Ion Transporters, Channelopathies, and Glucose Disorders
by
 ,
,
Huseyin Demirbilek
1, 
Sonya Galcheva
2,
Dogus Vuralli
1,
Sara Al-Khawaga
3,4 and
Khalid Hussain
3,* 1
Department of Paediatric Endocrinology, Hacettepe University Faculty of Medicine, 06230 Ankara, Turkey
2
Department of Paediatrics, Varna Medical University/University Hospital “St. Marina”, Varna 9002, Bulgaria
3
Department of Paediatric Medicine, Division of Endocrinology, Sidra Medicine, Doha, Qatar
4
College of Health & Life Sciences, Hamad Bin Khalifa University, Qatar Foundation, Education City, Doha, Qatar
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2019, 20(10), 2590; https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20102590
Submission received: 24 April 2019
/
Revised: 21 May 2019
/
Accepted: 22 May 2019
/
Published: 27 May 2019
(This article belongs to the Special Issue Ion Transporters and Channels in Physiology and Pathophysiology 2.0: from Molecules to Whole Bodies)
Abstract
:Ion channels and transporters play essential roles in excitable cells including cardiac, skeletal and smooth muscle cells, neurons, and endocrine cells. In pancreatic beta-cells, for example, potassium KATP channels link the metabolic signals generated inside the cell to changes in the beta-cell membrane potential, and ultimately regulate insulin secretion. Mutations in the genes encoding some ion transporter and channel proteins lead to disorders of glucose homeostasis (hyperinsulinaemic hypoglycaemia and different forms of diabetes mellitus). Pancreatic KATP, Non-KATP, and some calcium channelopathies and MCT1 transporter defects can lead to various forms of hyperinsulinaemic hypoglycaemia (HH). Mutations in the genes encoding the pancreatic KATP channels can also lead to different types of diabetes (including neonatal diabetes mellitus (NDM) and Maturity Onset Diabetes of the Young, MODY), and defects in the solute carrier family 2 member 2 (SLC2A2) leads to diabetes mellitus as part of the Fanconi–Bickel syndrome. Variants or polymorphisms in some ion channel genes and transporters have been reported in association with type 2 diabetes mellitus.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
Ion channels and transporters are membrane-embedded proteins which play a key role in transporting ions and biomolecules across cell membranes. Although both ion channels and transporters are membrane-bound proteins, they have some differences. Ion channels are typically formed by the assembly of several different proteins and they function to allow the movement of specific ions (selectivity). The flow of ions across the channel (transmembrane flux) creates an electrical signal or action potential which is essential for the function of the particular channel [1]. Another property of ion channels is gating, which allows the channels to open and close in response to certain specific stimuli. An example of an ion channel is the adenosine triphosphate (ATP)-dependent potassium channel (KATP) that allows the rapid and selective flow of K+ ions across the cell membrane, and thus generates electrical signals in cells. Distinct from ion channels, some ion transporters may function as ion pumps, thereby moving ions across the plasma membrane against the electrochemical gradient.
Ion channels and transporters play essential roles in excitable cells including cardiac, skeletal and smooth muscle cells, neurons, and endocrine cells. In pancreatic beta-cells, KATP channels link the metabolic signals generated inside the cell to changes in the beta-cell membrane potential and ultimately, insulin secretion. In addition, ion channels and transporters have roles in non-excitable tissues such as the liver and skeletal muscle.
Mutations in the genes encoding the ion transporter and channel proteins lead to several diseases [2]. Disorders of glucose homeostasis (hypoglycaemia and different forms of diabetes mellitus (DM)) have been linked to several channelopathies and ion transporter defects. For example, pancreatic KATP channelopathies and defects in transporters such as GLUT1 and MCT1 are associated with different forms of hypoglycaemia and DM. In addition, Genome-Wide Association Studies (GWAS) have identified variants or polymorphisms in channel and ion transporter genes associated with type 2 diabetes mellitus (T2DM) [3,4]. For example, the non-synonymous E23K variant in the KCNJ11 was the first robustly replicating signal to emerge as a link to T2DM.
In this review, we focus on ion channel and transporter function in relation to glucose physiology. We will firstly describe the role of ion channels and transporters in relation to insulin secretion from the pancreatic beta-cell and then describe mechanistic insights into how defects in ion channels and transporters lead to hyperinsulinaemic hypoglycemia and diabetes mellitus.
2. Ion Channel Defects and Hyperinsulinaemic Hypoglycaemia
Hyperinsulinaemic Hypoglycaemia (HH), is a clinically and genetically heterogeneous group of disorders characterised by inappropriate insulin secretion from the beta cell, despite low blood glucose [5,6,7]. HH is the most common cause of severe, persistent hypoglycaemia in neonates and infants with an increased risk of permanent brain damage. The most severe forms of HH are caused by mutations in the genes involved in the regulation of insulin secretion from the pancreatic beta-cell [5,6,7,8].
2.1. Pancreas Beta-Cell Physiology
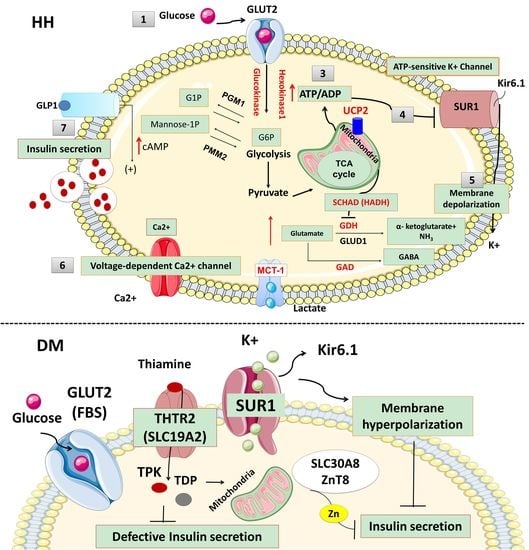
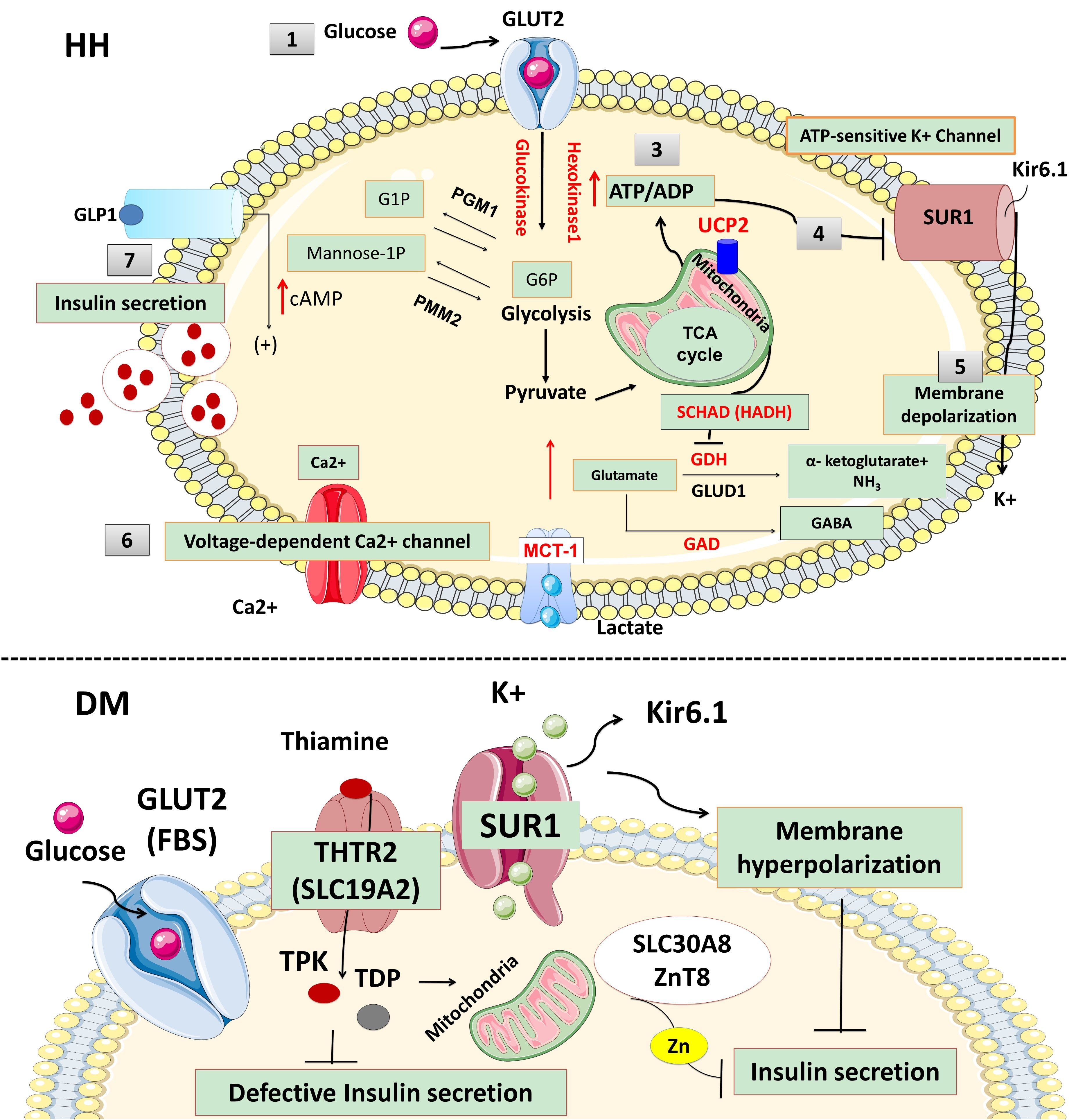
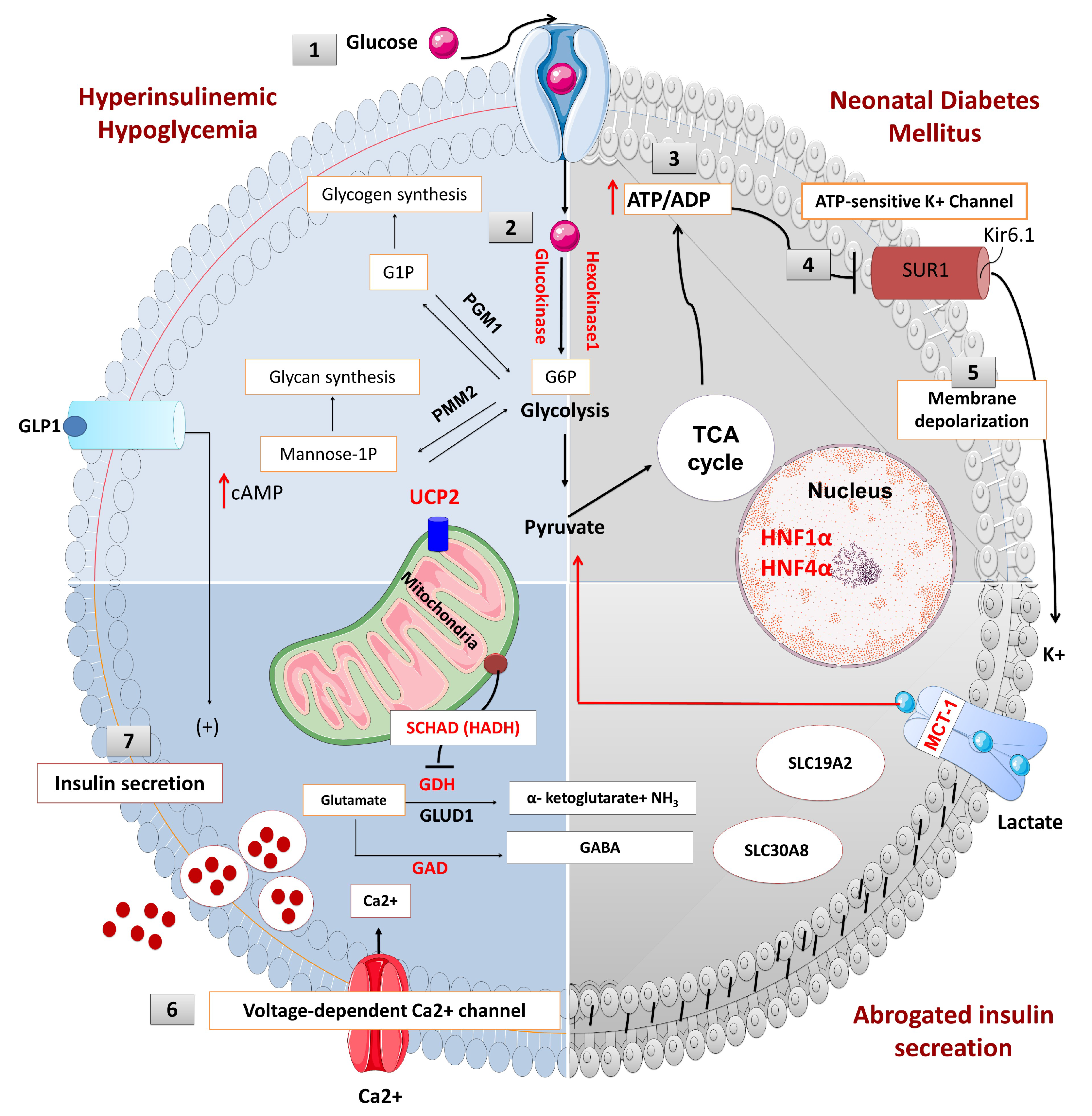
Pancreatic beta-cells play a crucial role in the regulation of insulin secretion. ATP-sensitive potassium channels (KATP) are localized in the pancreatic beta-cell membrane and allow K+ ion efflux across the beta-cell membrane. Glucose enters the beta cell through a facilitative glucose transporter, glucose transporter 2 (GLUT 2), and is converted to glucose-6-phosphate (G6P) by the enzyme glucokinase [9]. GLUT2 is a low-affinity transporter for glucose which allows glucose transport into the cell in proportion to the blood glucose concentrations [10]. The utilization of G6P from glycolysis generates high-energy molecules, adenosine triphosphate (ATP) and increases the ratio of ATP:ADP (adenosine diphosphate), which closes the ATP-sensitive potassium channels (KATP). Closure of this channel generates a membrane potential which triggers the opening of L-type voltage-gated calcium channels allowing calcium influx into the beta cell. Increased intracellular Ca2+ stimulates insulin-stored granules to release insulin through exocytosis (Figure 1). Recently, it has been shown that leucine-rich repeat-containing protein 8A (LRRC8A), a subunit of volume-regulated anion channels (VRAC) enhances the beta-cell glucose sensing and insulin secretion [11].
2.2. ATP-Sensitive Potassium (KATP) Channels and Insulin Secretion
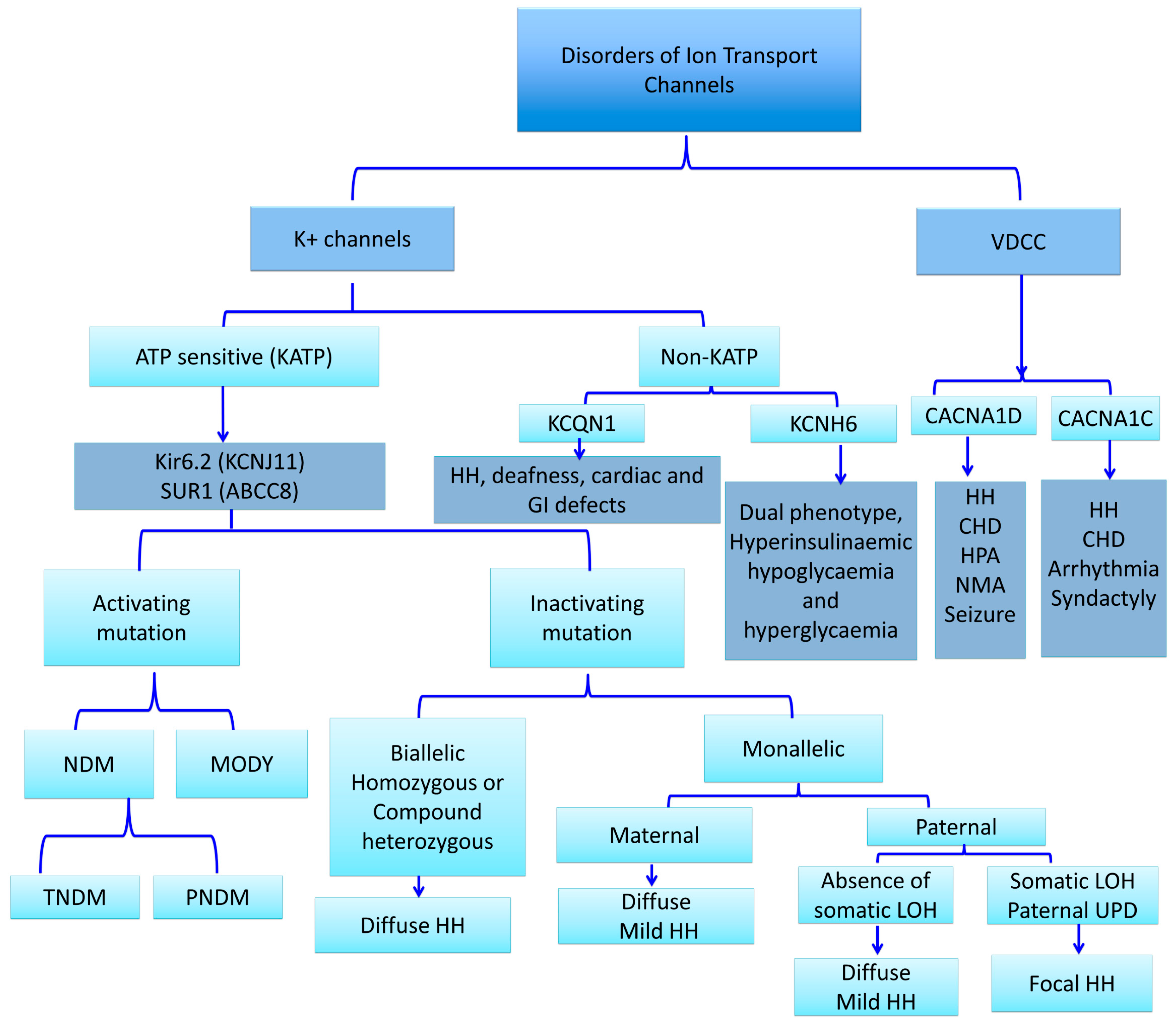
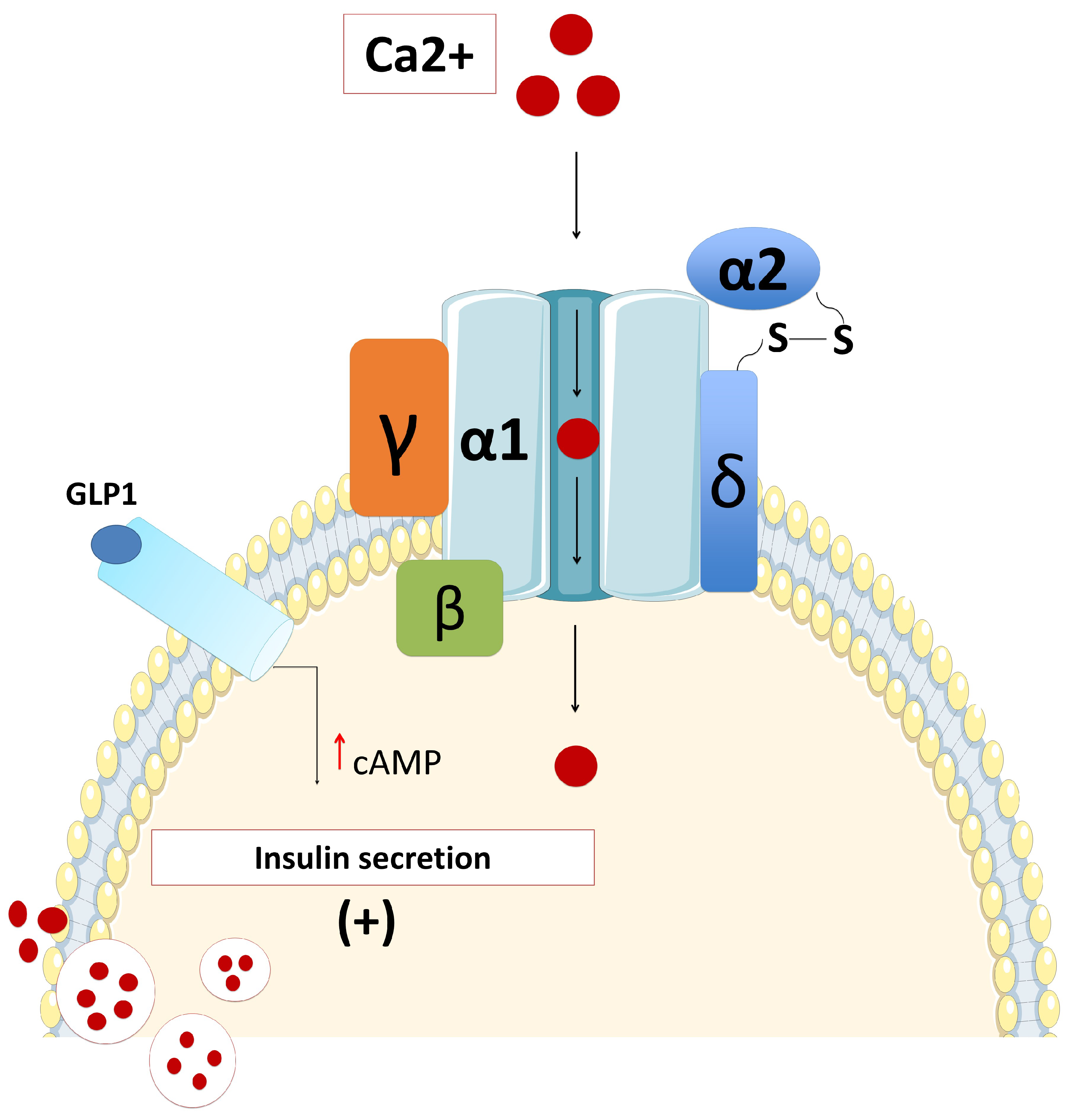
KATP channels, members of the inwardly rectifying potassium channel family, are large octameric complexes which permit the flux of potassium ions across the cell membrane [12,13,14,15]. They are composed of four pore-forming subunits (Kir6.1 or Kir6.2, encoded by KCNJ8 and KCNJ11, respectively) and four regulatory sulphonylurea receptors (SUR1, SUR2A or SUR2B) which are encoded by ATP-binding cassette subfamily C member 8 (ABCC8) [13,14,15]. The channel composition and its physiological roles differ based on their tissue expression, e.g., in the cardiac myocytes, pancreatic beta-cells, skeletal or vascular smooth muscles, neurons, and the kidneys [16,17,18]. One of the best-characterized potassium channels are the KATP channels in the beta-cell membrane [19]. They have an essential role in the glucose homeostasis and regulate the process of stimulated insulin release by linking the metabolism of the cell to its membrane excitability [6,19,20,21,22,23]. Defects in the genes (KCNJ11 and ABCC8) encoding KATP channel proteins can lead to abnormal glucose homeostasis. Thus, the “loss-of-function” or inactivating mutations in SUR1 and Kir6.2 encoding genes lead to the most common and severe forms of HH (Figure 2) [24].
2.3. KATP Channel Defects (ABCC8 and KCNJ11 Gene Mutations) and HH
Defects in the subunits of the pancreatic beta-cell KATP channels due to inactivating mutations in ABCC8 and KCNJ11 genes are the most common genetic cause of HH [7,25]. Recessive and dominant mutations have been described in about 50% of HH patients [26,27,28]. These mutations affect the functioning of KATP channels either by altering their surface expression due to defects in trafficking or by impairing stimulation by MgADP [26]. As a result, in both cases the pancreatic beta-cell membrane will be depolarized with dysregulated insulin release, despite severely low blood glucose [29].
Recessive homozygous or compound heterozygous inactivating mutations in ABCC8 and KCNJ11 usually cause the most severely diffuse forms of HH, typically unresponsive to diazoxide treatment and often requiring a resection of the pancreas [28]. However, there are compound heterozygous mutations that may be milder and may respond to diazoxide [30].
Dominant inactivating mutations in ABCC8 and KCNJ11 genes are associated with normal KATP channel assembly and trafficking to the cell membrane, but the majority of the channel complexes have an impaired function [31]. These mutations are not so common, they are generally milder and often present later in life, although the clinical manifestations may vary in severity [32]. Dominant inactivating ABCC8/KCNJ11 mutations typically respond to diazoxide treatment [32,33]. However, medically unresponsive forms have also been found due to a dominant negative trafficking defect [27,34].
The finding of a single, paternally-inherited KATP channel mutation with a post-zygotic loss of the corresponding maternal chromosomal region usually results in a focal adenomatous hyperplasia affecting one or more areas of the pancreas, with dysregulated insulin secretion within the lesion(s), constituting up to 30%–40% of the HH cases [35,36].
2.4. Non-ATP-Sensitive Potassium Channel Defects and HH (Non-KATP-HH)
2.4.1. Non-ATP-Sensitive (Non-KATP) Potassium Channel and Insulin Secretion
The role of the non-ATP-sensitive potassium channels (non-KATP) in the regulation of pancreatic insulin secretion is not well-characterized compared to KATP-sensitive channels. Human beta-cells also express other non-KATP channels such as delayed rectifying (K(V)2.1/2.2) and large-conductance Ca(2+)-activated K(+) (BK) channels [24]. However, no human hypoglycaemia or diabetes phenotype has been described due to mutations in genes encoding these proteins.
2.4.2. Kv11.2 Potassium Channel and HH
The voltage-gated potassium channel (Kv11.2) is encoded by the potassium voltage-gated channel subfamily H member 6 (KCNH6) gene. In a recent study [25] it was shown that this channel might have an important role in regulating insulin secretion in humans and mice. The study reported a multigenerational family with diabetes and neonatal HH due to mutations in the KCNH6 gene. In addition, KCNH6 knockout (KO) and p.P235L knockin (KI) mice were shown to mimic a phenotype of hyperinsulinaemia and hypoinsulinaemia [37] similar to that in humans. Further support for the role of KCNH6 in potentially causing HH is provided by another study [38]. Proverbio et al. [38] performed whole exome sequencing in a group of patients with HH and identified various single nucleotide polymorphism (SNP) in the genes involved in the regulation of insulin secretion [38]. Of them, the heterozygous p.V532F mutation in the KCNH6 has been found as a possible aetiology of HH in an Italian family [38].
2.4.3. KCNQ1 Channels Mutations and HH
The KCNQ1 gene encodes the Kv7.1 protein, a voltage-activated potassium channel α-subunit, which forms a homotetrameric channel located in the myocardium, inner ear, stomach, colon, and pancreatic beta-cells. It is critical for ion homeostasis in these tissues. The phenotype of individuals harbouring mutations in this gene includes inherited cardiac arrhythmias (long-QT syndrome (LQTS)), deafness, and gastrointestinal defects [39]. A recent report has presented evidence of HH in individuals with LQTS caused by mutations in KCNQ1 [40]. Although the mechanism underlying the involvement of Kv7.1 in the regulation of glucose homeostasis is not entirely elucidated, the evidence suggests that it may play a role in the regulation of insulin release by regulating plasma membrane repolarization.
2.5. Defects in Calcium Channels and HH
2.5.1. Voltage-Gated Calcium Channel and Insulin Secretion
Voltage-gated calcium (Ca2+) channels are ubiquitously expressed membrane channels that play a signal transducer role for membrane potential changes, leading to increased intracellular Ca2+ which initiate many physiological events. These include processes such as myofibrillar contraction, hormonal secretion, neurotransmission, enzyme regulation, protein phosphorylation/dephosphorylation, and gene transcription [41,42].
Human beta-cells comprise the L-type (Ca(V)1.3), P/Q-type (Ca(V)2.1), and T-type (Ca(V)3.2) calcium channels, while there are no N- or R-type Ca2+ channels [43]. The dominant subtype of Ca2+ channels involved in the regulation of glucose-induced insulin secretion are L-type (Ca(V)1.3) channels (Figure 3) [43]. Therefore, mutations in the gene that encodes for the L-type (Ca(V)1.3) channels might lead to dysregulated glucose-stimulated insulin release [43,44,45]. However, defects in the activity of T- and P/Q-type Ca2+ channels cause a partial (about 70%–80%) reduction in the glucose-induced insulin secretion. Membrane potential recordings suggested that L- and T-type Ca2+ channels participate in the action potential generation, while exocytosis of insulin-containing granules is principally triggered by Ca2+ influx through P/Q-type Ca2+ channels [43]. Although the critical role of voltage-gated calcium channels in the regulation of insulin secretion has been studied extensively, particularly in experimental studies, in clinical practice there are very few case reports with disorders in glucose metabolism as a direct consequence of defects in genes encoding the voltage-dependent calcium channels.
2.5.2. CACNA1D Mutations and HH
Calcium Voltage-Gated Channel Subunit Alpha1 D (CACNA1D) is highly expressed in pancreatic beta-cells and encodes an L-type voltage-gated calcium channel that plays a pivotal role in the regulation of insulin secretion from the pancreatic beta-cells [6,45]. Activating germline mutations in the CACNA1D gene have previously been reported in patients with primary hyperaldosteronism, congenital heart defects (such as biventricular hypertrophy and ventricular septal defect), seizures, neuromuscular abnormalities, and transient diazoxide-responsive hypoglycaemia [46]. In the same report, another de novo germline mutation (p.Ile770Met) in the CACNA1D gene was found in a patient with hyperaldosteronism with the absence of HH [46]. Recently, a heterozygous de novo c.1208G>A (p.G403D) mutation has been found in the CACNA1D gene in a patient with diazoxide-responsive HH, heart defects (prenatal bradycardia, mild aortic insufficiency), umbilical hernia, hypermetropia, severe axial hypotonia, limb spasticity, and seizures [44]. This germline gain-of-function mutation was thought to cause an increase in the sensitivity of the L-type voltage-gated calcium channel and lead to the channel remaining open at a lower membrane potential, thereby resulting in dysregulated insulin secretion [6,46].
In electrophysiological studies, it was shown that the c.1208G>A (p.G403D) mutation leads to premature activation of the L-type voltage-gated calcium channel at a lower membrane potential, while the p.Ile770Met mutation impairs the inhibition of the channel [46]. Interestingly, CACNA1D mutations were also described in almost 9.3% of cases with aldosterone-producing adenoma [47]. Therefore, further investigations are required to confirm whether the CACNA1D gene mutations should be considered as the underlying molecular aetiology of HH.
2.5.3. CACNA1C Mutations and HH (Timothy Syndrome)
Calcium Channel, Voltage-Dependent, L Type, alpha 1C Subunit (CACNA1C) is mapped on the chromosome 12p13.33 and encodes for the voltage-dependent L-type Ca-channel, Ca(V)1.2. Missense mutations of this gene cause a syndromic form of HH, Timothy syndrome, which is characterized with multi-system disorders including lethal cardiac arrhythmias, congenital heart defects, syndactyly, immune deficiency, intermittent HH, intellectual disability, autism, and autistic spectrum disorders [48].
3. Membrane Transporters Defects and HH
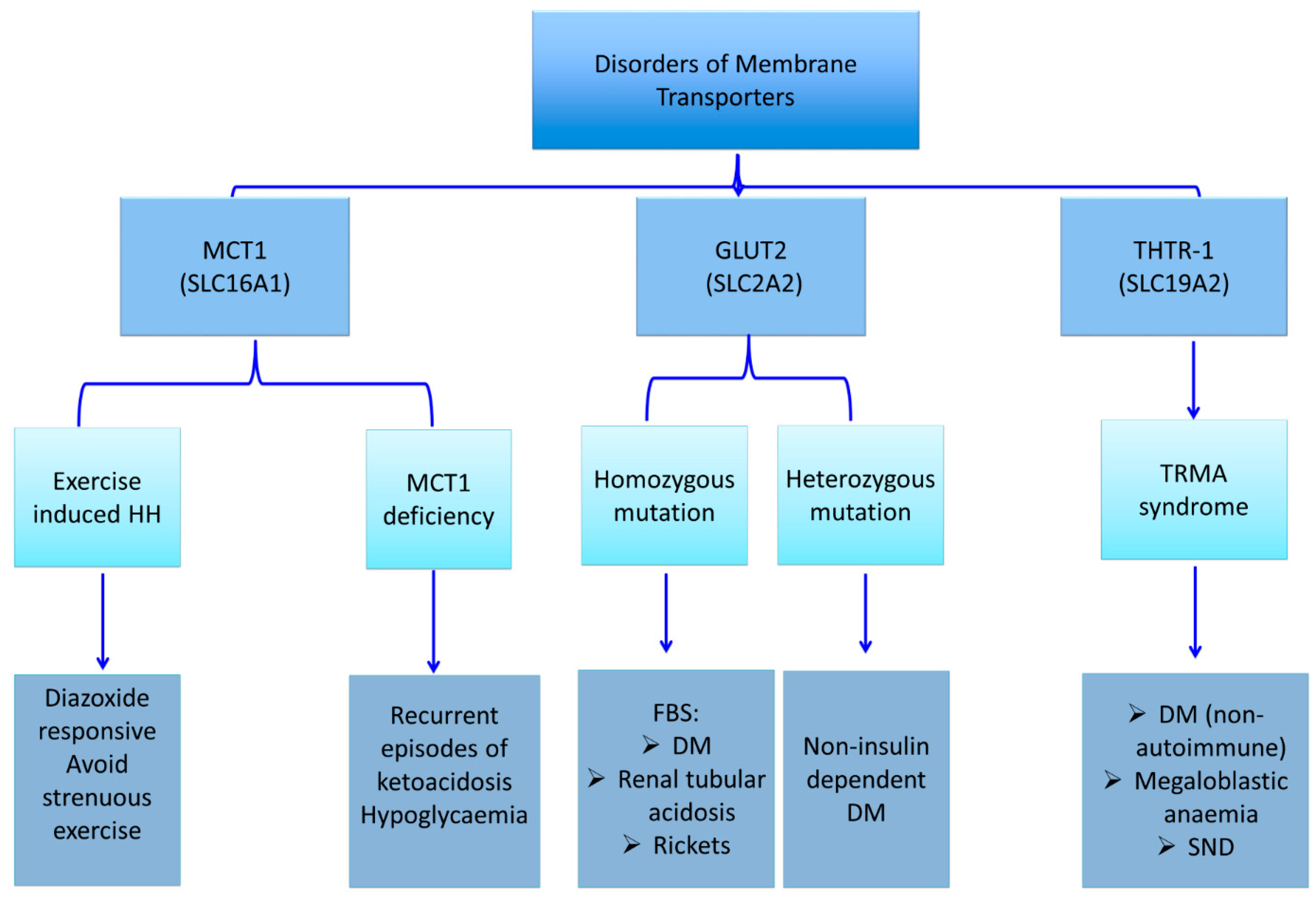
3.1. Monocarboxylate Transporter 1 (MCT1)
The solute carrier family 16, member 1 (SLC16A1) gene is localized on chromosome 1p13.2-1p12, spanning approximately 44 kb, and is organised as five exons intervened by four introns and encodes for a transporter protein, monocarboxylate transporter 1 (MCT1) [49]. MCT1 is a proton-linked monocarboxylate transporter that mediates the import and export of lactate, pyruvate, branched-chain oxo acids derived from leucine, valine, and isoleucine, and the ketone bodies such as acetoacetate, beta-hydroxybutyrate, and acetate through the cell membrane [50,51,52].
Lactate and pyruvate are potent insulin secretagogues. Under normal physiological conditions, lactate and pyruvate concentrations are low, and thus they are unable to trigger beta-cell insulin release. Although SLC16A1 gene is highly expressed in other tissues, it is transcriptionally and cell-specifically silenced in the pancreatic beta-cells [53,54,55]. Dominant activating mutations in the SLC16A1 gene promoter cause enhanced MCT1 expression in the pancreatic islets [56,57,58,59]. This increased expression leads to an increased uptake of pyruvate and its metabolism in the Krebs cycle, with a subsequently increased production of ATP and insulin secretion [56]. There are three distinct clinical conditions that have been attributed to the mutation of the SLC16A1 gene: exercise-induced hyperinsulinaemic hypoglycaemia due to an activating mutation, erythrocyte lactate transporter defect, and MCT1 deficiency due to inactivating mutations (Figure 4) [56,57,58,59,60,61,62]. The latter two disorders do not primarily affect the glucose metabolism. Therefore, they are not in the scope of the present article.
3.2. Exercise-Induced Hyperinsulinaemic Hypoglycaemia due to SLC16A1 Mutations
Activating mutations in the promoter region of the SLC16A1 gene increases MCT1 expression in beta cells, thereby increasing the influx of monocarboxylates (lactate and pyruvate) into beta cells (Figure 4) [56,57,58,59]. This, in turn, triggers inappropriate insulin release from beta cells and results in HH. Patients with a mutation in SLC16A1 develop HH following strenuous anaerobic exercises. The affected individuals typically become hypoglycaemic within 30–45 min following a strenuous anaerobic exercise due to pyruvate and lactate accumulation, which act as insulin secretagogues [58]. Although, the majority of mutations detected in SLC16A1 have been reported in the promoter region, recently, the first intragenic heterozygous mutation (c.556C>G, p.L186V) in SLC16A1 was reported in a patient with HH [63].
4. Ion Channel and Membrane Transporter Defects in Diabetes Mellitus
4.1. Neonatal Diabetes Mellitus
Neonatal diabetes mellitus (NDM) is a monogenic form of diabetes which presents within the first six months of life [64,65]. NDM is autoantibody-negative type of diabetes that is caused by mutations in the genes encoding for transcription factors that regulate pancreatic development or are involved in either insulin synthesis or secretion [64,65,66]. The worldwide incidence rate has been reported at a broad range of 1/260.000 and 1/400.000 to 1/80.000-90.000 [67,68,69,70,71]. However, in the populations with a high-rate of consanguinity, the incidence of NDM has been reported at a higher rate of 1/30.000 and 1/21.000 [72,73].
NDM patients can present with a wide range of clinical manifestations as a consequence of intrauterine (low birth weight) or postnatal insulin deficiency (growth retardation, weight loss or poor weight gain, polyuria, signs and symptoms of dehydration) and in case of a delay in the diagnosis, diabetic ketoacidosis may eventually develop [64]. NDM can be transient (TNDM) or permanent (PNDM) [66,74]. TNDM remits within the first few months of life, but in the majority of these cases, diabetes relapses later in life, particularly at puberty [66,74]. However, PNDM patients require life-long therapy [66,74].
KATP Channel Defects (ABCC8 and KCNJ11 Mutations) and NDM
Mutations in the genes ABCC8 and KCNJ11 are the most frequent cause of NDM in the Western world. Interestingly, adult-onset diabetes due to potassium channel mutations can also develop or PNDM can present later in adolescence or during pregnancy [75,76,77].
Heterozygous activating KCNJ11 mutations are the main cause of PNDM, but in rare cases can also cause TNDM (Figure 2) [78]. These mutations affect the ATP binding site to Kir6.2 (binding mutations) or indirectly reduce the ATP inhibition of the channel activity (gating mutations). Activating heterozygous or homozygous missense mutations in the ABCC8 gene are found mainly in TNDM, but also in PNDM [76,79].
The mutations lead to transient or permanent NDM due to the over-activation of the channels insensitive to the ATP inhibition, which enhances the stimulatory effect of the MgADP and alters the beta-cell electrical activity followed by insufficient pancreatic insulin release [75,79,80,81,82]. In about 20%–30% of the patients, the potassium channel mutations (mainly Kir6.2 mutations) also affect the KATP channel complexes in multiple neurons in the brain [83] leading to deactivation of the inhibitory neurons and more severe syndromes involving neurological and psychological manifestations in addition to NDM [76,83,84,85], such as developmental delay and epilepsy (DEND syndrome), intermediate DEND syndrome without epilepsy, ataxia, muscle weakness, attention deficit hyperactivity syndrome, anxiety, autism, or sleeping disorders [84,86,87,88].
The main role of the pancreatic KATP channel activation in the pathogenesis of NDM has led to the use of sulfonylureas as a potential treatment option in 90%–95% of patients [89,90]. They stimulate insulin secretion by binding to the high-affinity SUR1 binding site, inhibiting the MgADP activation of the channels, and unmasking the ATP inhibition on Kir6.2 [79,81,90,91]. Thus, sulfonylureas can normalize the secretion of insulin and lead to improved glycaemic control in the majority of the NDM cases, especially among SUR1 patients [86,90]. However, in patients with severe channel defects and neurological deficits, sulfonylurea treatment can be partially effective or ineffective [59,61,62,90,92,93]. Based on these findings, the international guidelines (The International Society for Pediatric and Adolescent Diabetes (ISPAD) clinical guidelines) suggest immediate genetic testing after a clinical diagnosis of NDM as insulin therapy in patients with KCNJ11 and ABCC8 mutations can be switched to oral sulfonylureas [66,94].
4.2. KATP Channel Defects (KCNJ11 and ABCC8 Mutations) and MODY
Maturity-onset diabetes of the young (MODY) is а group of non-autoimmune diabetes caused by a single gene mutation that leads to a defect in the glucose-stimulated insulin secretion from the pancreatic beta-cells. It is characterized by hyperglycaemia, usually manifested before 25 years of age, and autosomal dominant inheritance among the third-generation family members. C-peptide levels are usually within the normal range, suggesting the presence of insulin secretion [94]. It is a rare form of diabetes representing less than 2% of childhood diabetes cases, with 14 different subtypes being identified up to now, with different prevalence, clinical manifestations, and treatment requirements [95].
Gain-of-function ABCC8 and KCNJ11 gene mutations have been reported to cause rare forms of MODY (ABCC8-MODY 12 and KCNJ11-MODY13) with variable clinical presentation, ranging from asymptomatic glucose intolerance to overt diabetes, with age [77]. Furthermore, several reports have demonstrated that some patients may develop dual phenotypes – neonatal HH and diabetes in adult-life [31,32,96,97]. The underlying mechanisms by which a mutation causes HH and diabetes later in life have been suggested as pancreatic beta-cell apoptosis due to overstimulation of insulin secretion, enhanced cell depolarization, and increased intracellular calcium influx [29,36]. Most of the ABCC8- and KCNJ11-MODY patients are responsive to sulfonylureas.
4.3. Other Rare Types of Monogenic Diabetes due to Membrane Transporter Defects
4.3.1. Glucose Transporter 2 (GLUT 2) Deficiency and Diabetes Mellitus (Fanconi-Bickel syndrome; Glycogen Storage Disease Type XI)
Glucose transporters (GLUTs) are members of a large group of the solute carrier transporter superfamily, encoded by the SLC2A genes [98,99]. There are 4 main subtypes of GLUTs expressed in different tissues and involved in the organ-specific glucose transport. GLUT-1 is involved in the basal non-insulin-induced glucose uptake into many cells. GLUT-2 plays a role in the glucose sensing mechanism by mediating facilitative glucose transport into the beta cells (Figure 1). GLUT-3 is mainly involved in the non-insulin-mediated glucose uptake into brain neurons and placenta. And finally, GLUT-4 is mostly expressed in the muscle and adipose tissue and thereby mediates the peripheral action of insulin.
Solute carrier family 2 member 2 (SLC2A2), also known as the GLUT2 gene, consists of 11 exons and 10 introns spanning approximately 30 kb, and encodes for a glucose transporter known as GLUT2 [100]. GLUT2 is a facilitative glucose transporter, expressed in the liver, pancreatic beta-cell, renal tubular, and intestinal epithelial cells. GLUT2 mediates the passive transport of intracellular glucose and galactose across the basolateral membrane and down the concentration gradient [101]. After a carbohydrate-rich feeding, GLUT2 transports glucose and galactose into the hepatocytes and plays a role in the release of glucose from the liver during the fasting state [102]. GLUT2 is located on the apical membrane and mediates the absorption of simple sugars as a result of its temporary expression on the apical membrane of the intestinal mucosa, in a process that is independent of sodium-glucose transporter 1 (SGLT1 or SLC5A1). GLUT2 is involved in glucose transport in the beta cells and thereby regulates glucose-stimulated insulin secretion. Autosomal-recessive mutations of the SLC2A2 gene lead to Fanconi–Bickel syndrome (FBS), while autosomal-dominant mutations result in non-insulin dependent DM [103,104,105,106,107,108].
Fanconi–Bickel syndrome is a rare autosomal recessive disorder of carbohydrate metabolism leading to the accumulation of glycogen in the liver and kidneys. Homozygous or compound heterozygous mutations of the SLC2A2 gene are responsible for the molecular basis of FBS [102,106,107]. Patients may present with hepatomegaly, glucose and galactose intolerance, proximal tubular nephropathy, severe growth retardation, and rickets due to the accumulation of glycogen [106,107]. The mechanism of glycogen accumulation is a defective transport of glucose from the intracellular compartment to the extracellular compartment during glycogenolysis, due to GLUT2 deficiency. This causes a markedly elevated intracellular glucose, and thereby, inhibition of glycogenolysis. These patients suffer from postprandial hyperglycaemia, fasting hypoglycaemia, and DM. Hyperglycaemia and hypergalactosemia occur after feeding in patients with FBS as a result of the defective transport and decreased uptake of monosaccharides by the liver, and hyperglycaemia is further aggravated by decreased glucose-stimulated insulin secretion in pancreatic beta-cells. Fasting hypoglycaemia develops due to an impaired glucose export in the hepatocytes when peripheral glucose sources are depleted [102]. Glycated haemoglobin (HbA1c) is usually within normal range due to recurrent hypoglycaemia episodes. Fasting hypoglycaemia and postprandial hyperglycaemia have been shown to improve over time [109].
Proximal renal tubular dysfunction is characterized by glucosuria, phosphaturia, generalized aminoaciduria, and urinary bicarbonate loss, or may cause refractory rickets, which can be the presenting feature of FBS in infancy [107,110]. Renal glucosuria, which may occur at the blood glucose level of below renal glucosuria threshold (180 mg/dl), results from the impaired glucose reabsorption at the proximal renal tubular basolateral membrane.
4.3.2. Thiamine-Responsive Megaloblastic Anaemia (TRMA) and Diabetes Mellitus
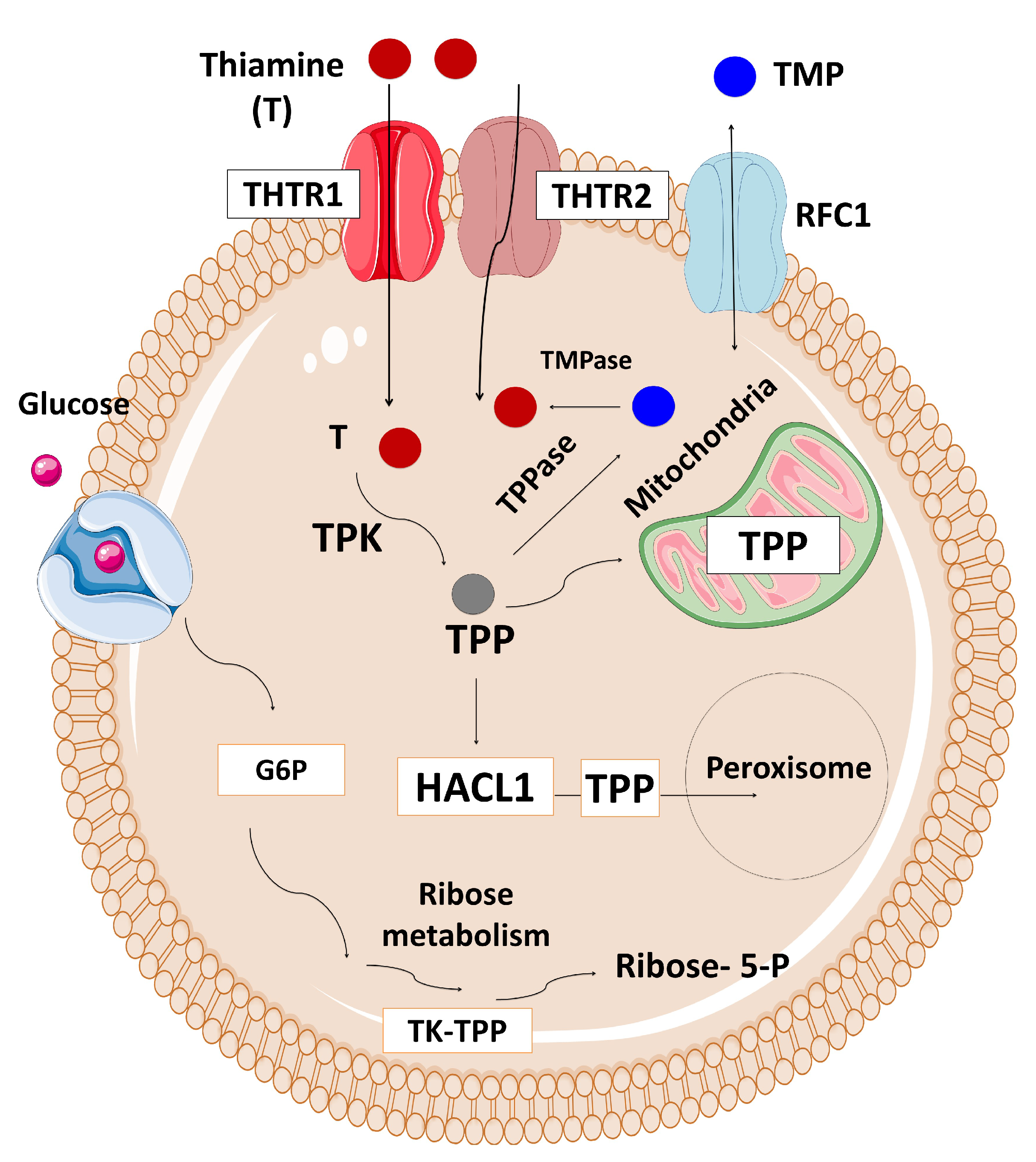
The Solute Carrier Family 19 Member 2 (SLC19A2) gene, located on chromosome 1q23.3, is composed of six exons and encodes for a high-affinity thiamine transporter 1 protein (THTR-1) containing 497 amino acids and 12 transmembrane domains [111,112,113,114,115]. SLC19A2 is expressed in various tissues, such as bone marrow, liver, colon, small intestine, pancreas, brain, retina, heart, skeletal muscle, kidney, lung, placenta, lymphocytes, and fibroblasts [111]. Thiamine plays a role in carbohydrate metabolism and energy production. There are two main thiamine transporter proteins which have high-affinity to thiamine located in the intestine, thiamine transporter 1 (THTR-1) which is encoded by the SLC19A2 gene, and thiamine transporter 2 (THTR-2) which is encoded by the SLC19A3 gene (Figure 5) [111,116,117]. Most tissues express both genes, although cochlear inner hair cells, pancreatic islet cells, and erythropoietic precursor cells express only SLC19A2. At high concentrations, thiamine is transported across the cell membrane via passive diffusion [118,119,120]. However, the active transport of thiamine through transporter mechanisms is impaired in thiamine-responsive megaloblastic anaemia (TRMA) syndrome resulting in intracellular thiamine deficiency. There are THTR-2-mediated compensatory mechanisms activated in most cells. However, three cell lines: cochlear inner hair cells, pancreatic islet cells, and erythropoietic precursor cells are THTR-1-dependent, and therefore they are mostly affected by thiamine deficiency.
Thiamine-responsive megaloblastic anaemia (TRMA), also known as Roger’s Syndrome, is an autosomal recessive disorder characterized by early-onset non-autoimmune DM, megaloblastic anaemia, and sensorineural deafness (SND) [113,121,122]. Other well-defined clinical features are congenital heart disease, arrhythmias, visual disturbances, retinal degeneration, optic atrophy, aminoaciduria, short stature, situs inversus, polycystic ovarian syndrome, stroke, and neurological disorders [123]. The disease can manifest at any time from infancy to adolescence, while cardinal manifestations may not be apparent at the initial presentation, but develop later over time.
As the TRMA is inherited in an autosomal recessive manner, it is more frequent in the consanguineous pedigrees [112]. However, TRMA due to compound heterozygous mutations has also been reported in non-consanguineous families [124,125]. To date, 51 mutations have been described in the SLC19A2 gene, the majority of which are missense and nonsense mutations (http://www.hgmd.cf.ac.uk/ac/gene.php?gene=SLC19A2). Most mutations in the SLC19A2 gene result in an abnormally truncated, nonfunctional THTR-1 protein. It has been demonstrated that some mutations cause single amino acid changes in THTR-1, resulting in an abnormal folding of the protein, or making them unable to traffic the transport protein to the cell surface which results in a defect in the intracellular transport of thiamine by THTR-1.
A high level of thiamine is required for the normal exocrine and endocrine functions of the pancreas [126]. Impaired insulin secretion has been demonstrated in the islet cells of thiamine-deficient mice [127]. Stagg et al. suggested that the primary disorder in TRMA is the deficiency of high-affinity thiamine transporters as low intracellular thiamine results in cellular death through apoptosis in the patient’s fibroblasts [128]. The insulin requirement physiologically increases in puberty, and pancreatic beta-cell reserves decline, possibly due to apoptosis, while the residual reserve fails to meet the insulin requirement in puberty [129]. In TRMA, thiamine replacement does not store insulin secretion at puberty. Therefore, these patients may have an insulin requirement despite thiamine replacement.
The goal of treatment for TRMA is symptom relief, and thiamine (vitamin B1) supplementation at pharmacological doses (50-100 mg/day) corrects haematological and endocrine manifestations, although neurological symptoms do not respond to the therapy [121,130]. Although the therapy corrects anaemia and hyperglycaemia, there are reports of patients requiring insulin therapy and regular blood transfusions in adulthood [115,131].
5. Variants in Ion Channel and Transporters Genes Associated with Type 2 Diabetes Mellitus
T2DM is rapidly increasing throughout the world and is characterized by hyperglycemia caused by defects in insulin secretion, insulin action, or both. The underlying genetic mechanisms of T2DM involve genetic and environmental factors. Genome-Wide Association Studies (GWAS) have identified a significant number of different loci, mostly in the non-coding regions of genes implicated in type 2 diabetes [3,4]. The non-synonymous E23K variant in the KCNJ11 was the first robustly replicating signal to emerge as a link to T2DM [132].
Mechanistic studies suggest that the E23K variant may have a diabetogenic effect by increasing the KATP channel activity in response to changes in the level of long-chain acyl CoAs which increase during fasting [133].
Two GWAS studies have identified polymorphisms in intron 15 and 11 of the potassium voltage-gated channel, KQT-like subfamily member 1 (KCNQ1) in association with T2DM [134,135]. The increased susceptibility for developing T2DM linked to polymorphisms in the KCNQ1 gene is likely to be caused by a reduction in insulin secretion. The pore-forming alpha subunit of the voltage-gated K+ channel (KvLQT1) (encoded by KCNQ1) and the regulatory beta subunit ISK (encoded by potassium channel, voltage-gated, ISK-related subfamily, member 1: KCNE1 gene) co-assemble to form the I(KS) potassium channel in the pancreas. Thus, there is a possibility that KCNQ1 polymorphisms alter the role of the I(KS) potassium channel, leading to the decreased insulin secretion [136].
The SLC30A8 gene encodes a zinc transporter family member 8 (ZnT8) in pancreatic beta-cells and is responsible for the accumulation of zinc in secretory granules. A non-synonymous polymorphism in SLC30A8 is associated with the risk of developing T2DM [137]. The precise mechanisms by which polymorphisms in the SLC30A8 are associated with T2DM are not fully understood but might involve a reduction in insulin secretion, increased insulin clearance, or changes in reactive oxygen species.
6. Conclusions
Ion channel and transporter defects lead to HH and various forms of DM. Defects in the pancreatic KATP channels can lead to HH, NDM (both transient and permanent), MODY, and variants in KCNJ11 and KCNQ1 channel genes are associated with T2DM. Understanding the molecular mechanisms of HH and different types of diabetes due to the ion channel and transporter defects has provided unique insights into the role of these proteins in normal physiology, especially in the pancreatic beta-cell. Further studies are required to develop pharmacological agents which can target defects in channel proteins and treat conditions such as severe HH. In the case of NDM, understanding the role of the pancreatic KATP channels in insulin secretion has transformed the lives of patients as they can now be treated with oral sulphonylureas.
Author Contributions
H.D.: Literature review, wrote the section on introduction, non-KATP channels, calcium channels and hyperinsulinism, membrane transporters and related disorders; neonatal diabetes, MODY and T2DM, conclusion statement and managed the citations and figures legends, integreted all the sections and made critical revision S.G.: literature review, wrote the section on KATP causes of hyperinsulinism and neonatal diabetes; S.A.-K: literature review, created all the figures; D.V.: literature review, wrote the section on membrane transporters and related disorders; exercise induced hyperinsulinism, FBS and TRMA syndrome, K.H: conception, planning, writing, organizing, checking and communicating.
Funding
The publication of this article was funded by the Qatar National Library.
Acknowledgments
The publication of this article was funded by the Qatar National Library.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| HH | Hyperinsulinaemic Hypoglycemia |
| DM | Diabetes Mellitus |
| T2DM | Type 2 Diabetes Mellitus |
| SUR1 | Sulphonylurea receptor 1 |
| KIR6.2 | Inward rectifier potassium channel subunit 2 |
| MCT1 | Monocarboxylate transporter 1 |
| MODY | Maturity-onset diabetes of the young |
| SLC2A2 | Solute carrier family 2 member 2 |
| GWAS | Genome-Wide Association Studies |
| GLUT 2 | Glucose transporter 2 |
| G6P | Glucose-6-phosphate |
| ATP | Adenosine triphosphate |
| ADP | Adenosine diphosphate |
| KATP | ATP-sensitive potassium channels |
| KCNJ11 | Potassium Voltage-Gated Channel Subfamily J Member 11 |
| ABCC8 | ATP-binding cassette subfamily C member 8 |
| KCNH6 | Potassium voltage-gated channel subfamily H member 6 |
| SNP | Single nucleotide polymorphism |
| LQTS | long-QT syndrome |
| CACNA1D | Calcium Voltage-Gated Channel Subunit Alpha1 D |
| CACNA1C | Calcium Channel, Voltage-Dependent, L Type, alpha 1C Subunit |
| SLC16A1 | Solute carrier family 16, member 1 |
| NDM | Neonatal diabetes mellitus |
| TNDM | Transient NDM |
| PNDM | Permanent NDM |
| DEND | Developmental delay and epilepsy syndrome |
| SLC2A2 | Solute carrier family 2 member 2 |
| FBS | Fanconi Bickel syndrome |
| HbA1c | Glycated haemoglobin |
| TRMA | Thiamine-responsive megaloblastic anaemia |
| THTR-1 | Thiamine transporter 1 protein |
| THTR-2 | Thiamine transporter 2 |
| SLC19A2 | The Solute Carrier Family 19 Member 2 |
| SND | Sensorineural deafness |
| ZnT8 | Zinc transporter family member 8 |
| RFC1 | Reduced folate carrier 1 |
| TMP | Thiamine monophosphate |
| TMPase | Thiamine monophosphatase |
| HACL1 | 2-Hydroxyacyl-CoA Lyase 1 |
| TPP | Thiamine pyrophosphate |
| TPPase | Thiamine pyrophosphatase |
| TK-TPP | Transketolase-Thiamine pirophosphate |
| TPK | Thiamine pyrophosphokinase |
| TDP | Thiamine diphosphate |
References
- Neverisky, D.L.; Abbott, G.W. Ion channel-transporter interactions. Crit. Rev. Biochem. Mol. Biol. 2015, 51, 257–267. [Google Scholar] [CrossRef] [PubMed]
- Hubner, C.A.; Jentsch, T.J. Ion channel diseases. Hum. Mol. Genet. 2002, 11, 2435–2445. [Google Scholar] [CrossRef] [Green Version]
- McCarthy, M.I.; Zeggini, E. Genome-wide association studies in type 2 diabetes. Curr. Diabetes Rep. 2009, 9, 164–171. [Google Scholar] [CrossRef]
- Fernandez-Tajes, J.; Gaulton, K.J.; van de Bunt, M.; Torres, J.; Thurner, M.; Mahajan, A.; Gloyn, A.L.; Lage, K.; McCarthy, M.I. Developing a network view of type 2 diabetes risk pathways through integration of genetic, genomic and functional data. Genome Med. 2019, 11, 19. [Google Scholar] [CrossRef] [PubMed]
- Demirbilek, H.; Hussain, K. Congenital Hyperinsulinism: Diagnosis and Treatment Update. J. Clin. Res. Pediatric Endocrinol. 2017, 9 (Suppl. 2), 69–87. [Google Scholar] [CrossRef]
- Shah, P.; Rahman, S.A.; Demirbilek, H.; Guemes, M.; Hussain, K. Hyperinsulinaemic hypoglycaemia in children and adults. Lancet Diabetes Endocrinol. 2017, 5, 729–742. [Google Scholar] [CrossRef]
- Stanley, C.A. Perspective on the Genetics and Diagnosis of Congenital Hyperinsulinism Disorders. J. Clin. Endocrinol. Metab. 2016, 101, 815–826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galcheva, S.; Al-Khawaga, S.; Hussain, K. Diagnosis and management of hyperinsulinaemic hypoglycaemia. Best Pract. Res. Clin. Endocrinol. Metab. 2018, 32, 551–573. [Google Scholar] [CrossRef]
- Johnson, J.H.; Newgard, C.B.; Milburn, J.L.; Lodish, H.F.; Thorens, B. The high Km glucose transporter of islets of Langerhans is functionally similar to the low affinity transporter of liver and has an identical primary sequence. J. Biol. Chem. 1990, 265, 6548–6551. [Google Scholar] [PubMed]
- Zhao, F.Q.; Keating, A.F. Functional properties and genomics of glucose transporters. Curr. Genom. 2007, 8, 113–128. [Google Scholar] [CrossRef]
- Stuhlmann, T.; Planells-Cases, R.; Jentsch, T.J. LRRC8/VRAC anion channels enhance β-cell glucose sensing and insulin secretion. Nat. Commun. 2018, 9, 1974. [Google Scholar] [CrossRef]
- Tinker, A.; Aziz, Q.; Li, Y.; Specterman, M. ATP-Sensitive Potassium Channels and Their Physiological and Pathophysiological Roles. Compr. Physiol. 2018, 8, 1463–1511. [Google Scholar] [PubMed]
- Aguilar-Bryan, L.; Bryan, J. Molecular biology of adenosine triphosphate-sensitive potassium channels. Endocr. Rev. 1999, 20, 101–135. [Google Scholar] [PubMed]
- Aguilar-Bryan, L.; Nichols, C.G.; Wechsler, S.W.; Clement, J.P.T.; Boyd, A.E., 3rd; Gonzalez, G.; Herrera-Sosa, H.; Nguy, K.; Bryan, J.; et al. Cloning of the beta cell high-affinity sulfonylurea receptor: A regulator of insulin secretion. Science 1995, 268, 423–426. [Google Scholar] [CrossRef] [PubMed]
- Clement, J.P.T.; Kunjilwar, K.; Gonzalez, G.; Schwanstecher, M.; Panten, U.; Aguilar-Bryan, L.; Bryan, J. Association and stoichiometry of K(ATP) channel subunits. Neuron 1997, 18, 827–838. [Google Scholar] [CrossRef]
- Seino, S.; Miki, T. Physiological and pathophysiological roles of ATP-sensitive K+ channels. Prog. Biophys. Mol. Biol. 2003, 81, 133–176. [Google Scholar] [CrossRef]
- Kefaloyianni, E.; Lyssand, J.S.; Moreno, C.; Delaroche, D.; Hong, M.; Fenyo, D.; Mobbs, C.V.; Neubert, T.A.; Coetzee, W.A. Comparative proteomic analysis of the ATP-sensitive K+ channel complex in different tissue types. Proteomics 2013, 13, 368–378. [Google Scholar] [CrossRef]
- Vivaudou, M.; Moreau, C.; Terzic, A. Structure and function of ATP-sensitive K+ channels. In Ion Channels: From Structure to Function, 1st ed.; Kew, J., Davies, C., Eds.; Oxford University Press: Oxford, UK, 2009; pp. 454–473. [Google Scholar]
- Cook, D.L.; Hales, C.N. Intracellular ATP directly blocks K+ channels in pancreatic B-cells. Nature 1984, 311, 271–273. [Google Scholar] [CrossRef]
- Ashcroft, F.M.; Harrison, D.E.; Ashcroft, S.J. Glucose induces closure of single potassium channels in isolated rat pancreatic beta-cells. Nature 1984, 312, 446–448. [Google Scholar] [CrossRef]
- Aguilar-Bryan, L.; Clement, J.P.; Gonzalez, G.; Kunjilwar, K.; Babenko, A.; Bryan, J. Toward understanding the assembly and structure of KATP channels. Physiol. Rev. 1998, 78, 227–245. [Google Scholar] [CrossRef]
- Lee, K.P.K.; Chen, J.; MacKinnon, R. Molecular structure of human KATP in complex with ATP and ADP. eLife 2017, 6, e32481. [Google Scholar] [CrossRef]
- Matsuo, M.; Kimura, Y.; Ueda, K. KATP channel interaction with adenine nucleotides. J. Mol. Cell. Cardiol. 2005, 38, 907–916. [Google Scholar] [CrossRef] [PubMed]
- Saint-Martin, C.; Arnoux, J.B.; de Lonlay, P.; Bellanne-Chantelot, C. KATP channel mutations in congenital hyperinsulinism. Semin. Pediatric Surg. 2011, 20, 18–22. [Google Scholar] [CrossRef] [PubMed]
- Ashcroft, F.M. ATP-sensitive potassium channelopathies: Focus on insulin secretion. J. Clin. Investig. 2005, 115, 2047–2058. [Google Scholar] [CrossRef]
- Flanagan, S.E.; Clauin, S.; Bellanne-Chantelot, C.; de Lonlay, P.; Harries, L.W.; Gloyn, A.L.; Ellard, S. Update of mutations in the genes encoding the pancreatic beta-cell K(ATP) channel subunits Kir6.2 (KCNJ11) and sulfonylurea receptor 1 (ABCC8) in diabetes mellitus and hyperinsulinism. Hum. Mutat. 2009, 30, 170–180. [Google Scholar] [CrossRef]
- Flanagan, S.E.; Kapoor, R.R.; Banerjee, I.; Hall, C.; Smith, V.V.; Hussain, K.; Ellard, S. Dominantly acting ABCC8 mutations in patients with medically unresponsive hyperinsulinaemic hypoglycaemia. Clin. Genet. 2011, 79, 582–587. [Google Scholar] [CrossRef] [PubMed]
- Flanagan, S.E.; Kapoor, R.R.; Hussain, K. Genetics of congenital hyperinsulinemic hypoglycemia. Semin. Pediatric Surg. 2011, 20, 13–17. [Google Scholar] [CrossRef] [PubMed]
- Nessa, A.; Rahman, S.A.; Hussain, K. Hyperinsulinemic Hypoglycemia—The Molecular Mechanisms. Front. Endocrinol. 2016, 7, 29. [Google Scholar] [CrossRef] [PubMed]
- Dekel, B.; Lubin, D.; Modan-Moses, D.; Quint, J.; Glaser, B.; Meyerovitch, J. Compound heterozygosity for the common sulfonylurea receptor mutations can cause mild diazoxide-sensitive hyperinsulinism. Clin. Pediatrics 2002, 41, 183–186. [Google Scholar] [CrossRef]
- Pinney, S.E.; MacMullen, C.; Becker, S.; Lin, Y.W.; Hanna, C.; Thornton, P.; Ganguly, A.; Shyng, S.L.; Stanley, C.A. Clinical characteristics and biochemical mechanisms of congenital hyperinsulinism associated with dominant KATP channel mutations. J. Clin. Investig. 2008, 118, 2877–2886. [Google Scholar] [CrossRef]
- Kapoor, R.R.; Flanagan, S.E.; James, C.T.; McKiernan, J.; Thomas, A.M.; Harmer, S.C.; Shield, J.P.; Tinker, A.; Ellard, S.; Hussain, K. Hyperinsulinaemic hypoglycaemia and diabetes mellitus due to dominant ABCC8/KCNJ11 mutations. Diabetologia 2011, 54, 2575–2583. [Google Scholar] [CrossRef] [Green Version]
- Nessa, A.; Aziz, Q.H.; Thomas, A.M.; Harmer, S.C.; Tinker, A.; Hussain, K. Molecular mechanisms of congenital hyperinsulinism due to autosomal dominant mutations in ABCC8. Hum. Mol. Genet. 2015, 24, 5142–5153. [Google Scholar] [CrossRef]
- Macmullen, C.M.; Zhou, Q.; Snider, K.E.; Tewson, P.H.; Becker, S.A.; Aziz, A.R.; Ganguly, A.; Shyng, S.L.; Stanley, C.A. Diazoxide-unresponsive congenital hyperinsulinism in children with dominant mutations of the beta-cell sulfonylurea receptor SUR1. Diabetes 2011, 60, 1797–1804. [Google Scholar] [CrossRef]
- Fournet, J.C.; Mayaud, C.; de Lonlay, P.; Gross-Morand, M.S.; Verkarre, V.; Castanet, M.; Devillers, M.; Rahier, J.; Brunelle, F.; Robert, J.J.; et al. Unbalanced expression of 11p15 imprinted genes in focal forms of congenital hyperinsulinism: Association with a reduction to homozygosity of a mutation in ABCC8 or KCNJ11. Am. J. Pathol. 2001, 158, 2177–2184. [Google Scholar] [CrossRef]
- Rahman, S.A.; Nessa, A.; Hussain, K. Molecular mechanisms of congenital hyperinsulinism. J. Mol. Endocrinol. 2015, 54, R119–R129. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.K.; Lu, J.; Yuan, S.S.; Asan; Cao, X.; Qiu, H.Y.; Shi, T.T.; Yang, F.Y.; Li, Q.; Liu, C.P.; et al. From Hyper- to Hypoinsulinemia and Diabetes: Effect of KCNH6 on Insulin Secretion. Cell Rep. 2018, 25, 3800.e6–3810.e6. [Google Scholar]
- Proverbio, M.C.; Mangano, E.; Gessi, A.; Bordoni, R.; Spinelli, R.; Asselta, R.; Valin, P.S.; Di Candia, S.; Zamproni, I.; Diceglie, C.; et al. Whole genome SNP genotyping and exome sequencing reveal novel genetic variants and putative causative genes in congenital hyperinsulinism. PLoS ONE 2013, 8, e68740. [Google Scholar] [CrossRef]
- Splawski, I.; Timothy, K.W.; Vincent, G.M.; Atkinson, D.L.; Keating, M.T. Molecular basis of the long-QT syndrome associated with deafness. N. Engl. J. Med. 1997, 336, 1562–1567. [Google Scholar] [CrossRef] [PubMed]
- Torekov, S.S.; Iepsen, E.; Christiansen, M.; Linneberg, A.; Pedersen, O.; Holst, J.J.; Kanters, J.K.; Hansen, T. KCNQ1 long QT syndrome patients have hyperinsulinemia and symptomatic hypoglycemia. Diabetes 2014, 63, 1315–1325. [Google Scholar] [CrossRef] [PubMed]
- Catterall, W.A. Voltage-gated calcium channels. Cold Spring Harb. Perspect. Biol. 2011, 3, a003947. [Google Scholar] [CrossRef]
- Yang, S.N.; Berggren, P.O. The role of voltage-gated calcium channels in pancreatic beta-cell physiology and pathophysiology. Endocr. Rev. 2006, 27, 621–676. [Google Scholar] [CrossRef] [PubMed]
- Braun, M.; Ramracheya, R.; Bengtsson, M.; Zhang, Q.; Karanauskaite, J.; Partridge, C.; Johnson, P.R.; Rorsman, P. Voltage-gated ion channels in human pancreatic beta-cells: Electrophysiological characterization and role in insulin secretion. Diabetes 2008, 57, 1618–1628. [Google Scholar] [CrossRef]
- Flanagan, S.E.; Vairo, F.; Johnson, M.B.; Caswell, R.; Laver, T.W.; Lango Allen, H.; Hussain, K.; Ellard, S. A CACNA1D mutation in a patient with persistent hyperinsulinaemic hypoglycaemia, heart defects, and severe hypotonia. Pediatric Diabetes 2017, 18, 320–323. [Google Scholar] [CrossRef]
- Iwashima, Y.; Pugh, W.; Depaoli, A.M.; Takeda, J.; Seino, S.; Bell, G.I.; Polonsky, K.S. Expression of calcium channel mRNAs in rat pancreatic islets and downregulation after glucose infusion. Diabetes 1993, 42, 948–955. [Google Scholar] [CrossRef]
- Scholl, U.I.; Goh, G.; Stolting, G.; de Oliveira, R.C.; Choi, M.; Overton, J.D.; Fonseca, A.L.; Korah, R.; Starker, L.F.; Kunstman, J.W.; et al. Somatic and germline CACNA1D calcium channel mutations in aldosterone-producing adenomas and primary aldosteronism. Nat. Genet. 2013, 45, 1050–1054. [Google Scholar] [CrossRef]
- Fernandes-Rosa, F.L.; Williams, T.A.; Riester, A.; Steichen, O.; Beuschlein, F.; Boulkroun, S.; Strom, T.M.; Monticone, S.; Amar, L.; Meatchi, T.; et al. Genetic spectrum and clinical correlates of somatic mutations in aldosterone-producing adenoma. Hypertension 2014, 64, 354–361. [Google Scholar] [CrossRef]
- Splawski, I.; Timothy, K.W.; Sharpe, L.M.; Decher, N.; Kumar, P.; Bloise, R.; Napolitano, C.; Schwartz, P.J.; Joseph, R.M.; Condouris, K.; et al. Ca(V)1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell 2004, 119, 19–31. [Google Scholar] [CrossRef]
- Cuff, M.A.; Shirazi-Beechey, S.P. The human monocarboxylate transporter, MCT1: Genomic organization and promoter analysis. Biochem. Biophys. Res. Commun. 2002, 292, 1048–1056. [Google Scholar] [CrossRef]
- Garcia, C.K.; Goldstein, J.L.; Pathak, R.K.; Anderson, R.G.; Brown, M.S. Molecular characterization of a membrane transporter for lactate, pyruvate, and other monocarboxylates: Implications for the Cori cycle. Cell 1994, 76, 865–873. [Google Scholar] [CrossRef]
- Halestrap, A.P.; Price, N.T. The proton-linked monocarboxylate transporter (MCT) family: Structure, function and regulation. Biochem. J. 1999, 343, 281–299. [Google Scholar] [CrossRef]
- Halestrap, A.P.; Meredith, D. The SLC16 gene family-from monocarboxylate transporters (MCTs) to aromatic amino acid transporters and beyond. Pflug. Arch. 2004, 447, 619–628. [Google Scholar] [CrossRef]
- Sekine, N.; Cirulli, V.; Regazzi, R.; Brown, L.J.; Gine, E.; Tamarit-Rodriguez, J.; Girotti, M.; Marie, S.; MacDonald, M.J.; Wollheim, C.B.; et al. Low lactate dehydrogenase and high mitochondrial glycerol phosphate dehydrogenase in pancreatic beta-cells. Potential role in nutrient sensing. J. Biol. Chem. 1994, 269, 4895–4902. [Google Scholar]
- Zhao, C.; Wilson, M.C.; Schuit, F.; Halestrap, A.P.; Rutter, G.A. Expression and distribution of lactate/monocarboxylate transporter isoforms in pancreatic islets and the exocrine pancreas. Diabetes 2001, 50, 361–366. [Google Scholar] [CrossRef] [PubMed]
- Pullen, T.J.; da Silva Xavier, G.; Kelsey, G.; Rutter, G.A. miR-29a and miR-29b contribute to pancreatic beta-cell-specific silencing of monocarboxylate transporter 1 (Mct1). Mol. Cell Biol. 2011, 31, 3182–3194. [Google Scholar] [CrossRef]
- Pullen, T.J.; Sylow, L.; Sun, G.; Halestrap, A.P.; Richter, E.A.; Rutter, G.A. Overexpression of monocarboxylate transporter-1 (SLC16A1) in mouse pancreatic beta-cells leads to relative hyperinsulinism during exercise. Diabetes 2012, 61, 1719–1725. [Google Scholar] [CrossRef]
- Meissner, T.; Otonkoski, T.; Feneberg, R.; Beinbrech, B.; Apostolidou, S.; Sipila, I.; Schaefer, F.; Mayatepek, E. Exercise induced hypoglycaemic hyperinsulinism. Arch. Dis. Child 2001, 84, 254–257. [Google Scholar] [CrossRef] [PubMed]
- Otonkoski, T.; Jiao, H.; Kaminen-Ahola, N.; Tapia-Paez, I.; Ullah, M.S.; Parton, L.E.; Schuit, F.; Quintens, R.; Sipila, I.; Mayatepek, E.; et al. Physical exercise-induced hypoglycemia caused by failed silencing of monocarboxylate transporter 1 in pancreatic beta cells. Am. J. Hum. Genet. 2007, 81, 467–474. [Google Scholar] [CrossRef] [PubMed]
- Otonkoski, T.; Kaminen, N.; Ustinov, J.; Lapatto, R.; Meissner, T.; Mayatepek, E.; Kere, J.; Sipila, I. Physical exercise-induced hyperinsulinemic hypoglycemia is an autosomal-dominant trait characterized by abnormal pyruvate-induced insulin release. Diabetes 2003, 52, 199–204. [Google Scholar] [CrossRef] [PubMed]
- Balasubramaniam, S.; Lewis, B.; Greed, L.; Meili, D.; Flier, A.; Yamamoto, R.; Bilic, K.; Till, C.; Sass, J.O. Heterozygous Monocarboxylate Transporter 1 (MCT1, SLC16A1) Deficiency as a Cause of Recurrent Ketoacidosis. JIMD Rep 2016, 29, 33–38. [Google Scholar]
- Fishbein, W.N. Lactate transporter defect: A new disease of muscle. Science 1986, 234, 1254–1256. [Google Scholar] [CrossRef] [PubMed]
- Merezhinskaya, N.; Fishbein, W.N.; Davis, J.I.; Foellmer, J.W. Mutations in MCT1 cDNA in patients with symptomatic deficiency in lactate transport. Muscle Nerve 2000, 23, 90–97. [Google Scholar] [CrossRef]
- Tosur, M.; Jeha, G.S. A Novel Intragenic SLC16A1 Mutation Associated With Congenital Hyperinsulinism. Glob. Pediatric Health 2017, 4, 2333794x17703462. [Google Scholar] [CrossRef] [PubMed]
- Sperling, M.A. Neonatal diabetes mellitus. In Pedaitric Endocrinology, 4th ed.; Saunders: Philadelphia, PA, USA, 2014; pp. 277–289. [Google Scholar]
- Von Muhlendahl, K.E.; Herkenhoff, H. Long-term course of neonatal diabetes. N. Engl. J. Med. 1995, 333, 704–708. [Google Scholar] [CrossRef] [PubMed]
- Rubio-Cabezas, O.; Ellard, S. Diabetes mellitus in neonates and infants: Genetic heterogeneity, clinical approach to diagnosis, and therapeutic options. Horm. Res. Paediatr. 2013, 80, 137–146. [Google Scholar] [CrossRef] [PubMed]
- Grulich-Henn, J.; Wagner, V.; Thon, A.; Schober, E.; Marg, W.; Kapellen, T.M.; Haberland, H.; Raile, K.; Ellard, S.; Flanagan, S.E.; et al. Entities and frequency of neonatal diabetes: Data from the diabetes documentation and quality management system (DPV). Diabet. Med. A J. Br. Diabet. Assoc. 2010, 27, 709–712. [Google Scholar] [CrossRef]
- Iafusco, D.; Massa, O.; Pasquino, B.; Colombo, C.; Iughetti, L.; Bizzarri, C.; Mammi, C.; Lo Presti, D.; Suprani, T.; Schiaffini, R.; et al. Minimal incidence of neonatal/infancy onset diabetes in Italy is 1:90,000 live births. Acta Diabetol. 2012, 49, 405–408. [Google Scholar] [CrossRef]
- Polak, M.; Cave, H. Neonatal diabetes mellitus: A disease linked to multiple mechanisms. Orphanet J. Rare Dis. 2007, 2, 12. [Google Scholar] [CrossRef]
- Nagashima, K.; Tanaka, D.; Inagaki, N. Epidemiology, clinical characteristics, and genetic etiology of neonatal diabetes in Japan. Pediatrics Int. Off. J. Jpn. Pediatric Soc. 2017, 59, 129–133. [Google Scholar] [CrossRef]
- Slingerland, A.S.; Shields, B.M.; Flanagan, S.E.; Bruining, G.J.; Noordam, K.; Gach, A.; Mlynarski, W.; Malecki, M.T.; Hattersley, A.T.; Ellard, S. Referral rates for diagnostic testing support an incidence of permanent neonatal diabetes in three European countries of at least 1 in 260,000 live births. Diabetologia 2009, 52, 1683–1685. [Google Scholar] [CrossRef] [Green Version]
- Habeb, A.M.; Al-Magamsi, M.S.; Eid, I.M.; Ali, M.I.; Hattersley, A.T.; Hussain, K.; Ellard, S. Incidence, genetics, and clinical phenotype of permanent neonatal diabetes mellitus in northwest Saudi Arabia. Pediatric Diabetes 2012, 13, 499–505. [Google Scholar] [CrossRef]
- Demirbilek, H.; Arya, V.B.; Ozbek, M.N.; Houghton, J.A.; Baran, R.T.; Akar, M.; Tekes, S.; Tuzun, H.; Mackay, D.J.; Flanagan, S.E.; et al. Clinical characteristics and molecular genetic analysis of 22 patients with neonatal diabetes from the South-Eastern region of Turkey: Predominance of non-KATP channel mutations. Eur. J. Endocrinol. 2015, 172, 697–705. [Google Scholar] [CrossRef]
- Rubio-Cabezas, O.; Klupa, T.; Malecki, M.T. Permanent neonatal diabetes mellitus—The importance of diabetes differential diagnosis in neonates and infants. Eur. J. Clin. Investig. 2011, 41, 323–333. [Google Scholar] [CrossRef]
- Flanagan, S.E.; Patch, A.M.; Mackay, D.J.; Edghill, E.L.; Gloyn, A.L.; Robinson, D.; Shield, J.P.; Temple, K.; Ellard, S.; Hattersley, A.T. Mutations in ATP-sensitive K+ channel genes cause transient neonatal diabetes and permanent diabetes in childhood or adulthood. Diabetes 2007, 56, 1930–1937. [Google Scholar] [CrossRef]
- Patch, A.M.; Flanagan, S.E.; Boustred, C.; Hattersley, A.T.; Ellard, S. Mutations in the ABCC8 gene encoding the SUR1 subunit of the KATP channel cause transient neonatal diabetes, permanent neonatal diabetes or permanent diabetes diagnosed outside the neonatal period. Diabetes Obes. Metab. 2007, 9 (Suppl. 2), 28–39. [Google Scholar] [CrossRef]
- Riveline, J.P.; Rousseau, E.; Reznik, Y.; Fetita, S.; Philippe, J.; Dechaume, A.; Hartemann, A.; Polak, M.; Petit, C.; Charpentier, G.; et al. Clinical and metabolic features of adult-onset diabetes caused by ABCC8 mutations. Diabetes Care 2012, 35, 248–251. [Google Scholar] [CrossRef]
- Edghill, E.L.; Flanagan, S.E.; Ellard, S. Permanent neonatal diabetes due to activating mutations in ABCC8 and KCNJ11. Rev. Endocr. Metab. Disord. 2010, 11, 193–198. [Google Scholar] [CrossRef]
- Babenko, A.P.; Polak, M.; Cave, H.; Busiah, K.; Czernichow, P.; Scharfmann, R.; Bryan, J.; Aguilar-Bryan, L.; Vaxillaire, M.; Froguel, P. Activating mutations in the ABCC8 gene in neonatal diabetes mellitus. N. Engl. J. Med. 2006, 355, 456–466. [Google Scholar] [CrossRef] [PubMed]
- Gloyn, A.L.; Pearson, E.R.; Antcliff, J.F.; Proks, P.; Bruining, G.J.; Slingerland, A.S.; Howard, N.; Srinivasan, S.; Silva, J.M.; Molnes, J.; et al. Activating mutations in the gene encoding the ATP-sensitive potassium-channel subunit Kir6.2 and permanent neonatal diabetes. N. Engl. J. Med. 2004, 350, 1838–1849. [Google Scholar] [CrossRef]
- Proks, P.; de Wet, H.; Ashcroft, F.M. Molecular mechanism of sulphonylurea block of K(ATP) channels carrying mutations that impair ATP inhibition and cause neonatal diabetes. Diabetes 2013, 62, 3909–3919. [Google Scholar] [CrossRef]
- Vedovato, N.; Cliff, E.; Proks, P.; Poovazhagi, V.; Flanagan, S.E.; Ellard, S.; Hattersley, A.T.; Ashcroft, F.M. Neonatal diabetes caused by a homozygous KCNJ11 mutation demonstrates that tiny changes in ATP sensitivity markedly affect diabetes risk. Diabetologia 2016, 59, 1430–1436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clark, R.H.; McTaggart, J.S.; Webster, R.; Mannikko, R.; Iberl, M.; Sim, X.L.; Rorsman, P.; Glitsch, M.; Beeson, D.; Ashcroft, F.M. Muscle dysfunction caused by a KATP channel mutation in neonatal diabetes is neuronal in origin. Science 2010, 329, 458–461. [Google Scholar] [CrossRef] [PubMed]
- Gloyn, A.L.; Diatloff-Zito, C.; Edghill, E.L.; Bellanne-Chantelot, C.; Nivot, S.; Coutant, R.; Ellard, S.; Hattersley, A.T.; Robert, J.J. KCNJ11 activating mutations are associated with developmental delay, epilepsy and neonatal diabetes syndrome and other neurological features. Eur. J. Hum. Genet. 2006, 14, 824–830. [Google Scholar] [CrossRef]
- Proks, P.; Shimomura, K.; Craig, T.J.; Girard, C.A.; Ashcroft, F.M. Mechanism of action of a sulphonylurea receptor SUR1 mutation (F132L) that causes DEND syndrome. Hum. Mol. Genet. 2007, 16, 2011–2019. [Google Scholar] [CrossRef] [Green Version]
- Rafiq, M.; Flanagan, S.E.; Patch, A.M.; Shields, B.M.; Ellard, S.; Hattersley, A.T. Effective treatment with oral sulfonylureas in patients with diabetes due to sulfonylurea receptor 1 (SUR1) mutations. Diabetes Care 2008, 31, 204–209. [Google Scholar] [CrossRef] [PubMed]
- Landmeier, K.A.; Lanning, M.; Carmody, D.; Greeley, S.A.W.; Msall, M.E. ADHD, learning difficulties and sleep disturbances associated with KCNJ11-related neonatal diabetes. Pediatric Diabetes 2017, 18, 518–523. [Google Scholar] [CrossRef] [PubMed]
- Bowman, P.; Hattersley, A.T.; Knight, B.A.; Broadbridge, E.; Pettit, L.; Reville, M.; Flanagan, S.E.; Shepherd, M.H.; Ford, T.J.; Tonks, J. Neuropsychological impairments in children with KCNJ11 neonatal diabetes. Diabet. Med. A J. Br. Diabet. Assoc. 2017, 34, 1171–1173. [Google Scholar] [CrossRef]
- Klupa, T.; Kowalska, I.; Wyka, K.; Skupien, J.; Patch, A.M.; Flanagan, S.E.; Noczynska, A.; Arciszewska, M.; Ellard, S.; Hattersley, A.T.; et al. Mutations in the ABCC8 (SUR1 subunit of the K(ATP) channel) gene are associated with a variable clinical phenotype. Clin. Endocrinol. 2009, 71, 358–362. [Google Scholar] [CrossRef] [PubMed]
- Pearson, E.R.; Flechtner, I.; Njolstad, P.R.; Malecki, M.T.; Flanagan, S.E.; Larkin, B.; Ashcroft, F.M.; Klimes, I.; Codner, E.; Iotova, V.; et al. Switching from insulin to oral sulfonylureas in patients with diabetes due to Kir6.2 mutations. N. Engl. J. Med. 2006, 355, 467–477. [Google Scholar] [CrossRef]
- Babiker, T.; Vedovato, N.; Patel, K.; Thomas, N.; Finn, R.; Mannikko, R.; Chakera, A.J.; Flanagan, S.E.; Shepherd, M.H.; Ellard, S.; Ashcroft, F.M.; Hattersley, A.T. Successful transfer to sulfonylureas in KCNJ11 neonatal diabetes is determined by the mutation and duration of diabetes. Diabetologia 2016, 59, 1162–1166. [Google Scholar] [CrossRef] [Green Version]
- Ashcroft, F.M.; Puljung, M.C.; Vedovato, N. Neonatal Diabetes and the KATP Channel: From Mutation to Therapy. Trends Endocrinol. Metab. 2017, 28, 377–387. [Google Scholar] [CrossRef] [PubMed]
- Beltrand, J.; Elie, C.; Busiah, K.; Fournier, E.; Boddaert, N.; Bahi-Buisson, N.; Vera, M.; Bui-Quoc, E.; Ingster-Moati, I.; Berdugo, M.; et al. Sulfonylurea Therapy Benefits Neurological and Psychomotor Functions in Patients With Neonatal Diabetes Owing to Potassium Channel Mutations. Diabetes Care 2015, 38, 2033–2041. [Google Scholar] [CrossRef] [Green Version]
- Rubio-Cabezas, O.; Hattersley, A.T.; Njolstad, P.R.; Mlynarski, W.; Ellard, S.; White, N.; Chi, D.V.; Craig, M.E. ISPAD Clinical Practice Consensus Guidelines 2014. The diagnosis and management of monogenic diabetes in children and adolescents. Pediatric Diabetes 2014, 15 (Suppl. 20), 47–64. [Google Scholar] [CrossRef]
- Lachance, C.H. Practical Aspects of Monogenic Diabetes: A Clinical Point of View. Can. J. Diabetes 2016, 40, 368–375. [Google Scholar] [CrossRef] [Green Version]
- Bonnefond, A.; Philippe, J.; Durand, E.; Dechaume, A.; Huyvaert, M.; Montagne, L.; Marre, M.; Balkau, B.; Fajardy, I.; Vambergue, A.; et al. Whole-exome sequencing and high throughput genotyping identified KCNJ11 as the thirteenth MODY gene. PLoS ONE 2012, 7, e37423. [Google Scholar] [CrossRef]
- Isik, E.; Demirbilek, H.; Houghton, J.A.L.; Ellard, S.; Flanagan, S.E.; Hussain, K. Congenital hyperinsulinism and evolution to sulfonylurea-responsive diabetes later in life due to a novel homozygous p.L171F ABCC8 mutation. J. Clin. Res. Pediatric Endocrinol. 2019, 11, 82–87. [Google Scholar] [CrossRef]
- Thorens, B.; Mueckler, M. Glucose transporters in the 21st Century. Am. J. Physiol. Endocrinol. Metab. 2010, 298, E141–E145. [Google Scholar] [CrossRef] [Green Version]
- Yan, N. A Glimpse of Membrane Transport through Structures-Advances in the Structural Biology of the GLUT Glucose Transporters. J. Mol. Biol. 2017, 429, 2710–2725. [Google Scholar] [CrossRef]
- Takeda, J.; Kayano, T.; Fukomoto, H.; Bell, G.I. Organization of the human GLUT2 (pancreatic beta-cell and hepatocyte) glucose transporter gene. Diabetes 1993, 42, 773–777. [Google Scholar] [CrossRef]
- Brown, G.K. Glucose transporters: Structure, function and consequences of deficiency. J. Inherit. Metab. Dis. 2000, 23, 237–246. [Google Scholar] [CrossRef]
- Santer, R.; Schneppenheim, R.; Dombrowski, A.; Gotze, H.; Steinmann, B.; Schaub, J. Mutations in GLUT2, the gene for the liver-type glucose transporter, in patients with Fanconi-Bickel syndrome. Nat. Genet. 1997, 17, 324–326. [Google Scholar] [CrossRef]
- Mannstadt, M.; Magen, D.; Segawa, H.; Stanley, T.; Sharma, A.; Sasaki, S.; Bergwitz, C.; Mounien, L.; Boepple, P.; Thorens, B.; et al. Fanconi-Bickel syndrome and autosomal recessive proximal tubulopathy with hypercalciuria (ARPTH) are allelic variants caused by GLUT2 mutations. J. Clin. Endocrinol. Metab. 2012, 97, E1978–E1986. [Google Scholar] [CrossRef]
- Moller, A.M.; Jensen, N.M.; Pildal, J.; Drivsholm, T.; Borch-Johnsen, K.; Urhammer, S.A.; Hansen, T.; Pedersen, O. Studies of genetic variability of the glucose transporter 2 promoter in patients with type 2 diabetes mellitus. J. Clin. Endocrinol. Metab. 2001, 86, 2181–2186. [Google Scholar] [CrossRef]
- Mueckler, M.; Kruse, M.; Strube, M.; Riggs, A.C.; Chiu, K.C.; Permutt, M.A. A mutation in the Glut2 glucose transporter gene of a diabetic patient abolishes transport activity. J. Biol. Chem. 1994, 269, 17765–17767. [Google Scholar]
- Sansbury, F.H.; Flanagan, S.E.; Houghton, J.A.; Shuixian Shen, F.L.; Al-Senani, A.M.; Habeb, A.M.; Abdullah, M.; Kariminejad, A.; Ellard, S.; Hattersley, A.T. SLC2A2 mutations can cause neonatal diabetes, suggesting GLUT2 may have a role in human insulin secretion. Diabetologia 2012, 55, 2381–2385. [Google Scholar] [CrossRef] [Green Version]
- Santer, R.; Steinmann, B.; Schaub, J. Fanconi-Bickel syndrome—A congenital defect of facilitative glucose transport. Curr. Mol. Med. 2002, 2, 213–227. [Google Scholar] [CrossRef]
- Al-Haggar, M. Fanconi-Bickel syndrome as an example of marked allelic heterogeneity. World J. Nephrol. 2012, 1, 63–68. [Google Scholar] [CrossRef]
- Taha, D.; Al-Harbi, N.; Al-Sabban, E. Hyperglycemia and hypoinsulinemia in patients with Fanconi-Bickel syndrome. J. Pediatr. Endocrinol. Metab. 2008, 21, 581–586. [Google Scholar]
- Berry, G.T.; Baker, L.; Kaplan, F.S.; Witzleben, C.L. Diabetes-like renal glomerular disease in Fanconi-Bickel syndrome. Pediatr. Nephrol. 1995, 9, 287–291. [Google Scholar] [CrossRef]
- Fleming, J.C.; Tartaglini, E.; Steinkamp, M.P.; Schorderet, D.F.; Cohen, N.; Neufeld, E.J. The gene mutated in thiamine-responsive anaemia with diabetes and deafness (TRMA) encodes a functional thiamine transporter. Nat. Genet. 1999, 22, 305–308. [Google Scholar] [CrossRef]
- Neufeld, E.J.; Mandel, H.; Raz, T.; Szargel, R.; Yandava, C.N.; Stagg, A.; Faure, S.; Barrett, T.; Buist, N.; Cohen, N. Localization of the gene for thiamine-responsive megaloblastic anemia syndrome, on the long arm of chromosome 1, by homozygosity mapping. Am. J. Hum. Genet. 1997, 61, 1335–1341. [Google Scholar] [CrossRef]
- Labay, V.; Raz, T.; Baron, D.; Mandel, H.; Williams, H.; Barrett, T.; Szargel, R.; McDonald, L.; Shalata, A.; Nosaka, K.; et al. Mutations in SLC19A2 cause thiamine-responsive megaloblastic anaemia associated with diabetes mellitus and deafness. Nat. Genet. 1999, 22, 300–304. [Google Scholar] [CrossRef]
- Dutta, B.; Huang, W.; Molero, M.; Kekuda, R.; Leibach, F.H.; Devoe, L.D.; Ganapathy, V.; Prasad, P.D. Cloning of the human thiamine transporter, a member of the folate transporter family. J. Biol. Chem. 1999, 274, 31925–31929. [Google Scholar] [CrossRef]
- Ricketts, C.J.; Minton, J.A.; Samuel, J.; Ariyawansa, I.; Wales, J.K.; Lo, I.F.; Barrett, T.G. Thiamine-responsive megaloblastic anaemia syndrome: Long-term follow-up and mutation analysis of seven families. Acta Paediatr. 2006, 95, 99–104. [Google Scholar] [CrossRef]
- Rajgopal, A.; Edmondnson, A.; Goldman, I.D.; Zhao, R. SLC19A3 encodes a second thiamine transporter ThTr2. Biochim. Biophys. Acta 2001, 1537, 175–178. [Google Scholar] [CrossRef] [Green Version]
- Pácal, L.; Kuricová, K.; Kaňková, K. Evidence for altered thiamine metabolism in diabetes: Is there a potential to oppose gluco- and lipotoxicity by rational supplementation? World J. Diabetes 2014, 5, 288–295. [Google Scholar] [CrossRef]
- Rindi, G.; Ferrari, G. Thiamine transport by human intestine in vitro. Experientia 1977, 33, 211–213. [Google Scholar] [CrossRef]
- Laforenza, U.; Patrini, C.; Alvisi, C.; Faelli, A.; Licandro, A.; Rindi, G. Thiamine uptake in human intestinal biopsy specimens, including observations from a patient with acute thiamine deficiency. Am. J. Clin. Nutr. 1997, 66, 320–326. [Google Scholar] [CrossRef]
- Hoyumpa, A.M., Jr.; Strickland, R.; Sheehan, J.J.; Yarborough, G.; Nichols, S. Dual system of intestinal thiamine transport in humans. J. Lab. Clin. Med. 1982, 99, 701–708. [Google Scholar]
- Porter, F.S.; Rogers, L.E.; Sidbury, J.B., Jr. Thiamine-responsive megaloblastic anemia. J. Pediatr. 1969, 74, 494–504. [Google Scholar]
- Viana, M.B.; Carvalho, R.I. Thiamine-responsive megaloblastic anemia, sensorineural deafness, and diabetes mellitus: A new syndrome? J. Pediatr. 1978, 93, 235–238. [Google Scholar] [CrossRef]
- Bergmann, A.K.; Sahai, I.; Falcone, J.F.; Fleming, J.; Bagg, A.; Borgna-Pignati, C.; Casey, R.; Fabris, L.; Hexner, E.; Mathews, L.; et al. Thiamine-responsive megaloblastic anemia: Identification of novel compound heterozygotes and mutation update. J. Pediatr. 2009, 155, 888e1–892e1. [Google Scholar] [CrossRef]
- Mozzillo, E.; Melis, D.; Falco, M.; Fattorusso, V.; Taurisano, R.; Flanagan, S.E.; Ellard, S.; Franzese, A. Thiamine responsive megaloblastic anemia: A novel SLC19A2 compound heterozygous mutation in two siblings. Pediatric Diabetes 2013, 14, 384–387. [Google Scholar] [CrossRef]
- Pichler, H.; Zeitlhofer, P.; Dworzak, M.N.; Diakos, C.; Haas, O.A.; Kager, L. Thiamine-responsive megaloblastic anemia (TRMA) in an Austrian boy with compound heterozygous SLC19A2 mutations. Eur. J. Pediatr. 2012, 171, 1711–1715. [Google Scholar] [CrossRef]
- Prasannan, K.G.; Sundaresan, R.; Venkatesan, D. Thiamine deficency and protein secretion by pancreatic slices in vitro. Experientia 1977, 33, 169–170. [Google Scholar] [CrossRef]
- Rathanaswami, P.; Pourany, A.; Sundaresan, R. Effects of thiamine deficiency on the secretion of insulin and the metabolism of glucose in isolated rat pancreatic islets. Biochem. Int. 1991, 25, 577–583. [Google Scholar]
- Stagg, A.R.; Fleming, J.C.; Baker, M.A.; Sakamoto, M.; Cohen, N.; Neufeld, E.J. Defective high-affinity thiamine transporter leads to cell death in thiamine-responsive megaloblastic anemia syndrome fibroblasts. J. Clin. Investig. 1999, 103, 723–729. [Google Scholar] [CrossRef] [Green Version]
- Valerio, G.; Franzese, A.; Poggi, V.; Tenore, A. Long-term follow-up of diabetes in two patients with thiamine-responsive megaloblastic anemia syndrome. Diabetes Care 1998, 21, 38–41. [Google Scholar] [CrossRef]
- Alzahrani, A.S.; Baitei, E.; Zou, M.; Shi, Y. Thiamine transporter mutation: An example of monogenic diabetes mellitus. Eur. J. Endocrinol. 2006, 155, 787–792. [Google Scholar] [CrossRef]
- Ghaemi, N.; Ghahraman, M.; Abbaszadegan, M.R.; Baradaran-Heravi, A.; Vakili, R. Novel mutation in the SLC19A2 gene in an Iranian family with thiamine-responsive megaloblastic anemia: A series of three cases. J. Clin. Res. Pediatr. Endocrinol. 2013, 5, 199–201. [Google Scholar]
- Gloyn, A.L.; Weedon, M.N.; Owen, K.R.; Turner, M.J.; Knight, B.A.; Hitman, G.; Walker, M.; Levy, J.C.; Sampson, M.; Halford, S.; et al. Large-scale association studies of variants in genes encoding the pancreatic beta-cell KATP channel subunits Kir6.2 (KCNJ11) and SUR1 (ABCC8) confirm that the KCNJ11 E23K variant is associated with type 2 diabetes. Diabetes 2003, 52, 568–572. [Google Scholar] [CrossRef]
- Riedel, M.J.; Boora, P.; Steckley, D.; de Vries, G.; Light, P.E. Kir6.2 polymorphisms sensitize beta-cell ATP-sensitive potassium channels to activation by acyl CoAs: A possible cellular mechanism for increased susceptibility to type 2 diabetes? Diabetes 2003, 52, 2630–2635. [Google Scholar] [CrossRef]
- Yasuda, K.; Miyake, K.; Horikawa, Y.; Hara, K.; Osawa, H.; Furuta, H.; Hirota, Y.; Mori, H.; Jonsson, A.; Sato, Y.; et al. Variants in KCNQ1 are associated with susceptibility to type 2 diabetes mellitus. Nat. Genet. 2008, 40, 1092–1097. [Google Scholar] [CrossRef]
- Unoki, H.; Takahashi, A.; Kawaguchi, T.; Hara, K.; Horikoshi, M.; Andersen, G.; Ng, D.P.; Holmkvist, J.; Borch-Johnsen, K.; Jorgensen, T.; et al. SNPs in KCNQ1 are associated with susceptibility to type 2 diabetes in East Asian and European populations. Nat. Genet. 2008, 40, 1098–1102. [Google Scholar] [CrossRef]
- Thevenod, F. Ion channels in secretory granules of the pancreas and their role in exocytosis and release of secretory proteins. Am. J. Physiol. Cell Physiol. 2002, 283, C651–C672. [Google Scholar] [CrossRef] [Green Version]
- Sladek, R.; Rocheleau, G.; Rung, J.; Dina, C.; Shen, L.; Serre, D.; Boutin, P.; Vincent, D.; Belisle, A.; Hadjadj, S.; et al. A genome-wide association study identifies novel risk loci for type 2 diabetes. Nature 2007, 445, 881–885. [Google Scholar] [CrossRef]
Figure 1.
The physiology of insulin secretion from pancreatic beta-cells. Glucose enters into the beta cells through facilitative transport mediated by glucose transporter 2 (GLUT 2) (1) and is converted to glucose-6-phosphate by the enzyme glucokinase (2). Generation of high energy molecules such as adenosine triphosphate (ATP), leads to an increase in the ratio of ATP/ADP (adenosine diphosphate) (3). Elevated ATP/ADP ratio closes the ATP-sensitive potassium channel (KATP). The Kir6.2 subunits of the KATP channels are responsible for K+ ion efflux from the pancreatic beta-cell, and thus maintains a steady state membrane potential (4). The closure of the KATP channels results in a depolarization of the pancreatic beta-cell membrane and the activation of voltage-gated calcium channels located on the beta-cell membrane (5). Calcium enters into beta cells through these voltage-gated calcium channels (6), and the increase in intracellular calcium triggers secretory granule exocytosis and insulin release (7). In congenital hyperinsulinism, defects in the KATP channel or energy metabolism leading to prolonged closure of KATP channels, create a membrane potential between the inner and outer sites of the beta cell (depolarization) which is followed by the opening of the voltage-gated calcium channel and calcium influx. Constantly closed KATP channels or uncontrolled ATP generation uncouples the insulin secretion from the blood glucose level and causes inappropriate insulin secretion, despite low blood glucose. On the contrary, in neonatal diabetes mellitus (NDM), the KATP channel remains open and a constant efflux of K+ ion hyperpolarizes the beta cell. Lack of depolarization does not allow for voltage-gated calcium channel opening and insulin secretion despite elevated blood glucose. (Ca2+: calcium ions, cAMP: cyclic adenosine monophosphate, G1P: glucose-1-phosphate, G6P: glucose 6-phosphate, GABA: γ-aminobutyric acid, GAD: glutamate decarboxylase enzyme, GLUD1: glutamate dehydrogenase 1, GDH: glutamate dehydrogenase, GLP1: glucagon-like peptide 1, GLUT2: glucose transporter 2, HADH: hydroxy acyl-CoA dehydrogenase, HNF1α: hepatocyte nuclear factor 1α, HNF4α: hepatocyte nuclear factor 4α, K+: potassium, Kir6.2: inward rectifier potassium channel 6.2, MCT1: monocarboxylate transporter 1, NH3: ammonia, PGM1: phosphoglucomutase 1, PMM2: phosphomannomutase 2, SUR1: sulfonylurea receptor 1, TCA: tricarboxylic acid, UCP2: mitochondrial uncoupling protein 2).
Figure 1.
The physiology of insulin secretion from pancreatic beta-cells. Glucose enters into the beta cells through facilitative transport mediated by glucose transporter 2 (GLUT 2) (1) and is converted to glucose-6-phosphate by the enzyme glucokinase (2). Generation of high energy molecules such as adenosine triphosphate (ATP), leads to an increase in the ratio of ATP/ADP (adenosine diphosphate) (3). Elevated ATP/ADP ratio closes the ATP-sensitive potassium channel (KATP). The Kir6.2 subunits of the KATP channels are responsible for K+ ion efflux from the pancreatic beta-cell, and thus maintains a steady state membrane potential (4). The closure of the KATP channels results in a depolarization of the pancreatic beta-cell membrane and the activation of voltage-gated calcium channels located on the beta-cell membrane (5). Calcium enters into beta cells through these voltage-gated calcium channels (6), and the increase in intracellular calcium triggers secretory granule exocytosis and insulin release (7). In congenital hyperinsulinism, defects in the KATP channel or energy metabolism leading to prolonged closure of KATP channels, create a membrane potential between the inner and outer sites of the beta cell (depolarization) which is followed by the opening of the voltage-gated calcium channel and calcium influx. Constantly closed KATP channels or uncontrolled ATP generation uncouples the insulin secretion from the blood glucose level and causes inappropriate insulin secretion, despite low blood glucose. On the contrary, in neonatal diabetes mellitus (NDM), the KATP channel remains open and a constant efflux of K+ ion hyperpolarizes the beta cell. Lack of depolarization does not allow for voltage-gated calcium channel opening and insulin secretion despite elevated blood glucose. (Ca2+: calcium ions, cAMP: cyclic adenosine monophosphate, G1P: glucose-1-phosphate, G6P: glucose 6-phosphate, GABA: γ-aminobutyric acid, GAD: glutamate decarboxylase enzyme, GLUD1: glutamate dehydrogenase 1, GDH: glutamate dehydrogenase, GLP1: glucagon-like peptide 1, GLUT2: glucose transporter 2, HADH: hydroxy acyl-CoA dehydrogenase, HNF1α: hepatocyte nuclear factor 1α, HNF4α: hepatocyte nuclear factor 4α, K+: potassium, Kir6.2: inward rectifier potassium channel 6.2, MCT1: monocarboxylate transporter 1, NH3: ammonia, PGM1: phosphoglucomutase 1, PMM2: phosphomannomutase 2, SUR1: sulfonylurea receptor 1, TCA: tricarboxylic acid, UCP2: mitochondrial uncoupling protein 2).

Figure 2.
Summary of ion transport channel defects and related disorders (K+: Potassium, VDCC: Voltage dependent calcium channels, KATP: ATP-sensitive potassium channels, KIR6.2: Inward rectifier potassium channel subunit 2, KCNJ11: Potassium Voltage-Gated Channel Subfamily J Member, SUR1: Sulphonylurea receptor 1, ABCC8: ATP-binding cassette subfamily C member 8, MODY: Maturity-onset diabetes of the young, CACNA1D: Calcium Voltage-Gated Channel Subunit Alpha1 D, CACNA1C: Calcium Channel, Voltage-Dependent, L Type, alpha 1C Subunit, KCNH6: Potassium voltage-gated channel subfamily H member 6, NDM: Neonatal diabetes mellitus, TNDM: Transient NDM, PNDM: permanent NDM, HH: Hyperinsulinaemic Hypoglycemia, CHD: Congenital heart diseases, HPA: Hyperaldosteronism, NMA: Neuromuscular abnormalities, LOH: Loss of heterozygosity, UPD: Uniparental isodisomy).
Figure 2.
Summary of ion transport channel defects and related disorders (K+: Potassium, VDCC: Voltage dependent calcium channels, KATP: ATP-sensitive potassium channels, KIR6.2: Inward rectifier potassium channel subunit 2, KCNJ11: Potassium Voltage-Gated Channel Subfamily J Member, SUR1: Sulphonylurea receptor 1, ABCC8: ATP-binding cassette subfamily C member 8, MODY: Maturity-onset diabetes of the young, CACNA1D: Calcium Voltage-Gated Channel Subunit Alpha1 D, CACNA1C: Calcium Channel, Voltage-Dependent, L Type, alpha 1C Subunit, KCNH6: Potassium voltage-gated channel subfamily H member 6, NDM: Neonatal diabetes mellitus, TNDM: Transient NDM, PNDM: permanent NDM, HH: Hyperinsulinaemic Hypoglycemia, CHD: Congenital heart diseases, HPA: Hyperaldosteronism, NMA: Neuromuscular abnormalities, LOH: Loss of heterozygosity, UPD: Uniparental isodisomy).

Figure 3.
Opening of the “Voltage-dependent calcium channel VDDC” located on the beta-cell membrane increases calcium influx and increased intracellular calcium trigger beta-cell insulin secretion through exocytosis. GLP1 contribute to the exocytosis process by increasing the cAMP (GLP1: Glucagon like peptide 1, cAMP: Cyclic adenosine monophosphate).
Figure 3.
Opening of the “Voltage-dependent calcium channel VDDC” located on the beta-cell membrane increases calcium influx and increased intracellular calcium trigger beta-cell insulin secretion through exocytosis. GLP1 contribute to the exocytosis process by increasing the cAMP (GLP1: Glucagon like peptide 1, cAMP: Cyclic adenosine monophosphate).

Figure 4.
Summary of membrane transporters defects and related disorders (MCT1: monocarboxylate transporter 1, SLC16A1: Solute carrier family 16, member 1, GLUT 2: Glucose transporter 2, SLC2A2: Solute carrier family 2 member 2, THTR-1: Thiamine transporter 1 protein, SLC19A2: The Solute Carrier Family 19 Member 2, HH: Hyperinsulinaemic hypoglycaemia, TRMA: Thiamine-responsive megaloblastic anaemia, DM: Diabetes Mellitus, FBS: Fanconi Bickel syndrome, SND: Sensorineural deafness).
Figure 4.
Summary of membrane transporters defects and related disorders (MCT1: monocarboxylate transporter 1, SLC16A1: Solute carrier family 16, member 1, GLUT 2: Glucose transporter 2, SLC2A2: Solute carrier family 2 member 2, THTR-1: Thiamine transporter 1 protein, SLC19A2: The Solute Carrier Family 19 Member 2, HH: Hyperinsulinaemic hypoglycaemia, TRMA: Thiamine-responsive megaloblastic anaemia, DM: Diabetes Mellitus, FBS: Fanconi Bickel syndrome, SND: Sensorineural deafness).

Figure 5.
The role of thiamine transporters, THTR-1 in glucose metabolism. (THTR-1: Thiamin transporter 1, THTR-2: Thiamin transporter 2, RFC1: Reduced folate carrier 1, TMP: Thiamine monophosphate, TMPase: Thiamine monophosphatase, HACL1: 2-Hydroxyacyl-CoA Lyase 1, TPP: Thiamine pyrophosphate, TPPase: Thiamine pyrophosphatase, TK-TPP: Transketolase-Thiamine pyrophosphate, TPK: Thiamine pyrophosphokinase, TDP: Thiamine diphosphate).
Figure 5.
The role of thiamine transporters, THTR-1 in glucose metabolism. (THTR-1: Thiamin transporter 1, THTR-2: Thiamin transporter 2, RFC1: Reduced folate carrier 1, TMP: Thiamine monophosphate, TMPase: Thiamine monophosphatase, HACL1: 2-Hydroxyacyl-CoA Lyase 1, TPP: Thiamine pyrophosphate, TPPase: Thiamine pyrophosphatase, TK-TPP: Transketolase-Thiamine pyrophosphate, TPK: Thiamine pyrophosphokinase, TDP: Thiamine diphosphate).

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Demirbilek, H.; Galcheva, S.; Vuralli, D.; Al-Khawaga, S.; Hussain, K. Ion Transporters, Channelopathies, and Glucose Disorders. Int. J. Mol. Sci. 2019, 20, 2590. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20102590
AMA Style
Demirbilek H, Galcheva S, Vuralli D, Al-Khawaga S, Hussain K. Ion Transporters, Channelopathies, and Glucose Disorders. International Journal of Molecular Sciences. 2019; 20(10):2590. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20102590
Chicago/Turabian StyleDemirbilek, Huseyin, Sonya Galcheva, Dogus Vuralli, Sara Al-Khawaga, and Khalid Hussain. 2019. "Ion Transporters, Channelopathies, and Glucose Disorders" International Journal of Molecular Sciences 20, no. 10: 2590. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20102590
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.