary thyroid cancer. Endocr. J. 2010, 57, 161–170. [Google Scholar] [CrossRef]
- Nussbaum, S.R.; Gaz, R.D.; Arnold, A. Hypercalcemia and ectopic secretion of parathyroid hormone by an ovarian carcinoma with rearrangement of the gene for parathyroid hormone. N. Engl. J. Med. 1990, 323, 1324–1328. [Google Scholar] [CrossRef]
- Yoshimoto, K.; Yamasaki, R.; Sakai, H.; Tezuka, U.; Takahashi, M.; Iizuka, M.; Sekiya, T.; Saito, S. Ectopic production of parathyroid hormone by small cell lung cancer in a patient with hypercalcemia. J. Clin. Endocrinol. Metab. 1989, 68, 976–981. [Google Scholar] [CrossRef] [PubMed]
- Strewler, G.J.; Budayr, A.A.; Clark, O.H.; Nissenson, R.A. Production of parathyroid hormone by a malignant nonparathyroid tumor in a hypercalcemic patient. J. Clin. Endocrinol. Metab. 1993, 76, 1373–1375. [Google Scholar] [PubMed]
- Schwartz, G.G. Prostate cancer, serum parathyroid hormone, and the progression of skeletal metastases. Cancer Epidemiol. Biomark. Prev. 2008, 17, 478–483. [Google Scholar] [CrossRef]
- Cooper, E.; Whelan, P.; Purves, D. Bone alkaline phosphatase and prostate-specific antigen in the monitoring of prostate cancer. Prostate 1994, 25, 236–242. [Google Scholar] [CrossRef]
- Kylmälä, T.; Tammela, T.; Risteli, L.; Risteli, J.; Kontturi, M.; Elomaa, I. Type I collagen degradation product (ICTP) gives information about the nature of bone metastases and has prognostic value in prostate cancer. Br. J. Cancer 1995, 71, 1061. [Google Scholar] [CrossRef] [PubMed]
- Philbrick, W.; Wysolmerski, J.; Galbraith, S.; Holt, E.; Orloff, J.; Yang, K.; Vasavada, R.; Weir, E.; Broadus, A.; Stewart, A. Defining the roles of parathyroid hormone-related protein in normal physiology. Physiol. Rev. 1996, 76, 127–173. [Google Scholar] [CrossRef]
- Blomme, E.A.; Dougherty, K.M.; Pienta, K.J.; Capen, C.C.; Rosol, T.J.; McCauley, L.K. Skeletal metastasis of prostate adenocarcinoma in rats: Morphometric analysis and role of parathyroid hormone-related protein. Prostate 1999, 39, 187–197. [Google Scholar] [CrossRef]
- Rabbani, S.A.; Gladu, J.; Harakidas, P.; Jamison, B.; Goltzman, D. Over-production of parathyroid hormone-related peptide results in increased osteolytic skeletal metastasis by prostate cancer cells in vivo. Int. J. Cancer 1999, 80, 257–264. [Google Scholar] [CrossRef]
- Liao, J.; Li, X.; Koh, A.J.; Berry, J.E.; Thudi, N.; Rosol, T.J.; Pienta, K.J.; McCauley, L.K. Tumor expressed PTHrP facilitates prostate cancer-induced osteoblastic lesions. Int. J. Cancer 2008, 123, 2267–2278. [Google Scholar] [CrossRef] [Green Version]
- Corey, E.; Brown, L.G.; Kiefer, J.A.; Quinn, J.E.; Pitts, T.E.; Blair, J.M.; Vessella, R.L. Osteoprotegerin in prostate cancer bone metastasis. Cancer Res. 2005, 65, 1710–1718. [Google Scholar] [CrossRef] [PubMed]
- Kiefer, J.A.; Vessella, R.L.; Quinn, J.E.; Odman, A.M.; Zhang, J.; Keller, E.T.; Kostenuik, P.J.; Dunstan, C.R.; Corey, E. The effect of osteoprotegerin administration on the intra-tibial growth of the osteoblastic LuCaP 23.1 prostate cancer xenograft. Clin. Exp. Metast. 2004, 21, 381–387. [Google Scholar] [CrossRef]
- Zhang, J.; Dai, J.; Qi, Y.; Lin, D.L.; Smith, P.; Strayhorn, C.; Mizokami, A.; Fu, Z.; Westman, J.; Keller, E.T. Osteoprotegerin inhibits prostate cancer-induced osteoclastogenesis and prevents prostate tumor growth in the bone. J. Clin. Investig. 2001, 107, 1235–1244. [Google Scholar] [CrossRef] [PubMed]
- Powell, G.J.; Southby, J.; Danks, J.A.; Stillwell, R.G.; Hayman, J.A.; Henderson, M.A.; Bennett, R.C.; Martin, T.J. Localization of parathyroid hormone-related protein in breast cancer metastases: Increased incidence in bone compared with other sites. Cancer Res. 1991, 51, 3059–3061. [Google Scholar] [PubMed]
- Ye, Y.; Li, S.L.; Ma, Y.Y.; Diao, Y.J.; Yang, L.; Su, M.Q.; Li, Z.; Ji, Y.; Wang, J.; Lei, L.; et al. Exosomal miR-141-3p regulates osteoblast activity to promote the osteoblastic metastasis of prostate cancer. Proc. Natl. Acad. Sci. USA 2017, 8, 94834–94849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yonou, H.; Horiguchi, Y.; Ohno, Y.; Namiki, K.; Yoshioka, K.; Ohori, M.; Hatano, T.; Tachibana, M. Prostate-specific antigen stimulates osteoprotegerin production and inhibits receptor activator of nuclear factor-kappaB ligand expression by human osteoblasts. Prostate 2007, 67, 840–848. [Google Scholar] [CrossRef] [PubMed]
- Katopodis, H.; Philippou, A.; Tenta, R.; Doillon, C.; Papachroni, K.K.; Papavassiliou, A.G.; Koutsilieris, M. MG-63 osteoblast-like cells enhance the osteoprotegerin expression of PC-3 prostate cancer cells. Anticancer Res. 2009, 29, 4013–4018. [Google Scholar] [PubMed]
- Armstrong, A.P.; Miller, R.E.; Jones, J.C.; Zhang, J.; Keller, E.T.; Dougall, W.C. RANKL acts directly on RANK-expressing prostate tumor cells and mediates migration and expression of tumor metastasis genes. Prostate 2008, 68, 92–104. [Google Scholar] [CrossRef]
- Whang, P.G.; Schwarz, E.M.; Gamradt, S.C.; Dougall, W.C.; Lieberman, J.R. The effects of RANK blockade and osteoclast depletion in a model of pure osteoblastic prostate cancer metastasis in bone. J. Orthop. Res. 2005, 23, 1475–1483. [Google Scholar] [CrossRef]
- Barrow, J.R.; Thomas, K.R.; Boussadia-Zahui, O.; Moore, R.; Kemler, R.; Capecchi, M.R.; McMahon, A.P. Ectodermal Wnt3/β-catenin signaling is required for the establishment and maintenance of the apical ectodermal ridge. Genes Dev. 2003, 17, 394–409. [Google Scholar] [CrossRef]
- Martinovic, S.; Borovecki, F.; Sampath, K.T.; Vukicevic, S. Biology of bone morphogenetic proteins. In Bone Morphogenetic Proteins; Springer: Berlin, Germany, 2002; pp. 87–119. [Google Scholar]
- Prins, G.S.; Korach, K.S. The role of estrogens and estrogen receptors in normal prostate growth and disease. Steroids 2008, 73, 233–244. [Google Scholar] [CrossRef] [Green Version]
- Leto, G.; Incorvaia, L.; Badalamenti, G.; Tumminello, F.M.; Gebbia, N.; Flandina, C.; Crescimanno, M.; Rini, G. Activin A circulating levels in patients with bone metastasis from breast or prostate cancer. Clin. Exp. Metast. 2006, 23, 117–122. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.C.; Gajdosik, M.S.; Josic, D.; Clifton, J.G.; Logothetis, C.; Yu-Lee, L.Y.; Gallick, G.E.; Maity, S.N.; Lin, S.H. Secretome analysis of an osteogenic prostate tumor identifies complex signaling networks mediating cross-talk of cancer and stromal cells within the tumor microenvironment. Mol. Cell Proteom. 2015, 14, 471–483. [Google Scholar] [CrossRef]
- Mishra, S.; Tang, Y.; Wang, L.; de Graffenried, L.; Yeh, I.T.; Werner, S.; Troyer, D.; Copland, J.A.; Sun, L.Z. Blockade of transforming growth factor-beta (TGFβ) signaling inhibits osteoblastic tumorigenesis by a novel human prostate cancer cell line. Prostate 2011, 71, 1441–1454. [Google Scholar] [CrossRef]
- Rafiei, S.; Komarova, S.V. Molecular signaling pathways mediating osteoclastogenesis induced by prostate cancer cells. BMC Cancer 2013, 13, 605. [Google Scholar] [CrossRef]
- AlShaibi, H.F.; Ahmed, F.; Buckle, C.; Fowles, A.C.M.; Awlia, J.; Cecchini, M.G.; Eaton, C.L. The BMP antagonist Noggin is produced by osteoblasts in response to the presence of prostate cancer cells. Biotechnol. Appl. Biochem. 2017. [Google Scholar] [CrossRef]
- Hogan, B. Bone morphogenetic proteins: Multifunctional regulators of vertebrate development. Genes Dev. 1996, 10, 1580–1594. [Google Scholar] [CrossRef]
- Bobinac, D.; Maric, I.; Zoricic, S.; Spanjol, J.; Dordevic, G.; Mustac, E.; Fuckar, Z. Expression of bone morphogenetic proteins in human metastatic prostate and breast cancer. Croat Med. J. 2005, 46, 389–396. [Google Scholar]
- Feeley, B.T.; Krenek, L.; Liu, N.; Hsu, W.K.; Gamradt, S.C.; Schwarz, E.M.; Huard, J.; Lieberman, J.R. Overexpression of noggin inhibits BMP-mediated growth of osteolytic prostate cancer lesions. Bone 2006, 38, 154–166. [Google Scholar] [CrossRef]
- Autzen, P.; Robson, C.N.; Bjartell, A.; Malcolm, A.J.; Johnson, M.I.; Neal, D.E.; Hamdy, F.C. Bone morphogenetic protein 6 in skeletal metastases from prostate cancer and other common human malignancies. Br. J. Cancer 1998, 78, 1219–1223. [Google Scholar] [CrossRef] [Green Version]
- Masuda, H.; Fukabori, Y.; Nakano, K.; Takezawa, Y.; T, C.S.; Yamanaka, H. Increased expression of bone morphogenetic protein-7 in bone metastatic prostate cancer. Prostate 2003, 54, 268–274. [Google Scholar] [CrossRef]
- Masuda, H.; Fukabori, Y.; Nakano, K.; Shimizu, N.; Yamanaka, H. Expression of bone morphogenetic protein-7 (BMP-7) in human prostate. Prostate 2004, 59, 101–106. [Google Scholar] [CrossRef]
- Dai, J.; Hall, C.L.; Escara-Wilke, J.; Mizokami, A.; Keller, J.M.; Keller, E.T. Prostate cancer induces bone metastasis through Wnt-induced bone morphogenetic protein-dependent and independent mechanisms. Cancer Res. 2008, 68, 5785–5794. [Google Scholar] [CrossRef]
- Li, Z.G.; Mathew, P.; Yang, J.; Starbuck, M.W.; Zurita, A.J.; Liu, J.; Sikes, C.; Multani, A.S.; Efstathiou, E.; Lopez, A.; et al. Androgen receptor-negative human prostate cancer cells induce osteogenesis in mice through FGF9-mediated mechanisms. J. Clin. Investig. 2008, 118, 2697–2710. [Google Scholar] [CrossRef]
- Ritchie, C.K.; Andrews, L.R.; Thomas, K.G.; Tindall, D.J.; Fitzpatrick, L.A. The effects of growth factors associated with osteoblasts on prostate carcinoma proliferation and chemotaxis: Implications for the development of metastatic disease. Endocrinology 1997, 138, 1145–1150. [Google Scholar] [CrossRef]
- Bratland, A.; Boender, P.J.; Hoifodt, H.K.; Ostensen, I.H.; Ruijtenbeek, R.; Wang, M.Y.; Berg, J.P.; Lilleby, W.; Fodstad, O.; Ree, A.H. Osteoblast-induced EGFR/ERBB2 signaling in androgen-sensitive prostate carcinoma cells characterized by multiplex kinase activity profiling. Clin. Exp. Metast. 2009, 26, 485–496. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.; Whang, Y.M.; Campbell, P.; Mulcrone, P.L.; Elefteriou, F.; Cho, S.W.; Park, S.I. Dual targeting c-met and VEGFR2 in osteoblasts suppresses growth and osteolysis of prostate cancer bone metastasis. Cancer Lett. 2018, 414, 205–213. [Google Scholar] [CrossRef]
- Azrina, A.; Khoo, H.E.; Idris, M.A.; Amin, I.; Razman, M.R. Major inorganic elements in tap water samples in Peninsular Malaysia. Malays. J. Nutr. 2011, 17, 271–276. [Google Scholar] [PubMed]
- Wheater, G.; Elshahaly, M.; Tuck, S.P.; Datta, H.K.; van Laar, J.M. The clinical utility of bone marker measurements in osteoporosis. J. Transl. Med. 2013, 11, 201. [Google Scholar] [CrossRef]
- Lu, Y.; Cai, Z.; Xiao, G.; Keller, E.T.; Mizokami, A.; Yao, Z.; Roodman, G.D.; Zhang, J. Monocyte chemotactic protein-1 mediates prostate cancer-induced bone resorption. Cancer Res. 2007, 67, 3646–3653. [Google Scholar] [CrossRef]
- Lu, Y.; Chen, Q.; Corey, E.; Xie, W.; Fan, J.; Mizokami, A.; Zhang, J. Activation of MCP-1/CCR2 axis promotes prostate cancer growth in bone. Clin. Exp. Metast. 2009, 26, 161–169. [Google Scholar] [CrossRef]
- Morrissey, C.; Lai, J.S.; Brown, L.G.; Wang, Y.C.; Roudier, M.P.; Coleman, I.M.; Gulati, R.; Vakar-Lopez, F.; True, L.D.; Corey, E.; et al. The expression of osteoclastogenesis-associated factors and osteoblast response to osteolytic prostate cancer cells. Prostate 2010, 70, 412–424. [Google Scholar] [CrossRef]
- Southby, J.; Kissin, M.W.; Danks, J.A.; Hayman, J.A.; Moseley, J.M.; Henderson, M.A.; Bennett, R.C.; Martin, T.J. Immunohistochemical localization of parathyroid hormone-related protein in human breast cancer. Cancer Res. 1990, 50, 7710–7716. [Google Scholar] [PubMed]
- Logothetis, C.J.; Lin, S.H. Osteoblasts in prostate cancer metastasis to bone. Nat. Rev. Cancer 2005, 5, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Maeda, H.; Koizumi, M.; Yoshimura, K.; Yamauchi, T.; Kawai, T.; Ogata, E. Correlation between bone metabolic markers and bone scan in prostatic cancer. J. Urol. 1997, 157, 539–543. [Google Scholar] [CrossRef]
- Sano, M.; Kushida, K.; Takahashi, M.; Ohishi, T.; Kawana, K.; Okada, M.; Inoue, T. Urinary pyridinoline and deoxypyridinoline in prostate carcinoma patients with bone metastasis. Br. J. Cancer 1994, 70, 701–703. [Google Scholar] [CrossRef] [Green Version]
- Revilla, M.; Arribas, I.; Sanchez-Chapado, M.; Villa, L.F.; Bethencourt, F.; Rico, H. Total and regional bone mass and biochemical markers of bone remodeling in metastatic prostate cancer. Prostate 1998, 35, 243–247. [Google Scholar] [CrossRef]
- Achbarou, A.; Kaiser, S.; Tremblay, G.; Ste-Marie, L.G.; Brodt, P.; Goltzman, D.; Rabbani, S.A. Urokinase overproduction results in increased skeletal metastasis by prostate cancer cells in vivo. Cancer Res. 1994, 54, 2372–2377. [Google Scholar] [PubMed]
- Lin, S.H.; Lee, Y.C.; Choueiri, M.B.; Wen, S.; Mathew, P.; Ye, X.; Do, K.A.; Navone, N.M.; Kim, J.; Tu, S.M.; et al. Soluble ErbB3 levels in bone marrow and plasma of men with prostate cancer. Clin. Cancer Res. 2008, 14, 3729–3736. [Google Scholar] [CrossRef]
- Liu, F.; Shen, W.; Qiu, H.; Hu, X.; Zhang, C.; Chu, T. Prostate cancer cells induce osteoblastic differentiation via semaphorin 3A. Prostate 2015, 75, 370–380. [Google Scholar] [CrossRef] [PubMed]
- Bosland, M.C.; Ford, H.; Horton, L. Induction at high incidence of ductal prostate adenocarcinomas in NBL/Cr and Sprague-Dawley Hsd:SD rats treated with a combination of testosterone and estradiol-17 beta or diethylstilbestrol. Carcinogenesis 1995, 16, 1311–1317. [Google Scholar] [CrossRef]
- Ricke, W.A.; McPherson, S.J.; Bianco, J.J.; Cunha, G.R.; Wang, Y.; Risbridger, G.P. Prostatic hormonal carcinogenesis is mediated by in situ estrogen production and estrogen receptor alpha signaling. FASEB J. 2008, 22, 1512–1520. [Google Scholar] [CrossRef]
- Khosla, S.; Oursler, M.J.; Monroe, D.G. Estrogen and the skeleton. Trends Endocrinol. Metab. 2012, 23, 576–581. [Google Scholar] [CrossRef] [Green Version]
- Streicher, C.; Heyny, A.; Andrukhova, O.; Haigl, B.; Slavic, S.; Schüler, C.; Kollmann, K.; Kantner, I.; Sexl, V.; Kleiter, M.; et al. Estrogen Regulates Bone Turnover by Targeting RANKL Expression in Bone Lining Cells. Sci. Rep. 2017, 7, 6460. [Google Scholar] [CrossRef] [PubMed]
- Mishra, S.; Tai, Q.; Gu, X.; Schmitz, J.; Poullard, A.; Fajardo, R.J.; Mahalingam, D.; Chen, X.; Zhu, X.; Sun, L.Z. Estrogen and estrogen receptor alpha promotes malignancy and osteoblastic tumorigenesis in prostate cancer. Oncotarget 2015, 6, 44388–44402. [Google Scholar] [CrossRef]
- Suva, L.J.; Washam, C.; Nicholas, R.W.; Griffin, R.J. Bone metastasis: Mechanisms and therapeutic opportunities. Nat. Rev. Endocrinol. 2011, 7, 208–218. [Google Scholar] [CrossRef]
- Smith, M.R.; Egerdie, B.; Hernandez Toriz, N.; Feldman, R.; Tammela, T.L.; Saad, F.; Heracek, J.; Szwedowski, M.; Ke, C.; Kupic, A.; et al. Denosumab in men receiving androgen-deprivation therapy for prostate cancer. N. Engl. J. Med. 2009, 361, 745–755. [Google Scholar] [CrossRef]
- Smith, M.R.; Eastham, J.; Gleason, D.M.; Shasha, D.; Tchekmedyian, S.; Zinner, N. Randomized controlled trial of zoledronic acid to prevent bone loss in men receiving androgen deprivation therapy for nonmetastatic prostate cancer. J. Urol. 2003, 169, 2008–2012. [Google Scholar] [CrossRef] [PubMed]
- Michaelson, M.D.; Kaufman, D.S.; Lee, H.; McGovern, F.J.; Kantoff, P.W.; Fallon, M.A.; Finkelstein, J.S.; Smith, M.R. Randomized controlled trial of annual zoledronic acid to prevent gonadotropin-releasing hormone agonist-induced bone loss in men with prostate cancer. J. Clin. Oncol. 2007, 25, 1038–1042. [Google Scholar] [CrossRef]
- Smith, M.R.; Saad, F.; Coleman, R.; Shore, N.; Fizazi, K.; Tombal, B.; Miller, K.; Sieber, P.; Karsh, L.; Damiao, R.; et al. Denosumab and bone-metastasis-free survival in men with castration-resistant prostate cancer: Results of a phase 3, randomised, placebo-controlled trial. Lancet 2012, 379, 39–46. [Google Scholar] [CrossRef]
- Hussain, M.; Smith, M.; Sweeney, C.; Corn, P.; Elfiky, A.; Gordon, M.; Haas, N.; Harzstark, A.; Kurzrock, R.; Lara, P. Cabozantinib (XL184) in metastatic castration-resistant prostate cancer (mCRPC): Results from a phase II randomized discontinuation trial. J. Clin. Oncol. 2011, 29, 4516. [Google Scholar] [CrossRef]
- Araujo, J.C.; Mathew, P.; Armstrong, A.J.; Braud, E.L.; Posadas, E.; Lonberg, M.; Gallick, G.E.; Trudel, G.C.; Paliwal, P.; Agrawal, S.; et al. Dasatinib combined with docetaxel for castration-resistant prostate cancer: Results from a phase 1-2 study. Cancer 2012, 118, 63–71. [Google Scholar] [CrossRef]
- Bagnato, A.; Spinella, F.; Rosano, L. The endothelin axis in cancer: The promise and the challenges of molecularly targeted therapy. Can. J. Physiol. Pharmacol. 2008, 86, 473–484. [Google Scholar] [CrossRef] [PubMed]
- Qiao, L.; Liang, Y.; Li, N.; Hu, X.; Luo, D.; Gu, J.; Lu, Y.; Zheng, Q. Endothelin-A receptor antagonists in prostate cancer treatment-a meta-analysis. Int. J. Clin. Exp. Med. 2015, 8, 3465–3473. [Google Scholar] [PubMed]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wong, S.K.; Mohamad, N.-V.; Giaze, T.R.; Chin, K.-Y.; Mohamed, N.; Ima-Nirwana, S. Prostate Cancer and Bone Metastases: The Underlying Mechanisms. Int. J. Mol. Sci. 2019, 20, 2587. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20102587
Wong SK, Mohamad N-V, Giaze TR, Chin K-Y, Mohamed N, Ima-Nirwana S. Prostate Cancer and Bone Metastases: The Underlying Mechanisms. International Journal of Molecular Sciences. 2019; 20(10):2587. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20102587
Chicago/Turabian StyleWong, Sok Kuan, Nur-Vaizura Mohamad, Tijjani Rabiu Giaze, Kok-Yong Chin, Norazlina Mohamed, and Soelaiman Ima-Nirwana. 2019. "Prostate Cancer and Bone Metastases: The Underlying Mechanisms" International Journal of Molecular Sciences 20, no. 10: 2587. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20102587