IDH1R132H Causes Resistance to HDAC Inhibitors by Increasing NANOG in Glioblastoma Cells

Abstract
:1. Introduction
2. Results and Discussion
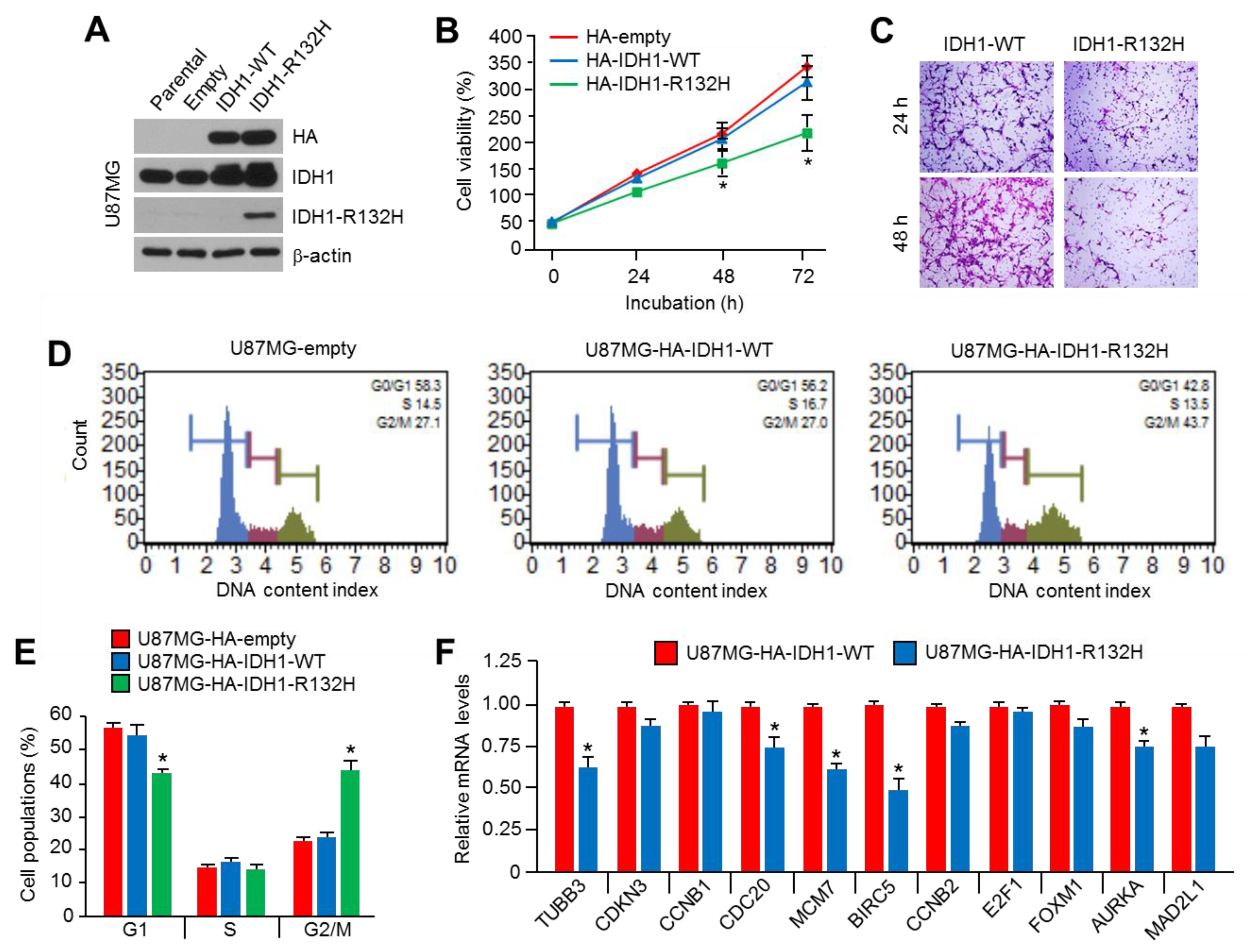
2.1. Overexpression of IDH1R132H Suppresses Viability, Motility, and Cell Cycle Progression in U87MG Glioblastoma Cells
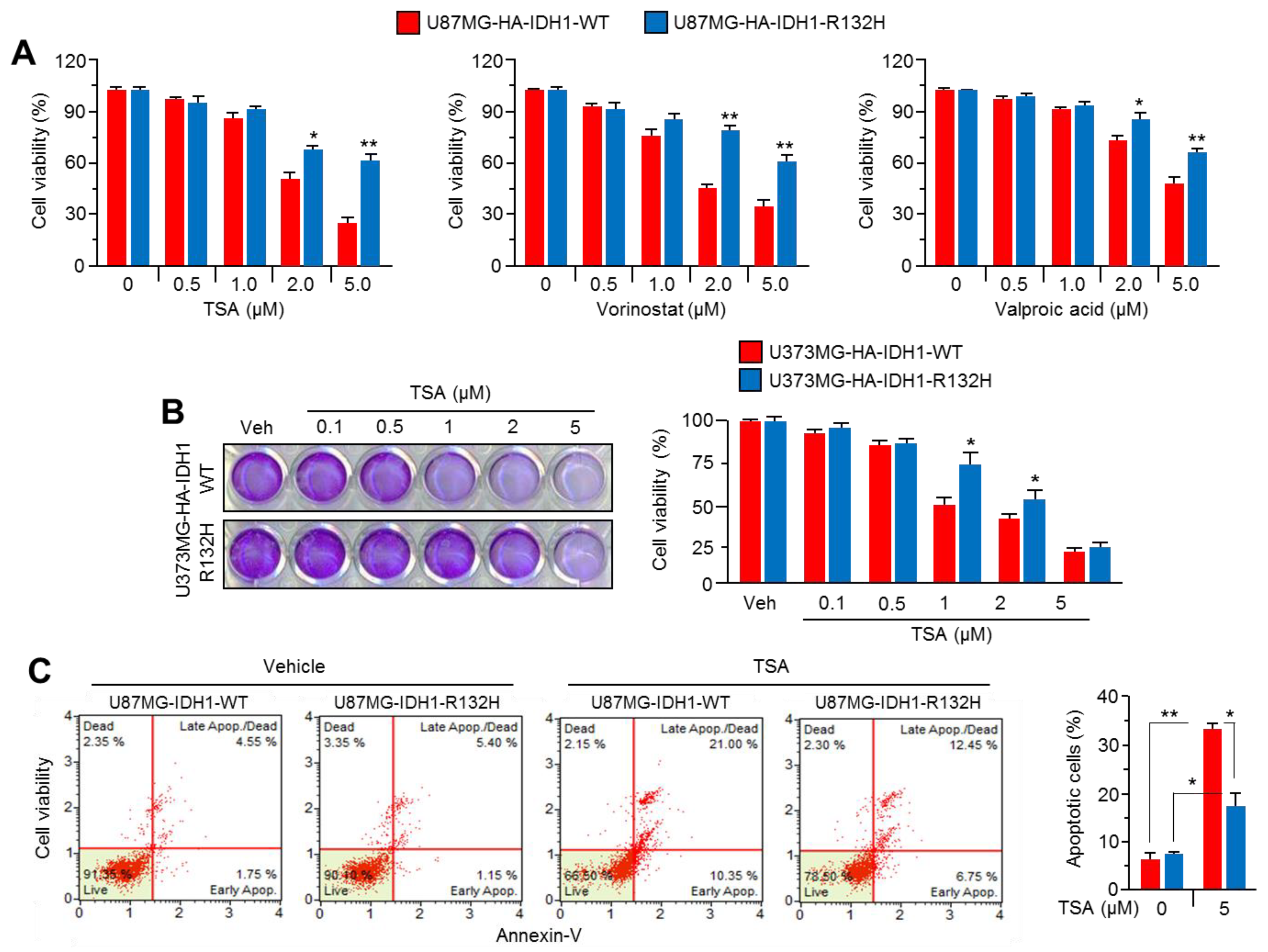
2.2. Overexpression of IDH1R132H Abolishes the Anti-Cancer Effect of HDAC Inhibitors
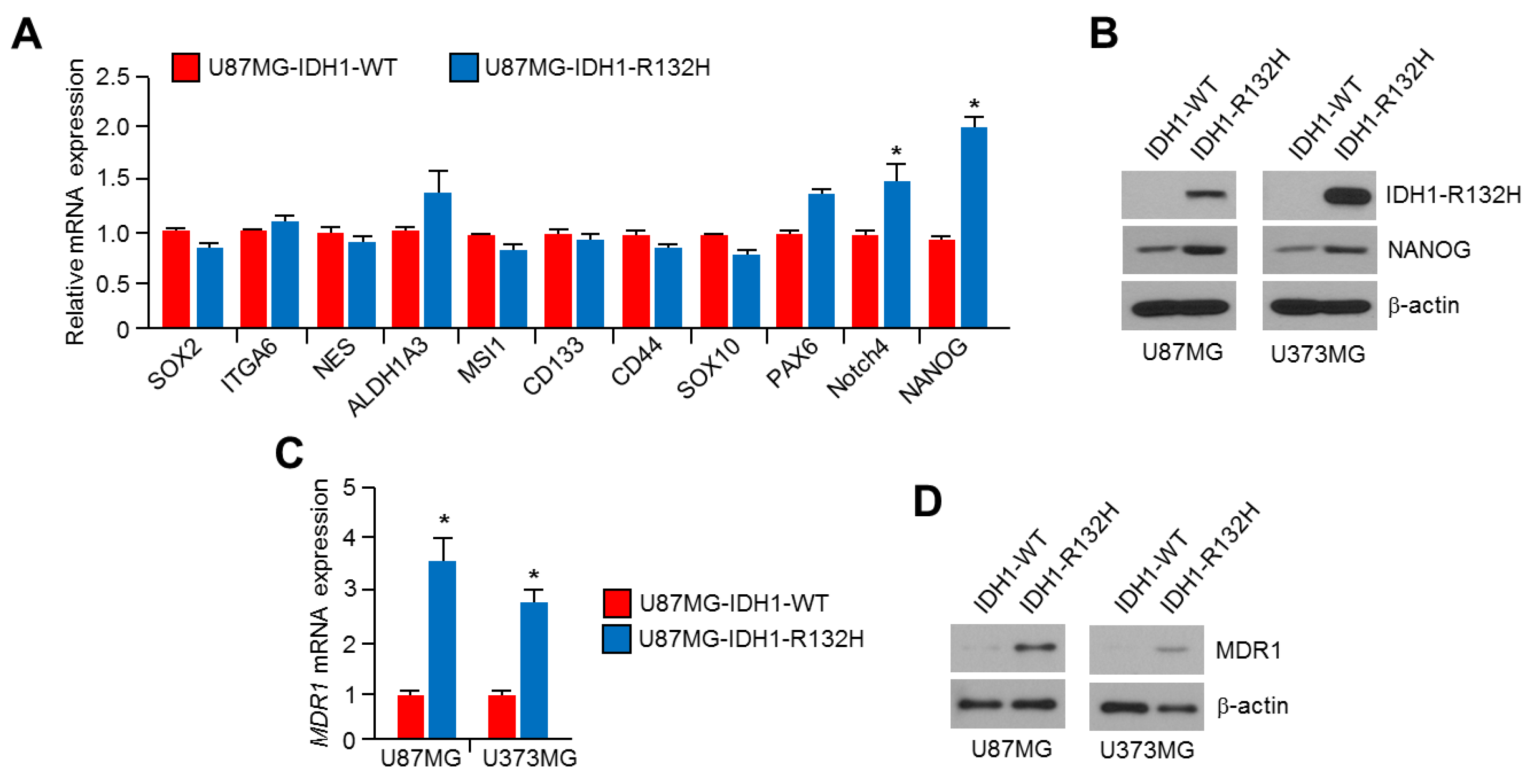
2.3. NANOG Is Increased in IDH1R132H-Overexpressing U87MG and U373MG Glioblastoma Cells
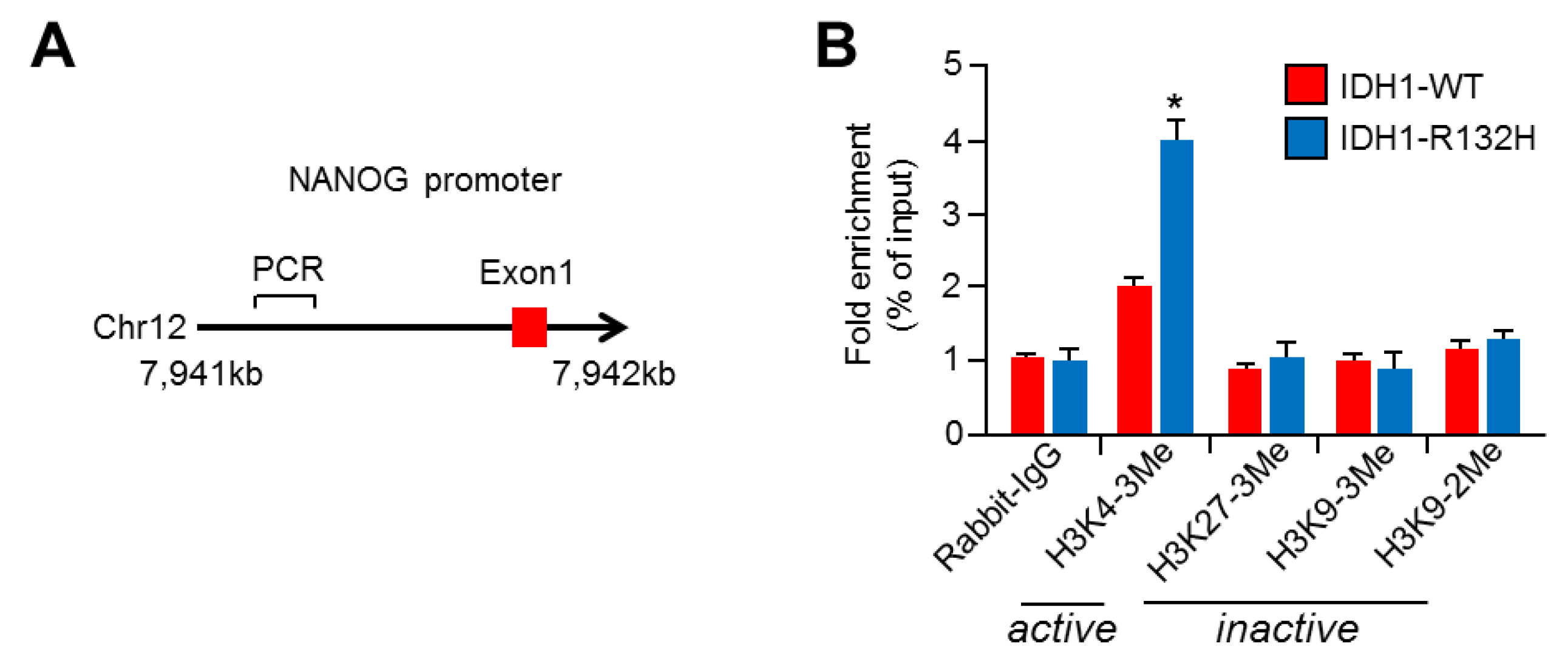
2.4. NANOG Promoter Is Activated with Increased H3K4-Trimethylation (H3K4-3Me) in IDH1R132H-Expressing U87MG Cells
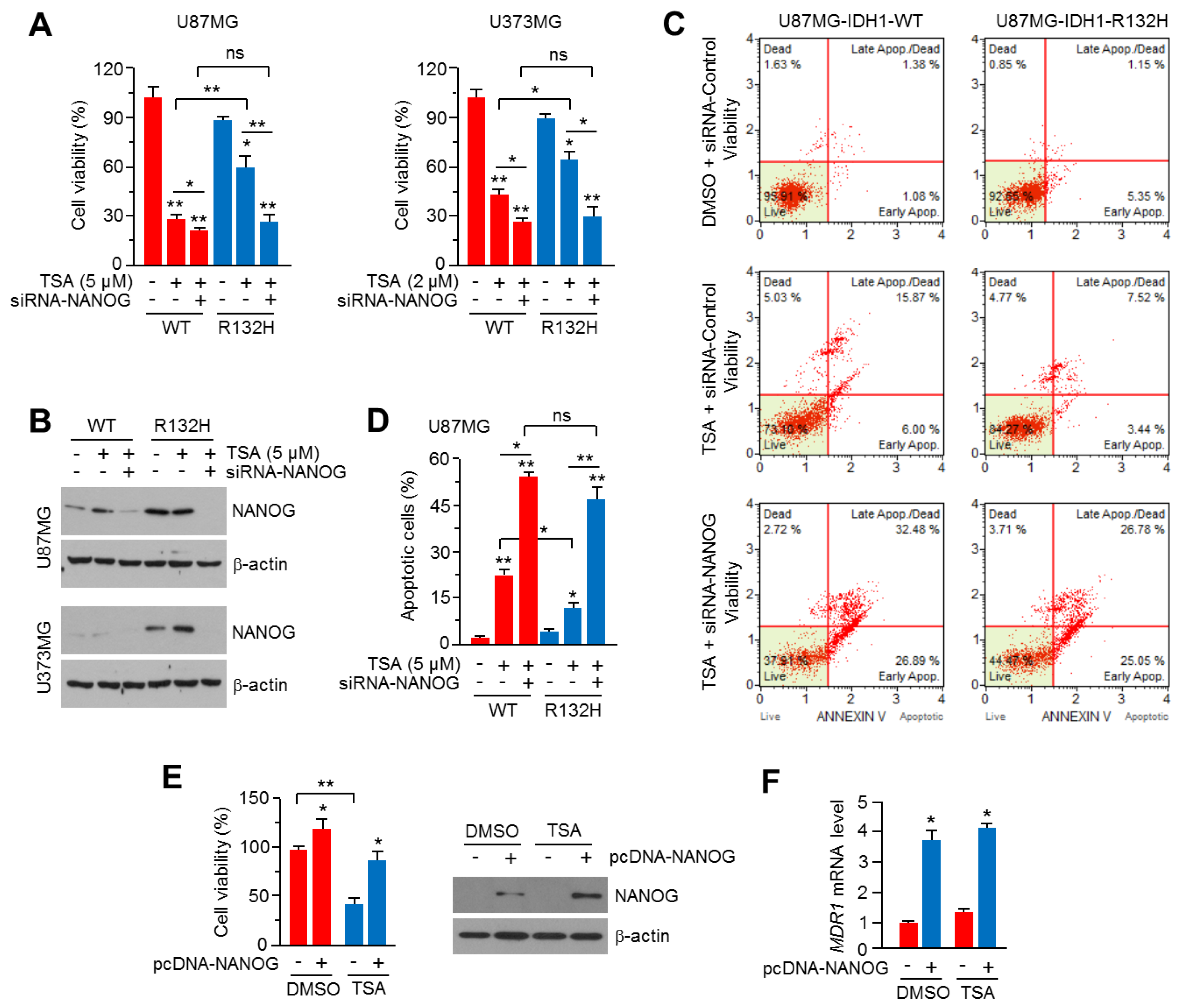
2.5. Knock-Down of NANOG Attenuates HDACi Resistance in IDH1R132H-Expressing U87MG and U373MG Glioblastoma Cells
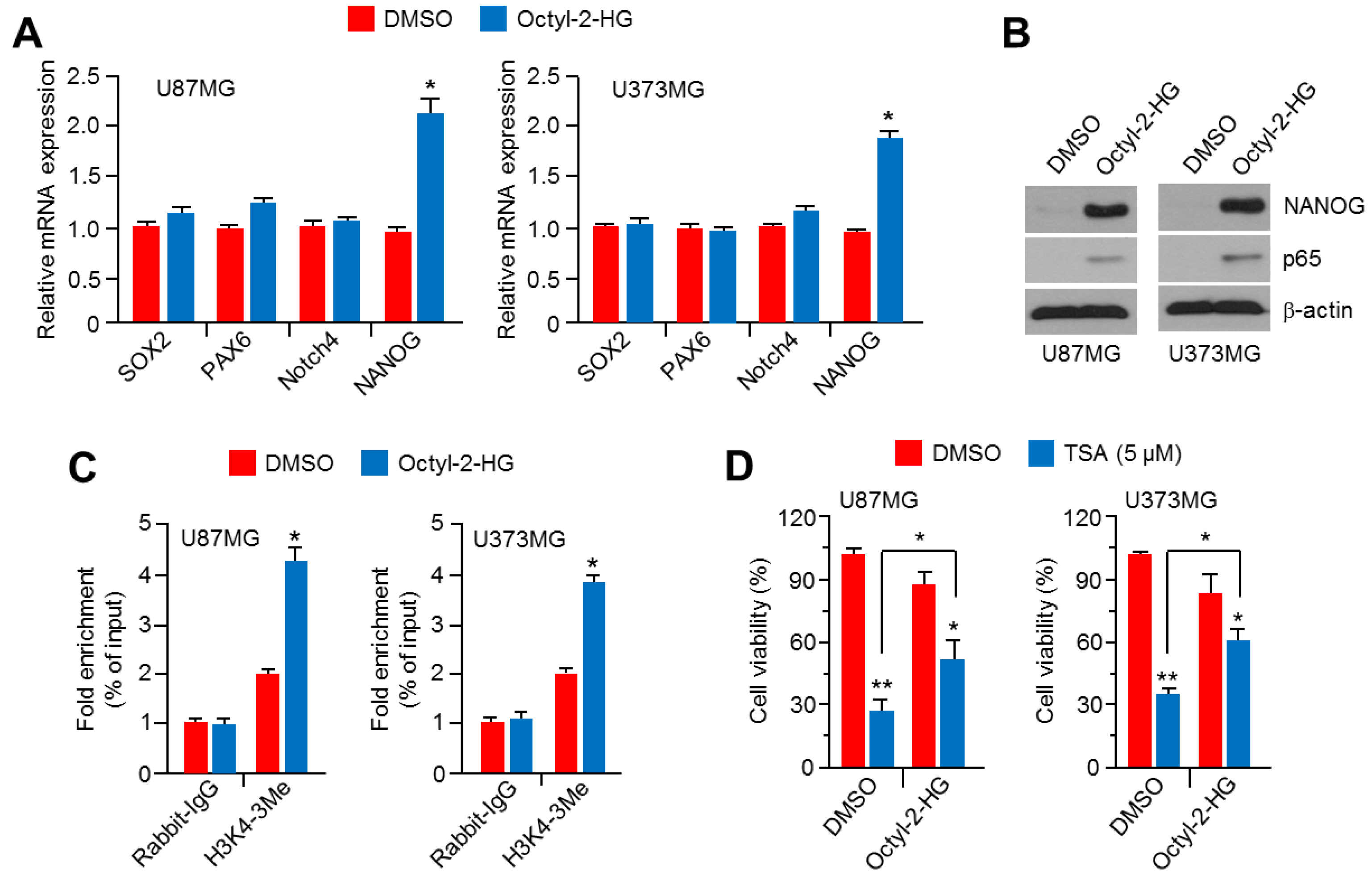
2.6. Octyl-2-Hydroxyglutarate (Octyl-2HG) Increases NANOG and HDACi Resistance in Glioblastoma Cells
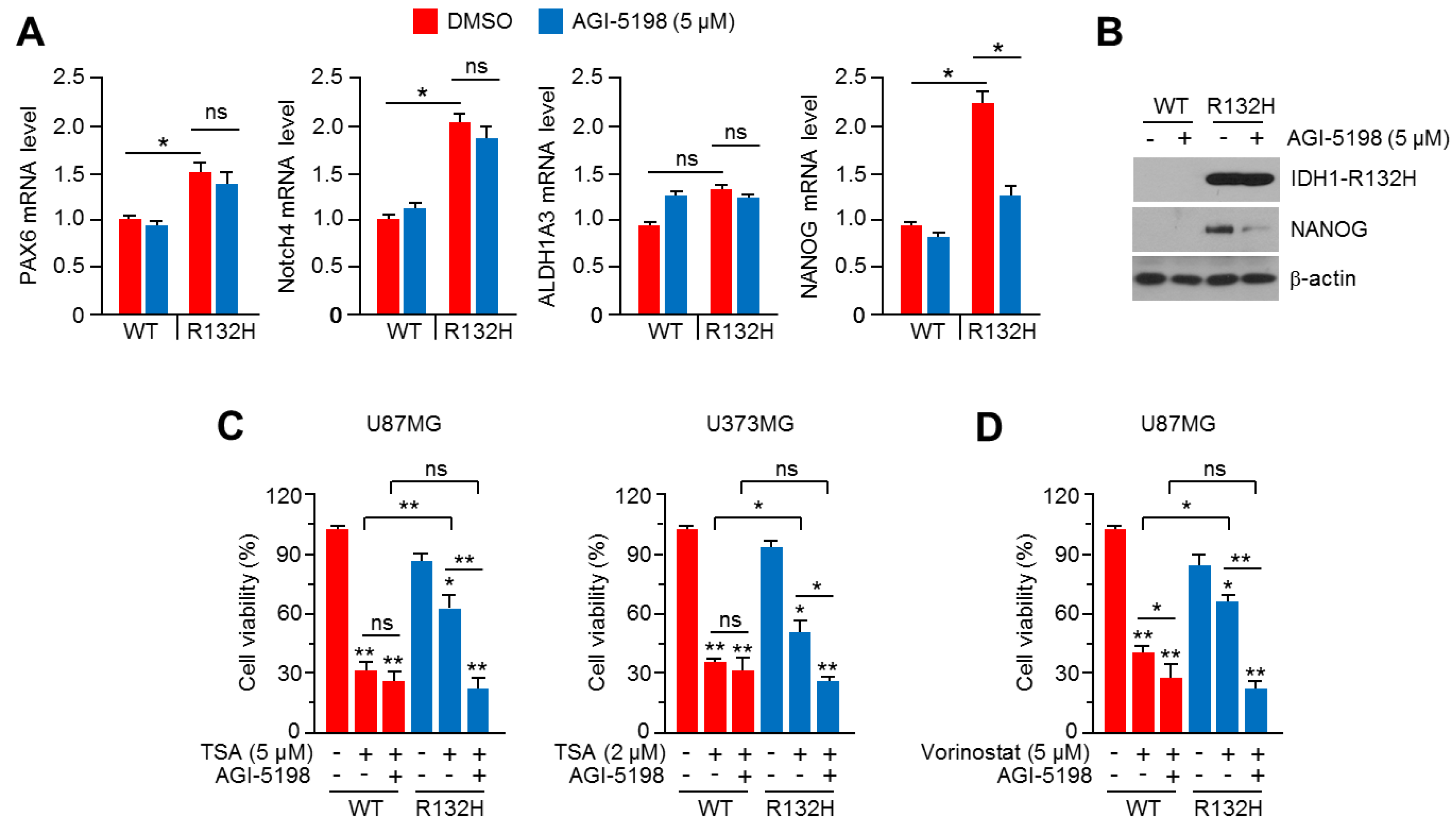
2.7. Pharmacological Inhibition of IDH1R132H Attenuates HDACi Resistance in U87MG and U373MG Glioblastoma Cells
3. Materials and Methods
3.1. Reagents and Antibodies
3.2. Cell Culture, Cell Viability Assay and Generation of Stable Cell Lines
3.3. Expression Constructs and Generation of Stable Cell Lines
3.4. Western Blotting
3.5. Quantitative Real-Time PCR
3.6. Cell Cycle Analysis and Apoptosis Assays
3.7. Chromatin Immunoprecipitation (ChIP) Assay
3.8. In Vitro Migration Assay
3.9. Statistical Analysis
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| IDH1 | Isocitrate dehydrogenase |
| GBM | Glioblastoma multiforme |
| HDAC | Histone deacetylase |
| R-2HG | (R)-2-hydroxyglutarate |
| AML | Acute myeloid leukemia |
| 5mC | 5-methylcytosine |
| KDM4C | Lysine demethylase 4C |
| EMT | Epithelial-mesenchymal transition |
| α-KG | α-ketoglutarate |
| TMZ | Temozolomide |
| HIF | Hypoxia-inducible factor |
References
- Ward, P.S.; Patel, J.; Wise, D.R.; Abdel-Wahab, O.; Bennett, B.D.; Coller, H.A.; Cross, J.R.; Fantin, V.R.; Hedvat, C.V.; Perl, A.E.; et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell 2010, 17, 225–234. [Google Scholar] [CrossRef]
- Gross, S.; Sasaki, M.; Jin, S.; Dang, L.; Cairns, R.A.; Minden, M.D.; Driggers, E.M.; Bittinger, M.A.; Jang, H.G.; Schenkein, D.P.; et al. Cancer-associated metabolite 2-hydroxyglutarate accumulates in acute myelogenous leukemia with isocitrate dehydrogenase 1 and 2 mutations. J. Exp. Med. 2010, 207, 339–344. [Google Scholar] [CrossRef] [PubMed]
- Dang, L.; White, D.W.; Gross, S.; Bennett, B.D.; Bittinger, M.A.; Driggers, E.M.; Fantin, V.R.; Jang, H.G.; Jin, S.; Keenan, M.C.; et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 2009, 462, 739–744. [Google Scholar] [CrossRef] [Green Version]
- Mardis, E.R.; Ding, L.; Dooling, D.J.; Larson, D.E.; McLellan, M.D.; Chen, K.; Koboldt, D.C.; Fulton, R.S.; Delehaunty, K.D.; McGrath, S.D.; et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N. Engl. J. Med. 2009, 361, 1058–1066. [Google Scholar] [CrossRef]
- Yan, H.; Parsons, D.W.; Jin, G.; McLendon, R.; Rasheed, B.A.; Yuan, W.; Kos, I.; Batinic-Haberle, I.; Jones, S.; Riggins, G.J.; et al. IDH1 and IDH2 mutations in gliomas. N. Engl. J. Med. 2009, 360, 765–773. [Google Scholar] [CrossRef] [PubMed]
- Parsons, D.W.; Jones, S.; Zhang, X.; Lin, J.C.-H.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Siu, I.-M.; Gallia, G.L.; et al. An integrated genomic analysis of human glioblastoma multiforme. Science 2008, 321, 1807–1812. [Google Scholar] [CrossRef] [PubMed]
- Waitkus, M.S.; Diplas, B.H.; Yan, H. Biological Role and Therapeutic Potential of IDH Mutations in Cancer. Cancer Cell 2018, 34, 186–195. [Google Scholar] [CrossRef] [Green Version]
- Yao, Q.; Cai, G.; Yu, Q.; Shen, J.; Gu, Z.; Chen, J.; Shi, W.; Shi, J. IDH1 mutation diminishes aggressive phenotype in glioma stem cells. Int J. Oncol. 2018, 52, 270–278. [Google Scholar] [CrossRef]
- Jiang, B.; Zhang, J.; Xia, J.; Zhao, W.; Wu, Y.; Shi, M.; Luo, L.; Zhou, H.; Chen, A.; Ma, H.-H.; et al. IDH1 Mutation Promotes Tumorigenesis by Inhibiting JNK Activation and Apoptosis Induced by Serum Starvation. Cell Rep. 2017, 19, 389–400. [Google Scholar] [CrossRef] [Green Version]
- Bardella, C.; Al-Dalahmah, O.; Krell, D.; Brazauskas, P.; Al-Qahtani, K.; Tomkova, M.; Adam, J.; Serres, S.; Lockstone, H.; Freeman-Mills, L.; et al. Expression of Idh1R132H in the Murine Subventricular Zone Stem Cell Niche Recapitulates Features of Early Gliomagenesis. Cancer Cell 2016, 30, 578–594. [Google Scholar] [CrossRef]
- Cui, D.; Ren, J.; Shi, J.; Feng, L.; Wang, K.; Zeng, T.; Jin, Y.; Gao, L. R132H mutation in IDH1 gene reduces proliferation, cell survival and invasion of human glioma by downregulating Wnt/β-catenin signaling. Int. J. Biochem. Cell Biol. 2016, 73, 72–81. [Google Scholar] [CrossRef]
- Yang, X.; Shen, L.; Guo, P.; Nie, Q.-M.; Lin, Y.-Y.; Guo, L.-M.; Que, S.-L.; Li, X.-X.; Ge, J.-W.; Wang, G.-S.; et al. IDH1R132H decreases the proliferation of U87 glioma cells through upregulation of microRNA-128a. Mol. Med. Rep. 2015, 12, 6695–6701. [Google Scholar]
- Kessler, J.; Güttler, A.; Wichmann, H.; Rot, S.; Kappler, M.; Bache, M.; Vordermark, D. IDH1(R132H) mutation causes a less aggressive phenotype and radiosensitizes human malignant glioma cells independent of the oxygenation status. Radiother. Oncol. 2015, 116, 381–387. [Google Scholar] [CrossRef]
- Zhang, C.; Moore, L.M.; Li, X.; Yung, W.K.; Zhang, W. IDH1/2 mutations target a key hallmark of cancer by deregulating cellular metabolism in glioma. Neuro-Oncology 2013, 15, 1114–1126. [Google Scholar] [CrossRef]
- Sasaki, M.; Knobbe, C.B.; Munger, J.C.; Lind, E.F.; Brenner, D.; Brüstle, A.; Harris, I.S.; Holmes, R.; Wakeham, A.; Haight, J.; et al. IDH1(R132H) mutation increases murine haematopoietic progenitors and alters epigenetics. Nature 2012, 488, 656–659. [Google Scholar] [CrossRef] [Green Version]
- Xu, W.; Yang, H.; Liu, Y.; Yang, Y.; Wang, P.; Kim, S.-H.; Ito, S.; Yang, C.; Wang, P.; Xiao, M.-T.; et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of α-ketoglutarate-dependent dioxygenases. Cancer Cell 2011, 19, 17–30. [Google Scholar] [CrossRef]
- Lu, C.; Ward, P.S.; Kapoor, G.S.; Rohle, D.; Turcan, S.; Abdel-Wahab, O.; Edwards, C.R.; Khanin, R.; Figueroa, M.E.; Melnick, A.; et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 2012, 483, 474–478. [Google Scholar] [CrossRef] [Green Version]
- Carey, B.W.; Finley, L.W.; Cross, J.R.; Allis, C.D.; Thompson, C.B. Intracellular α-ketoglutarate maintains the pluripotency of embryonic stem cells. Nature 2015, 518, 413–416. [Google Scholar] [CrossRef]
- Koivunen, P.; Lee, S.; Duncan, C.G.; Lopez, G.; Lu, G.; Ramkissoon, S.; Losman, J.A.; Joensuu, P.; Bergmann, U.; Gross, S.; et al. Transformation by the (R)-enantiomer of 2-hydroxyglutarate linked to EGLN activation. Nature 2012, 483, 484–488. [Google Scholar] [CrossRef]
- Colvin, H.; Nishida, N.; Konno, M.; Haraguchi, N.; Takahashi, H.; Nishimura, J.; Hata, T.; Kawamoto, K.; Asai, A.; Tsunekuni, K.; et al. Oncometabolite D-2-Hydroxyglutarate Directly Induces Epithelial-Mesenchymal Transition and is Associated with Distant Metastasis in Colorectal Cancer. Sci. Rep. 2016, 6, 36289. [Google Scholar] [CrossRef]
- Chen, J.Y.; Lai, Y.S.; Tsai, H.J.; Kuo, C.C.; Yen, B.L.; Yeh, S.P.; Sun, H.S.; Hung, W.C. The oncometabolite R-2-hydroxyglutarate activates NF-kB-dependent tumor-promoting stromal niche for acute myeloid leukemia cells. Sci. Rep. 2016, 6, 32428. [Google Scholar] [CrossRef]
- Fu, X.; Chin, R.M.; Vergnes, L.; Hwang, H.; Deng, G.; Xing, Y.; Pai, M.Y.; Li, S.; Ta, L.; Fazlollahi, F.; et al. 2-Hydroxyglutarate Inhibits ATP Synthase and mTOR Signaling. Cell Metab. 2015, 22, 508–515. [Google Scholar] [CrossRef] [Green Version]
- Su, R.; Dong, L.; Li, C.; Nachtergaele, S.; Wunderlich, M.; Qing, Y.; Deng, X.; Wang, Y.; Weng, X.; Hu, C.; et al. R-2HG Exhibits Anti-tumor Activity by Targeting FTO/m6A/MYC/CEBPA Signaling. Cell 2018, 172, 90–105. [Google Scholar] [CrossRef] [PubMed]
- Molenaar, R.J.; Maciejewski, J.P.; Wilmink, J.W.; Van Noorden, C.J.F. Wild-type and mutated IDH1/2 enzymes and therapy responses. Oncogene 2018, 37, 1949–1960. [Google Scholar] [CrossRef] [Green Version]
- Rohle, D.; Popovici-Muller, J.; Palaskas, N.; Turcan, S.; Grommes, C.; Campos, C.; Tsoi, J.; Clark, O.; Oldrini, B.; Komisopoulou, E.; et al. An Inhibitor of Mutant IDH1 Delays Growth and Promotes Differentiation of Glioma Cells. Science 2013, 340, 626–630. [Google Scholar] [CrossRef] [Green Version]
- Kopinja, J.; Sevilla, R.S.; Levitan, D.; Dai, D.; Vanko, A.; Spooner, E.; Ware, C.; Forget, R.; Hu, K.; Kral, A.; et al. A Brain Penetrant Mutant IDH1 Inhibitor Provides In Vivo Survival Benefit. Sci. Rep. 2017, 7, 13853. [Google Scholar] [CrossRef] [PubMed]
- Molenaar, R.J.; Botman, D.; Smits, M.A.; Hira, V.V.; Van Lith, S.A.; Stap, J.; Henneman, P.; Khurshed, M.; Lenting, K.; Mul, A.N.; et al. Radioprotection of IDH1-mutated cancer cells by the IDH1-mutant inhibitor AGI-5198. Cancer Res. 2015, 75, 4790–4802. [Google Scholar] [CrossRef] [PubMed]
- Sulkowski, P.L.; Corso, C.D.; Robinson, N.D.; Scanlon, S.E.; Purshouse, K.R.; Bai, H.; Liu, Y.; Sundaram, R.K.; Hegan, D.C.; Fons, N.R.; et al. 2-Hydroxyglutarate produced by neomorphic IDH mutations suppresses homologous recombination and induces PARP inhibitor sensitivity. Sci. Transl. Med. 2017, 9, eaal2463. [Google Scholar] [CrossRef]
- Molenaar, R.J.; Radivoyevitch, T.; Nagata, Y.; Khurshed, M.; Przychodzen, B.; Makishima, H.; Xu, M.; Bleeker, F.E.; Wilmink, J.W.; Carraway, H.E.; et al. IDH1/2Mutations Sensitize Acute Myeloid Leukemia to PARP Inhibition and This Is Reversed by IDH1/2-Mutant Inhibitors. Clin. Cancer Res. 2018, 24, 1705–1715. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.-B.; Dong, D.-F.; Wang, M.-D.; Gao, K. IDH1 Overexpression Induced Chemotherapy Resistance and IDH1 Mutation Enhanced Chemotherapy Sensitivity in Glioma Cells in Vitro and in Vivo. Asian Pac. J. Cancer Prev. 2014, 15, 427–432. [Google Scholar] [CrossRef] [Green Version]
- Houillier, C.; Wang, X.; Kaloshi, G.; Mokhtari, K.; Guillevin, R.; Laffaire, J.; Paris, S.; Boisselier, B.; Idbaih, A.; Laigle-Donadey, F.; et al. IDH1 or IDH2 mutations predict longer survival and response to temozolomide in low-grade gliomas. Neurology 2010, 75, 1560–1566. [Google Scholar] [CrossRef] [PubMed]
- Khurshed, M.; Aarnoudse, N.; Hulsbos, R.; Hira, V.V.V.; Van Laarhoven, H.W.M.; Wilmink, J.W.; Molenaar, R.J.; Van Noorden, C.J.F. IDH1-mutant cancer cells are sensitive to cisplatin and an IDH1-mutant inhibitor counteracts this sensitivity. FASEB J. 2018, 32, 6344–6352. [Google Scholar] [CrossRef] [PubMed]
- Cuyàs, E.; Fernández-Arroyo, S.; Corominas-Faja, B.; Rodríguez-Gallego, E.; Bosch-Barrera, J.; Martin-Castillo, B.; Llorens, R.D.; Joven, J.; Menendez, J.A. Oncometabolite mutation IDH1 R132H confers a metformin hypersensitive phenotype. Oncotarget 2015, 6, 12279–12296. [Google Scholar] [CrossRef]
- Li, S.; Chou, A.P.; Chen, W.; Chen, R.; Deng, Y.; Phillips, H.S.; Selfridge, J.; Zurayk, M.; Lou, J.J.; Everson, R.G.; et al. Overexpression of isocitrate dehydrogenase mutant proteins renders glioma cells more sensitive to radiation. Neuro-Oncology 2013, 15, 57–68. [Google Scholar] [CrossRef]
- Eckschlager, T.; Plch, J.; Stiborova, M.; Hrabeta, J. Histone Deacetylase Inhibitors as Anticancer Drugs. Int. J. Mol. Sci. 2017, 18, 1414. [Google Scholar] [CrossRef]
- Bezecny, P. Histone deacetylase inhibitors in glioblastoma: Pre-clinical and clinical experience. Med. Oncol. 2014, 31, 985. [Google Scholar] [CrossRef] [PubMed]
- Cavaleri, F.; Schöler, H.R. Nanog: A new recruit to the embryonic stem cell orchestra. Cell 2003, 113, 551–552. [Google Scholar] [CrossRef]
- Xie, X.; Old, M.; Teknos, T.N.; Pan, Q.; Santaliz-Ruiz, L.E.; Santaliz-Ruiz, L.E.I.; Santaliz-Ruiz, L.E.I. Emerging Role of Nanog in Tumorigenesis and Cancer Stem Cells. Int. J. Cancer 2014, 135, 2741–2748. [Google Scholar]
- Zhou, J.-J.; Deng, X.-G.; He, X.-Y.; Zhou, Y.; Yu, M.; Gao, W.-C.; Zeng, B.; Zhou, Q.-B.; Li, Z.-H.; Chen, R.-F. Knockdown of NANOG enhances chemosensitivity of liver cancer cells to doxorubicin by reducing MDR1 expression. Int. J. Oncol. 2014, 44, 2034–2040. [Google Scholar] [CrossRef] [Green Version]
- Song, K.-H.; Choi, C.H.; Lee, H.-J.; Oh, S.J.; Woo, S.R.; Hong, S.-O.; Noh, K.H.; Cho, H.; Chung, E.J.; Kim, J.-H.; et al. HDAC1 upregulation by NANOG promotes multidrug resistance and a stem-like phenotype in immune edited tumor cells. Cancer Res. 2017, 77, 5039–5053. [Google Scholar] [CrossRef]
- Cai, M.-H.; Xu, X.-G.; Yan, S.-L.; Sun, Z.; Ying, Y.; Wang, B.-K.; Tu, Y.-X. Depletion of HDAC1, 7 and 8 by Histone Deacetylase Inhibition Confers Elimination of Pancreatic Cancer Stem Cells in Combination with Gemcitabine. Sci. Rep. 2018, 8, 1621. [Google Scholar] [CrossRef] [Green Version]
- West, A.C.; Johnstone, R.W. New and emerging HDAC inhibitors for cancer treatment. J. Clin. Investig. 2014, 124, 30–39. [Google Scholar] [CrossRef] [Green Version]
- Brown, D.V.; Filiz, G.; Daniel, P.M.; Hollande, F.; Dworkin, S.; Amiridis, S.; Kountouri, N.; Ng, W.; Morokoff, A.P.; Mantamadiotis, T. Expression of CD133 and CD44 in glioblastoma stem cells correlates with cell proliferation, phenotype stability and intra-tumor heterogeneity. PLoS ONE 2017, 12, e0172791. [Google Scholar] [CrossRef]
- Garros-Regulez, L.; Garcia, I.; Carrasco-Garcia, E.; Lantero, A.; Aldaz, P.; Moreno-Cugnon, L.; Arrizabalaga, O.; Undabeitia, J.; Torres-Bayona, S.; Villanua, J.; et al. Targeting SOX2 as a Therapeutic Strategy in Glioblastoma. Front. Oncol. 2016, 6, 222. [Google Scholar] [CrossRef]
- Ben-Porath, I.; Thomson, M.W.; Carey, V.J.; Ge, R.; Bell, G.W.; Regev, A.; Weinberg, R.A. An embryonic stem cell–like gene expression signature in poorly differentiated aggressive human tumors. Nat. Genet. 2008, 40, 499–507. [Google Scholar] [CrossRef]
- Zbinden, M.; Duquet, A.; Ngwabyt, S.-N.; Borges, I.; I Altaba, A.R.; Lorente-Trigos, A.; Lorente-Trigos, A. NANOG regulates glioma stem cells and is essential in vivo acting in a cross-functional network with GLI1 and p53. EMBO J. 2010, 29, 2659–2674. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.-L.; Chiou, S.-H.; Wu, C.-W.; Wu, C.-W. (Ken) Targeting cancer stem cells: Emerging role of Nanog transcription factor. OncoTargets Ther. 2013, 6, 1207–1220. [Google Scholar]
- Raineri, S.; Mellor, J. IDH1: Linking Metabolism and Epigenetics. Front. Genet. 2018, 9, 493. [Google Scholar] [CrossRef] [Green Version]
- Turcan, S.; Makarov, V.; Taranda, J.; Wang, Y.; Fabius, A.W.M.; Wu, W.; Zheng, Y.; El-Amine, N.; Haddock, S.; Nanjangud, G.; et al. Mutant-IDH1-dependent chromatin state reprogramming, reversibility, and persistence. Nat. Genet. 2018, 50, 62–72. [Google Scholar] [CrossRef]
- Kim, Y.-S.; Lee, Y.-M.; Oh, T.-I.; Shin, D.H.; Kim, G.-H.; Kan, S.-Y.; Kang, H.; Kim, J.H.; Kim, B.M.; Yim, W.J.; et al. Emodin Sensitizes Hepatocellular Carcinoma Cells to the Anti-Cancer Effect of Sorafenib through Suppression of Cholesterol Metabolism. Int. J. Mol. Sci. 2018, 19, 3127. [Google Scholar] [CrossRef]
- Chou, B.-K.; Mali, P.; Huang, X.; Ye, Z.; Dowey, S.N.; Resar, L.M.; Zou, C.; Zhang, Y.A.; Tong, J.; Cheng, L. Efficient human iPS cell derivation by a non-integrating plasmid from blood cells with unique epigenetic and gene expression signatures. Cell Res. 2011, 21, 518–529. [Google Scholar] [CrossRef] [PubMed]
- Lu, G.; Li, Y.; Ma, Y.; Lu, J.; Chen, Y.; Jiang, Q.; Qin, Q.; Zhao, L.; Huang, Q.; Luo, Z.; et al. Long noncoding RNA LINC00511 contributes to breast cancer tumorigenesis and stemness by inducing the miR-185-3p/E2F1/Nanog axis. J. Exp. Clin. Cancer Res. 2018, 37, 289. [Google Scholar] [CrossRef] [PubMed]
- Park, E.-J.; Lee, Y.-M.; Oh, T.-I.; Kim, B.M.; Lim, B.-O.; Lim, J.-H.; Morikawa, T. Vanillin Suppresses Cell Motility by Inhibiting STAT3-Mediated HIF-1α mRNA Expression in Malignant Melanoma Cells. Int. J. Mol. Sci. 2017, 18, 532. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward Primer | Reverse Primer |
|---|---|---|
| TUBB3 | AGCAAGAACAGCAGCTACTTCGT | GATGAAGGTGGAGGACATCTTGA |
| CDKN3 | TCCAGTAGCTGCTTGTCTCCTACTATA | TCTTAGGTCTCGCAGGCTGTCT |
| CCNB1 | AGCTGCTGCCTGGTGAAGAG | GCCATGTTGATCTTCGCCTTA |
| CDC20 | GCCCACCAAGAAGGAACATC | TTTTCCACTGAGCCGAAGGA |
| MCM7 | GGAAATATCCCTCGTAGTATCAC | CTGAGAGTAAACCCTGTACC |
| BIRC5 | CGAGGCTGGCTTCATCCACT | ACGGCGCACTTTCTTCGCA |
| CCNB2 | CCCAACTCCCTCTACCCTTGA | TCTGTCTCCCTCCCTCACTTTC |
| E2F1 | CCCAACTCCCTCTACCCTTGA | TCTGTCTCCCTCCCTCACTTTC |
| FOXM1 | TGCCCAGCAGTCTCTTACCT | CTACCCACCTTCTGGCAGTC |
| AURKA | GGAGAGCTTAAAATTGCAGATTTTG | GCTCCAGAGATCCACCTTCTCAT |
| MAD2L1 | ACTTAAATATCTCCCTACCTATACTGAGTCAA | TAGTAACTGTAGATGGAAAAACTTGTGCTA |
| SOX2 | CACATGAAGGAGCACCCGGATTAT | GTTCATGTGCGCGTAACTGTCCAT |
| ITGA6 | GCTGGTTATAATCCTTCAATATCAATTGT | TTGGGCTCAGAACCTTGGTTT |
| NES | AGGCTGAGAACTCTCGCTTGC | GGTGCTGGTCCTCTGGTATCC |
| ALDH1A3 | GCATGAGCCCATTGGTGTCT | CGCAGGCTTCAGGACCAT |
| MSI1 | CTCCAAAACAATTGACCCTAAGGT | GACAGCCCCCCCACAAAG |
| CD133 | AGAGCTTGCACCAACAAAGTACAC | AAGCACAGAGGGTCATTGAGAGA |
| CD44 | TGCCGCTTTGCAGGTGTAT | GGCCTCCGTCCGAGAGA |
| SOX10 | ACTTCGGCAACGTGGACATT | CAGCCACATCAAAGGTCTCCAT |
| PAX6 | TTCAGAGCCCCATATTCGAG | GTTGGACACCTGCAGAAT |
| Notch4 | AACTCCTCCCCAGGAATCTG | CCTCCATCCAGCAGAGGTT |
| NANOG | CCTCAGCCTCCAGCAGATGC | CCGCTTGCACTTCACCCTTTG |
| MDR1 | TGACATTTATTCAAAGTTAAAAGCA | TAGACACTTTATGCAAACATTTCAA |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, G.-H.; Choi, S.Y.; Oh, T.-I.; Kan, S.-Y.; Kang, H.; Lee, S.; Oh, T.; Ko, H.M.; Lim, J.-H. IDH1R132H Causes Resistance to HDAC Inhibitors by Increasing NANOG in Glioblastoma Cells. Int. J. Mol. Sci. 2019, 20, 2679. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20112679
Kim G-H, Choi SY, Oh T-I, Kan S-Y, Kang H, Lee S, Oh T, Ko HM, Lim J-H. IDH1R132H Causes Resistance to HDAC Inhibitors by Increasing NANOG in Glioblastoma Cells. International Journal of Molecular Sciences. 2019; 20(11):2679. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20112679
Chicago/Turabian StyleKim, Geon-Hee, So Young Choi, Taek-In Oh, Sang-Yeon Kan, Hyeji Kang, Sujin Lee, Taerim Oh, Hyun Myung Ko, and Ji-Hong Lim. 2019. "IDH1R132H Causes Resistance to HDAC Inhibitors by Increasing NANOG in Glioblastoma Cells" International Journal of Molecular Sciences 20, no. 11: 2679. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20112679