ERK Pathway in Activated, Myofibroblast-Like, Hepatic Stellate Cells: A Critical Signaling Crossroad Sustaining Liver Fibrosis


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction: Fibrogenic Progression of Chronic Liver Diseases
- chronic infection by hepatothropic viruses like hepatitis C virus (HCV), globally distributed, and hepatitis B virus (HBV) being predominant in Asia;
- non-alcoholic fatty liver disease (NAFLD), an obesity and diabetes type II-related CLD whose incidence and prevalence is dramatically growing worldwide, particularly in western countries;
- excess ethanol consumption, responsible for alcoholic liver disease (ALD), relevant in western countries;
- autoimmune-mediated form of CLD, including either conditions affecting the biliary tree such as primary biliary cholangitis (PBC) and primary sclerosing cholangitis (PSC), or autoimmune hepatitis (AIH);
- a number of more rare hereditary diseases including Wilson’s disease (WD), α1-anti-trypsin (α1-AT) deficiency, and the different genetic variants of hemochromatosis.
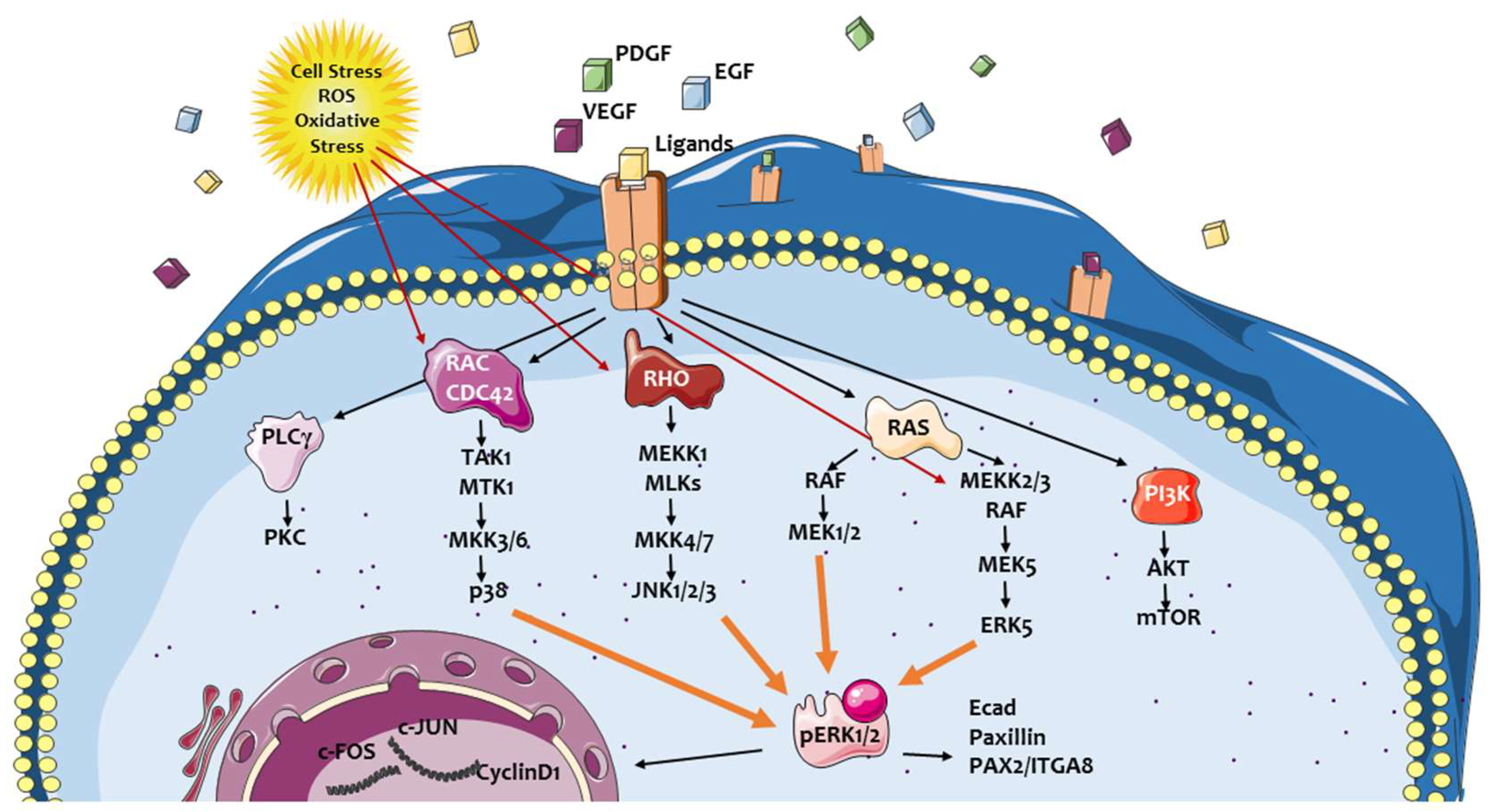
2. ERK Signaling Pathway: A Crossroad Conveying Multiple Signals and Modulating Different Cellular Responses
- ERK1/2 cascade can regulate immediate early-genes following extracellular stimulation (i.e., by growth factors) through the regulation of a number of transcription factors; a classic example is represented by activation of Elk1, a nuclear ETS domain transcription factor, which is rapidly phosphorylated following direct binding of the CD/CRS domain of ERK1/2 with the D-domain of Elk1; activation of Elk1 leads to the induction of c-Fos, which is critical for proper progression of cell proliferation and differentiation;
- ERK1/2 can regulate transcriptional suppression, as is the case of ETS2 repressor factor Erf1, which is known, in its dephosphorylated and nuclear located form, to suppress transcription in resting and/or serum starved cells; following activation by a mitogen, Erf1 is phosphorylated by ERK1/2 and exported from the nucleus then alleviating its role in suppressing transcription. Prevention of Erf1 phosphorylation has been reported also to arrest fibroblast proliferation in the G0/G1 phase of the cell cycle. Additionally, ERK1/2 suppressing function can result also by the direct interaction (particularly of ERK2) with DNA, through specific binding to the DNA sequence C/CAAAG/C independently on its own catalytic activity [35];
- ERK1/2 cascade is involved in the chromatin remodeling which is relevant, following proper stimulation, to allow proteins (mainly transcription factors) to access and bind to their specific DNA sequences. ERK1/2 cascade has a role in histone deacetylation, phosphorylation of specific chromatin-rearranging protein histones H3 and H4, or by non-conventional influence on PolyADP ribose-polymerase 1 (PARP1);
- ERK1/2 cascade can finally regulate the general nuclear import machinery by interactions with the so-called nuclear pore complexes that control nuclear-cytoplasmic exchange of different molecules.
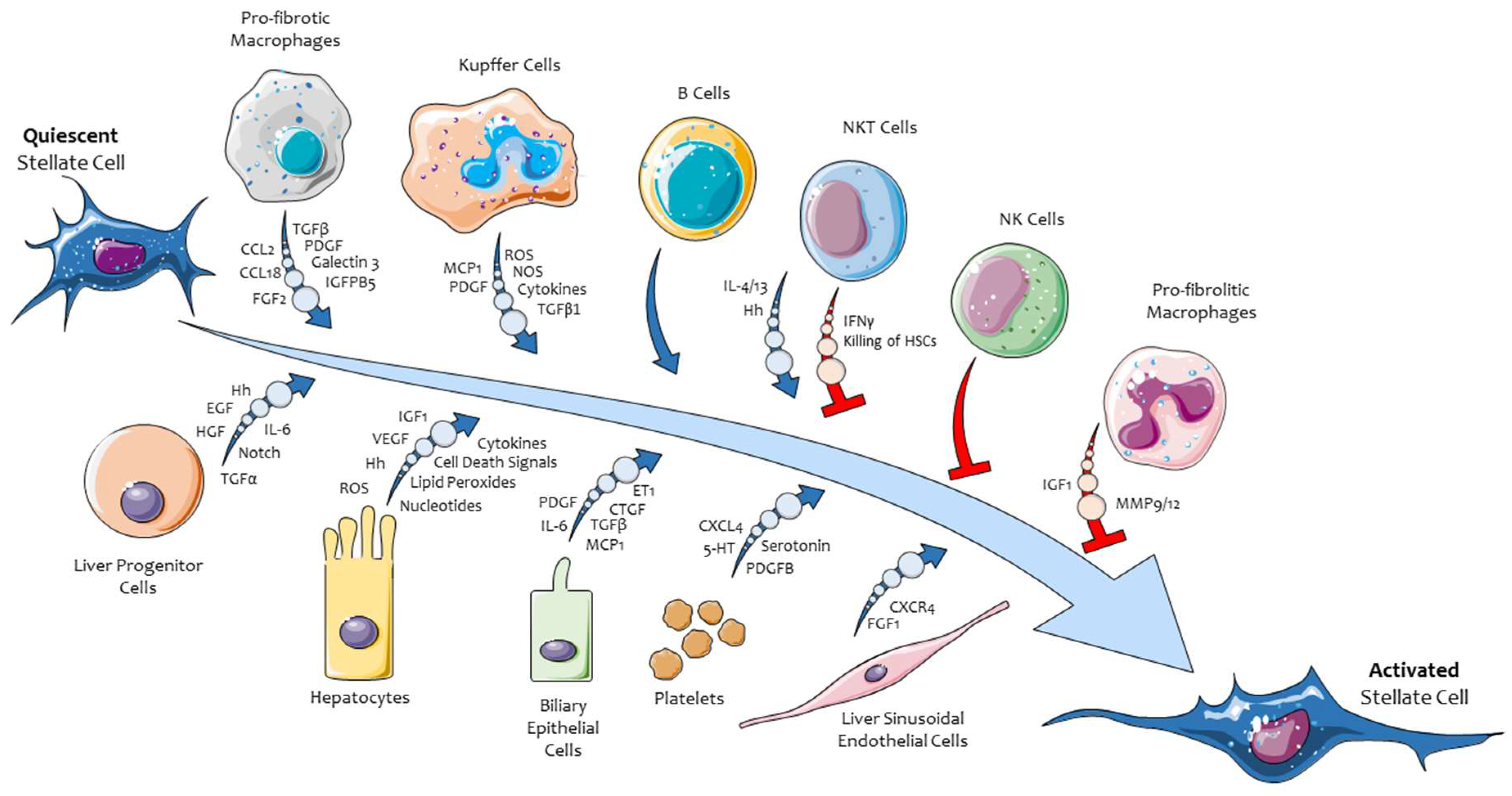
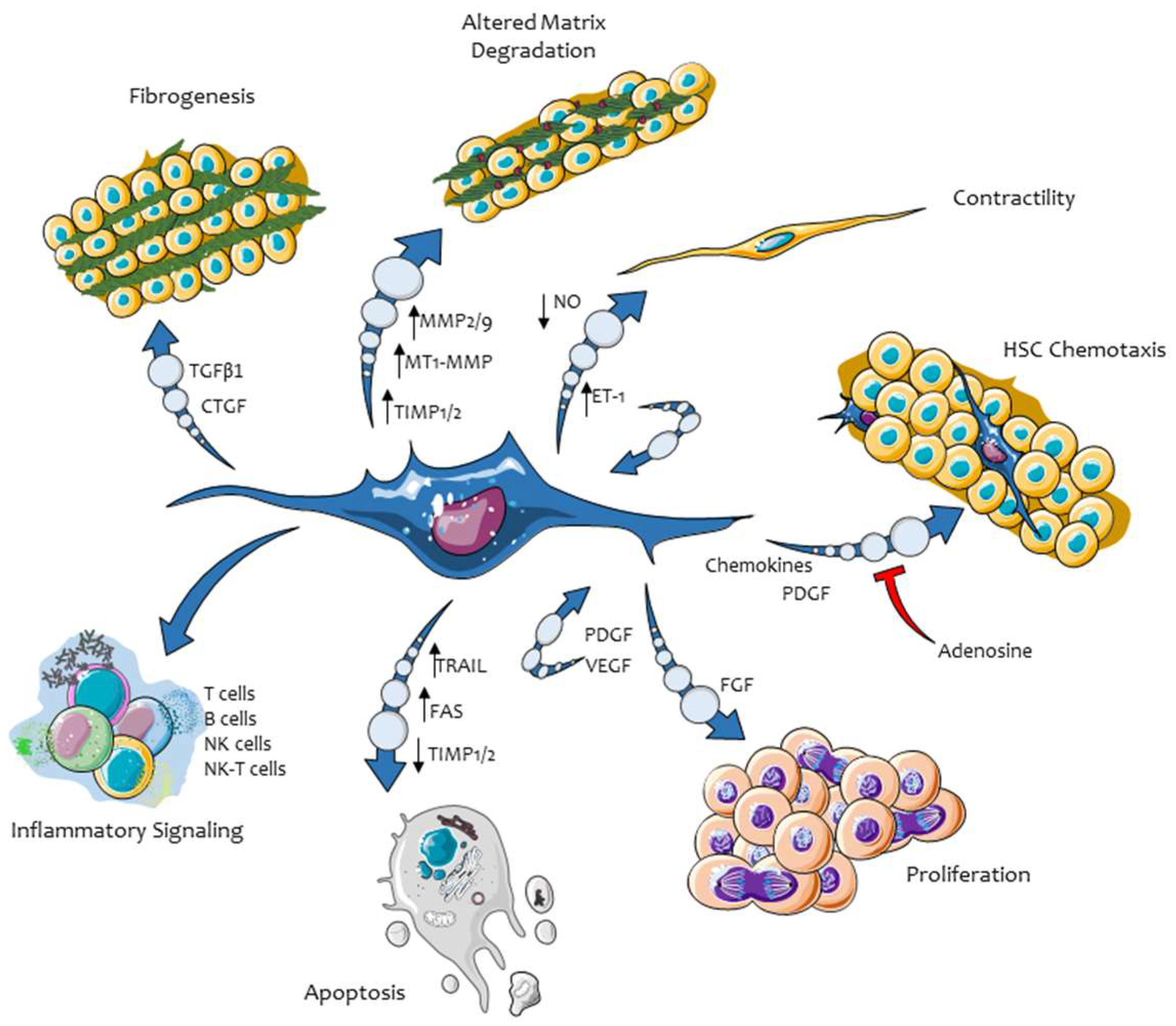
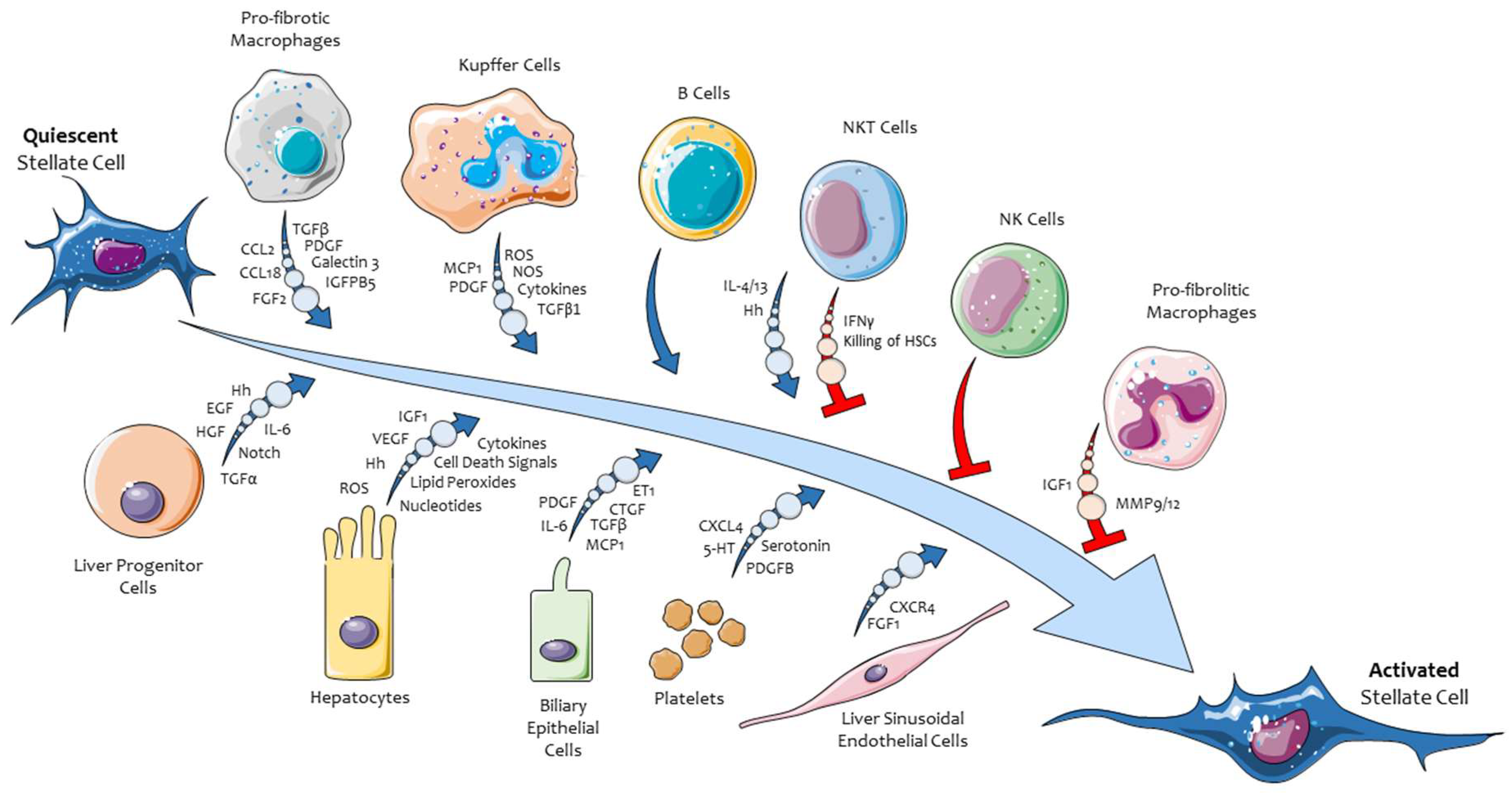
3. Hepatic Myofibroblasts, Their Pro-Fibrogenic Phenotypic Responses, and the Role of ERK Signaling
3.1. Proliferation and Survival of Hepatic MFs
3.2. Synthesis and Remodelling of Extracellular Matrix (ECM) Components
3.3. Migration in Response to Chemoattractants and Reactive Oxygen Species (ROS)
3.4. Pro-Inflammatory and Immune-Modulatory Role
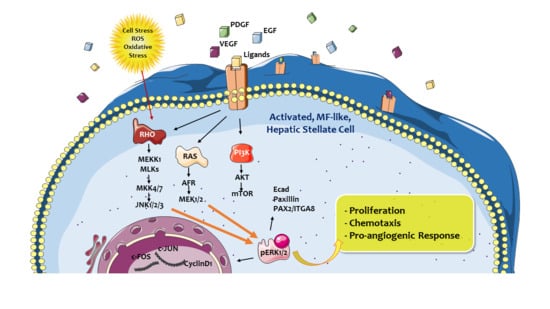
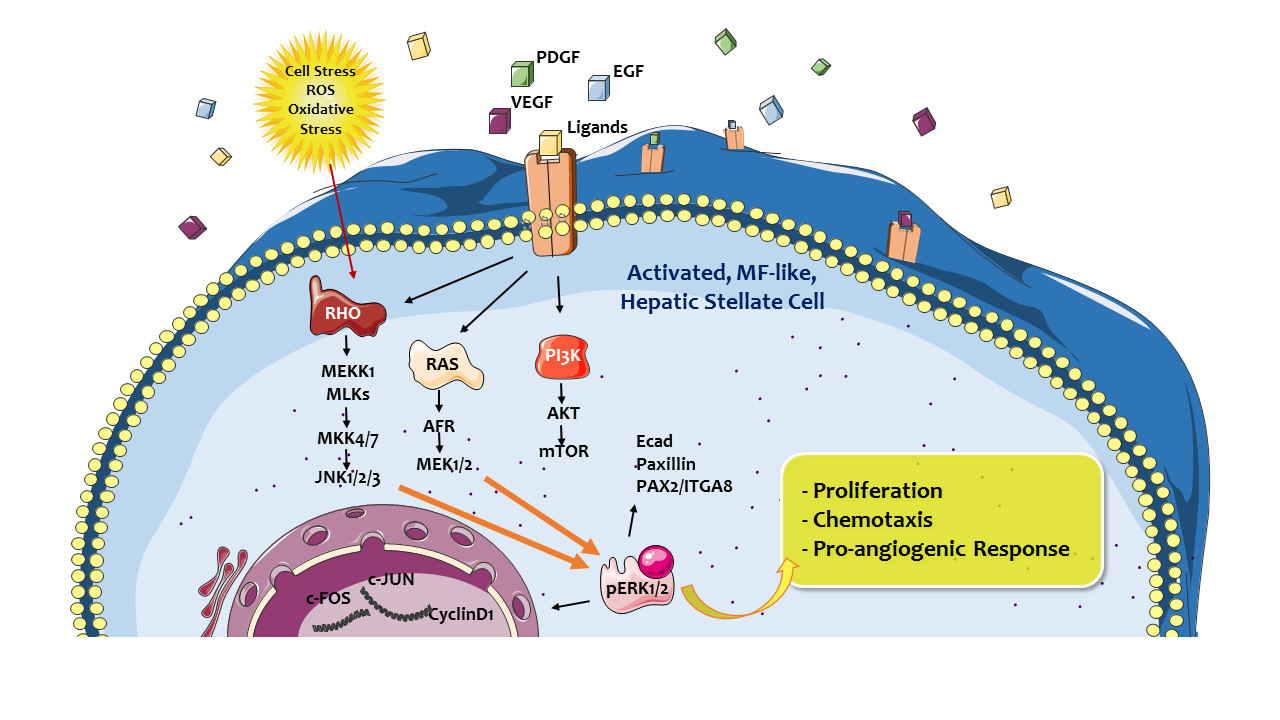
3.5. Proangiogenic Role
4. Therapeutic Anti-Fibrotic Strategies
4.1. General Concepts on Antifibrotic Strategies and Pathogenic Therapeutic Targets to Affect Chronic Liver Disease (CLD) Progression
4.2. Drugs or Strategies Designed to Minimize Parenchymal Liver Injury
4.3. Drugs or Strategies Designed to Target Activated Macrophages
4.4. Drugs or Strategies Designed to Target MFs
- To interfere with MF-mediated crosslinking of collagens and elastins by the use of the humanized anti-LOXL2 antibody Simtuzumab (G6-6624);
- to interfere with mechanisms resulting in dysregulation of critical molecular pathways in activated HSC or MFs; this approach is by far the most interesting, with several studies dedicated to either blocking pathways elicited by ligand-receptor interactions (we will focus mainly on these approaches that directly or indirectly target Ras/Raf/MEK/ERK pathway), including those elicited by TGFβ1, PDGF, ligand-receptor-induced signalling pathways, HGF, VEGF/VEGFR, Wnt/β-catenin, EGF/EGFR, Hedgehog, endotelins, cannabinoids, adipokines, retinoid, and vitamin D receptors, integrins, and toll-like receptors (TLRs) [5,6,7,22];
- to interfere with nuclear receptor transcription factors expressed by HSC and MFs, including peroxisome proliferator-activated receptor (PPAR)-γ and PPAR-δ, farnesoid X receptor (FXR), liver X receptor (LXR), vitamin D receptor (VDR), nuclear receptor subfamily 4 group A member 1 (NR4A1), and nuclear receptor subfamily 1 group D member 1 (REV-ERBα) [5,22];
- to interfere with transcription factors that positively contribute to HSC and MF activation, including myocardin-related transcription factor A (MRTF-A), sex-determining region Y-box 9 (SOX9), aryl hydrocarbon receptor (AhR), Yes associated protein (YAP), and Gα-interacting vesicle-associated protein (GIV); similarly, to interfere with transcription factors that negatively affect pro-fibrogenic genes and HSC activation like Kruppel-like factors (KLF6 and -2), GATA binding protein 4 (GATA4), NR4A1, and NR4A2 [5,6,7,22];
- to interfere with epigenetic transcriptional dysregulation, particularly with profibrogenic miRNAs either overexpressed in activated HSC (miR-21, miR-27, mirR-125, miR-195, miR-199a, miR-199b, miR-221, and miR-222), and able to sustain MF phenotypic responses, as well as on antifibrotic miRNAs that are down-regulated in activated HSC (miR15b, miR-16, miR-29, miR-122, miR-133b, and miR-200a) [61].
4.5. To Promote Fibrosis Resolution
- by inducing selective elimination, reversion, or senescence of MFs; selective killing of activated MFs has been reported in preclinical studies employing gliotoxin, the nuclear factor-κB (NF-κB) inhibitor BAY 11-7082, or the proteasome inhibitors MG-132 and bortezomib as well as by employing the histone-deacetylase inhibitor nilotinib [5,50]; senescence of activated HSC has been obtained using cysteine-rich protein 61 (CCN-Cyr61), curcumin, or OSU03012, a celecoxib derivative deactivation; reversion of MFs to HSC has been observed following withdrawal of etiological agents;
- by increasing ECM degradation, obtained by either using the LOXL2 inhibitor Simtuzumab, an approach effective in preclinical studies but abandoned since it was found ineffective in clinical trials; alternatively, one may transplant bone marrow-derived cells, particularly “resolutive macrophages”, an attempt under evaluation and designed to promote fibrillary ECM degradation and eventually favour regeneration [5,6,7,22].
5. Therapeutic Antifibrotic Strategies Designed to Affect Phenotypic Responses of Hepatic MFs that Operate by Involving Ras/Raf/MEK/ERK Signalling Pathway
5.1. Antifibrogenic Drug and Strategies Directly Targeting Ras/Raf/MEK/ERK Cascade in Hepatic MFs
5.1.1. Pentoxifylline
5.1.2. N-Acetyl Cysteine and Curcumin
5.1.3. Raf-Kinase Inhibitor Protein
5.1.4. MAPK Tumour Progression Locus 2
5.1.5. Embryonic Stem Cell-Expressed RAS
5.1.6. Direct Inhibition of RAS and ERK
5.1.7. SIRT2 Inhibition
5.2. Strategies Designed to Target Signalling Pathways and/or ROS Intracellular Generation Upstream to the Activation of Ras/Raf/MEK/ERK Cascade
5.2.1. Strategies to Target Platelet-Derived Growth Factor (PDGF) Signalling Pathway
- to target the PDGFR-β, either by using an antisense strategy [93], a dominant–negative soluble PDGFR-β [94], or by using PDGFR tyrosine kinase inhibitors [88]. For the latter option, several RTK inhibitors have been used either in vivo in preclinical studies or in vitro, including: Imatinib mesylate (imatinib, STI571, or Gleevec), an inhibitor of tyrosine kinases active on both PDGFR-β and –α, that can also affect the bcr-abl fusion protein c-kit and Flt3 [95,96]; Sorafenib, a potent inhibitor of VEGF receptor 2 (VEGFR-2), PDGFR-β, and Raf kinases [97]; Nilotinib, a second generation RTK inhibitor, approximately 20 times more potent than imatinib mesylate, able to affect multiple mechanisms, both in vitro and in vivo, including induction of HSC apoptosis, inhibition of PDGF, TGF-β, and other signal pathways, as well as suppression of neo-angiogenesis [98,99,100]. No one of these drugs, effective in preclinical studies, has been translated and/or approved for anti-fibrotic treatment of CLD, although sorafenib is currently employed to treat patients with advanced HCC.
- To use endogenous inhibitors of PDGF signalling (reviewed in reference [88]). Along these lines, a strategy able to reduce liver fibrosis in vivo (preclinical studies) has been designed and tested in order to down-regulate the expression of secreted protein acidic and is rich in cysteine (SPARC), an ECM protein that can represent low-affinity docking sites or reservoirs for the PDGF growth factors [105,106].
5.2.2. Strategies Designed to Target ROS Production by NADPH-Oxidase Isoforms
6. Summary
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| α1-AT | α1-anti-trypsin |
| AhR | Aryl hydrocarbon receptor |
| AIH | Autoimmune hepatitis |
| ALD | Alcoholic liver disease |
| bFGF | Basic fibroblast growth factor |
| CCL2 | C-C Chemokine motif chemokine ligand 2 |
| CCl4 | Carbon tetrachloride |
| CCNCyr61 | Cysteine-rich protein 61 |
| CLD | Chronic liver disease |
| DAA | Direct antiviral agents |
| DOX | Dual oxidase |
| ECM | Extracellular matrix |
| EGF | Epidermal growth factor |
| EMT | Epithelial mesenchymal transition |
| ERAS | Embryonic stem cell-expressed RAS |
| ERK | Extracellular signal-regulated kinase |
| ET-1 | Endothelin-1 |
| FTS | Farnesylthiosalicylic acid |
| FXR | Farnesoid X receptor |
| GATA4 | GATA binding protein 4 |
| GIV | Gα-interacting vesicle-associated protein |
| GPCRs | G-protein-coupled receptors |
| GRB2 | Growth factor receptor-bound protein 2 |
| HCB | Hepatitis virus C |
| HCC | Hepatocellular carcinoma |
| HCV | Hepatitis virus C |
| HDAC | Histone deacetylase |
| HNE | 4-hydroxy-nonenal |
| HPC | Hepatic progenitor cells |
| HSC | Hepatic stellate cells |
| IKK | IKB kinase |
| KC | Kupffer cell |
| KLF6/KLF2 | Kruppel-like factors |
| LXR | Liver X receptor |
| MAPK | Mitogen-activated protein kinase |
| MCD | Methionine-choline-deficient |
| MFs | Myofibroblasts |
| MMPs | Metalloproteinases |
| MMT | Mesothelial mesenchymal transition |
| MRTF-A | Myocardin-related transcription factor A |
| NAFLD | Non-alcoholic fatty liver disease |
| NF-κB | Nuclear factor κB |
| NOX | NADPH-oxidase |
| NR4A1 | Nuclear receptor subfamily 4 group A member 1 |
| PARP1 | PolyADP ribose-polymerase 1 |
| PBC | Primary biliary cholangitis |
| PDGF | Plateled-derived growth factor |
| PI3K | Phosphoinositide-3-kinase |
| PKB | Protein kinase B |
| PKC | Protein kinase C |
| PSC | Primary sclerosing cholangitis |
| PTF | Pentoxifillyne |
| REV-ERBα | Nuclear receptor subfamily 1 group D member 1 |
| RKIP | Raf kinase inhibitor protein |
| ROS | Reactive oxygen species |
| SEC | Sinusoidal endothelial cells |
| Sir2 | Silent information regulator 2 |
| SIRT2 | Sirtuin 2 |
| SOS | Son of Sevenless |
| SOX9 | Sex-determining region Y-box 9 |
| TGFα | Transforming growth factor α |
| TGFβ1 | Transforming growth factor- β1 |
| TIMPs | Tissue inhibitor of MMPs |
| TLRs | Toll-like receptors |
| TNF | Tumor necrosis factor |
| Tpl2 | Tumor progression locus 2 |
| VDR | Vitamin D receptor |
| VEGF-A | Vascular endothelial growth factor A |
| VEGFR-2 | VEGF receptor- 2 |
| WD | Wilson’s disease |
| YAP | Yes associated protein |
References
- Trautwein, C.; Friedman, S.L.; Schuppan, D.; Pinzani, M. Hepatic fibrosis: Concept to treatment. J. Hepatol. 2015, 62 (Suppl. 1), S15–S24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.A.; Wallace, M.C.; Friedman, S.L. Pathobiology of liver fibrosis: A translational success story. Gut 2015, 64, 830–841. [Google Scholar] [CrossRef] [PubMed]
- Seki, E.; Schwabe, R.F. Hepatic inflammation and fibrosis: Functional links and key pathways. Hepatology 2015, 61, 1066–1079. [Google Scholar] [CrossRef] [PubMed]
- Koyama, Y.; Brenner, D.A. Liver inflammation and fibrosis. J. Clin. Investig. 2017, 127, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Higashi, T.; Friedman, S.L.; Hoshida, Y. Hepatic stellate cells as key target in liver fibrosis. Adv. Drug Deliv. Rev. 2017, 121, 27–42. [Google Scholar] [CrossRef]
- Cannito, S.; Novo, E.; Parola, M. Therapeutic pro-fibrogenic signaling pathways in fibroblasts. Adv. Drug Deliv. Rev. 2017, 121, 57–84. [Google Scholar] [CrossRef]
- Parola, M.; Pinzani, M. Liver fibrosis: Pathophysiology, pathogenetic targets and clinical issues. Mol. Asp. Med. 2019, 65, 37–55. [Google Scholar] [CrossRef] [PubMed]
- Bocca, C.; Novo, E.; Miglietta, A.; Parola, M. Angiogenesis and Fibrogenesis in Chronic Liver Diseases. Cell Mol. Gastroenterol. Hepatol. 2015, 1, 477–488. [Google Scholar] [CrossRef] [Green Version]
- Lemoinne, S.; Cadoret, A.; El Mourabit, H.; Thabut, D.; Housset, C. Origins and functions of liver myofibroblasts. Biochim. Biophys. Acta 2013, 1832, 948–954. [Google Scholar] [CrossRef] [Green Version]
- Rosselli, M.; MacNaughtan, J.; Jalan, R.; Pinzani, M. Beyond scoring: A modern interpretation of disease progression in chronic liver disease. Gut 2013, 62, 1234–1241. [Google Scholar] [CrossRef]
- El-Serag, H.B. Hepatocellular carcinoma. N. Engl. J. Med. 2011, 365, 1118–1127. [Google Scholar] [CrossRef]
- McGlynn, K.A.; Petrick, J.L.; London, W.T. Global epidemiology of hepatocellular carcinoma: An emphasis on demographic and regional variability. Clin. Liver Dis. 2015, 19, 223–238. [Google Scholar] [CrossRef]
- Byass, P. The global burden of liver disease: A challenge for methods and for public health. BMC Med. 2014, 12, 159. [Google Scholar] [CrossRef] [PubMed]
- Marcellin, P.; Kutala, B.K. Liver diseases: A major, neglected global public health problem requiring urgent actions and large-scale screening. Liver Int. 2018, 38 (Suppl. 1), 2–6. [Google Scholar] [CrossRef] [Green Version]
- Thrift, A.P.; El-Serag, H.B.; Kanwal, F. Global epidemiology and burden of HCV infection and HCV-related disease. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 122–132. [Google Scholar] [CrossRef]
- Arndtz, K.; Hirschfield, G.M. The Pathogenesis of Autoimmune Liver Disease. Dig. Dis. 2016, 34, 327–333. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.; Anstee, Q.M.; Marietti, M.; Hardy, T.; Henry, L.; Eslam, M.; George, J.; Bugianesi, E. Global burden of NAFLD and NASH: Trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 11–20. [Google Scholar] [CrossRef]
- Weiskirchen, R.; Weiskirchen, S.; Tacke, F. Organ and tissue fibrosis: Molecular signals, cellular mechanisms and translational implications. Mol. Asp. Med. 2019, 65, 2–15. [Google Scholar] [CrossRef] [PubMed]
- Böttcher, K.; Pinzani, M. Pathophysiology of liver fibrosis and the methodological barriers to the development of anti-fibrogenic agents. Adv. Drug Del. Rev. 2017, 121, 3–8. [Google Scholar] [CrossRef]
- Pellicoro, A.; Ramachandran, P.; Iredale, J.P.; Fallowfield, J.A. Liver fibrosis and repair: Immune regulation of wound healing in a solid organ. Nat. Rev. Immunol. 2014, 14, 181–194. [Google Scholar] [CrossRef]
- Friedman, S.L. Hepatic stellate cells: Protean, multifunctional, and enigmatic cells of the liver. Physiol. Rev. 2008, 88, 125–172. [Google Scholar] [CrossRef]
- Tsuchida, T.; Friedman, S.L. Mechanisms of hepatic stellate cell activation. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 397–411. [Google Scholar] [CrossRef] [PubMed]
- Wells, R.G.; Schwabe, R.F. Origin and function of myofibroblasts in the liver. Semin. Liver Dis. 2015, 35, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Forbes, S.J.; Parola, M. Liver fibrogenic cells. Best Pract. Res. Clin. Gastroenterol. 2011, 25, 207–217. [Google Scholar] [CrossRef] [PubMed]
- Xie, G.; Diehl, A.M. Evidence for and against epithelial-to-mesenchymal transition in the liver. Am. J. Physiol. Gastrointest. Liver Physiol. 2013, 305, G881–G890. [Google Scholar] [CrossRef] [PubMed]
- Munker, S.; Wu, Y.L.; Ding, H.G.; Liebe, R.; Weng, H.L. Can a fibrotic liver afford epithelial mesenchymal transition? World J. Gastroenterol. 2017, 23, 4661–4668. [Google Scholar] [CrossRef]
- Li, Y.; Wang, J.; Asahina, K. Mesothelial cells give rise to hepatic stellate cells and myofibroblasts via mesothelial-mesenchymal transition in liver injury. Proc. Natl. Acad. Sci. USA 2013, 110, 2324–2329. [Google Scholar] [CrossRef]
- Schaul, Y.D.; Seger, R. The MEK/ERK cascade: From signaling specificity to diverse functions. Biochim. Biophys. Acta 2007, 1773, 1213–1226. [Google Scholar] [CrossRef] [Green Version]
- Raman, M.; Chen, W.; Cobb, M.H. Differential regulation and properties of MAPKs. Oncogene 2007, 26, 3100–3112. [Google Scholar] [CrossRef] [Green Version]
- Pimienta, G.; Pascual, J. Canonical and alternative MAPK signaling. Cell Cycle 2007, 6, 2628–2632. [Google Scholar] [CrossRef]
- Plotnikov, A.; Zehorai, E.; Procaccia, S.; Seger, R. The MAPK cascades: Signaling components, nuclear roles and mechanisms of nuclear translocation. Biochim. Biophys. Acta 2011, 1813, 1619–1633. [Google Scholar] [CrossRef] [Green Version]
- Von Kriegsheim, A.; Baiocchi, D.; Birtwistle, M.; Sumpton, D.; Bienvenut, W.; Morrice, N.; Yamada, K.; Lamond, A.; Kalna, G.; Orton, R.; et al. Cell fate decisions are specified by the dynamic ERK interactome. Nat. Cell Biol. 2009, 11, 1458–1464. [Google Scholar] [CrossRef] [Green Version]
- Buscà, R.; Pouysségur, J.; Lenormand, P. ERK1 and ERK2 Map Kinases: Specific roles or functional redundancy? Front. Cell Dev. Biol. 2016, 4, 53. [Google Scholar] [CrossRef]
- Seger, R.; Krebs, E.G. The MAPK signaling cascade. FASEB J. 1995, 9, 726–735. [Google Scholar] [CrossRef]
- Hu, S.; Xie, Z.; Onishi, A.; Yu, X.; Jiang, L.; Lin, J.; Rho, H.S.; Woodard, C.; Wang, H.; Jeong, J.S.; et al. Profiling the human protein-DNA interactome reveals ERK2 as a transcriptional repressor of interferon signaling. Cell 2009, 139, 610–622. [Google Scholar] [CrossRef]
- Yoon, S.; Seger, R. The extracellular signal-regulated kinase: Multiple substrates regulate diverse cellular functions. Growth Factors 2006, 24, 21–44. [Google Scholar] [CrossRef]
- Kocabayoglu, P.; Lade, A.; Lee, Y.A.; Dragomir, A.C.; Sun, X.; Fiel, M.I.; Thung, S.; Aloman, C.; Soriano, P.; Hoshida, Y.; et al. β-PDGF receptor expressed by hepatic stellate cells regulates fibrosis in murine liver injury, but not carcinogenesis. J. Hepatol. 2015, 63, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Lemmon, M.A.; Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 2010, 141, 1117–1134. [Google Scholar] [CrossRef]
- Regad, T. Targeting RTK Signaling Pathways in Cancer. Cancers 2015, 7, 1758–1784. [Google Scholar] [CrossRef]
- Novo, E.; Marra, F.; Zamara, E.; Valfrè di Bonzo, L.; Monitillo, L.; Cannito, S.; Petrai, I.; Mazzocca, A.; Bonacchi, A.; De Franco, R.S.; et al. Overexpression of Bcl-2 by activated human hepatic stellate cells: Resistance to apoptosis as a mechanism of progressive hepatic fibrogenesis in humans. Gut 2006, 55, 1174–1182. [Google Scholar] [CrossRef]
- Novo, E.; Marra, F.; Zamara, E.; Valfrè di Bonzo, L.; Caligiuri, A.; Cannito, S.; Antonaci, C.; Colombatto, S.; Pinzani, M.; Parola, M. Dose dependent and divergent effects of superoxide anion on cell death, proliferation, and migration of activated human hepatic stellate cells. Gut 2006, 55, 90–97. [Google Scholar] [CrossRef] [PubMed]
- Parola, M.; Pinzani, M.; Casini, A.; Albano, E.; Poli, G.; Gentilini, A.; Gentilini, P.; Dianzani, M.U. Stimulation of lipid peroxidation or 4-hydroxynonenal treatment increases procollagen alpha 1 (I) gene expression in human liver fat-storing cells. Biochem. Biophys. Res. Commun. 1993, 194, 1044–1050. [Google Scholar] [CrossRef]
- Zamara, E.; Novo, E.; Marra, F.; Gentilini, A.; Romanelli, R.G.; Caligiuri, A.; Robino, G.; Tamagno, E.; Aragno, M.; Danni, O.; et al. 4-Hydroxynonenal as a selective pro-fibrogenic stimulus for activated human hepatic stellate cells. J. Hepatol. 2004, 40, 60–68. [Google Scholar] [CrossRef]
- Parola, M.; Robino, G.; Marra, F.; Pinzani, M.; Bellomo, G.; Leonarduzzi, G.; Chiarugi, P.; Camandola, S.; Poli, G.; Waeg, G.; et al. HNE interacts directly with JNK isoforms in human hepatic stellate cells. J. Clin. Investig. 1998, 102, 1942–1950. [Google Scholar] [CrossRef]
- Wynn, T.A. Cellular and molecular mechanisms of fibrosis. J. Pathol. 2008, 214, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Novo, E.; Busletta, C.; Bonzo, L.V.; Povero, D.; Paternostro, C.; Mareschi, K.; Ferrero, I.; David, E.; Bertolani, C.; Caligiuri, A.; et al. Intracellular reactive oxygen species are required for directional migration of resident and bone marrow-derived hepatic pro-fibrogenic cells. J. Hepatol. 2011, 54, 964–974. [Google Scholar] [CrossRef]
- Novo, E.; Povero, D.; Busletta, C.; Paternostro, C.; di Bonzo, L.V.; Cannito, S.; Compagnone, A.; Bandino, A.; Marra, F.; Colombatto, S.; et al. The biphasic nature of hypoxia-induced directional migration of activated human hepatic stellate cells. J. Pathol. 2012, 226, 588–597. [Google Scholar] [CrossRef]
- Novo, E.; Cannito, S.; Paternostro, C.; Bocca, C.; Miglietta, A.; Parola, M. Cellular and molecular mechanisms in liver fibrogenesis. Arch. Biochem. Biophys. 2014, 548, 20–37. [Google Scholar] [CrossRef]
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD development and therapeutic strategies. Nat. Med. 2018, 24, 908–922. [Google Scholar] [CrossRef] [PubMed]
- Weiskirchen, R. Hepatoprotective and Anti-fibrotic Agents: It’s Time to Take the Next Step. Front. Pharmacol. 2016, 6, 303. [Google Scholar] [CrossRef]
- Luangmonkong, T.; Suriguga, S.; Mutsaers, H.A.M.; Groothuis, G.M.M.; Olinga, P.; Boersema, M. Targeting Oxidative Stress for the Treatment of Liver Fibrosis. Rev. Physiol. Biochem. Pharmacol. 2018, 175, 71–102. [Google Scholar] [CrossRef]
- Barreyro, F.J.; Holod, S.; Finocchietto, P.V.; Camino, A.M.; Aquino, J.B.; Avagnina, A.; Carreras, M.C.; Poderoso, J.J.; Gores, G.J. The pan-caspase inhibitor Emricasan (IDN-6556) decreases liver injury and fibrosis in a murine model of non-alcoholic steatohepatitis. Liver Int. 2015, 35, 953–966. [Google Scholar] [CrossRef]
- Eguchi, A.; Koyama, Y.; Wree, A.; Johnson, C.D.; Nakamura, R.; Povero, D.; Kneiber, D.; Tameda, M.; Contreras, P.; Spada, A.; et al. Emricasan, a pan-caspase inhibitor, improves survival and portal hypertension in a murine model of common bile-duct ligation. J. Mol. Med. 2018, 96, 575–583. [Google Scholar] [CrossRef] [PubMed]
- Pockros, P.J.; Schiff, E.R.; Shiffman, M.L.; McHutchison, J.G.; Gish, R.G.; Afdhal, N.H.; Makhviladze, M.; Huyghe, M.; Hecht, D.; Oltersdorf, T.; et al. Oral IDN-6556, an antiapoptotic caspase inhibitor, may lower aminotransferase activity in patients with chronic hepatitis C. Hepatology 2007, 46, 324–329. [Google Scholar] [CrossRef] [PubMed]
- Shiffman, M.L.; Pockros, P.; McHutchison, J.G.; Schiff, E.R.; Morris, M.; Burgess, G. Clinical trial: The efficacy and safety of oral PF-03491390, a pancaspase inhibitor - a randomized placebo-controlled study in patients with chronic hepatitis C. Aliment. Pharmacol. Ther. 2010, 31, 969–978. [Google Scholar] [CrossRef]
- Tacke, F. Targeting hepatic macrophages to treat liver diseases. J. Hepatol. 2017, 66, 1300–1312. [Google Scholar] [CrossRef]
- Krenkel, O.; Tacke, F. Liver macrophages in tissue homeostasis and disease. Nat. Rev. Immunol. 2017, 17, 306–321. [Google Scholar] [CrossRef] [PubMed]
- Loomba, R.; Lawitz, E.; Mantry, P.S.; Jayakumar, S.; Caldwell, S.H.; Arnold, H.; Diehl, A.M.; Djedjos, C.S.; Han, L.; Myers, R.P.; et al. The ASK1 inhibitor selonsertib in patients with nonalcoholic steatohepatitis: A randomized, phase 2 trial. Hepatology 2018, 67, 549–559. [Google Scholar] [CrossRef]
- Krenkel, O.; Puengel, T.; Govaere, O.; Abdallah, A.T.; Mossanen, J.C.; Kohlhepp, M.; Liepelt, A.; Lefebvre, E.; Luedde, T.; Hellerbrand, C.; et al. Therapeutic inhibition of inflammatory monocyte recruitment reduces steatohepatitis and liver fibrosis. Hepatology 2018, 67, 1270–1283. [Google Scholar] [CrossRef]
- Friedman, S.L.; Ratziu, V.; Harrison, S.A.; Abdelmalek, M.F.; Aithal, G.P.; Caballeria, J.; Francque, S.; Farrell, G.; Kowdley, K.V.; Craxi, A.; et al. A randomized, placebo-controlled trial of cenicriviroc for treatment of nonalcoholic steatohepatitis with fibrosis. Hepatology 2018, 67, 1754–1767. [Google Scholar] [CrossRef]
- Coll, M.; EI Taghdouini, A.; Perea, L.; Mannaerts, I.; Vila-Casadesús, M.; Blaya, D.; Rodrigo-Torres, D.; Affò, S.; Morales-Ibanez, O.; Graupera, I.; et al. Integrative miRNA and Gene Expression Profiling Analysis of Human Quiescent Hepatic Stellate Cells. Sci. Rep. 2015, 5, 11549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marra, F.; Arrighi, M.C.; Fazi, M.; Caligiuri, A.; Pinzani, M.; Romanelli, R.G.; Efsen, E.; Laffi, G.; Gentilini, P. Extracellular signal-regulated kinase activation differentially regulates platelet-derived growth factor′s actions in hepatic stellate cells, and is induced by in vivo liver injury in the rat. Hepatology 1999, 30, 951–958. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.L.; Liu, J.M.; Yang, C.C.; Zheng, Y.L.; Liu, L.; Wang, Z.K.; Jiang, H.Q. Dynamic expression of extracellular signal-regulated kinase in rat liver tissue during hepatic fibrogenesis. World J. Gastroenterol. 2006, 12, 6376–6381. [Google Scholar] [CrossRef]
- Pinzani, M.; Marra, F.; Caligiuri, A.; DeFranco, R.; Gentilini, A.; Failli, P.; Gentilini, P. Inhibition by pentoxifylline of extracellular signal-regulated kinase activation by platelet-derived growth factor in hepatic stellate cells. Br. J. Pharmacol. 1996, 119, 1117–1124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peterson, T. Pentoxifylline prevents fibrosis in an animal model and inhibits platelet-derived growth factor-driven proliferation of fibroblasts. Hepatology 1993, 17, 486–493. [Google Scholar] [CrossRef] [PubMed]
- Romanelli, R.G.; Caligiuri, A.; Carloni, V.; De Franco, R.; Montalto, P.; Ceni, E.; Casini, A.; Gentilini, P.; Pinzani, M. Effect of pentoxifylline on the degradation of procollagen type I produced by human hepatic stellate cells in response to transforming growth factor-beta 1. Br. J. Pharmacol. 1997, 122, 1047–1054. [Google Scholar] [CrossRef]
- Rotman, Y.; Sanyal, A.J. Current and upcoming pharmacotherapy for non-alcoholic fatty liver disease. Gut 2017, 66, 180–190. [Google Scholar] [CrossRef] [PubMed]
- Parola, M.; Robino, G. Oxidative stress-related molecules and liver fibrosis. J. Hepatol. 2001, 35, 297–306. [Google Scholar] [CrossRef]
- Novo, E.; Parola, M. Redox mechanisms in hepatic chronic wound healing and fibrogenesis. Fibrogenesis Tissue Repair 2008, 13, 5. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.Y.; Rhim, T.; Choi, I.; Kim, S.S. N-acetylcysteine induces cell cycle arrest in hepatic stellate cells through its reducing activity. J. Biol. Chem. 2001, 276, 40591–40598. [Google Scholar] [CrossRef]
- Trappoliere, M.; Caligiuri, A.; Schmid, M.; Bertolani, C.; Failli, P.; Vizzutti, F.; Novo, E.; di Manzano, C.; Marra, F.; Loguercio, C.; et al. Silybin, a component of sylimarin, exerts anti-inflammatory and anti-fibrogenic effects on human hepatic stellate cells. J. Hepatol. 2009, 50, 1102–1111. [Google Scholar] [CrossRef]
- Zhou, Y.; Zheng, S.; Lin, J.; Zhang, Q.J.; Chen, A. The interruption of the PDGF and EGF signaling pathways by curcumin stimulates gene expression of PPARgamma in rat activated hepatic stellate cell in vitro. Lab. Investig. 2007, 87, 488–498. [Google Scholar] [CrossRef]
- Yeung, K.; Seitz, T.; Li, S.; Janosch, P.; McFerran, B.; Kaiser, C.; Fee, F.; Katsanakis, K.D.; Rose, D.W.; Mischak, H.; et al. Suppression of Raf-1 kinase activity and MAP kinase signalling by RKIP. Nature 1999, 401, 173–177. [Google Scholar] [CrossRef] [Green Version]
- Lorenz, K.; Lohse, M.J.; Quitterer, U. Protein kinase C switches the Raf kinase inhibitor from Raf-1 to GRK-2. Nature 2003, 426, 574–5799. [Google Scholar] [CrossRef] [PubMed]
- Corbit, K.C.; Trakul, N.; Eves, E.M.; Diaz, B.; Marshall, M.; Rosner, M.R. Activation of Raf-1 signaling by protein kinase C through a mechanism involving Raf kinase inhibitory protein. J. Biol. Chem. 2003, 278, 13061–13068. [Google Scholar] [CrossRef]
- Ma, J.; Li, F.; Liu, L.; Cui, D.; Wu, X.; Jiang, X.; Jiang, H. Raf kinase inhibitor protein inhibits cell proliferation but promotes cell migration in rat hepatic stellate cells. Liver Int. 2009, 29, 567–574. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Liang, C.; Wei, L.; Nie, J.; Lu, S.; Lu, C.; Zhuo, L.; Lu, Z.; Lin, X. Raf Kinase Inhibitory Protein Down-Expression Exacerbates Hepatic Fibrosis In Vivo and In Vitro. Cell Physiol. Biochem. 2016, 40, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Bai, F.; Nie, J.; Lu, S.; Lu, C.; Zhu, X.; Wei, J.; Lu, Z.; Huang, Q. Didymin Alleviates Hepatic Fibrosis Through Inhibiting ERK and PI3K/Akt Pathways via Regulation of Raf Kinase Inhibitor Protein. Cell Physiol. Biochem. 2016, 40, 1422–1432. [Google Scholar] [CrossRef]
- Vougioukalaki, M.; Kanellis, D.C.; Gkouskou, K.; Eliopoulos, A.G. Tpl2 kinase signal transduction in inflammation and cancer. Cancer Lett. 2011, 304, 80–89. [Google Scholar] [CrossRef] [PubMed]
- Salmeron, A.; Ahmad, T.B.; Carlile, G.W.; Pappin, D.; Narsimhan, R.P.; Ley, S.C. Activation of MEK-1 and SEK-1 by Tpl-2 proto-oncoprotein, a novel MAP kinase kinase kinase. EMBO J. 1996, 15, 817–826. [Google Scholar] [CrossRef]
- Perugorria, M.J.; Murphy, L.B.; Fullard, N.; Chakraborty, J.B.; Vyrla, D.; Wilson, C.L.; Oakley, F.; Mann, J.; Mann, D.A. Tumor progression locus 2/Cot is required for activation of extracellular regulated kinase in liver injury and toll-like receptor-induced TIMP-1 gene transcription in hepatic stellate cells in mice. Hepatology 2013, 57, 1238–1249. [Google Scholar] [CrossRef] [PubMed]
- Nakhaei-Rad, S.; Nakhaeizadeh, H.; Götze, S.; Kordes, C.; Sawitza, I.; Hoffmann, M.J.; Franke, M.; Schulz, W.A.; Scheller, J.; Piekorz, R.P.; et al. The role of rmbryonic stem cell-expressed RAS (ERAS) in the maintenance of quiescent hepatic stellate cells. J. Biol. Chem. 2016, 291, 8399–8413. [Google Scholar] [CrossRef] [PubMed]
- Reif, S.; Weisz, B.; Gana-Weisz, M.; Aeed, H.; Zaidel, Y.; Avni, Y.; Kloog, Y.; Bruck, R. The Ras antagonist, farnesylthiosalicylic acid (FTS), inhibits experimentally-induced liver cirrhosis in rats. J. Hepatol. 1999, 31, 1053–1061. [Google Scholar] [CrossRef]
- Reif, S.; Aeed, H.; Shilo, Y.; Reich, R.; Kloog, Y.; Kweon, Y.O.; Bruck, R. Treatment of thioacetamide-induced liver cirrhosis by the Ras antagonist, farnesylthiosalicylic acid. J. Hepatol. 2004, 41, 235–241. [Google Scholar] [CrossRef] [PubMed]
- Zhong, W.; Shen, W.F.; Ning, B.F.; Hu, P.F.; Lin, Y.; Yue, H.Y.; Yin, C.; Hou, J.L.; Chen, Y.X.; Zhang, J.P.; et al. Inhibition of extracellular signal-regulated kinase 1 by adenovirus mediated small interfering RNA attenuates hepatic fibrosis in rats. Hepatology 2009, 50, 1524–1536. [Google Scholar] [CrossRef]
- Van Beneden, K.; Mannaerts, I.; Pauwels, M.; Van den Branden, C.; van Grunsven, L.A. HDAC inhibitors in experimental liver and kidney fibrosis. Fibrogenesis Tissue Repair 2013, 6, 1. [Google Scholar] [CrossRef]
- Arteaga, M.; Shang, N.; Ding, X.; Yong, S.; Cotler, S.J.; Denning, M.F.; Shimamura, T.; Breslin, P.; Lüscher, B.; Qiu, W. Inhibition of SIRT2 suppresses hepatic fibrosis. Am. J. Physiol. Gastrointest. Liver Physiol. 2016, 310, G1155–G1168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borkham-Kamphorst, E.; Weiskirchen, R. The PDGF system and its antagonists in liver fibrosis. Cytokine Growth Factor Rev. 2016, 28, 53–61. [Google Scholar] [CrossRef]
- Wong, L.; Yamasaki, G.; Johnson, R.J.; Friedman, S.L. Induction of beta-platelet-derived growth factor receptor in rat hepatic lipocytes during cellular activation in vivo and in culture. J. Clin. Investig. 1994, 94, 1563–1569. [Google Scholar] [CrossRef]
- Pinzani, M.; Gentilini, A.; Caligiuri, A.; De Franco, R.; Pellegrini, G.; Milani, S.; Marra, F.; Gentilini, P. Transforming growth factor-beta 1 regulates platelet-derived growth factor receptor beta subunit in human liver fat-storing cells. Hepatology 1995, 21, 232–239. [Google Scholar] [CrossRef]
- Borkham-Kamphorst, E.; van Roeyen, C.R.; Ostendorf, T.; Floege, J.; Gressner, A.M.; Weiskirchen, R. Pro-fibrogenic potential of PDGF-D in liver fibrosis. J. Hepatol. 2007, 46, 1064–1074. [Google Scholar] [CrossRef] [PubMed]
- Borkham-Kamphorst, E.; Meurer, S.K.; Van de Leur, E.; Haas, U.; Tihaa, L.; Weiskirchen, R. PDGF-D signaling in portal myofibroblasts and hepatic stellate cells proves identical to PDGF-B via both PDGF receptor type α and β. Cell Signal. 2015, 27, 1305–1314. [Google Scholar] [CrossRef] [PubMed]
- Borkham-Kamphorst, E.; Stoll, D.; Gressner, A.M.; Weiskirchen, R. Antisense strategy against PDGF B-chain proves effective in preventing experimental liver fibrogenesis. Biochem. Biophys. Res. Commun. 2004, 321, 413–423. [Google Scholar] [CrossRef] [PubMed]
- Borkham-Kamphorst, E.; Herrmann, J.; Stoll, D.; Treptau, J.; Gressner, A.M.; Weiskirchen, R. Dominant-negative soluble PDGF-beta receptor inhibits hepatic stellate cell activation and attenuates liver fibrosis. Lab Investig. 2004, 84, 766–777. [Google Scholar] [CrossRef] [PubMed]
- Yoshiji, H.; Noguchi, R.; Kuriyama, S.; Ikenaka, Y.; Yoshii, J.; Yanase, K.; Namisaki, T.; Kitade, M.; Masaki, T.; Fukui, H. Imatinib mesylate (STI-571) attenuates liver fibrosis development in rats. Am. J. Physiol. Gastrointest Liver Physiol. 2005, 288, G907–G913. [Google Scholar] [CrossRef] [PubMed]
- Neef, M.; Ledermann, M.; Saegesser, H.; Schneider, V.; Widmer, N.; Decosterd, L.A.; Rochat, B.; Reichen, J. Oral imatinib treatment reduces early fibrogenesis but does not prevent progression in the long term. J. Hepatol. 2006, 44, 167–175. [Google Scholar] [CrossRef]
- Wang, Y.; Gao, J.; Zhang, D.; Zhang, J.; Ma, J.; Jiang, H. New insights into the antifibrotic effects of sorafenib on hepatic stellate cells and liver fibrosis. J. Hepatol. 2010, 53, 132–144. [Google Scholar] [CrossRef]
- Shaker, M.E.; Salem, H.A.; Shiha, G.E.; Ibrahim, T.M. Nilotinib counteracts thioacetamide-induced hepatic oxidative stress and attenuates liver fibrosis progression. Fundam. Clin. Pharmacol. 2011, 25, 248–257. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, Z.; Kwong, S.Q.; Lui, E.L.; Friedman, S.L.; Li, F.R.; Lam, R.W.; Zhang, G.C.; Zhang, H.; Ye, T. Inhibition of PDGF, TGF-β, and Abl signaling and reduction of liver fibrosis by the small molecule Bcr-Abl tyrosine kinase antagonist Nilotinib. J. Hepatol. 2011, 55, 612–625. [Google Scholar] [CrossRef]
- Shaker, M.E.; Ghani, A.; Shiha, G.E.; Ibrahim, T.M.; Mehal, W.Z. Nilotinib induces apoptosis and autophagic cell death of activated hepatic stellate cells via inhibition of histone deacetylases. Biochim. Biophys. Acta 2013, 1833, 1992–2003. [Google Scholar] [CrossRef] [Green Version]
- Ogawa, S.; Ochi, T.; Shimada, H.; Inagaki, K.; Fujita, I.; Nii, A.; Moffat, M.A.; Katragadda, M.; Violand, B.N.; Arch, R.H.; et al. Anti-PDGF-B monoclonal antibody reduces liver fibrosis development. Hepatol. Res. 2010, 40, 1128–1141. [Google Scholar] [CrossRef] [PubMed]
- Kuai, J.; Mosyak, L.; Brooks, J.; Cain, M.; Carven, G.J.; Ogawa, S.; Ishino, T.; Tam, M.; Lavallie, E.R.; Yang, Z.; et al. Characterization of binding mode of action of a blocking anti-platelet-derived growth factor (PDGF)-B monoclonal antibody, MOR8457, reveals conformational flexibility and avidity needed for PDGF-BB to bind PDGF receptor-β. Biochemistry 2015, 54, 1918–1929. [Google Scholar] [CrossRef]
- Chen, S.W.; Zhang, X.R.; Wang, C.Z.; Chen, W.Z.; Xie, W.F.; Chen, Y.X. RNA interference targeting the platelet-derived growth factor receptor beta subunit ameliorates experimental hepatic fibrosis in rats. Liver Int. 2008, 28, 1446–1457. [Google Scholar] [CrossRef]
- Chen, S.W.; Chen, Y.X.; Zhang, X.R.; Qian, H.; Chen, W.Z.; Xie, W.F. Targeted inhibition of platelet-derived growth factor receptor-beta subunit in hepatic stellate cells ameliorates hepatic fibrosis in rats. Gene Ther. 2008, 15, 1424–1435. [Google Scholar] [CrossRef]
- Camino, A.M.; Atorrasagasti, C.; Maccio, D.; Prada, F.; Salvatierra, E.; Rizzo, M.; Alaniz, L.; Aquino, J.B.; Podhajcer, O.L.; Silva, M.; et al. Adenovirus-mediated inhibition of SPARC attenuates liver fibrosis in rats. J. Gene Med. 2008, 10, 993–1004. [Google Scholar] [CrossRef]
- Atorrasagasti, C.; Aquino, J.B.; Hofman, L.; Alaniz, L.; Malvicini, M.; Garcia, M.; Benedetti, L.; Friedman, S.L.; Podhajcer, O.; Mazzolini, G. SPARC downregulation attenuates the profibrogenic response of hepatic stellate cells induced by TGF-β1 and PDGF. Am. J. Physiol. Gastrointest Liver Physiol. 2011, 300, G739–G748. [Google Scholar] [CrossRef]
- Paik, Y.H.; Kim, J.; Aoyama, T.; De Minicis, S.; Bataller, R.; Brenner, D.A. Role of NADPH oxidases in liver fibrosis. Antioxid. Redox Signal. 2014, 20, 2854–2872. [Google Scholar] [CrossRef] [PubMed]
- Liang, S.; Kisseleva, T.; Brenner, D.A. The role of NADPH oxidases (NOXs) in liver fibrosis and the activation of myofibroblasts. Front. Physiol. 2016, 7, 17. [Google Scholar] [CrossRef] [PubMed]
- Bedard, K.; Krause, K.H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef]
- Aoyama, T.; Paik, Y.H.; Watanabe, S.; Laleu, B.; Gaggini, F.; Fioraso-Cartier, L.; Molango, S.; Heitz, F.; Merlot, C.; Szyndralewiez, C.; et al. Nicotinamide adenine dinucleotide phosphate oxidase in experimental liver fibrosis: GKT137831 as a novel potential therapeutic agent. Hepatology 2012, 56, 2316–2327. [Google Scholar] [CrossRef] [Green Version]
- Bettaieb, A.; Jiang, J.X.; Sasaki, Y.; Chao, T.I.; Kiss, Z.; Chen, X.; Tian, J.; Katsuyama, M.; Yabe-Nishimura, C.; Xi, Y.; et al. Hepatocyte Nicotinamide Adenine Dinucleotide Phosphate Reduced Oxidase 4 Regulates Stress Signaling, Fibrosis, and Insulin Sensitivity During Development of Steatohepatitis in Mice. Gastroenterology 2015, 149, 468–480. [Google Scholar] [CrossRef] [PubMed]
- Lan, T.; Kisseleva, T.; Brenner, D.A. Deficiency of NOX1 or NOX4 Prevents Liver Inflammation and Fibrosis in Mice through Inhibition of Hepatic Stellate Cell Activation. PLoS ONE 2015, 10, e0129743. [Google Scholar] [CrossRef]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Foglia, B.; Cannito, S.; Bocca, C.; Parola, M.; Novo, E. ERK Pathway in Activated, Myofibroblast-Like, Hepatic Stellate Cells: A Critical Signaling Crossroad Sustaining Liver Fibrosis. Int. J. Mol. Sci. 2019, 20, 2700. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20112700
Foglia B, Cannito S, Bocca C, Parola M, Novo E. ERK Pathway in Activated, Myofibroblast-Like, Hepatic Stellate Cells: A Critical Signaling Crossroad Sustaining Liver Fibrosis. International Journal of Molecular Sciences. 2019; 20(11):2700. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20112700
Chicago/Turabian StyleFoglia, Beatrice, Stefania Cannito, Claudia Bocca, Maurizio Parola, and Erica Novo. 2019. "ERK Pathway in Activated, Myofibroblast-Like, Hepatic Stellate Cells: A Critical Signaling Crossroad Sustaining Liver Fibrosis" International Journal of Molecular Sciences 20, no. 11: 2700. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20112700