IRTKS Promotes Insulin Signaling Transduction through Inhibiting SHIP2 Phosphatase Activity

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
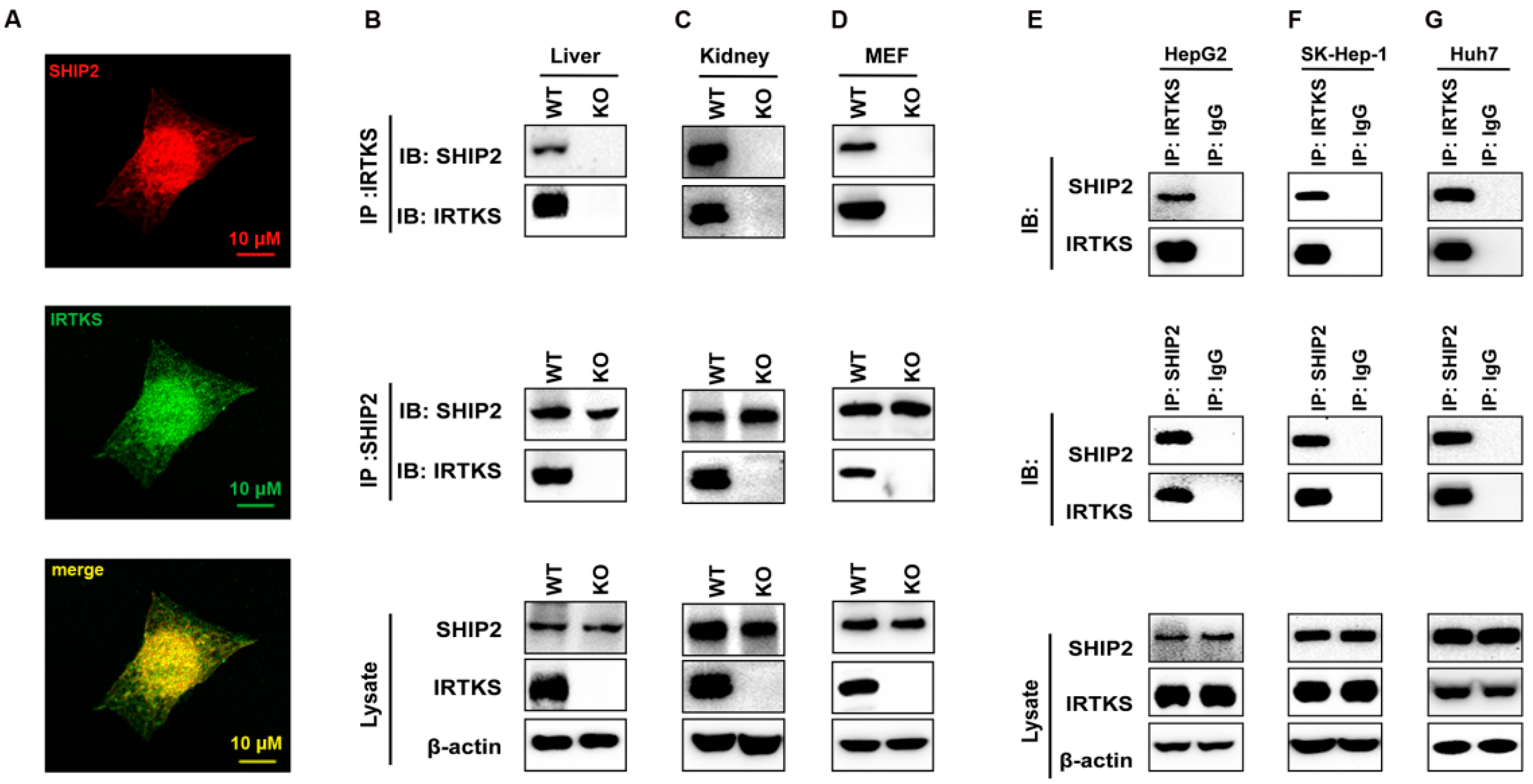
2.1. IRTKS Interacts with SHIP2
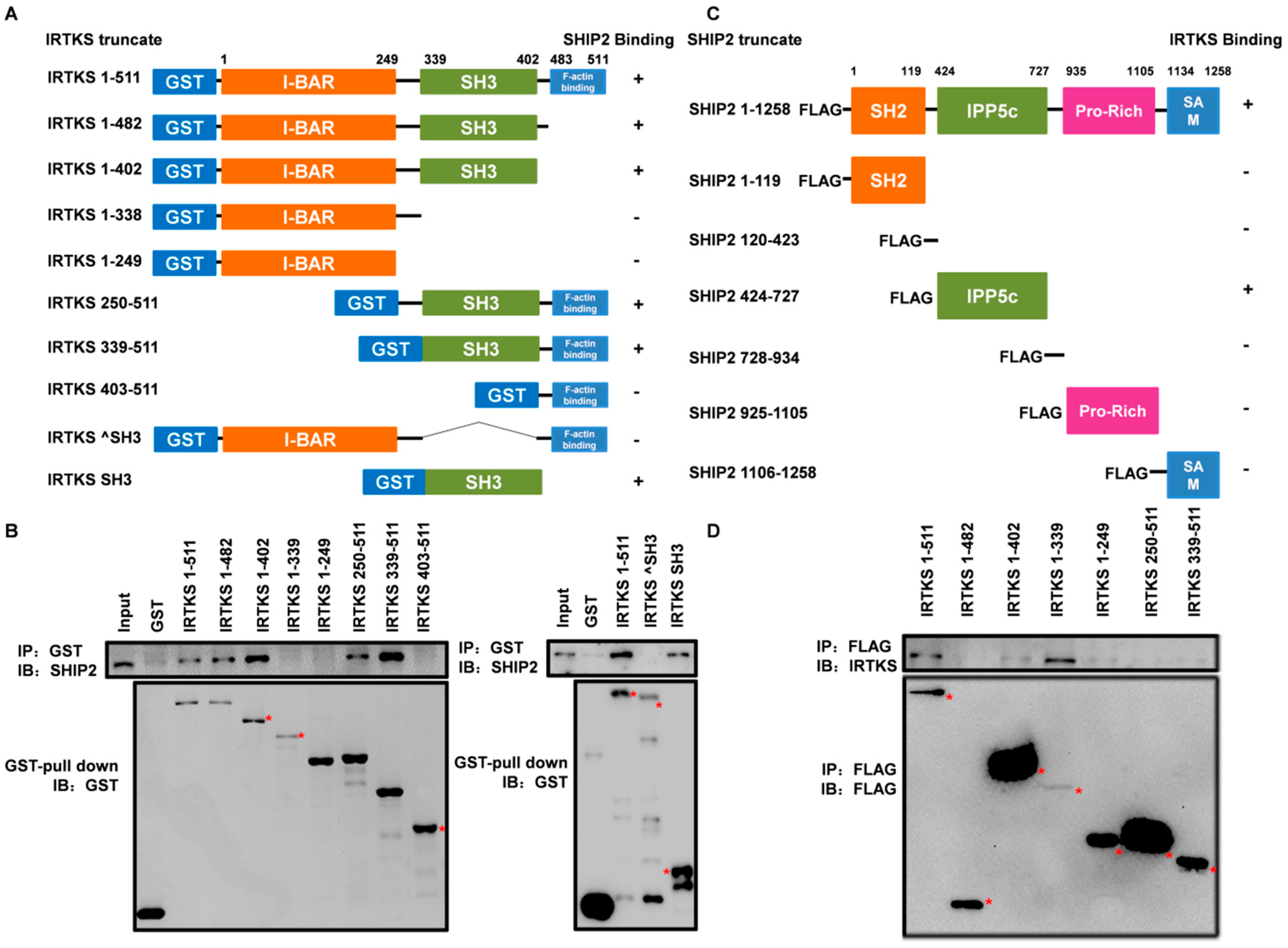
2.2. The Interaction between IRTKS and SHIP2 Is Mediated by the SH3 and IPP5c Domains
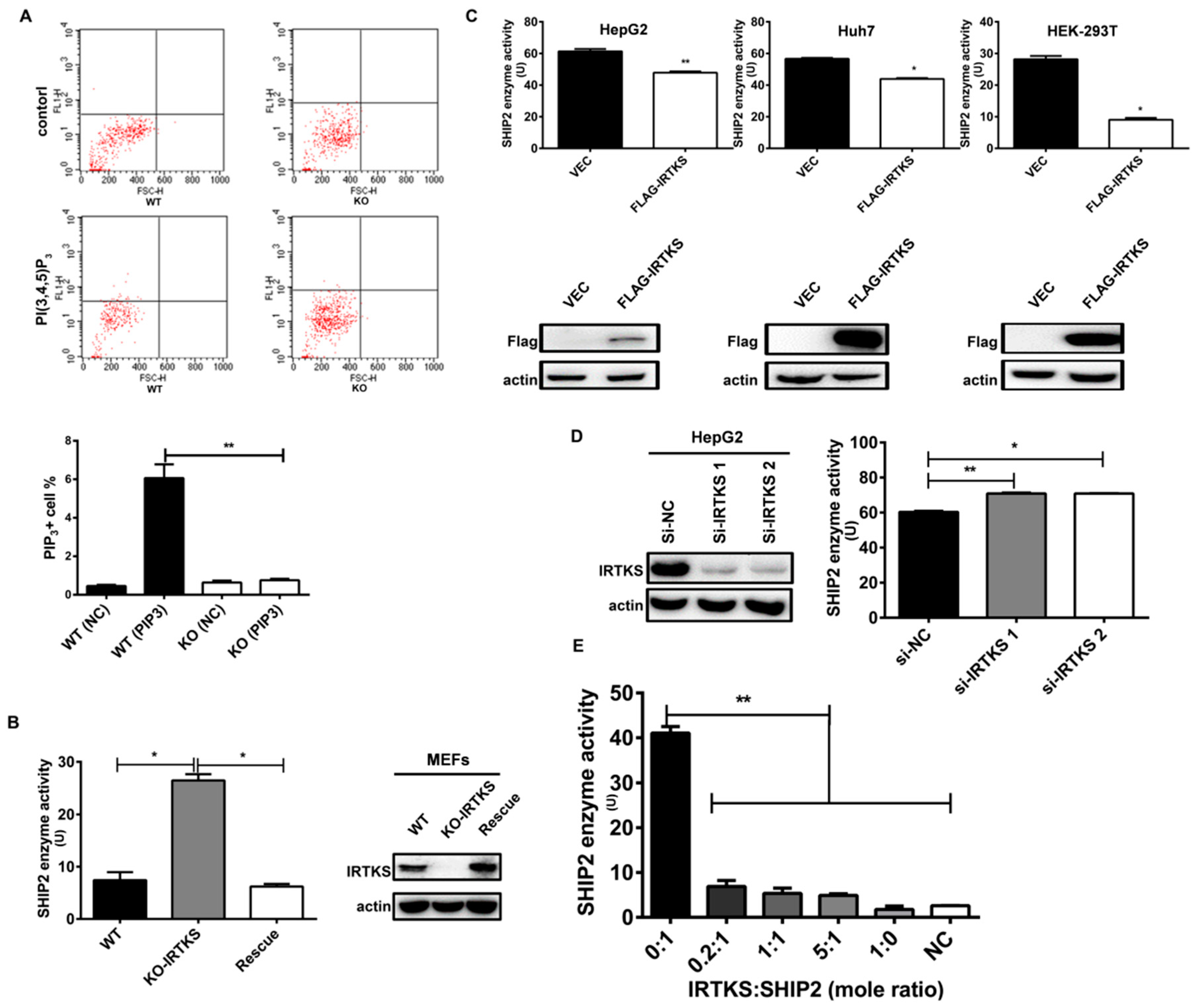
2.3. IRTKS Inhibits SHIP2 Enzyme Activity
2.4. Interaction between IRTKS and SHIP2 Is Dynamically Regulated by Insulin
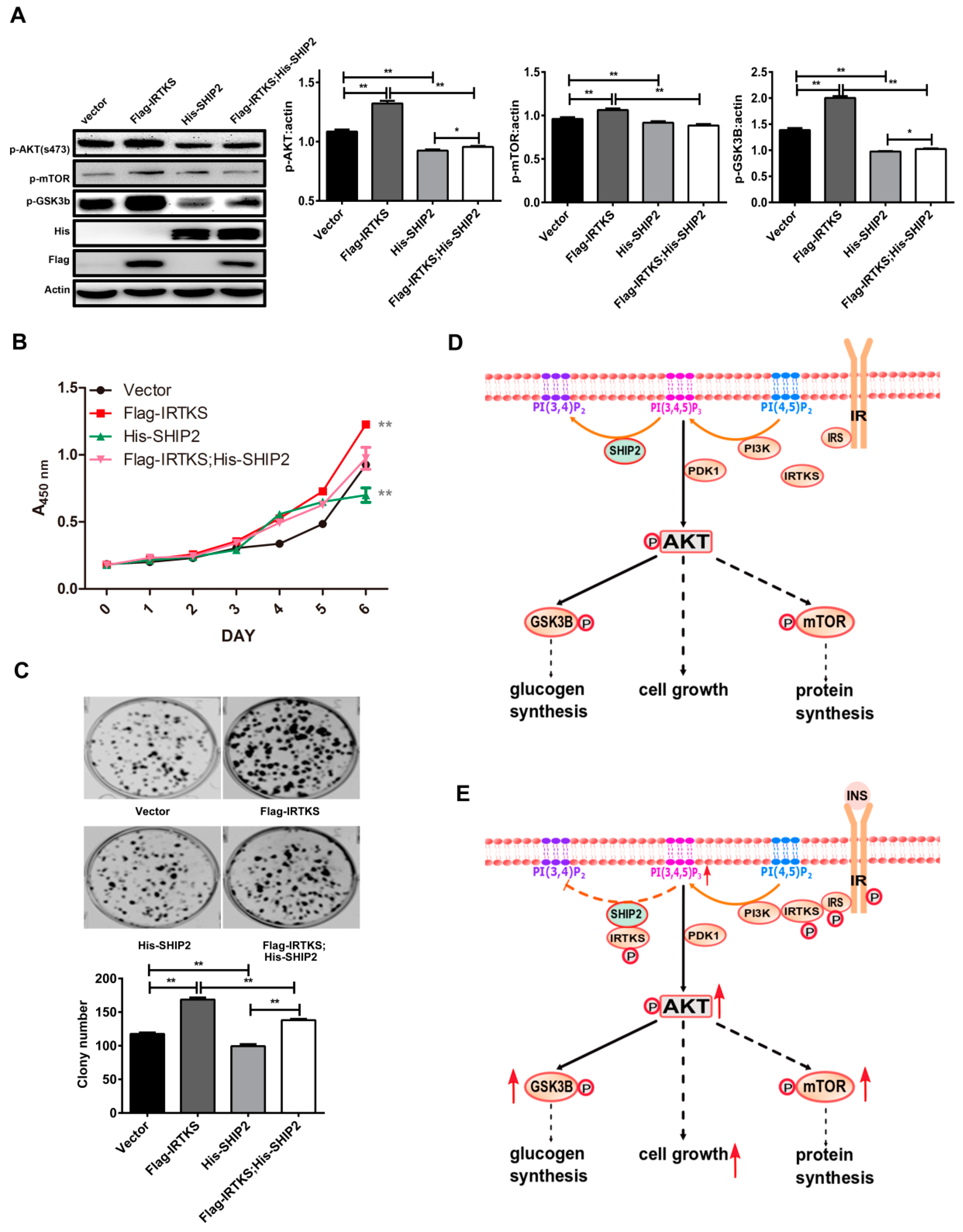
2.5. IRTKS Promotes Insulin Signaling and Cell Proliferation Partially Due to Inhibiting SHIP2 Activity
3. Discussion
4. Materials and Methods
4.1. Cells and Reagents
4.2. siRNA, Plasmids, and Transient Transfection
4.3. Immunoprecipitation Assay
4.4. GST Pull-Down Assay
4.5. Western Blot Assay
4.6. SHIP2 Enzyme Activity Assay
4.7. PIP3 Determinate Assay
4.8. Cell Proliferation Curve
4.9. Cell Colony Formation Assay
4.10. Quantification and Statistical Analyses
Author Contributions
Funding
Conflicts of Interest
References
- Hu, R.M.; Han, Z.G.; Song, H.D.; Peng, Y.D.; Huang, Q.H.; Ren, S.X.; Gu, Y.J.; Huang, C.H.; Li, Y.B.; Jiang, C.L.; et al. Gene expression profiling in the human hypothalamus-pituitary-adrenal axis and full-length cDNA cloning. Proc. Natl. Acad. Sci. USA 2000, 97, 9543–9548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simunovic, M.; Voth, G.A.; Callan-Jones, A.; Bassereau, P. When Physics Takes Over: BAR Proteins and Membrane Curvature. Trends Cell Biol. 2015, 25, 780–792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heung, M.Y.; Visegrady, B.; Futterer, K.; Machesky, L.M. Identification of the insulin-responsive tyrosine phosphorylation sites on IRSp53. Eur. J. Cell Biol. 2008, 87, 699–708. [Google Scholar] [CrossRef] [PubMed]
- Burette, A.C.; Park, H.; Weinberg, R.J. Postsynaptic distribution of IRSp53 in spiny excitatory and inhibitory neurons. J. Comp. Neurol. 2014, 522, 2164–2178. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.Y.; Wang, X.; Cui, X.F.; Li, H.; Zhao, J.; Wu, C.C.; Min, L.; Zhou, Z.; Wan, L.; Wang, Y.P.; et al. IRTKS is correlated with progression and survival time of patients with gastric cancer. Gut 2017. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.Y.; Wang, Y.P.; Wei, B.F.; Yang, J.; Wang, J.Q.; Wu, B.H.; Zhang, Z.Z.; Hou, Y.Y.; Sun, W.M.; Hu, R.M.; et al. Deficiency of IRTKS as an adaptor of insulin receptor leads to insulin resistance. Cell Res. 2013, 23, 1310–1321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorgani-Firuzjaee, S.; Adeli, K.; Meshkani, R. Inhibition of SH2-domain-containing inositol 5-phosphatase (SHIP2) ameliorates palmitate induced-apoptosis through regulating Akt/FOXO1 pathway and ROS production in HepG2 cells. Biochem. Biophys. Res. Commun. 2015, 464, 441–446. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Ge, Y.M.; Xiao, M.M.; Guo, L.M.; Li, Q.; Hao, J.Q.; Da, J.; Hu, W.L.; Zhang, X.D.; Xu, J.; et al. Suppression of SHIP2 contributes to tumorigenesis and proliferation of gastric cancer cells via activation of Akt. J. Gastroenterol. 2016, 51, 230–240. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Scozzaro, S.; Ramos, A.R.; Delcambre, S.; Chevalier, C.; Krejci, P.; Erneux, C. Inhibition of SHIP2 activity inhibits cell migration and could prevent metastasis in breast cancer cells. J. Cell Sci. 2018, 131. [Google Scholar] [CrossRef] [PubMed]
- Manna, P.; Jain, S.K. Decreased hepatic phosphatidylinositol-3,4,5-triphosphate (PIP3) levels and impaired glucose homeostasis in type 1 and type 2 diabetic rats. Cell. Physiol. Biochem. 2012, 30, 1363–1370. [Google Scholar] [CrossRef] [PubMed]
- Arnold, S.E.; Arvanitakis, Z.; Macauley-Rambach, S.L.; Koenig, A.M.; Wang, H.Y.; Ahima, R.S.; Craft, S.; Gandy, S.; Buettner, C.; Stoeckel, L.E.; et al. Brain insulin resistance in type 2 diabetes and Alzheimer disease: Concepts and conundrums. Nat. Rev. Neurol. 2018, 14, 168–181. [Google Scholar] [CrossRef] [PubMed]
- Linkner, J.; Witte, G.; Zhao, H.; Junemann, A.; Nordholz, B.; Runge-Wollmann, P.; Lappalainen, P.; Faix, J. The inverse BAR domain protein IBARa drives membrane remodeling to control osmoregulation, phagocytosis and cytokinesis. J. Cell Sci. 2014, 127, 1279–1292. [Google Scholar] [CrossRef] [PubMed]
- Li, L.S.; Liu, H.Y.; Baxter, S.S.; Gu, N.; Ji, M.; Zhan, X. The SH3 domain distinguishes the role of I-BAR proteins IRTKS and MIM in chemotactic response to serum. Biochem. Biophys. Res. Commun. 2016, 479, 787–792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, M.P.; Erneux, C.; Potter, B.V. SHIP2: Structure, Function and Inhibition. Chembiochem. A Eur. J. Chem. Biol. 2017, 18, 233–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deneubourg, L.; Vanderwinden, J.M.; Erneux, C. Regulation of SHIP2 function through plasma membrane interaction. Adv. Enzym. Regul. 2010, 50, 262–271. [Google Scholar] [CrossRef] [PubMed]
- Mills, S.J.; Persson, C.; Cozier, G.; Thomas, M.P.; Tresaugues, L.; Erneux, C.; Riley, A.M.; Nordlund, P.; Potter, B.V. A synthetic polyphosphoinositide headgroup surrogate in complex with SHIP2 provides a rationale for drug discovery. ACS Chem. Biol. 2012, 7, 822–828. [Google Scholar] [CrossRef] [PubMed]
- He, K.; Jang, S.W.; Joshi, J.; Yoo, M.H.; Ye, K. Akt-phosphorylated PIKE-A inhibits UNC5B-induced apoptosis in cancer cell lines in a p53-dependent manner. Mol. Biol. Cell 2011, 22, 1943–1954. [Google Scholar] [CrossRef] [PubMed]
- Fruman, D.A.; Chiu, H.; Hopkins, B.D.; Bagrodia, S.; Cantley, L.C.; Abraham, R.T. The PI3K Pathway in Human Disease. Cell 2017, 170, 605–635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, K.S.; Chen, G.; Shen, H.L.; Li, T.T.; Chen, F.; Wang, Q.W.; Wang, Z.Q.; Han, Z.G.; Zhang, X. Insulin receptor tyrosine kinase substrate enhances low levels of MDM2-mediated p53 ubiquitination. PLoS ONE 2011, 6, e23571. [Google Scholar] [CrossRef] [PubMed]
- Cheng, N.; Li, Y.; Han, Z.G. Argonaute2 promotes tumor metastasis by way of up-regulating focal adhesion kinase expression in hepatocellular carcinoma. Hepatology 2013, 57, 1906–1918. [Google Scholar] [CrossRef] [PubMed]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, C.; Cui, X.; Huang, L.; Shang, X.; Wu, B.; Wang, N.; He, K.; Han, Z. IRTKS Promotes Insulin Signaling Transduction through Inhibiting SHIP2 Phosphatase Activity. Int. J. Mol. Sci. 2019, 20, 2834. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20112834
Wu C, Cui X, Huang L, Shang X, Wu B, Wang N, He K, Han Z. IRTKS Promotes Insulin Signaling Transduction through Inhibiting SHIP2 Phosphatase Activity. International Journal of Molecular Sciences. 2019; 20(11):2834. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20112834
Chicago/Turabian StyleWu, Chongchao, Xiaofang Cui, Liyu Huang, Xueying Shang, Binghao Wu, Na Wang, Kunyan He, and Zeguang Han. 2019. "IRTKS Promotes Insulin Signaling Transduction through Inhibiting SHIP2 Phosphatase Activity" International Journal of Molecular Sciences 20, no. 11: 2834. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20112834