The Cytoskeletal Protein Cyclase-Associated Protein 1 (CAP1) in Breast Cancer: Context-Dependent Roles in Both the Invasiveness and Proliferation of Cancer Cells and Underlying Cell Signals

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. CAP1 as a Versatile Actin-Regulating Protein That Promotes Actin Filament Turnover
1.1. CAP, Identified as a Protein That Interacts with Adenylyl Cyclase in Yeast, Is a Conserved Actin-Regulating Protein across Eukaryotes
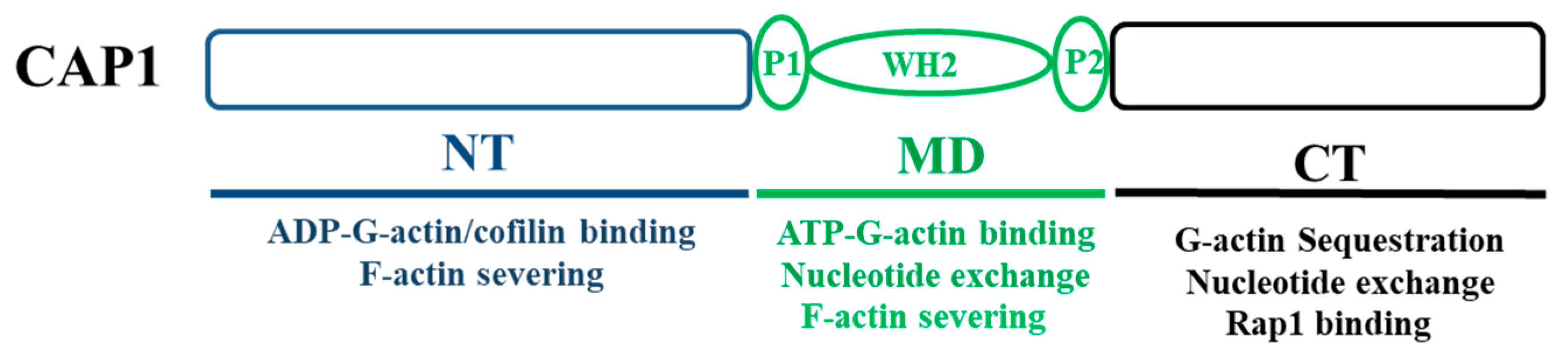
1.2. CAP Promotes Actin Filament Turnover through Multiple Mechanisms That Are Mediated by All of Its Structural Domains
1.3. Loss of CAP1 Functions Universally Leads to Enhanced Stress Fibers in Mammalian Cells
2. Cell Context-Dependent Roles for CAP1 in Cell Migration, and the Invasiveness of Human Cancers
3. A Novel Function for CAP1 in Regulating ERK and the Proliferation of Breast Cancer Cells
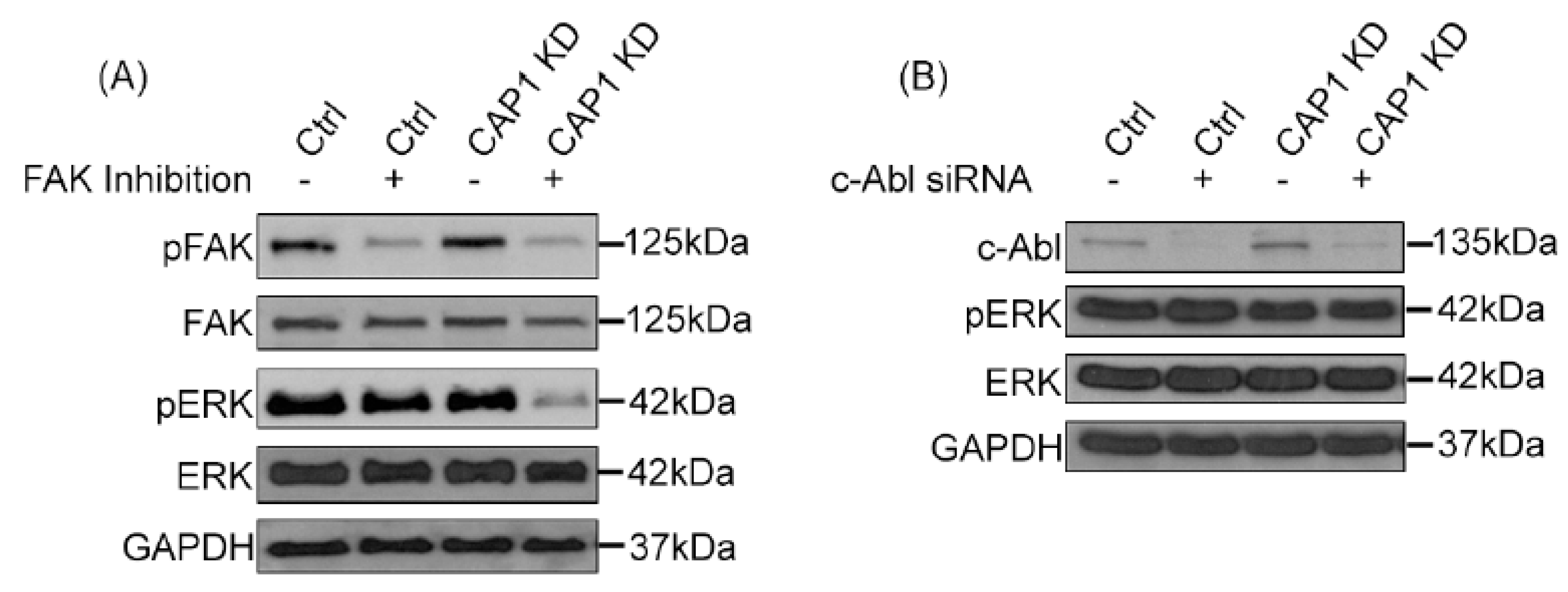
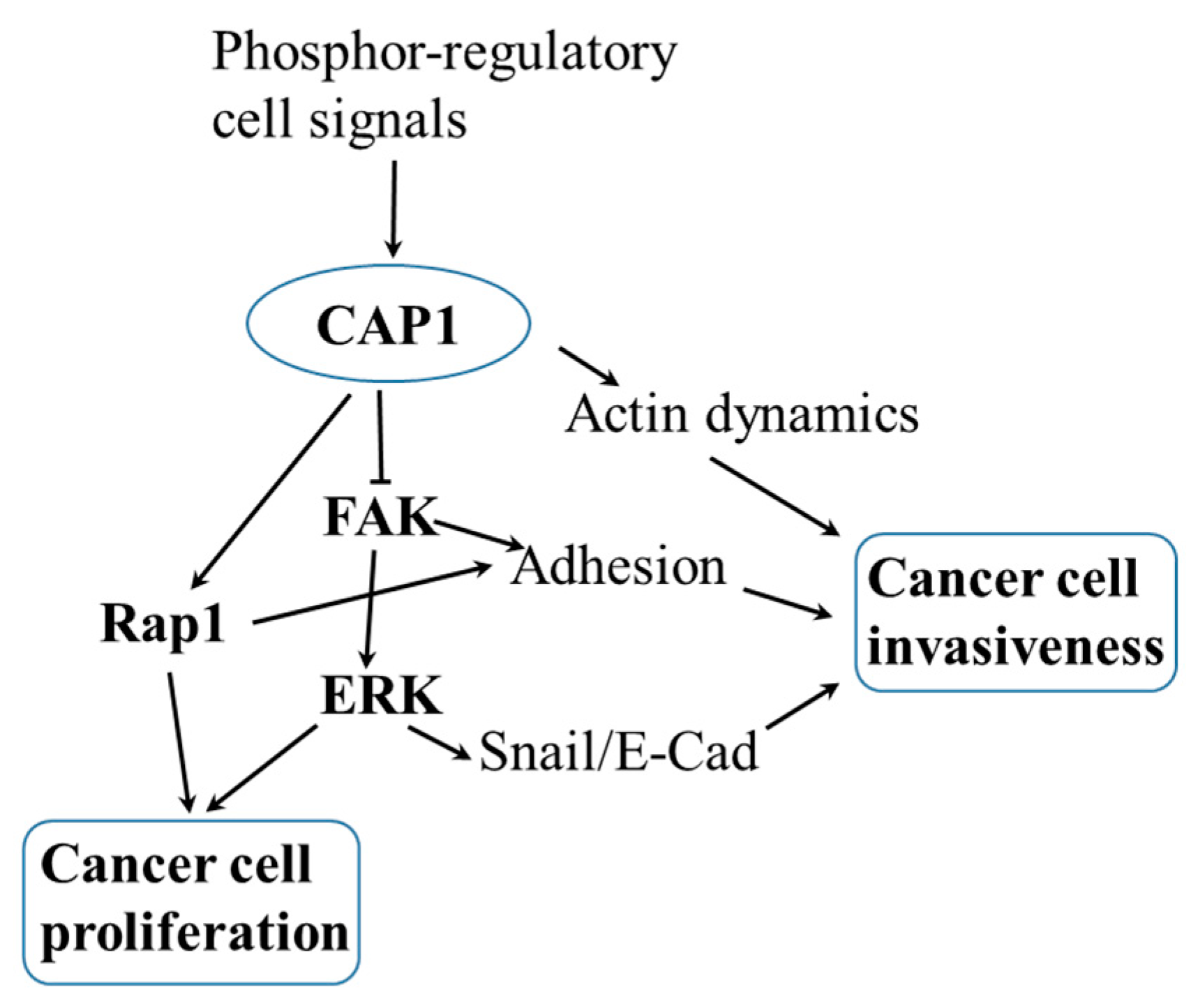
4. A Signaling Molecule That Links CAP1 to the Regulation of ERK
5. Molecular Mechanisms That May Underlie the Regulation of CAP1 Function in Cell Adhesion
6. Relevance of These Mechanistic Insights to Realizing the Translational Potential of CAP1 in Breast Cancer
7. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| CAP1 | adenylyl Cyclase-Associated Protein |
| ERK | External signal-Regulated Kinase |
| FAK | Focal Adhesion Kinase |
| GSK3 | Glycogen Synthase Kinase 3 |
| SRV2 | Suppressor of the activated RAS2Val-19 allele |
| WH2 | WASP Homology domain 2 |
| SH3 | Src Homology 3 |
| Rap1 | Ras-related protein 1 |
References
- Field, J.; Vojtek, A.; Ballester, R.; Bolger, G.; Colicelli, J.; Ferguson, K.; Gerst, J.; Kataoka, T.; Michaeli, T.; Powers, S.; et al. Cloning and characterization of cap, the s. Cerevisiae gene encoding the 70 kd adenylyl cyclase-associated protein. Cell 1990, 61, 319–327. [Google Scholar] [CrossRef]
- Fedor-Chaiken, M.; Deschenes, R.J.; Broach, J.R. Srv2, a gene required for ras activation of adenylate cyclase in yeast. Cell 1990, 61, 329–340. [Google Scholar] [CrossRef]
- Mintzer, K.A.; Field, J. Interactions between adenylyl cyclase, cap and ras from saccharomyces cerevisiae. Cell. Signal. 1994, 6, 681–694. [Google Scholar] [CrossRef]
- Nishida, Y.; Shima, F.; Sen, H.; Tanaka, Y.; Yanagihara, C.; Yamawaki-Kataoka, Y.; Kariya, K.; Kataoka, T. Coiled-coil interaction of n-terminal 36 residues of cyclase-associated protein with adenylyl cyclase is sufficient for its function in saccharomyces cerevisiae ras pathway. J. Biol. Chem. 1998, 273, 28019–28024. [Google Scholar] [CrossRef] [PubMed]
- Shima, F.; Okada, T.; Kido, M.; Sen, H.; Tanaka, Y.; Tamada, M.; Hu, C.D.; Yamawaki-Kataoka, Y.; Kariya, K.; Kataoka, T. Association of yeast adenylyl cyclase with cyclase-associated protein cap forms a second ras-binding site which mediates its ras-dependent activation. Mol. Cell. Biol. 2000, 20, 26–33. [Google Scholar] [CrossRef]
- Kawamukai, M.; Gerst, J.; Field, J.; Riggs, M.; Rodgers, L.; Wigler, M.; Young, D. Genetic and biochemical analysis of the adenylyl cyclase-associated protein, cap, in schizosaccharomyces pombe. Mol. Biol. Cell 1992, 3, 167–180. [Google Scholar] [CrossRef] [PubMed]
- Bahn, Y.S.; Sundstrom, P. Cap1, an adenylate cyclase-associated protein gene, regulates bud-hypha transitions, filamentous growth, and cyclic amp levels and is required for virulence of candida albicans. J. Bacteriol. 2001, 183, 3211–3223. [Google Scholar] [CrossRef]
- Noegel, A.A.; Blau-Wasser, R.; Sultana, H.; Muller, R.; Israel, L.; Schleicher, M.; Patel, H.; Weijer, C.J. The cyclase-associated protein cap as regulator of cell polarity and camp signaling in dictyostelium. Mol. Biol. Cell 2004, 15, 934–945. [Google Scholar] [CrossRef]
- Gerst, J.E.; Ferguson, K.; Vojtek, A.; Wigler, M.; Field, J. Cap is a bifunctional component of the saccharomyces cerevisiae adenylyl cyclase complex. Mol. Cell. Biol. 1991, 11, 1248–1257. [Google Scholar] [CrossRef] [PubMed]
- Freeman, N.L.; Chen, Z.; Horenstein, J.; Weber, A.; Field, J. An actin monomer binding activity localizes to the carboxyl-terminal half of the saccharomyces cerevisiae cyclase-associated protein. J. Biol. Chem. 1995, 270, 5680–5685. [Google Scholar] [CrossRef]
- Wegner, A. Head to tail polymerization of actin. J. Mol. Biol. 1976, 108, 139–150. [Google Scholar] [CrossRef]
- Carlier, M.F.; Pantaloni, D. Control of actin dynamics in cell motility. J. Mol. Biol. 1997, 269, 459–467. [Google Scholar] [CrossRef] [Green Version]
- Matviw, H.; Yu, G.; Young, D. Identification of a human cdna encoding a protein that is structurally and functionally related to the yeast adenylyl cyclase-associated cap proteins. Mol. Cell. Biol. 1992, 12, 5033–5040. [Google Scholar] [CrossRef]
- Vojtek, A.B.; Cooper, J.A. Identification and characterization of a cdna encoding mouse cap: A homolog of the yeast adenylyl cyclase associated protein. J. Cell Sci. 1993, 105, 777–785. [Google Scholar] [PubMed]
- Zelicof, A.; Gatica, J.; Gerst, J.E. Molecular cloning and characterization of a rat homolog of cap, the adenylyl cyclase-associated protein from saccharomyces cerevisiae. J. Biol. Chem. 1993, 268, 13448–13453. [Google Scholar] [PubMed]
- Yu, G.; Swiston, J.; Young, D. Comparison of human cap and cap2, homologs of the yeast adenylyl cyclase-associated proteins. J. Cell Sci. 1994, 107, 1671–1678. [Google Scholar]
- Gottwald, U.; Brokamp, R.; Karakesisoglou, I.; Schleicher, M.; Noegel, A.A. Identification of a cyclase-associated protein (cap) homologue in dictyostelium discoideum and characterization of its interaction with actin. Mol. Biol. Cell 1996, 7, 261–272. [Google Scholar] [CrossRef]
- Kawai, M.; Aotsuka, S.; Uchimiya, H. Isolation of a cotton cap gene: A homologue of adenylyl cyclase-associated protein highly expressed during fiber elongation. Plant Cell Physiol. 1998, 39, 1380–1383. [Google Scholar] [CrossRef]
- Zhou, G.L.; Miyazaki, Y.; Nakagawa, T.; Tanaka, K.; Shishido, K.; Matsuda, H.; Kawamukai, M. Identification of a cap (adenylyl-cyclase-associated protein) homologous gene in lentinus edodes and its functional complementation of yeast cap mutants. Microbiology 1998, 144, 1085–1093. [Google Scholar] [CrossRef]
- Freeman, N.L.; Field, J. Mammalian homolog of the yeast cyclase associated protein, cap/srv2p, regulates actin filament assembly. Cell Motil. Cytoskelet. 2000, 45, 106–120. [Google Scholar] [CrossRef]
- Baum, B.; Li, W.; Perrimon, N. A cyclase-associated protein regulates actin and cell polarity during drosophila oogenesis and in yeast. Curr. Biol. 2000, 10, 964–973. [Google Scholar] [CrossRef]
- Barrero, R.A.; Umeda, M.; Yamamura, S.; Uchimiya, H. Arabidopsis cap regulates the actin cytoskeleton necessary for plant cell elongation and division. Plant Cell 2002, 14, 149–163. [Google Scholar] [CrossRef]
- Deeks, M.J.; Rodrigues, C.; Dimmock, S.; Ketelaar, T.; Maciver, S.K.; Malho, R.; Hussey, P.J. Arabidopsis cap1—A key regulator of actin organisation and development. J. Cell Sci. 2007, 120, 2609–2618. [Google Scholar] [CrossRef]
- Zou, H.; Fang, H.M.; Zhu, Y.; Wang, Y. Candida albicans cyr1, cap1 and g-actin form a sensor/effector apparatus for activating camp synthesis in hyphal growth. Mol. Microbiol. 2010, 75, 579–591. [Google Scholar] [CrossRef]
- Nomura, K.; Ono, K.; Ono, S. Cas-1, a c. Elegans cyclase-associated protein, is required for sarcomeric actin assembly in striated muscle. J. Cell Sci. 2012, 125, 4077–4089. [Google Scholar] [CrossRef] [PubMed]
- Swiston, J.; Hubberstey, A.; Yu, G.; Young, D. Differential expression of cap and cap2 in adult-rat tissues. Gene 1995, 165, 273–277. [Google Scholar] [CrossRef]
- Hubberstey, A.V.; Mottillo, E.P. Cyclase-associated proteins: Capacity for linking signal transduction and actin polymerization. FASEB J. 2002, 16, 487–499. [Google Scholar] [CrossRef]
- Ono, S. The role of cyclase-associated protein in regulating actin filament dynamics—More than a monomer-sequestration factor. J. Cell Sci. 2013, 126, 3249–3258. [Google Scholar] [CrossRef] [PubMed]
- Chaudhry, F.; Little, K.; Talarico, L.; Quintero-Monzon, O.; Goode, B.L. A central role for the wh2 domain of srv2/cap in recharging actin monomers to drive actin turnover in vitro and in vivo. Cytoskeleton 2010, 67, 120–133. [Google Scholar] [CrossRef]
- Paunola, E.; Mattila, P.K.; Lappalainen, P. Wh2 domain: A small, versatile adapter for actin monomers. FEBS Lett. 2002, 513, 92–97. [Google Scholar] [CrossRef]
- Moriyama, K.; Yahara, I. Human cap1 is a key factor in the recycling of cofilin and actin for rapid actin turnover. J. Cell Sci. 2002, 115, 1591–1601. [Google Scholar]
- Quintero-Monzon, O.; Jonasson, E.M.; Bertling, E.; Talarico, L.; Chaudhry, F.; Sihvo, M.; Lappalainen, P.; Goode, B.L. Reconstitution and dissection of the 600-kda srv2/cap complex: Roles for oligomerization and cofilin-actin binding in driving actin turnover. J. Biol. Chem. 2009, 284, 10923–10934. [Google Scholar] [CrossRef]
- Makkonen, M.; Bertling, E.; Chebotareva, N.A.; Baum, J.; Lappalainen, P. Mammalian and malaria parasite cyclase-associated proteins catalyze nucleotide exchange on g-actin through a conserved mechanism. J. Biol. Chem. 2013, 288, 984–994. [Google Scholar] [CrossRef]
- Normoyle, K.P.; Brieher, W.M. Cyclase-associated protein (cap) acts directly on f-actin to accelerate cofilin-mediated actin severing across the range of physiological ph. J. Biol. Chem. 2012, 287, 35722–35732. [Google Scholar] [CrossRef]
- Bertling, E.; Hotulainen, P.; Mattila, P.K.; Matilainen, T.; Salminen, M.; Lappalainen, P. Cyclase-associated protein 1 (cap1) promotes cofilin-induced actin dynamics in mammalian nonmuscle cells. Mol. Biol. Cell 2004, 15, 2324–2334. [Google Scholar] [CrossRef]
- Zhang, H.; Ghai, P.; Wu, H.; Wang, C.; Field, J.; Zhou, G.L. Mammalian adenylyl cyclase-associated protein 1 (cap1) regulates cofilin function, the actin cytoskeleton, and cell adhesion. J. Biol. Chem. 2013, 288, 20966–20977. [Google Scholar] [CrossRef]
- Zhou, G.L.; Zhang, H.; Wu, H.; Ghai, P.; Field, J. Phosphorylation of the cytoskeletal protein cap1 controls its association with cofilin and actin. J. Cell Sci. 2014. [Google Scholar] [CrossRef]
- Zhou, G.L.; Zhang, H.; Field, J. Mammalian cap (cyclase-associated protein) in the world of cell migration: Roles in actin filament dynamics and beyond. Cell Adhes. Migr. 2014, 8, 55–59. [Google Scholar] [CrossRef]
- Tojkander, S.; Gateva, G.; Lappalainen, P. Actin stress fibers—Assembly, dynamics and biological roles. J. Cell Sci. 2012, 125, 1855–1864. [Google Scholar] [CrossRef]
- Zhang, H.; Zhou, G.L. Cap1 (cyclase-associated protein 1) exerts distinct functions in the proliferation and metastatic potential of breast cancer cells mediated by erk. Sci. Rep. 2016, 6, 25933. [Google Scholar] [CrossRef]
- Zhang, X.F.; Cao, S.F.; Barila, G.; Edreira, M.M.; Hong, K.; Wankhede, M.; Naim, N.; Buck, M.; Altschuler, D.L. Cyclase-associated protein 1 (cap1) is a prenyl-binding partner of rap1 gtpase (vol 293, pg 7659, 2018). J. Biol. Chem. 2018, 293, 13849. [Google Scholar] [CrossRef] [PubMed]
- Bos, J.L.; de Rooij, J.; Reedquist, K.A. Rap1 signalling: Adhering to new models. Nat. Rev. Mol. Cell Biol. 2001, 2, 369–377. [Google Scholar] [CrossRef] [PubMed]
- Boettner, B.; Van Aelst, L. Control of cell adhesion dynamics by rap1 signaling. Curr. Opin. Cell Biol. 2009, 21, 684–693. [Google Scholar] [CrossRef] [PubMed]
- Ilic, D.; Furuta, Y.; Kanazawa, S.; Takeda, N.; Sobue, K.; Nakatsuji, N.; Nomura, S.; Fujimoto, J.; Okada, M.; Yamamoto, T. Reduced cell motility and enhanced focal adhesion contact formation in cells from fak-deficient mice. Nature 1995, 377, 539–544. [Google Scholar]
- Mitra, S.K.; Hanson, D.A.; Schlaepfer, D.D. Focal adhesion kinase: In command and control of cell motility. Nat. Rev. Mol. Cell Biol. 2005, 6, 56–68. [Google Scholar] [CrossRef]
- Tilghman, R.W.; Slack-Davis, J.K.; Sergina, N.; Martin, K.H.; Iwanicki, M.; Hershey, E.D.; Beggs, H.E.; Reichardt, L.F.; Parsons, J.T. Focal adhesion kinase is required for the spatial organization of the leading edge in migrating cells. J. Cell Sci. 2005, 118, 2613–2623. [Google Scholar] [CrossRef] [Green Version]
- Klein, E.A.; Assoian, R.K. Transcriptional regulation of the cyclin d1 gene at a glance. J. Cell Sci. 2008, 121, 3853–3857. [Google Scholar] [CrossRef] [PubMed]
- Lavoie, J.N.; L’Allemain, G.; Brunet, A.; Muller, R.; Pouyssegur, J. Cyclin d1 expression is regulated positively by the p42/p44mapk and negatively by the p38/hogmapk pathway. J. Biol. Chem. 1996, 271, 20608–20616. [Google Scholar] [CrossRef]
- Dong, C.; Wu, Y.; Yao, J.; Wang, Y.; Yu, Y.; Rychahou, P.G.; Evers, B.M.; Zhou, B.P. G9a interacts with snail and is critical for snail-mediated e-cadherin repression in human breast cancer. J. Clin. Investig. 2012, 122, 1469–1486. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Tam, W.L.; Shibue, T.; Kaygusuz, Y.; Reinhardt, F.; Ng Eaton, E.; Weinberg, R.A. Distinct emt programs control normal mammary stem cells and tumour-initiating cells. Nature 2015, 525, 256–260. [Google Scholar] [CrossRef]
- Yamazaki, K.; Takamura, M.; Masugi, Y.; Mori, T.; Du, W.; Hibi, T.; Hiraoka, N.; Ohta, T.; Ohki, M.; Hirohashi, S.; et al. Adenylate cyclase-associated protein 1 overexpressed in pancreatic cancers is involved in cancer cell motility. Lab. Investig. 2009, 89, 425–432. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Cui, X.; Hu, B.; Lu, C.; Huang, X.; Cai, J.; He, S.; Lv, L.; Cong, X.; Liu, G.; et al. Upregulated expression of cap1 is associated with tumor migration and metastasis in hepatocellular carcinoma. Pathol. Res. Pract. 2013. [Google Scholar] [CrossRef]
- Tan, M.; Song, X.; Zhang, G.; Peng, A.; Li, X.; Li, M.; Liu, Y.; Wang, C. Overexpression of adenylate cyclase-associated protein 1 is associated with metastasis of lung cancer. Oncol. Rep. 2013, 30, 1639–1644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, X.F.; Ni, Q.C.; Chen, J.P.; Xu, J.F.; Jiang, Y.; Yang, S.Y.; Ma, J.; Gu, X.L.; Wang, H.; Wang, Y.Y. Knocking down the expression of adenylate cyclase-associated protein 1 inhibits the proliferation and migration of breast cancer cells. Exp. Mol. Pathol. 2014, 96, 188–194. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Yao, N.; Qian, J.; Huang, H. High expression and prognostic role of cap1 and ctbp2 in breast carcinoma: Associated with e-cadherin and cell proliferation. Med. Oncol. 2014, 31, 878. [Google Scholar] [CrossRef]
- Hua, M.H.; Yan, S.J.; Deng, Y.; Xi, Q.H.; Liu, R.; Yang, S.Y.; Liu, J.; Tang, C.H.; Wang, Y.Y.; Zhong, J.X. Cap1 is overexpressed in human epithelial ovarian cancer and promotes cell proliferation. Int. J. Mol. Med. 2015, 35, 941–949. [Google Scholar] [CrossRef]
- Xie, S.S.; Tan, M.; Lin, H.Y.; Xu, L.; Shen, C.X.; Yuan, Q.; Song, X.L.; Wang, C.H. Overexpression of adenylate cyclase-associated protein 1 may predict brain metastasis in non-small cell lung cancer. Oncol. Rep. 2015, 33, 363–371. [Google Scholar] [CrossRef]
- Bao, Z.; Qiu, X.; Wang, D.; Ban, N.; Fan, S.; Chen, W.; Sun, J.; Xing, W.; Wang, Y.; Cui, G. High expression of adenylate cyclase-associated protein 1 accelerates the proliferation, migration and invasion of neural glioma cells. Pathol. Res. Pract. 2016, 212, 264–273. [Google Scholar] [CrossRef]
- Fan, Y.C.; Cui, C.C.; Zhu, Y.S.; Zhang, L.; Shi, M.; Yu, J.S.; Bai, J.; Zheng, J.N. Overexpression of cap1 and its significance in tumor cell proliferation, migration and invasion in glioma. Oncol. Rep. 2016, 36, 1619–1625. [Google Scholar] [CrossRef]
- Kakurina, G.V.; Kondakova, I.V.; Cheremisina, O.V.; Shishkin, D.A.; Choinzonov, E.L. Adenylyl cyclase-associated protein 1 in the development of head and neck squamous cell carcinomas. Bull. Exp. Biol. Med. 2016, 160, 695–697. [Google Scholar] [CrossRef]
- Xie, S.; Liu, Y.; Li, X.; Tan, M.; Wang, C.; Field, J.; Zhou, G.L. Phosphorylation of the cytoskeletal protein cap1 regulates non-small cell lung cancer survival and proliferation by gsk3beta. J. Cancer 2018, 9, 2825–2833. [Google Scholar] [CrossRef]
- Wu, H.; Hasan, R.; Zhang, H.; Gray, J.; Williams, D.; Miller, M.; Allen, F.; Lee, V.; Kelly, T.; Zhou, G.L. Phosphorylation regulates cap1 (cyclase-associated protein 1) functions in the motility and invasion of pancreatic cancer cells. Sci. Rep. 2019, 9, 4925. [Google Scholar] [CrossRef]
- Effendi, K.; Yamazaki, K.; Mori, T.; Masugi, Y.; Makino, S.; Sakamoto, M. Involvement of hepatocellular carcinoma biomarker, cyclase-associated protein 2 in zebrafish body development and cancer progression. Exp. Cell Res. 2013, 319, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Li, M.; Wu, D.C.; Liu, L.L.; Chen, S.L.; Yun, J.P. Increased expression of cap2 indicates poor prognosis in hepatocellular carcinoma. Transl. Oncol. 2015, 8, 400–406. [Google Scholar] [CrossRef]
- Masugi, Y.; Tanese, K.; Emoto, K.; Yamazaki, K.; Effendi, K.; Funakoshi, T.; Mori, M.; Sakamoto, M. Overexpression of adenylate cyclase-associated protein 2 is a novel prognostic marker in malignant melanoma. Pathol. Int. 2015, 65, 627–634. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Zhou, G.L.; Vedantam, S.; Li, P.; Field, J. Mitochondrial shuttling of cap1 promotes actin- and cofilin-dependent apoptosis. J. Cell Sci. 2008, 121, 2913–2920. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Zhou, G.L.; Yamamoto, T.; Ozoe, F.; Yano, D.; Tanaka, K.; Matsuda, H.; Kawamukai, M. Identification of a 14-3-3 protein from lentinus edodes that interacts with cap (adenylyl cyclase-associated protein), and conservation of this interaction in fission yeast. Biosci. Biotechnol. Biochem. 2000, 64, 149–159. [Google Scholar] [CrossRef]
- Yamamoto, T.; Kobayashi-Ooka, Y.; Zhou, G.L.; Kawamukai, M. Identification and characterization of csh3 as an sh3 protein that interacts with fission yeast cap1. Fems Yeast Res. 2015, 15, fov097. [Google Scholar] [CrossRef]
- Wills, Z.; Emerson, M.; Rusch, J.; Bikoff, J.; Baum, B.; Perrimon, N.; Van Vactor, D. A drosophila homolog of cyclase-associated proteins collaborates with the abl tyrosine kinase to control midline axon pathfinding. Neuron 2002, 36, 611–622. [Google Scholar] [CrossRef]
- Mizuchi, D.; Kurosu, T.; Kida, A.; Jin, Z.H.; Jin, A.; Arai, A.; Miura, O. Bcr/abl activates rap1 and b-raf to stimulate the mek/erk signaling pathway in hematopoietic cells. Biochem. Biophys. Res. Commun. 2005, 326, 645–651. [Google Scholar] [CrossRef]
- Freeman, N.L.; Lila, T.; Mintzer, K.A.; Chen, Z.; Pahk, A.J.; Ren, R.; Drubin, D.G.; Field, J. A conserved proline-rich region of the saccharomyces cerevisiae cyclase-associated protein binds sh3 domains and modulates cytoskeletal localization. Mol. Cell. Biol. 1996, 16, 548–556. [Google Scholar] [CrossRef]
- Barberis, L.; Wary, K.K.; Fiucci, G.; Liu, F.; Hirsch, E.; Brancaccio, M.; Altruda, F.; Tarone, G.; Giancotti, F.G. Distinct roles of the adaptor protein shc and focal adhesion kinase in integrin signaling to erk. J. Biol. Chem. 2000, 275, 36532–36540. [Google Scholar] [CrossRef]
- Giancotti, F.G.; Ruoslahti, E. Integrin signaling. Science 1999, 285, 1028–1032. [Google Scholar] [CrossRef]
- Legate, K.R.; Wickstrom, S.A.; Fassler, R. Genetic and cell biological analysis of integrin outside-in signaling. Genes Dev. 2009, 23, 397–418. [Google Scholar] [CrossRef] [Green Version]
- Neve, R.M.; Chin, K.; Fridlyand, J.; Yeh, J.; Baehner, F.L.; Fevr, T.; Clark, L.; Bayani, N.; Coppe, J.P.; Tong, F.; et al. A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell 2006, 10, 515–527. [Google Scholar] [CrossRef] [Green Version]
- Luo, J. Glycogen synthase kinase 3beta (gsk3beta) in tumorigenesis and cancer chemotherapy. Cancer Lett. 2009, 273, 194–200. [Google Scholar] [CrossRef]
- Sulzmaier, F.J.; Jean, C.; Schlaepfer, D.D. Fak in cancer: Mechanistic findings and clinical applications. Nat. Rev. Cancer 2014, 14, 598–610. [Google Scholar] [CrossRef]
- Luo, M.; Guan, J.L. Focal adhesion kinase: A prominent determinant in breast cancer initiation, progression and metastasis. Cancer Lett. 2010, 289, 127–139. [Google Scholar] [CrossRef] [Green Version]
- McLean, G.W.; Carragher, N.O.; Avizienyte, E.; Evans, J.; Brunton, V.G.; Frame, M.C. The role of focal-adhesion kinase in cancer—A new therapeutic opportunity. Nat. Rev. Cancer 2005, 5, 505–515. [Google Scholar] [CrossRef]
- Provenzano, P.P.; Inman, D.R.; Eliceiri, K.W.; Keely, P.J. Matrix density-induced mechanoregulation of breast cell phenotype, signaling and gene expression through a fak-erk linkage. Oncogene 2009, 28, 4326–4343. [Google Scholar] [CrossRef] [PubMed]
- Roberts, P.J.; Der, C.J. Targeting the raf-mek-erk mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene 2007, 26, 3291–3310. [Google Scholar] [CrossRef] [PubMed]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hasan, R.; Zhou, G.-L. The Cytoskeletal Protein Cyclase-Associated Protein 1 (CAP1) in Breast Cancer: Context-Dependent Roles in Both the Invasiveness and Proliferation of Cancer Cells and Underlying Cell Signals. Int. J. Mol. Sci. 2019, 20, 2653. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20112653
Hasan R, Zhou G-L. The Cytoskeletal Protein Cyclase-Associated Protein 1 (CAP1) in Breast Cancer: Context-Dependent Roles in Both the Invasiveness and Proliferation of Cancer Cells and Underlying Cell Signals. International Journal of Molecular Sciences. 2019; 20(11):2653. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20112653
Chicago/Turabian StyleHasan, Rokib, and Guo-Lei Zhou. 2019. "The Cytoskeletal Protein Cyclase-Associated Protein 1 (CAP1) in Breast Cancer: Context-Dependent Roles in Both the Invasiveness and Proliferation of Cancer Cells and Underlying Cell Signals" International Journal of Molecular Sciences 20, no. 11: 2653. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20112653