Identification and Analysis of Micro-Exon Genes in the Rice Genome


 ,
,
Abstract
:1. Introduction
2. Results
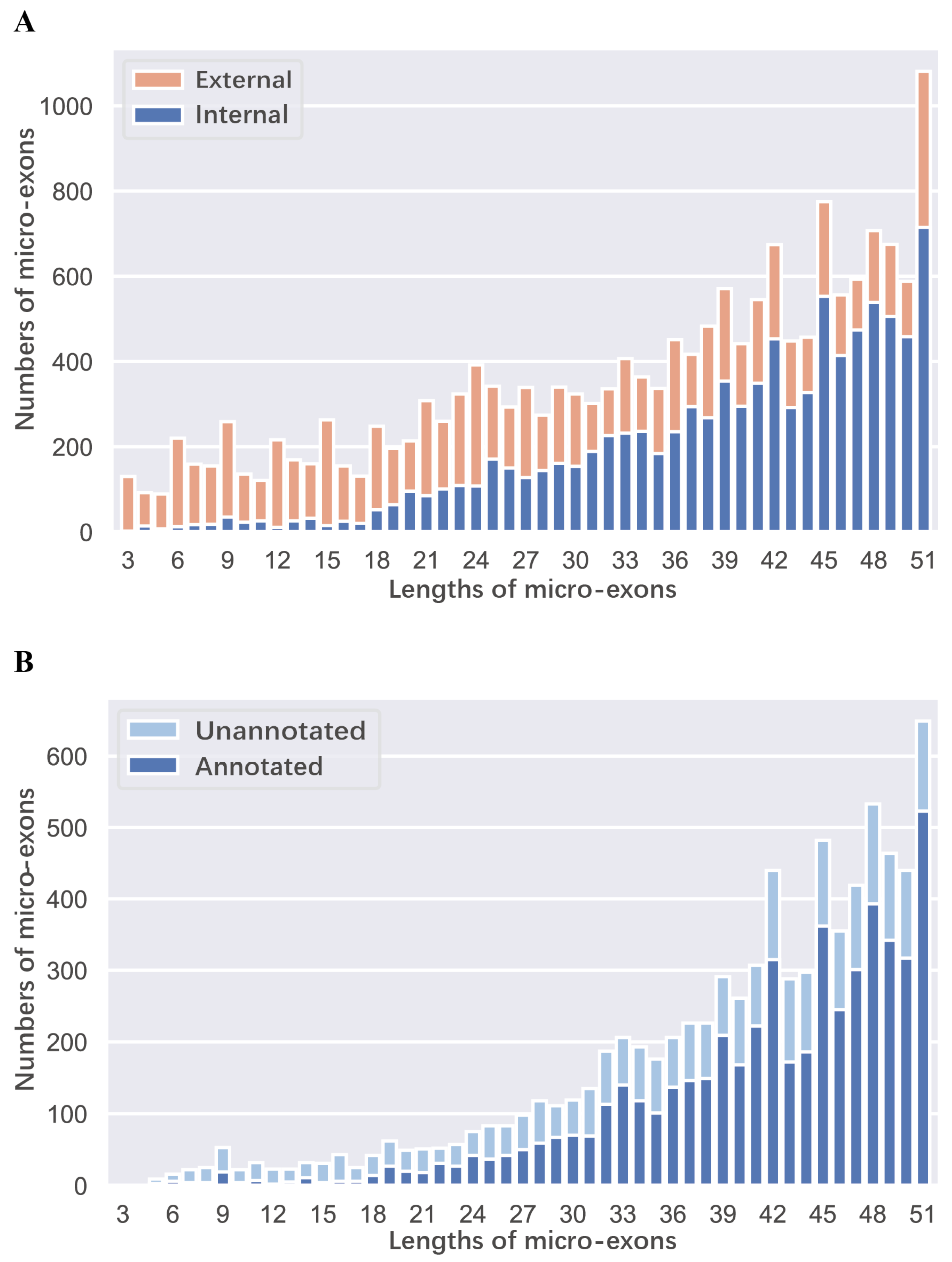
2.1. Identification and Inclusion Ratio of Micro-Exons in ZS97 and MH63
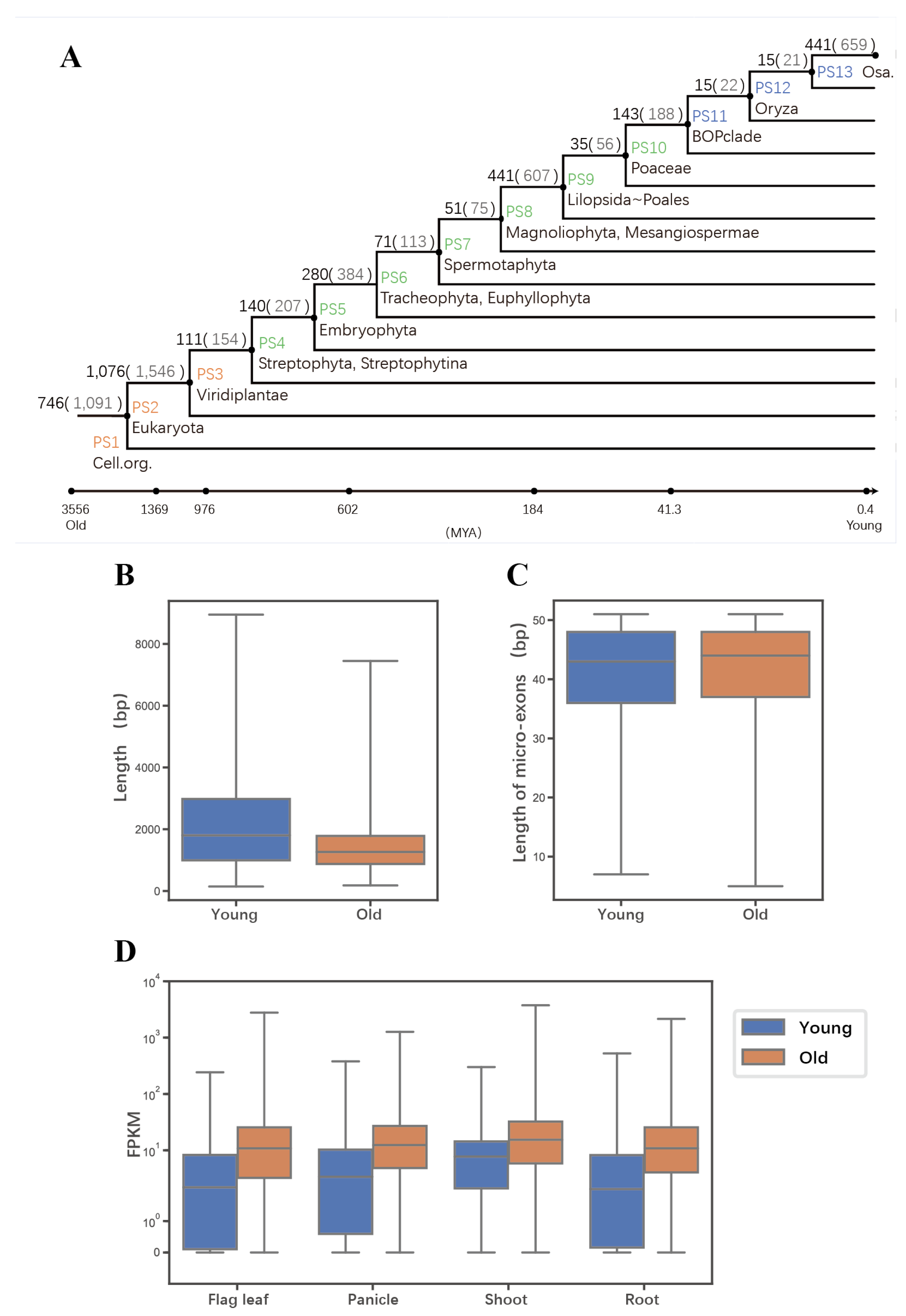
2.2. Evolutionary Age of Genes Containing Micro-Exons
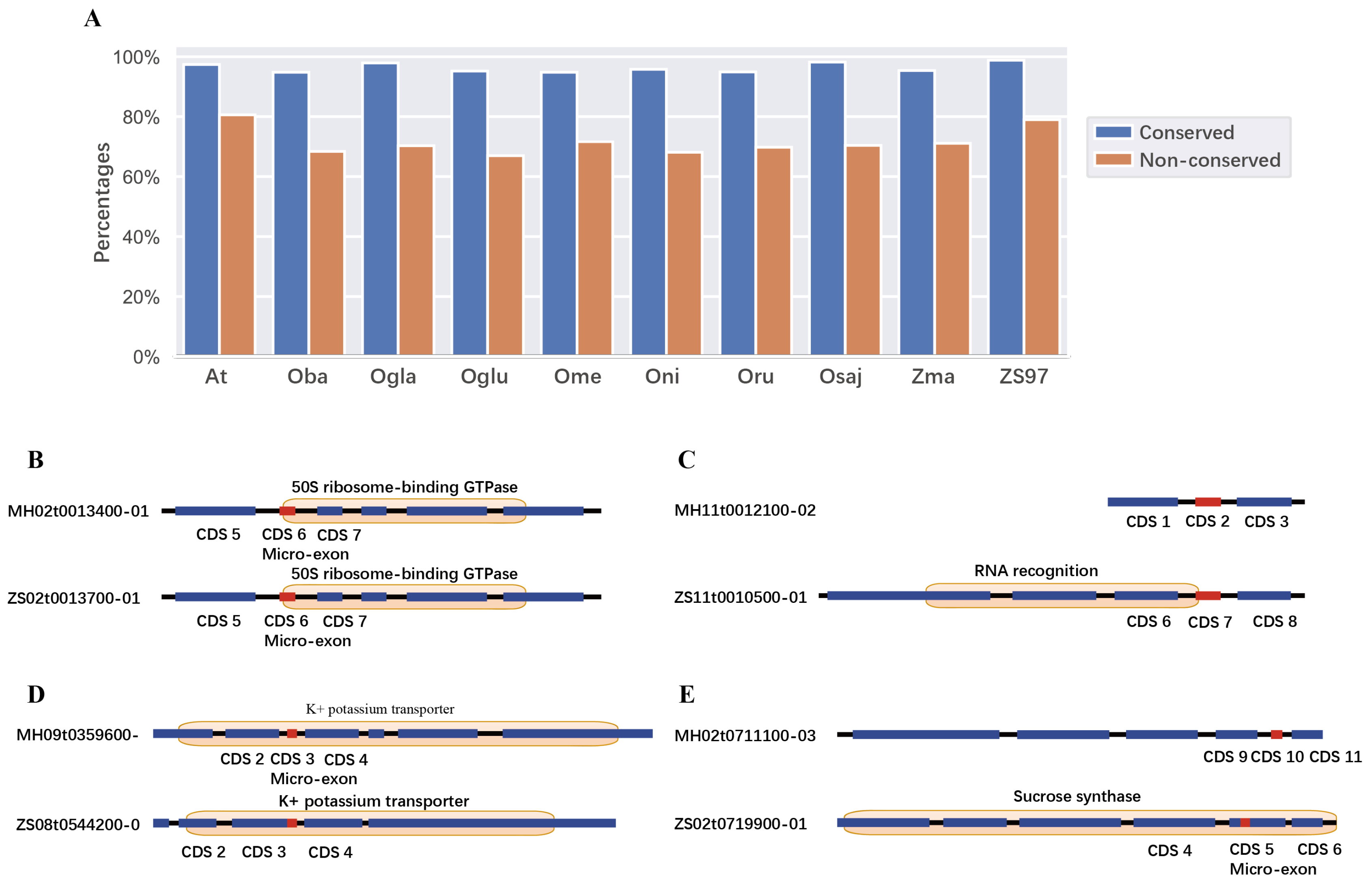
2.3. Conservation of Domains and Micro-Exons
2.4. Quantification of Micro-Exon Usage and Gene Ontology Enrichment Analysis
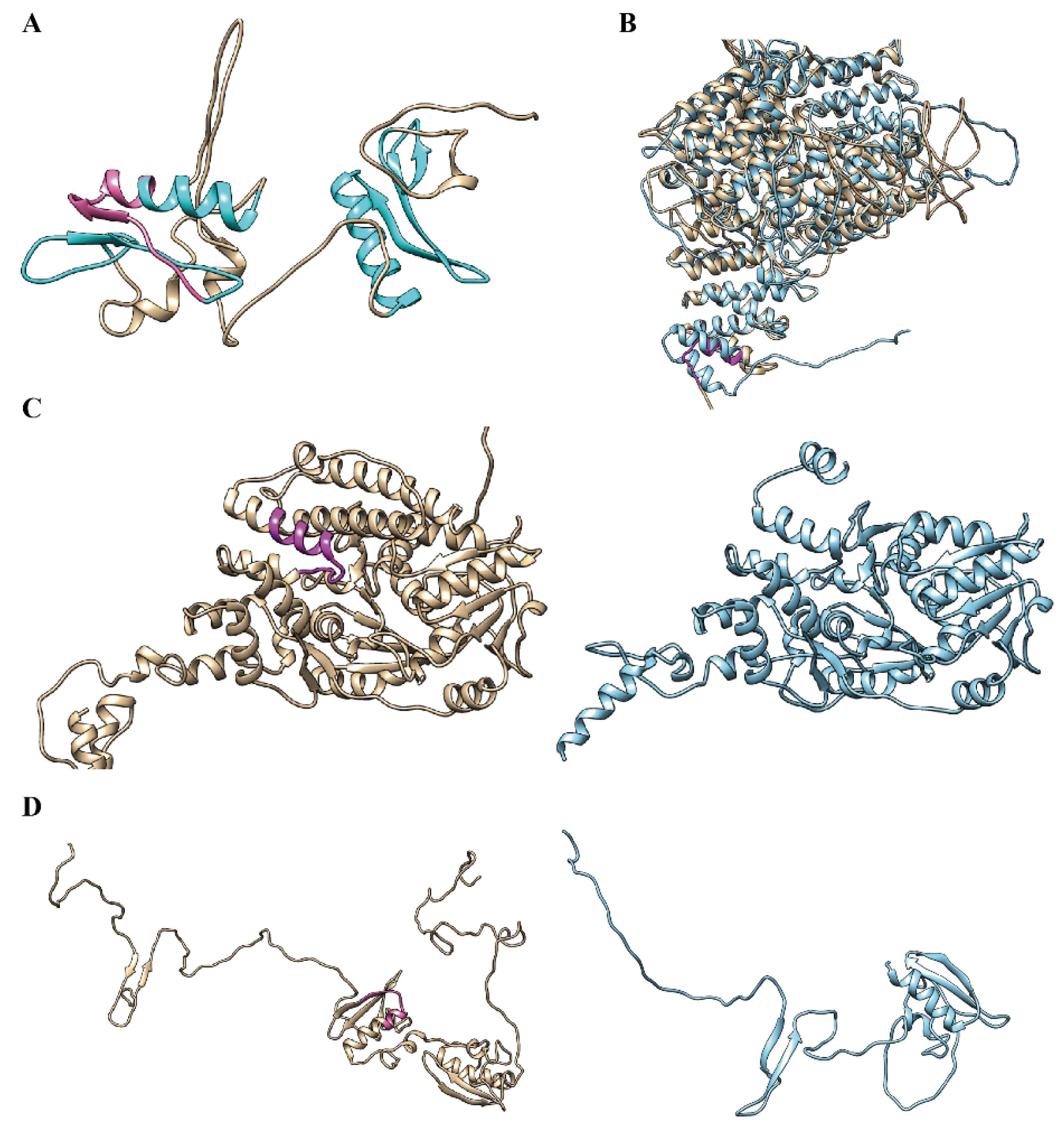
2.5. Structure and Functional Analyses of Alternative Spliced Micro-Exons
3. Discussion
4. Materials and Methods
4.1. Materials and Data Preparation
4.2. Identification of Micro-Exons
4.3. Evolutionary Age of Genes Containing Micro-Exons
4.4. Conservation of Micro-Exons and Domains
4.5. Percent Splice-In (PSI) Index of Micro-Exons
4.6. Functional Annotation of Micro-Exon Genes
4.7. Structures and Domains of Micro-Exons
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ESE | Exonic splicing enhancers |
| ISE | Intronic splicing enhancers |
| RBP | RNA-binding proteins |
| GO | Gene ontology |
| ORF | Open reading frames |
| NR | Non-redundant |
| PS | Phylostratum |
| PSI | Percent spliced-in |
| AP2 | Apetala 2 |
| SNB | Supernumerary bract |
| P3DB | Plant Protein Phosphorylation DataBase |
| AS | Alternatively spliced |
| CS | Constitutively spliced |
References
- Irimia, M.; Weatheritt, R.J.; Ellis, J.D.; Parikshak, N.N.; Gonatopoulos-Pournatzis, T.; Babor, M.; Quesnel-Vallières, M.; Tapial, J.; Raj, B.; O’Hanlon, D.; et al. A Highly Conserved Program of Neuronal Microexons Is Misregulated in Autistic Brains. Cell 2014, 159, 1511–1523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McAllister, L. Alternative Splicing of Micro-Exons Creates Multiple Forms of the Insect Cell Adhesion Molecule Fasciclin I. J. Neurosci. 1992, 12, 895–905. [Google Scholar] [CrossRef]
- Bournay, A.-S.; Hedley, P.E.; Maddison, A.; Waugh, R.; Machray, G.C. Exon Skipping Induced by Cold Stress in a Potato Invertase Gene Transcript. Nucleic Acids Res. 1996, 24, 2347–2351. [Google Scholar] [CrossRef] [Green Version]
- Volfovsky, N. Computational Discovery of Internal Micro-Exons. Genome Res. 2003, 13, 1216–1221. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.I.; Sanchez-Pulido, L.; Haerty, W.; Ponting, C.P. RBFOX and PTBP1 proteins regulate the alternative splicing of micro-exons in human brain transcripts. Genome Res. 2015, 25, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Romero, P.R.; Zaidi, S.; Fang, Y.Y.; Uversky, V.N.; Radivojac, P.; Oldfield, C.J.; Cortese, M.S.; Sickmeier, M.; Legall, T.; Obradovic, Z. Alternative splicing in concert with protein intrinsic disorder enables increased functional diversity in multicellular organisms. Proc. Natl. Acad. Sci. USA 2006, 103, 8390–8395. [Google Scholar] [CrossRef] [Green Version]
- Black, D.L. Mechanisms of Alternative Pre-Messenger RNA Splicing. Annu. Rev. Biochem. 2003, 72, 291–336. [Google Scholar] [CrossRef] [Green Version]
- Lykke-Andersen, S.; Jensen, T.H. Nonsense-mediated mRNA decay: An intricate machinery that shapes transcriptomes. Nat. Rev. Mol. Cell Biol. 2015, 16, 665–677. [Google Scholar] [CrossRef]
- Quesnel-Vallières, M.; Irimia, M.; Cordes, S.P.; Blencowe, B.J. Essential roles for the splicing regulator nSR100/SRRM4 during nervous system development. Genes Dev. 2015, 29, 746–759. [Google Scholar] [CrossRef] [Green Version]
- Simpson, C.G.; Hedley, P.E.; Watters, J.A.; Clark, G.P.; Mcquade, C.; Machray, G.C.; Brown, J.W. Requirements for mini-exon inclusion in potato invertase mRNAs provides evidence for exon-scanning interactions in plants. RNA 2000, 6, 422. [Google Scholar] [CrossRef] [PubMed]
- Simpson, C.G.; Thow, G.; Clark, G.P.; Jennings, S.N.; Watters, J.A.; Brown, J.W. Mutational analysis of a plant branchpoint and polypyrimidine tract required for constitutive splicing of a mini-exon. RNA 2002, 8, 47. [Google Scholar] [CrossRef] [PubMed]
- Ustianenko, D.; Weyn-Vanhentenryck, S.M.; Zhang, C. Microexons: Discovery, regulation, and function: Microexons: Discovery, regulation, and function. Wiley Interdiscip. Rev. RNA 2017, 8, e1418. [Google Scholar] [CrossRef] [PubMed]
- Huntsman, M.M.; Tran, B.-V.; Potkin, S.G.; Bunney, W.E.; Jones, E.G. Altered ratios of alternatively spliced long and short 2 subunit mRNAs of the -amino butyrate type A receptor in prefrontal cortex of schizophrenics. Proc. Natl. Acad. Sci. USA 1998, 95, 15066–15071. [Google Scholar] [CrossRef]
- Miyoshi, K.; Ito, Y.; Serizawa, A.; Kurata, N. OsHAP3 genes regulate chloroplast biogenesis in rice. Plant J. 2010, 36, 532–540. [Google Scholar] [CrossRef]
- Fu, F.-F.; Xue, H.-W. Coexpression Analysis Identifies Rice Starch Regulator1, a Rice AP2/EREBP Family Transcription Factor, as a Novel Rice Starch Biosynthesis Regulator. PLANT Physiol. 2010, 154, 927–938. [Google Scholar] [CrossRef]
- Zhang, J.; Chen, L.-L.; Xing, F.; Kudrna, D.A.; Yao, W.; Copetti, D.; Mu, T.; Li, W.; Song, J.-M.; Xie, W.; et al. Extensive sequence divergence between the reference genomes of two elite indica rice varieties Zhenshan 97 and Minghui 63. Proc. Natl. Acad. Sci. USA 2016, 113, E5163–E5171. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Mauleon, R.; Hu, Z.; Chebotarov, D.; Tai, S.; Wu, Z.; Li, M.; Zheng, T.; Fuentes, R.R.; Zhang, F.; et al. Genomic variation in 3,010 diverse accessions of Asian cultivated rice. Nature 2018, 557, 43–49. [Google Scholar] [CrossRef]
- Domazet-Lošo, T.; Brajković, J.; Tautz, D. A phylostratigraphy approach to uncover the genomic history of major adaptations in metazoan lineages. Trends Genet. 2007, 23, 533–539. [Google Scholar] [CrossRef]
- Wang, Y.; Tang, H.; DeBarry, J.D.; Tan, X.; Li, J.; Wang, X.; Lee, T.-H.; Jin, H.; Marler, B.; Guo, H.; et al. MCScanX: A toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012, 40, e49. [Google Scholar] [CrossRef]
- Wang, P.; Yan, B.; Guo, J.; Hicks, C.; Xu, Y. Structural genomics analysis of alternative splicing and application to isoform structure modeling. Proc. Natl. Acad. Sci. USA 2005, 102, 18920–18925. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Soltis, P.S.; Wall, K.; Soltis, D.E. Phylogeny and Domain Evolution in the APETALA2-like Gene Family. Mol. Biol. Evol. 2006, 23, 107–120. [Google Scholar] [CrossRef]
- Fowler, S. GIGANTEA: A circadian clock-controlled gene that regulates photoperiodic flowering in Arabidopsis and encodes a protein with several possible membrane-spanning domains. EMBO J. 1999, 18, 4679–4688. [Google Scholar] [CrossRef]
- Tuteja, N.; Tarique, M.; Tuteja, R. Rice SUV3 is a bidirectional helicase that binds both DNA and RNA. BMC Plant Biol. 2014, 14. [Google Scholar] [CrossRef]
- Lee, D.-Y.; An, G. Two AP2 family genes, SUPERNUMERARY BRACT (SNB) and OsINDETERMINATE SPIKELET 1 (OsIDS1), synergistically control inflorescence architecture and floral meristem establishment in rice: SNB and OsIDS1 control rice inflorescence architecture and floral meristem. Plant J. 2012, 69, 445–461. [Google Scholar]
- Buljan, M.; Chalancon, G.; Eustermann, S.; Wagner, G.P.; Fuxreiter, M.; Bateman, A.; Babu, M.M. Tissue-specific splicing of disordered segments that embed binding motifs rewires protein interaction networks. Mol. Cell 2012, 46, 871–883. [Google Scholar] [CrossRef]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 2016, 11, 1650. [Google Scholar] [CrossRef]
- AltschuP, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic Local Alignment Search Tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Jones, P.; Binns, D.; Chang, H.-Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G.; et al. InterProScan 5: Genome-scale protein function classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.-C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Durbin, R.; Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754-1760. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Du, Z.; Zhou, X.; Ling, Y.; Zhang, Z.; Su, Z. agriGO: A GO analysis toolkit for the agricultural community. Nucleic Acids Res. 2010, 38, W64–W70. [Google Scholar] [CrossRef] [PubMed]
- Källberg, M.; Wang, H.; Wang, S.; Peng, J.; Wang, Z. Template-based protein structure modeling using the RaptorX web server. Nat. Protoc. 2012, 7, 1511–1522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species (Paired with MH63) | Total Gene Pairs | Gene Pairs with Micro-Exons | Gene Pairs with Conserved Micro-Exons |
|---|---|---|---|
| Arabidopsis thaliana | 1828 | 253 | 214 (197) |
| Oryza barthii | 27,306 | 3821 | 3183 (3470) |
| Oryza glaberrima | 25,755 | 3559 | 2949 (3571) |
| Oryza glumipatula | 27,314 | 3731 | 3111 (3434) |
| Oryza meridionalis | 21,571 | 3156 | 2691 (2975) |
| Oryza nivara | 27,192 | 3638 | 3089 (3422) |
| Oryza rufipogon | 29,323 | 3864 | 3208 (3523) |
| Oryza sativa | 26,237 | 3875 | 3217 (3561) |
| Zea mays | 21,953 | 3524 | 2386 (2509) |
| ZS97 | 38,649 | 3747 | 3161 (4251) |
| Rank | Domains Including Micro-Exons | Domains Excluding Micro-Exons |
|---|---|---|
| 1 | Protein kinase domain (PF00069, 66) | WD domain, G-beta repeat (PF00400, 163) |
| 2 | RNA recognition motif (PF00076, 42) | RNA recognition motif (PF00076, 78) |
| 3 | AP2 domain (PF00847, 36) | Protein kinase domain (PF00069, 60) |
| 4 | K-box region (PF01486, 28) | IQ calmodulin-binding motif (PF00612, 46) |
| 5 | Glycosyl hydrolase family 1 (PF00232, 24) | PPR repeat family (PF13041, 44) |
| 6 | Protein tyrosine kinase (PF07714, 23) | PPR repeat (PF01535, 39) |
| 7 | Myb-like DNA-binding domain (PF00249, 20) | SRF-type transcription factor (PF00319, 32) |
| 8 | Serine carboxypeptidase (PF00450, 20) | C2 domain (PF00168, 28) |
| 9 | WD domain, G-beta repeat (PF00400, 17) | Helicase conserved C-terminal domain (PF00271, 28) |
| 10 | Major facilitator superfamily (PF07690, 16) | Gelsolin repeat (PF00626, 25) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, Q.; Lv, F.; Tahir ul Qamar, M.; Xing, F.; Zhou, R.; Li, H.; Chen, L.-L. Identification and Analysis of Micro-Exon Genes in the Rice Genome. Int. J. Mol. Sci. 2019, 20, 2685. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20112685
Song Q, Lv F, Tahir ul Qamar M, Xing F, Zhou R, Li H, Chen L-L. Identification and Analysis of Micro-Exon Genes in the Rice Genome. International Journal of Molecular Sciences. 2019; 20(11):2685. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20112685
Chicago/Turabian StyleSong, Qi, Fang Lv, Muhammad Tahir ul Qamar, Feng Xing, Run Zhou, Huan Li, and Ling-Ling Chen. 2019. "Identification and Analysis of Micro-Exon Genes in the Rice Genome" International Journal of Molecular Sciences 20, no. 11: 2685. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20112685