NAFLD in Some Common Endocrine Diseases: Prevalence, Pathophysiology, and Principles of Diagnosis and Management



Abstract
:1. Introduction
2. Polycystic Ovary Syndrome
2.1. Epidemiology and Diagnosis of PCOS
2.2. Epidemiological Evidence Linking PCOS to NAFLD
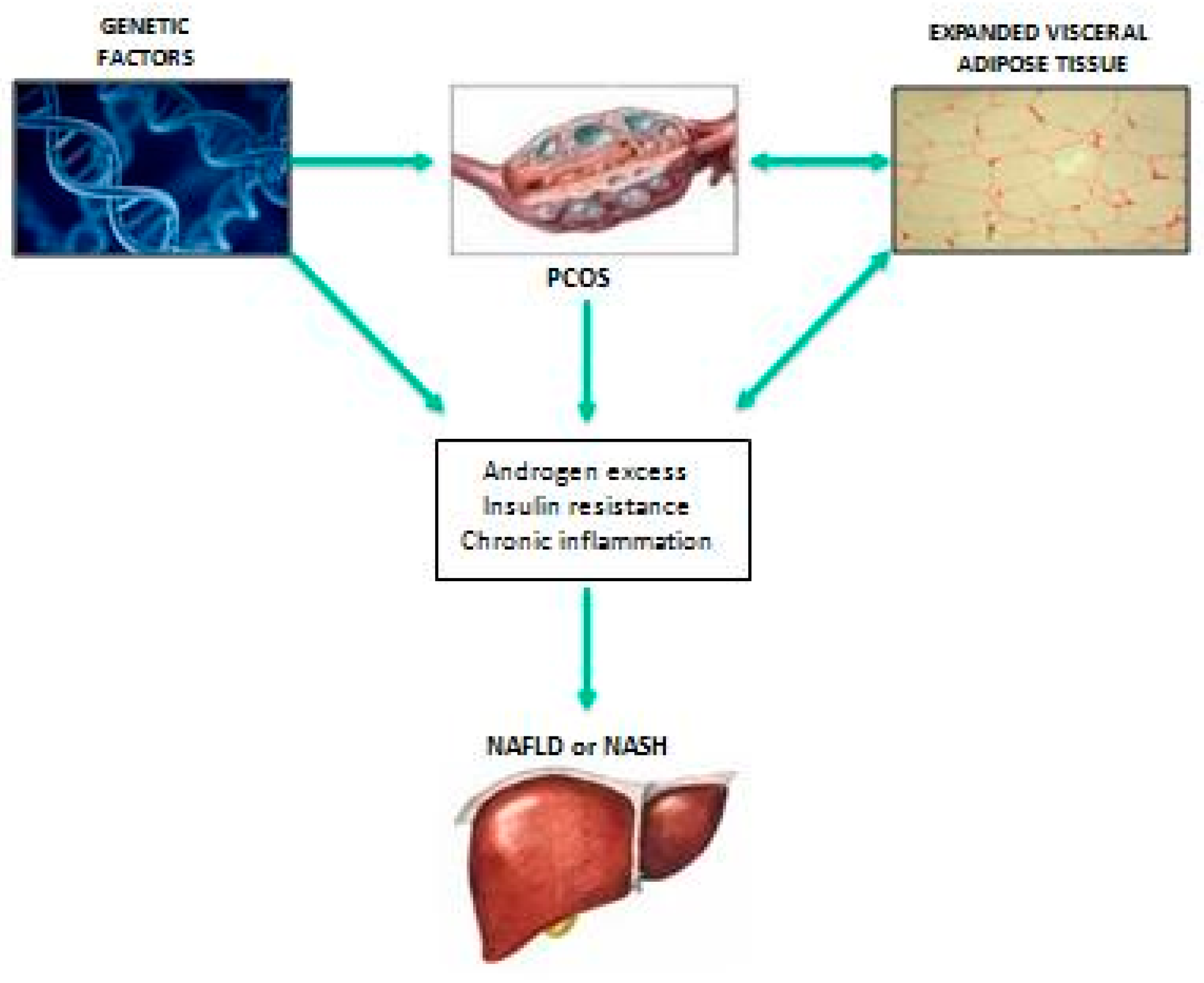
2.3. Putative Pathophysiological Mechanisms Linking PCOS with NAFLD
2.4. Screening and Therapeutic Approaches to NAFLD in PCOS
3. Hypothyroidism
3.1. Diagnosis of Primary Hypothyroidism and Epidemiological Evidence Linking Hypothyroidism with NAFLD
3.2. Putative Pathophysiological Mechanisms Linking Hypothyroidism with NAFLD
3.3. Diagnosis and Management of NAFLD in Patients with Hypothyroidism
4. Hypogonadism
4.1. Definition and Epidemiological Grounds
4.2. Pathophysiological Mechanisms Linking Hypogonadism with NAFLD
4.3. Principles of Diagnosis and Management
4.3.1. Diagnosis
4.3.2. Management
5. GH Deficiency
5.1. Epidemiology of GH Deficiency
5.2. Pathophysiological Mechanisms Linking GHD with NAFLD
5.3. Principles of Diagnosis and Management
6. Conclusions
Funding
Conflicts of Interest
Abbreviations
| ACTH | adrenocorticotropic hormone |
| AGHD | adult growth hormone deficiency |
| AMPK | AMP-activated protein kinase |
| apo | apolipoprotein |
| BAAT index | body mass index, age, ALT, and triglyceride |
| BMI | body mass index |
| CCL3 | C–C motif chemokine ligand 3 |
| CI | confidence interval |
| ERK | extracellular signal-regulated kinase |
| FT4 | free thyroxine |
| GH | growth hormone |
| GHD | growth hormone deficiency |
| GHRH | GH-releasing hormone |
| GHRLD | liver-specific deletion of GH receptor |
| HDL | high-density lipoprotein |
| hypertension | arterial hypertension |
| IGF-1 | insulin-like growth factor-1 |
| JAK-2 | Janus kinase 2 |
| JAK2L | liver-specific JAK2-deficient |
| JNK | c-Jun N-terminal kinase |
| LDL | low-density lipoprotein |
| LH | luteinizing hormone |
| MAPK | mitogen-activated protein kinase |
| MKK3 | MAP kinase-activated protein kinase-3 |
| MKK-4 | MAP kinase-activated protein kinase-4 |
| MRI | magnetic resonance imaging |
| MRS | magnetic resonance spectroscopy |
| NAFLD | nonalcoholic fatty liver disease |
| NASH | nonalcoholic steatohepatitis |
| OR | odds ratio |
| PCOS | polycystic ovary syndrome |
| PI3K | phosphoinositide 3-kinase |
| PKC | protein kinase C |
| PPARα | peroxisome proliferator-activated receptor-α |
| ROS | reactive oxygen species |
| SREBP-1c | sterol regulatory element-binding |
| STAT-5 | signal transducer and activator of transcription 5 |
| TNF-α | tumor necrosis factor-α |
| TSH | thyroid-stimulating hormone |
| VLDL | very low-density lipoprotein |
References
- Petäjä, E.M.; Yki-Järvinen, H. Definitions of Normal Liver Fat and the Association of Insulin Sensitivity with Acquired and Genetic NAFLD-A Systematic Review. Int. J. Mol. Sci. 2016, 17, 633. [Google Scholar]
- Italian Association for the Study of the Liver (AISF). AISF position paper on nonalcoholic fatty liver disease (NAFLD): Updates and future directions. Dig. Liver Dis. 2017, 49, 471–483. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.; Tacke, F.; Arrese, M.; Sharma, B.C.; Mostafa, I.; Bugianesi, E.; Wong, V.W.; Yilmaz, Y.; George, J.; Fan, J.; et al. Global Perspectives on Non-alcoholic Fatty Liver Disease and Non-alcoholic Steatohepatitis. Hepatology 2019, 69, 2672–2682. [Google Scholar] [CrossRef] [PubMed]
- Fazel, Y.; Koenig, A.B.; Sayiner, M.; Goodman, Z.D.; Younossi, Z.M. Epidemiology and natural history of non-alcoholic fatty liver disease. Metabolism 2016, 65, 1017–1025. [Google Scholar] [CrossRef] [Green Version]
- Vilar-Gomez, E.; Calzadilla-Bertot, L.; Wai-Sun Wong, V.; Castellanos, M.; Aller-de la Fuente, R.; Metwally, M.; Eslam, M.; Gonzalez-Fabian, L.; Alvarez-Quiñones Sanz, M.; Conde-Martin, A.F.; et al. Fibrosis Severity as a Determinant of Cause-Specific Mortality in Patients With Advanced Nonalcoholic Fatty Liver Disease: A Multi-National Cohort Study. Gastroenterology 2018, 155, 443–457. [Google Scholar] [CrossRef] [PubMed]
- European Association for the Study of the Liver (EASL); European Association for the Study of Diabetes (EASD); European Association for the Study of Obesity (EASO). EASL-EASD-EASO Clinical Practice Guidelines for the management of non-alcoholic fatty liver disease. J. Hepatol. 2016, 64, 1388–1402. [Google Scholar] [CrossRef] [PubMed]
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Charlton, M.; Cusi, K.; Rinella, M.; Harrison, S.A.; Brunt, E.M.; Sanyal, A.J. The diagnosis and management of nonalcoholic fatty liver disease: Practice guidance from the American Association for the Study of Liver Diseases. Hepatology 2018, 67, 328–357. [Google Scholar] [CrossRef] [PubMed]
- Lonardo, A.; Ballestri, S.; Marchesini, G.; Angulo, P.; Loria, P. Nonalcoholic fatty liver disease: a precursor of the metabolic syndrome. Dig. Liver Dis. 2015, 47, 181–190. [Google Scholar] [CrossRef] [PubMed]
- Iwen, K.A.; Oelkrug, R.; Kalscheuer, H.; Brabant, G. Metabolic Syndrome in Thyroid Disease. Front. Horm. Res. 2018, 49, 48–66. [Google Scholar]
- Rastrelli, G.; Filippi, S.; Sforza, A.; Maggi, M.; Corona, G. Metabolic Syndrome in Male Hypogonadism. Front. Horm. Res. 2018, 49, 131–155. [Google Scholar]
- Dwyer, A.A.; Quinton, R. The Metabolic Syndrome in Central Hypogonadotrophic Hypogonadism. Front. Horm. Res. 2018, 49, 156–169. [Google Scholar] [PubMed]
- Pasquali, R. Metabolic Syndrome in Polycystic Ovary Syndrome. Front. Horm. Res. 2018, 49, 114–130. [Google Scholar] [PubMed]
- Miljić, D.; Popovic, V. Metabolic Syndrome in Hypopituitarism. Front. Horm. Res. 2018, 49, 1–19. [Google Scholar] [PubMed]
- Lonardo, A.; Nascimbeni, F.; Mantovani, A.; Targher, G. Hypertension, diabetes, atherosclerosis and NASH: Cause or consequence? J. Hepatol. 2018, 68, 335–352. [Google Scholar] [CrossRef] [PubMed]
- Angulo, P. Nonalcoholic fatty liver disease. N. Engl. J. Med. 2002, 346, 1221–1231. [Google Scholar] [CrossRef] [PubMed]
- Lonardo, A.; Carani, C.; Carulli, N.; Loria, P. ‘Endocrine NAFLD’ a hormonocentric perspective of nonalcoholic fatty liver disease pathogenesis. J. Hepatol. 2006, 44, 1196–1207. [Google Scholar] [CrossRef] [PubMed]
- Loria, P.; Carulli, L.; Bertolotti, M.; Lonardo, A. Endocrine and liver interaction: the role of endocrine pathways in NASH. Nat. Rev. Gastroenterol. Hepatol. 2009, 6, 236–247. [Google Scholar] [CrossRef] [PubMed]
- Manka, P.; Bechmann, L.; Best, J.; Sydor, S.; Claridge, L.C.; Coombes, J.D.; Canbay, A.; Moeller, L.; Gerken, G.; Wedemeyer, H.; et al. Low Free Triiodothyronine Is Associated with Advanced Fibrosis in Patients at High Risk for Nonalcoholic Steatohepatitis. Dig. Dis. Sci. 2019. [Google Scholar] [CrossRef]
- Lonardo, A.; Ballestri, S.; Mantovani, A.; Nascimbeni, F.; Lugari, S.; Targher, G. Pathogenesis of hypothyroidism-induced NAFLD: Evidence for a distinct disease entity? Dig. Liver Dis. 2019, 5, 462–470. [Google Scholar] [CrossRef]
- McCartney, C.R.; Marshall, J.C. Clinical practice. Polycystic ovary syndrome. N. Engl. J. Med. 2016, 375, 54–64. [Google Scholar] [CrossRef]
- Anagnostis, P.; Tarlatzis, B.C.; Kauffman, R.P. Polycystic ovarian syndrome (PCOS): long-term metabolic consequences. Metabolism 2018, 86, 33–43. [Google Scholar] [CrossRef]
- Targher, G.; Zoppini, G.; Bonora, E.; Moghetti, P. Hemostatic and fibrinolytic abnormalities in polycystic ovary syndrome. Semin. Thromb. Hemost. 2014, 40, 600–618. [Google Scholar]
- Stein, I.F.; Leventhal, M.L. Amenorrhea associated with bilateral polycystic ovaries. Am. J. Obstet. Gynecol. 1935, 29, 181–191. [Google Scholar] [CrossRef]
- The Rotterdam ESHRE/ASRM-sponsored PCOS consensus workshop group. Revised 2003 consensus on diagnostic criteria and long-term health risks related to polycystic ovary syndrome (PCOS). Hum. Reprod. 2004, 19, 41–47. [Google Scholar] [CrossRef] [Green Version]
- Sirmans, S.M.; Pate, K.A. Epidemiology, diagnosis, and management of polycystic ovary syndrome. Clin. Epidemiol. 2014, 6, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Yildiz, B.O.; Bozdag, G.; Yapici, Z.; Esinler, I.; Yarali, H. Prevalence, phenotype and cardiometabolic risk of polycystic ovary syndrome under different diagnostic criteria. Hum. Reprod. 2012, 27, 3067–3073. [Google Scholar] [CrossRef] [PubMed]
- Targher, G.; Rossini, M.; Lonardo, A. Evidence that non-alcoholic fatty liver disease and polycystic ovary syndrome are associated by necessity rather than chance: a novel hepato-ovarian axis? Endocrine 2016, 51, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Setji, T.L.; Holland, N.D.; Sanders, L.L.; Pereira, K.C.; Diehl, A.M.; Brown, A.J. Nonalcoholic steatohepatitis and nonalcoholic fatty liver disease in young women with polycystic ovary syndrome. J. Clin. Endocrinol. Metab. 2006, 91, 1741–1747. [Google Scholar] [CrossRef] [PubMed]
- Brzozowska, M.M.; Ostapowicz, G.; Weltman, M.D. An association between non-alcoholic fatty liver disease and polycystic ovarian syndrome. J. Gastroenterol. Hepatol. 2009, 24, 243–247. [Google Scholar] [CrossRef]
- Hossain, N.; Stepanova, M.; Afendy, A.; Nader, F.; Younossi, Y.; Rafiq, N.; Goodman, Z.; Younossi, Z.M. Non-alcoholic steatohepatitis (NASH) in patients with polycystic ovarian syndrome (PCOS). Scand. J. Gastroenterol. 2011, 46, 479–484. [Google Scholar] [CrossRef]
- Wu, J.; Yao, X.Y.; Shi, R.X.; Liu, S.F.; Wang, X.Y. A potential link between polycystic ovary syndrome and non-alcoholic fatty liver disease: an update meta-analysis. Reprod. Health 2018, 15, 77. [Google Scholar] [CrossRef] [PubMed]
- Jones, H.; Sprung, V.S.; Pugh, C.J.; Daousi, C.; Irwin, A.; Aziz, N.; Adams, V.L.; Thomas, E.L.; Bell, J.D.; Kemp, G.J.; et al. Polycystic ovary syndrome with hyperandrogenism is characterized by an increased risk of hepatic steatosis compared to non-hyperandrogenic PCOS phenotypes and healthy controls, independent of obesity and insulin resistance. J. Clin. Endocrinol. Metab. 2012, 97, 3709–3716. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.J.; Kim, D.; Yim, J.Y.; Kang, J.H.; Han, K.H.; Kim, S.M.; Hwang, K.R.; Ku, S.Y.; Suh, C.S.; Kim, S.H.; et al. Polycystic ovary syndrome with hyperandrogenism as a risk factor for non-obese non-alcoholic fatty liver disease. Aliment Pharmacol. Ther. 2017, 45, 1403–1412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumarendran, B.; O’Reilly, M.W.; Manolopoulos, K.N.; Toulis, K.A.; Gokhale, K.M.; Sitch, A.J.; Wijeyaratne, C.N.; Coomarasamy, A.; Arlt, W.; Nirantharakumar, K. Polycystic ovary syndrome, androgen excess, and the risk of nonalcoholic fatty liver disease in women: a longitudinal study based on a United Kingdom primary care database. PLoS Med. 2018, 15, e1002542. [Google Scholar] [CrossRef] [PubMed]
- Vassilatou, E.; Lafoyianni, S.; Vryonidou, A.; Ioannidis, D.; Kosma, L.; Katsoulis, K.; Papavassiliou, E.; Tzavara, I. Increased androgen bioavailability is associated with non-alcoholic fatty liver disease in women with polycystic ovary syndrome. Hum. Reprod. 2010, 25, 212–220. [Google Scholar] [CrossRef] [PubMed]
- Cussons, A.J.; Watts, G.F.; Mori, T.A.; Stuckey, B.G. Omega-3 fatty acid supplementation decreases liver fat content in polycystic ovary syndrome: a randomized controlled trial employing proton magnetic resonance spectroscopy. J. Clin. Endocrinol. Metab. 2009, 94, 3842–3848. [Google Scholar] [CrossRef]
- Dawson, A.J.; Sathyapalan, T.; Smithson, J.A.; Vince, R.V.; Coady, A.M.; Ajjan, R.; Kilpatrick, E.S.; Atkin, S.L. A comparison of cardiovascular risk indices in patients with polycystic ovary syndrome with and without coexisting nonalcoholic fatty liver disease. Clin. Endocrinol. 2014, 80, 843–849. [Google Scholar] [CrossRef] [PubMed]
- Byrne, C.D.; Patel, J.; Scorletti, E.; Targher, G. Tests for diagnosing and monitoring non-alcoholic fatty liver disease in adults. BMJ 2018, 362, k2734. [Google Scholar] [CrossRef] [Green Version]
- Baptiste, C.G.; Battista, M.C.; Trottier, A.; Baillargeon, J.P. Insulin and hyperandrogenism in women with polycystic ovary syndrome. J. Steroid Biochem. Mol. Biol. 2010, 122, 42–52. [Google Scholar] [CrossRef]
- Munir, I.; Yen, H.W.; Geller, D.H.; Torbati, D.; Bierden, R.M.; Weitsman, S.R.; Agarwal, S.K.; Magoffin, D.A. Insulin augmentation of 17alpha-hydroxylase activity is mediated by phosphatidyl inositol 3- kinase but not extracellular signal-regulated kinase-1/2 in human ovarian theca cells. Endocrinology 2004, 145, 175–183. [Google Scholar] [CrossRef]
- Wickenheisser, J.K.; Nelson-DeGrave, V.L.; McAllister, J.M. Human ovarian theca cells in culture. Trends Endocrinol. Metab. 2006, 17, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Abbott, D.H.; Dumesic, D.A.; Levine, J.E. Hyperandrogenic origins of polycystic ovary syndrome—Implications for pathophysiology and therapy. Expert Rev. Endocrinol. Metab. 2019, 14, 131–143. [Google Scholar] [CrossRef] [PubMed]
- Schiffer, L.; Kempegowda, P.; Arlt, W.; O’Reilly, M.W. MECHANISMS IN ENDOCRINOLOGY: The sexually dimorphic role of androgens in human metabolic disease. Eur. J. Endocrinol. 2017, 177, R125–R143. [Google Scholar] [CrossRef] [PubMed]
- Du, Q.; Wang, Y.J.; Yang, S.; Wu, B.; Han, P.; Zhao, Y.Y. A systematic review and meta-analysis of randomized controlled trials comparing pioglitazone versus metformin in the treatment of polycystic ovary syndrome. Curr. Med. Res. Opin. 2012, 28, 723–730. [Google Scholar] [CrossRef] [PubMed]
- Tzotzas, T.; Karras, S.N.; Katsiki, N. Glucagon-like peptide-1 (GLP-1) receptor agonists in the treatment of obese women with polycystic ovary syndrome. Curr. Vasc. Pharmacol. 2017, 15, 218–229. [Google Scholar] [CrossRef] [PubMed]
- Frøssing, S.; Nylander, M.; Chabanova, E.; Frystyk, J.; Holst, J.J.; Kistorp, C.; Skouby, S.O.; Faber, J. Effect of liraglutide on ectopic fat in polycystic ovary syndrome: A randomized clinical trial. Diabetes Obes. Metab. 2018, 20, 215–218. [Google Scholar] [CrossRef] [PubMed]
- Chaker, L.; Bianco, A.C.; Jonklaas, J.; Peeters, R.P. Hypothyroidism. Lancet 2017, 390, 1550–1562. [Google Scholar] [CrossRef]
- Sinha, R.A.; Singh, B.K.; Yen, P.M. Thyroid hormone regulation of hepatic lipid and carbohydrate metabolism. Trends Endocrinol. Metab. 2014, 25, 538–545. [Google Scholar] [CrossRef] [PubMed]
- Sinha, R.A.; Singh, B.K.; Yen, P.M. Direct effects of thyroid hormones on hepatic lipid metabolism. Nat. Rev. Endocrinol. 2018, 14, 259–269. [Google Scholar] [CrossRef]
- Kar, K.; Sinha, S. Variations of adipokines and insulin resistance in primary hypothyroidism. J. Clin. Diagn Res. 2017, 11, BC07–BC09. [Google Scholar] [CrossRef] [PubMed]
- Leonhardt, U.; Ritzel, U.; Schäfer, G.; Becker, W.; Ramadori, G. Serum leptin levels in hypo and hyperthyroidism. J. Endocrinol. 1998, 157, 75–79. [Google Scholar] [CrossRef]
- Mantovani, A.; Grani, G. Thyroid Dysfunction and Nonalcoholic Fatty Liver Disease: We Need New Larger and Well-Designed Longitudinal Studies. Dig. Dis. Sci. 2018, 63, 1970–1976. [Google Scholar] [CrossRef]
- Liangpunsakul, S.; Chalasani, N. Is hypothyroidism a risk factor for non-alcoholic steatohepatitis? J. Clin. Gastroenterol. 2003, 37, 340–343. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Xu, L.; Yu, C.; Miao, M.; Li, Y. Association between thyroid function and nonalcoholic fatty liver disease in euthyroid elderly Chinese. Clin. Endocrinol. (Oxf.) 2011, 75, 240–246. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Sun, H.; Chen, L.; Zheng, J.; Hu, X.; Wang, S.; Chen, T. Relationship between serum TSH level with obesity and NAFLD in euthyroid subjects. J. Huazhong Univ. Sci. Technol. Med. Sci. 2012, 32, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Ittermann, T.; Haring, R.; Wallaschofski, H.; Baumeister, S.E.; Nauck, M.; Dörr, M.; Lerch, M.M.; Meyer zu Schwabedissen, H.E.; Rosskopf, D.; Völzke, H. Inverse association between serum free thyroxine levels and hepatic steatosis: Results from the Study of Health in Pomerania. Thyroid 2012, 22, 568–574. [Google Scholar] [CrossRef] [PubMed]
- Pagadala, M.R.; Zein, C.O.; Dasarathy, S.; Yerian, L.M.; Lopez, R.; McCullough, A.J. Prevalence of hypothyroidism in nonalcoholic fatty liver disease. Dig. Dis. Sci. 2012, 57, 528–534. [Google Scholar] [CrossRef] [PubMed]
- Chung, G.E.; Kim, D.; Kim, W.; Yim, J.Y.; Park, M.J.; Kim, Y.J.; Yoon, J.H.; Lee, H.S. Non-alcoholic fatty liver disease across the spectrum of hypothyroidism. J. Hepatol. 2012, 57, 150–156. [Google Scholar] [CrossRef]
- Xu, L.; Ma, H.; Miao, M.; Li, Y. Impact of subclinical hypothyroidism on the development of non-alcoholic fatty liver disease: A prospective case-control study. J. Hepatol. 2012, 57, 1153–1154. [Google Scholar] [CrossRef] [Green Version]
- Carulli, L.; Ballestri, S.; Lonardo, A.; Lami, F.; Violi, E.; Losi, L.; Bonilauri, L.; Verrone, A.M.; Odoardi, M.R.; Scaglioni, F.; et al. Is nonalcoholic steatohepatitis associated with a high-though-normal thyroid stimulating hormone level and lower cholesterol levels? Intern. Emerg. Med. 2013, 8, 297–305. [Google Scholar] [CrossRef] [PubMed]
- Eshraghian, A.; Dabbaghmanesh, M.H.; Eshraghian, H.; Fattahi, M.R.; Omrani, G.R. Nonalcoholic fatty liver disease in a cluster of Iranian population: Thyroid status and metabolic risk factors. Arch. Iran. Med. 2013, 16, 584–589. [Google Scholar] [PubMed]
- Posadas-Romero, C.; Jorge-Galarza, E.; Posadas-Sánchez, R.; Acuña-Valerio, J.; Juárez-Rojas, J.G.; Kimura-Hayama, E.; Medina-Urrutia, A.; Cardoso-Saldaña, G.C. Fatty liver largely explains associations of subclinical hypothyroidism with insulin resistance, metabolic syndrome, and subclinical coronary atherosclerosis. Eur. J. Endocrinol. 2014, 171, 319–325. [Google Scholar] [CrossRef] [Green Version]
- Tao, Y.; Gu, H.; Wu, J.; Sui, J. Thyroid function is associated with non-alcoholic fatty liver disease in euthyroid subjects. Endocr. Res. 2015, 40, 74–78. [Google Scholar] [CrossRef]
- Liu, G.; Zheng, X.; Guan, L.; Jiang, Z.; Lin, H.; Jiang, Q.; Zhang, N.; Zhang, Y.; Zhang, X.; Yu, C.; et al. Free triiodothyronine levels are positively associated with non-alcoholic fatty liver disease in euthyroid middle-aged subjects. Endocr. Res. 2015, 40, 188–193. [Google Scholar] [CrossRef] [PubMed]
- Ding, W.J.; Wang, M.M.; Wang, G.S.; Shen, F.; Qin, J.J.; Fan, J.G. Thyroid function is associated with non-alcoholic fatty liver disease in chronic hepatitis B-infected subjects. J. Gastroenterol. Hepatol. 2015, 30, 1753–1758. [Google Scholar] [CrossRef]
- Parikh, P.; Phadke, A.; Sawant, P. Prevalence of hypothyroidism in nonalcoholic fatty liver disease in patients attending a tertiary hospital in western India. Indian. J. Gastroenterol. 2015, 34, 169–173. [Google Scholar]
- Ludwig, U.; Holzner, D.; Denzer, C.; Greinert, A.; Haenle, M.M.; Oeztuerk, S.; Koenig, W.; Boehm, B.O.; Mason, R.A.; Kratzer, W.; et al. Subclinical and clinical hypothyroidism and non-alcoholic fatty liver disease: A cross-sectional study of a random population sample aged 18 to 65 years. BMC Endocr. Disord. 2015, 15, 41. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.W.; Bang, K.B.; Rhee, E.J.; Kwon, H.J.; Lee, M.Y.; Cho, Y.K. Impact of hypothyroidism on the development of non-alcoholic fatty liver disease: A 4-year retrospective cohort study. Clin. Mol. Hepatol. 2015, 21, 372–378. [Google Scholar] [CrossRef] [PubMed]
- Bril, F.; Kadiyala, S.; Portillo Sanchez, P.; Sunny, N.E.; Biernacki, D.; Maximos, M.; Kalavalapalli, S.; Lomonaco, R.; Suman, A.; Cusi, K. Plasma thyroid hormone concentration is associated with hepatic triglyceride content in patients with type 2 diabetes. J. Investig. Med. 2016, 64, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Bano, A.; Chaker, L.; Plompen, E.P.; Hofman, A.; Dehghan, A.; Franco, O.H.; Janssen, H.L.; Darwish Murad, S.; Peeters, R.P. Thyroid function and the risk of nonalcoholic fatty liver disease: The Rotterdam study. J. Clin. Endocrinol. Metab. 2016, 101, 3204–3211. [Google Scholar] [CrossRef] [PubMed]
- Gökmen, F.Y.; Ahbab, S.; Ataoğlu, H.E.; Türker, B.Ç.; Çetin, F.; Türker, F.; Mamaç, R.Y.; Yenigün, M. FT3/FT4 ratio predicts non-alcoholic fatty liver disease independent of metabolic parameters in patients with euthyroidism and hypothyroidism. Clinics (Sao Paulo) 2016, 71, 221–225. [Google Scholar] [CrossRef]
- van den Berg, E.H.; van Tienhoven-Wind, L.J.M.; Amini, M.; Schreuder, T.C.; Faber, K.N.; Blokzijl, H.; Dullaart, R.P. Higher free triiodothyronine is associated with non-alcoholic fatty liver disease in euthyroid subjects: The Lifelines Cohort Study. Metabolism 2017, 67, 62–71. [Google Scholar] [CrossRef] [PubMed]
- Lingad-Sayas, R.C.; Montano, C.N.; Isidro, M.J.C. Prevalence of elevated TSH and its association with dyslipidemia and NAFLD among Filipino adult executive check-up patients in a tertiary hospital. Philipp. J. Intern. Med. 2017, 55, 1–8. [Google Scholar]
- Mohanty, R.; Das, S.N.; Jena, A.K.; Behera, S.; Sahu, N.C.; Mohanty, B.; Suna, S.P.; Thatoi, P.K. Prevalence of non-alcoholic fatty liver disease in hypothyroidism in a tertiary care hospital in eastern India. J. Evol. Med. Dent. Sci. 2017, 6, 5589–5593. [Google Scholar] [CrossRef]
- Kim, D.; Kim, W.; Joo, S.K.; Bae, J.M.; Kim, J.H.; Ahmed, A. Subclinical hypothyroidism and low-normal thyroid function are associated with nonalcoholic steatohepatitis and fibrosis. Clin. Gastroenterol. Hepatol. 2018, 16, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Ha, J.; Jo, K.; Lim, D.J.; Lee, J.M.; Chang, S.A.; Kang, M.I.; Cha, B.Y.; Kim, M.H. Male-specific association between subclinical hypothyroidism and the risk of non-alcoholic fatty liver disease estimated by hepatic steatosis index: Korea National Health and Nutrition Examination Survey 2013 to 2015. Sci. Rep. 2018, 8, 15145. [Google Scholar] [CrossRef] [PubMed]
- Jaruvongvanich, V.; Sanguankeo, A.; Upala, S. Nonalcoholic fatty liver disease is not associated with thyroid hormone levels and hypothyroidism: A systematic review and meta-analysis. Eur. Thyroid J. 2017, 6, 208–215. [Google Scholar] [CrossRef] [PubMed]
- He, W.; An, X.; Li, L.; Shao, X.; Li, Q.; Yao, Q.; Zhang, J.A. Relationship between hypothyroidism and non-alcoholic fatty liver disease: A systematic review and meta-analysis. Front. Endocrinol. 2017, 8, 335. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Nascimbeni, F.; Lonardo, A.; Zoppini, G.; Bonora, E.; Mantzoros, C.S.; Targher, G. Association Between Primary Hypothyroidism and Nonalcoholic Fatty Liver Disease: A Systematic Review and Meta-Analysis. Thyroid 2018, 28, 1270–1284. [Google Scholar] [CrossRef]
- Guo, Z.; Li, M.; Han, B.; Qi, X. Association of non-alcoholic fatty liver disease with thyroid function: A systematic review and meta-analysis. Dig. Liver Dis. 2018, 50, 1153–1162. [Google Scholar] [CrossRef] [PubMed]
- Lugari, S.; Mantovani, A.; Nascimbeni, F.; Lonardo, A. Hypothyroidism and nonalcoholic fatty liver disease-a chance association? Horm. Mol. Biol. Clin. Investig. 2018. [Google Scholar] [CrossRef] [PubMed]
- Byrne, C.D.; Targher, G. NAFLD: a multisystem disease. J. Hepatol. 2015, 62, S47–S64. [Google Scholar] [CrossRef] [PubMed]
- Yan, F.; Wang, Q.; Lu, M.; Chen, W.; Song, Y.; Jing, F.; Guan, Y.; Wang, L.; Lin, Y.; Bo, T.; et al. Thyrotropin increases hepatic triglyceride content through upregulation of SREBP-1c activity. J. Hepatol. 2014, 61, 1358–1364. [Google Scholar] [CrossRef] [PubMed]
- Bril, F.; Kadiyala, S.; Cusi, K. Re: “Association Between Primary Hypothyroidism and Nonalcoholic Fatty Liver Disease: A Systematic Review and Meta-Analysis” by Mantovani et al. (Thyroid 2018;28:1270-1284). Thyroid 2019, 29, 452. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Yu, Y.; Zhao, M.; Zheng, D.; Zhang, X.; Guan, Q.; Xu, C.; Gao, L.; Zhao, J.; Zhang, H. Benefits of levothyroxine replacement therapy on nonalcoholic fatty liver disease in subclinical hypothyroidism patients. Int. J. Endocrinol. 2017, 2017, 5753039. [Google Scholar] [CrossRef] [PubMed]
- Bruinstroop, E.; Dalan, R.; Yang, C.; Bee, Y.M.; Chandran, K.; Cho, L.W.; Soh, S.B.; Teo, E.K.; Toh, S.A.; Leow, M.K.S.; et al. Low dose levothyroxine reduces intrahepatic lipid content in patients with type 2 diabetes mellitus and NAFLD. J. Clin. Endocrinol. Metab. 2018, 103, 2698–2706. [Google Scholar] [CrossRef] [PubMed]
- Mintziori, G.; Poulakos, P.; Tsametis, C.; Goulis, D.G. Hypogonadism and non-alcoholic fatty liver disease. Minerva Endocrinol. 2017, 42, 145–150. [Google Scholar] [PubMed]
- Haider, A.; Gooren, L.J.; Padungtod, P.; Saad, F. Improvement of the metabolic syndrome and of non-alcoholic liver steatosis upon treatment of hypogonadal elderly men with parenteral testosterone undecanoate. Exp. Clin. Endocrinol. Diabetes 2010, 118, 167–171. [Google Scholar] [CrossRef] [PubMed]
- Seo, N.K.; Koo, H.S.; Haam, J.H.; Kim, H.Y.; Kim, M.J.; Park, K.C.; Park, K.S.; Kim, Y.S. Prediction of prevalent but not incident non-alcoholic fatty liver disease by levels of serum testosterone. J. Gastroenterol. Hepatol. 2015, 30, 1211–1216. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.B.; She, F.; Xie, L.F.; Yan, W.H.; Ouyang, J.Z.; Wang, B.A.; Ma, H.Y.; Zang, L.; Mu, Y.M. Evaluation of Basal Serum Adrenocorticotropic Hormone and Cortisol Levels and Their Relationship with Nonalcoholic Fatty Liver Disease in Male Patients with Idiopathic Hypogonadotropic Hypogonadism. Chin. Med. J. 2016, 129, 1147–1153. [Google Scholar] [CrossRef]
- Barbonetti, A.; Caterina Vassallo, M.R.; Cotugno, M.; Felzani, G.; Francavilla, S.; Francavilla, F. Low testosterone and non-alcoholic fatty liver disease: Evidence for their independent association in men with chronic spinal cord injury. J. Spinal Cord Med. 2016, 39, 443–449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gild, P.; Cole, A.P.; Krasnova, A.; Dickerman, B.A.; von Landenberg, N.; Sun, M.; Mucci, L.A.; Lipsitz, S.R.; Chun, F.K.; Nguyen, P.L.; et al. Liver Disease in Men Undergoing Androgen Deprivation Therapy for Prostate Cancer. J. Urol. 2018, 200, 573–581. [Google Scholar] [CrossRef] [PubMed]
- Elsheikh, M.; Hodgson, H.J.; Wass, J.A.; Conway, G.S. Hormone replacement therapy may improve hepatic function in women with Turner’s syndrome. Clin. Endocrinol. 2001, 55, 227–231. [Google Scholar] [CrossRef]
- Bruno, S.; Maisonneuve, P.; Castellana, P.; Rotmensz, N.; Rossi, S.; Maggioni, M.; Persico, M.; Colombo, A.; Monasterolo, F.; Casadei-Giunchi, D.; et al. Incidence and risk factors for non-alcoholic steatohepatitis: prospective study of 5408 women enrolled in Italian tamoxifen chemoprevention trial. BMJ 2005, 330, 932. [Google Scholar] [CrossRef] [PubMed]
- McKenzie, J.; Fisher, B.M.; Jaap, A.J.; Stanley, A.; Paterson, K.; Sattar, N. Effects of HRT on liver enzyme levels in women with type 2 diabetes: a randomized placebo-controlled trial. Clin. Endocrinol. 2006, 65, 40–44. [Google Scholar] [CrossRef] [PubMed]
- Koulouri, O.; Ostberg, J.; Conway, G.S. Liver dysfunction in Turner’s syndrome: prevalence, natural history and effect of exogenous oestrogen. Clin. Endocrinol. 2008, 69, 306–310. [Google Scholar] [CrossRef] [PubMed]
- El-Mansoury, M.; Berntorp, K.; Bryman, I.; Hanson, C.; Innala, E.; Karlsson, A.; Landin-Wilhelmsen, K. Elevated liver enzymes in Turner syndrome during a 5-year follow-up study. Clin. Endocrinol. 2008, 68, 485–490. [Google Scholar] [CrossRef]
- Gutierrez-Grobe, Y.; Ponciano-Rodríguez, G.; Ramos, M.H.; Uribe, M.; Méndez-Sánchez, N. Prevalence of non alcoholic fatty liver disease in premenopausal, posmenopausal and polycystic ovary syndrome women. The role of estrogens. Ann. Hepatol. 2010, 9, 402–409. [Google Scholar]
- Yang, J.D.; Abdelmalek, M.F.; Pang, H.; Guy, C.D.; Smith, A.D.; Diehl, A.M.; Suzuki, A. Gender and menopause impact severity of fibrosis among patients with nonalcoholic steatohepatitis. Hepatology 2014, 59, 1406–1414. [Google Scholar] [CrossRef]
- Hanew, K. Women with Turner syndrome are at high risk of lifestyle-related disease—From questionnaire surveys by the Foundation for Growth Science in Japan. Endocr. J. 2016, 63, 449–456. [Google Scholar] [CrossRef]
- Yang, Y.J.; Kim, K.M.; An, J.H.; Lee, D.B.; Shim, J.H.; Lim, Y.S.; Lee, H.C.; Lee, Y.S.; Ahn, J.H.; Jung, K.H.; et al. Clinical significance of fatty liver disease induced by tamoxifen and toremifene in breast cancer patients. Breast 2016, 28, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Klair, J.S.; Yang, J.D.; Abdelmalek, M.F.; Guy, C.D.; Gill, R.M.; Yates, K.; Unalp-Arida, A.; Lavine, J.E.; Clark, J.M.; Diehl, A.M.; et al. Nonalcoholic Steatohepatitis Clinical Research Network. A longer duration of estrogen deficiency increases fibrosis risk among postmenopausal women with nonalcoholic fatty liver disease. Hepatology 2016, 64, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Lonardo, A.; Nascimbeni, F.; Ballestri, S.; Fairweather, D.L.; Win, S.; Than, T.A.; Manal, F.; Abdelmalek, M.F.; Suzuki, A. Sex Differences in NAFLD: State of the Art and Identification of Research Gaps. Hepatology 2019. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, K.; Gualtieri, M.R.; Cahoon, S.S.; Jung, C.E.; Paulson, R.J.; Shoupe, D.; Muderspach, L.I.; Wakatsuki, A.; Wright, J.D.; Roman, L.D. Surgical menopause and increased risk of nonalcoholic fatty liver disease in endometrial cancer. Menopause 2016, 23, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Xu, M.; Hu, Z.; Shrestha, U.K. Prevalence of nonalcoholic fatty liver disease and its metabolic risk factors in women of different ages and body mass index. Menopause 2015, 22, 667–673. [Google Scholar] [CrossRef] [PubMed]
- Venetsanaki, V.; Polyzos, S.A. Menopause and non-alcoholic fatty liver disease: A review focusing on therapeutic perspective. Curr. Vasc. Pharmacol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Kamada, Y.; Kiso, S.; Yoshida, Y.; Chatani, N.; Kizu, T.; Hamano, M.; Tsubakio, M.; Takemura, T.; Ezaki, H.; Hayashi, N.; et al. Estrogen deficiency worsens steatohepatitis in mice fed high-fat and high-cholesterol diet. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 301, G1031–G1043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, M.; Shi, H. Sex Hormones and Their Receptors Regulate Liver Energy Homeostasis. Int. J. Endocrinol. 2015, 2015, 294278. [Google Scholar] [CrossRef]
- Sakr, H.F.; Hussein, A.M.; Eid, E.A.; AIKhateeb, M. Possible mechanisms underlying fatty liver in a rat model of male hypogonadism: A protective role for testosterone. Steroids 2018, 135, 21–30. [Google Scholar] [CrossRef]
- Maffei, L.; Murata, Y.; Rochira, V.; Tubert, G.; Aranda, C.; Vazquez, M.; Clyne, C.D.; Davis, S.; Simpson, E.R.; Carani, C. Dysmetabolic syndrome in a man with a novel mutation of the aromatase gene: effects of testosterone, alendronate, and estradiol treatment. J. Clin. Endocrinol. Metab. 2004, 89, 61–70. [Google Scholar] [CrossRef]
- Maffei, L.; Rochira, V.; Zirilli, L.; Antunez, P.; Aranda, C.; Fabre, B.; Simone, M.L.; Pignatti, E.; Simpson, E.R.; Houssami, S.; et al. A novel compound heterozygous mutation of the aromatase gene in an adult man: reinforced evidence on the relationship between congenital oestrogen deficiency, adiposity and the metabolic syndrome. Clin. Endocrinol. 2007, 67, 218–224. [Google Scholar] [CrossRef] [PubMed]
- Kander, M.C. Gender difference in oxidative stress: a new look at the mechanisms for cardiovascular diseases. J. Cell Mol. Med. 2017, 21, 1024–1032. [Google Scholar] [CrossRef] [PubMed]
- Chukijrungroat, N. Hepatic FGF21 mediates sex differences in high-fat high-fructose diet-induced fatty liver. Am. J. Physiol. Endocrinol. Metab. 2017, 313, E203–E212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harada, N. Hypogonadism alters cecal and fecal microbiota in male mice. Gut Microbes 2016, 7, 533–539. [Google Scholar] [CrossRef] [PubMed]
- Charlton, M.; Angulo, P.; Chalasani, N.; Merriman, R.; Viker, K.; Charatcharoenwitthaya, P.; Sanderson, S.; Gawrieh, S.; Krishnan, A.; Lindor, K. Low circulating levels of dehydroepiandrosterone in histologically advanced nonalcoholic fatty liver disease. Hepatology 2008, 47, 484–492. [Google Scholar] [CrossRef] [PubMed]
- Sumida, Y.; Yonei, Y.; Kanemasa, K.; Hara, T.; Inada, Y.; Sakai, K.; Imai, S.; Hibino, S.; Yamaguchi, K.; Mitsuyoshi, H.; et al. Lower circulating levels of dehydroepiandrosterone, independent of insulin resistance, is an important determinant of severity of non-alcoholic steatohepatitis in Japanese patients. Hepatol. Res. 2010, 40, 901–910. [Google Scholar] [CrossRef]
- Koga, M.; Saito, H.; Mukai, M.; Saibara, T.; Kasayama, S. Serum dehydroepiandrosterone sulphate levels in patients with non-alcoholic fatty liver disease. Intern. Med. 2011, 50, 1657–1661. [Google Scholar] [CrossRef]
- Koehler, E.; Swain, J.; Sanderson, S.; Krishnan, A.; Watt, K.; Charlton, M. Growth hormone, dehydroepiandrosterone and adiponectin levels in non-alcoholic steatohepatitis: an endocrine signature for advanced fibrosis in obese patients. Liver Int. 2012, 32, 279–286. [Google Scholar] [CrossRef]
- Tokushige, K.; Hashimoto, E.; Kodama, K.; Tobari, M.; Matsushita, N.; Kogiso, T.; Taniai, M.; Torii, N.; Shiratori, K.; Nishizaki, Y.; et al. Serum metabolomic profile and potential biomarkers for severity of fibrosis in nonalcoholic fatty liver disease. J. Gastroenterol. 2013, 48, 1392–1400. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.H.; Cai, J.J.; She, Z.G.; Li, H.L. Noninvasive evaluation of nonalcoholic fatty liver disease: Current evidence and practice. World J. Gastroenterol. 2019, 25, 1307–1326. [Google Scholar] [CrossRef]
- Nascimbeni, F.; Pais, R.; Bellentani, S.; Day, C.P.; Ratziu, V.; Loria, P.; Lonardo, A. From NAFLD in clinical practice to answers from guidelines. J. Hepatol. 2013, 59, 859–871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nascimbeni, F.; Ballestri, S.; Machado, M.V.; Mantovani, A.; Cortez-Pinto, H.; Targher, G.; Lonardo, A. Clinical relevance of liver histopathology and different histological classifications of NASH in adults. Expert Rev. Gastroenterol. Hepatol. 2018, 12, 351–367. [Google Scholar] [CrossRef] [PubMed]
- Khera, M.; Broderick, G.A.; Carson, C.C.; Dobs, A.S.; Faraday, M.M.; Goldstein, I.; Hakim, L.S.; Hellstrom, W.J.; Kacker, R.; Köhler, T.S.; et al. Adult-Onset Hypogonadism. Mayo Clin. Proc. 2016, 91, 908–926. [Google Scholar] [CrossRef] [Green Version]
- Russell, N.; Grossmann, M. MECHANISMS IN ENDOCRINOLOGY: Estradiol as a male hormone. Eur. J. Endocrinol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Sigalos, J.T.; Pastuszak, A.W.; Khera, M. Hypogonadism: Therapeutic Risks, Benefits, and Outcomes. Med. Clin. N. Am. 2018, 102, 361–372. [Google Scholar] [CrossRef] [PubMed]
- Klein, K.O.; Rosenfield, R.L.; Santen, R.J.; Gawlik, A.M.; Backeljauw, P.F.; Gravholt, C.H.; Sas, T.C.J.; Mauras, N. Estrogen Replacement in Turner Syndrome: Literature Review and Practical Considerations. J. Clin. Endocrinol. Metab. 2018, 103, 1790–1803. [Google Scholar] [CrossRef]
- Ballestri, S.; Mantovani, A.; Nascimbeni, F.; Lugari, S.; Lonardo, A. Extra-hepatic manifestations and complications of NAFLD. Future Med. Chem. (In Press).
- Takahashi, Y. The Role of Growth Hormone and Insulin-Like Growth Factor-I in the Liver. Int. J. Mol. Sci. 2017, 18, 1447. [Google Scholar] [CrossRef] [PubMed]
- Vijayakumar, A.; Novosyadlyy, R.; Wu, Y.; Yakar, S.; LeRoith, D. Biological effects of growth hormone on carbohydrate and lipid metabolism. Growth Horm. IGF Res. 2010, 20, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.H.; Park, M.J. Effects of growth hormone on glucose metabolism and insulin resistance in human. Ann. Pediatr. Endocrinol. Metab. 2017, 22, 145–152. [Google Scholar] [CrossRef] [Green Version]
- Le Roith, D.; Bondy, C.; Yakar, S.; Liu, J.L.; Butler, A. The somatomedin hypothesis: 2001. Endocr. Rev. 2001, 22, 53–74. [Google Scholar] [CrossRef] [PubMed]
- Savage, M.O. Insulin-like growth factors, nutrition and growth. World Rev. Nutr. Diet. 2013, 106, 52–59. [Google Scholar] [PubMed]
- Thomas, J.D.; Monson, J.P. Adult GH deficiency throughout lifetime. Eur. J. Endocrinol. 2009, 1, S97–S106. [Google Scholar] [CrossRef] [PubMed]
- Ichikawa, T.; Nakao, K.; Hamasaki, K.; Furukawa, R.; Tsuruta, S.; Ueda, Y.; Taura, N.; Shibata, H.; Fujimoto, M.; Toriyama, K.; et al. Role of growth hormone, insulin-like growth factor 1 and insulin-like growth factor-binding protein 3 in development of non-alcoholic fatty liver disease. Hepatol. Int. 2007, 1, 287–294. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.; Xu, C.; Yu, C.; Miao, M.; Zhang, X.; Zhu, Z.; Ding, X.; Li, Y. Association between serum growth hormone levels and nonalcoholic fatty liver disease: a cross-sectional study. PLoS ONE 2012, 7, e44136. [Google Scholar] [CrossRef]
- Lonardo, A.; Loria, P.; Leonardi, F.; Ganazzi, D.; Carulli, N. Growth hormone plasma levels in nonalcoholic fatty liver disease. Am. J. Gastroenterol. 2002, 97, 1071–1072. [Google Scholar] [CrossRef] [PubMed]
- Arturi, F.; Succurro, E.; Procopio, C.; Pedace, E.; Mannino, G.C.; Lugarà, M.; Procopio, T.; Andreozzi, F.; Sciacqua, A.; Hribal, M.L.; et al. Nonalcoholic fatty liver disease is associated with low circulating levels of insulin-like growth factor-I. J. Clin. Endocrinol. Metab. 2011, 96, E1640–E1644. [Google Scholar] [CrossRef]
- Liang, S.; Yu, Z.; Song, X.; Wang, Y.; Li, M.; Xue, J. Reduced Growth Hormone Secretion is Associated with Nonalcoholic Fatty Liver Disease in Obese Children. Horm. Metab. Res. 2018, 50, 250–256. [Google Scholar] [CrossRef]
- Sumida, Y.; Yonei, Y.; Tanaka, S.; Mori, K.; Kanemasa, K.; Imai, S.; Taketani, H.; Hara, T.; Seko, Y.; Ishiba, H.; et al. Lower levels of insulin-like growth factor-1 standard deviation score are associated with histological severity of non-alcoholic fatty liver disease. Hepatol. Res. 2015, 45, 771–781. [Google Scholar] [CrossRef]
- Dichtel, L.E.; Corey, K.E.; Misdraji, J.; Bredella, M.A.; Schorr, M.; Osganian, S.A.; Young, B.J.; Sung, J.C.; Miller, K.K. The association between IGF-1 levels and the histological severity of Nonalcoholic Fatty Liver Disease. Clin. Transl. Gastroenterol. 2017, 8, e217. [Google Scholar] [CrossRef]
- Chishima, S.; Kogiso, T.; Matsushita, N.; Hashimoto, E.; Tokushige, K. The Relationship between the Growth Hormone/Insulin-like Growth Factor System and the Histological Features of Nonalcoholic Fatty Liver Disease. Intern. Med. 2017, 56, 473–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rufinatscha, K.; Ress, C.; Folie, S.; Haas, S.; Salzmann, K.; Moser, P.; Dobner, J.; Weiss, G.; Iruzubieta, P.; Arias-Loste, M.T.; et al. Metabolic effects of reduced growth hormone action in fatty liver disease. Hepatol. Int. 2018, 12, 474–481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, L.A.; Feldstein, A.; Lindor, K.D.; Angulo, P. Nonalcoholic fatty liver disease among patients with hypothalamic and pituitary dysfunction. Hepatology 2004, 39, 909–914. [Google Scholar] [CrossRef] [PubMed]
- Nishizawa, H.; Iguchi, G.; Murawaki, A.; Fukuoka, H.; Hayashi, Y.; Kaji, H.; Yamamoto, M.; Suda, K.; Takahashi, M.; Seo, Y.; et al. Nonalcoholic fatty liver disease in adult hypopituitary patients with GH deficiency and the impact of GH replacement therapy. Eur. J. Endocrinol. 2012, 167, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Ichikawa, T.; Hamasaki, K.; Ishikawa, H.; Ejima, E.; Eguchi, K.; Nakao, K. Non-alcoholic steatohepatitis and hepatic steatosis in patients with adult onset growth hormone deficiency. Gut 2003, 52, 914. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.W.; Kim, J.Y.; Kim, Y.E.; Lee, E.J. Metabolic parameters and nonalcoholic fatty liver disease in hypopituitary men. Horm. Metab. Res. 2011, 43, 48–54. [Google Scholar] [CrossRef] [PubMed]
- Gardner, C.J.; Irwin, A.J.; Daousi, C.; McFarlane, I.A.; Joseph, F.; Bell, J.D.; Thomas, E.L.; Adams, V.L.; Kemp, G.J.; Cuthbertson, D.J. Hepatic steatosis, GH deficiency and the effects of GH replacement: a Liverpool magnetic resonance spectroscopy study. Eur. J. Endocrinol. 2012, 166, 993–1002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meienberg, F.; Yee, M.; Johnston, D.; Cox, J.; Robinson, S.; Bell, J.D.; Thomas, E.L.; Taylor-Robinson, S.D.; Godsland, I. Liver fat in adults with GH deficiency: comparison to matched controls and the effect of GH replacement. Clin. Endocrinol. 2016, 85, 76–84. [Google Scholar] [CrossRef]
- Gilliland, T.; Dufour, S.; Shulman, G.I.; Petersen, K.F.; Emre, S.H. Resolution of non-alcoholic steatohepatitis after growth hormone replacement in a pediatric liver transplant patient with panhypopituitarism. Pediatr. Transplant. 2016, 20, 1157–1163. [Google Scholar] [CrossRef]
- de la Garza, R.G.; Morales-Garza, L.A.; Martin-Estal, I.; Castilla-Cortazar, I. Insulin-Like Growth Factor-1 Deficiency and Cirrhosis Establishment. J. Clin. Med. Res. 2017, 9, 233–247. [Google Scholar] [CrossRef] [Green Version]
- Adamek, A.; Kasprzak, A. Insulin-Like Growth Factor (IGF) System in Liver Diseases. Int. J. Mol. Sci. 2018, 19, 1308. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Fang, X.; Tajima, A.; Geng, X.; Ranganathan, S.; Dong, H.; Trucco, M.; Sperling, M.A. Evolution of hepatic steatosis to fibrosis and adenoma formation in liver-specific growth hormone receptor knockout mice. Front. Endocrinol. 2014, 5, 218. [Google Scholar] [CrossRef] [PubMed]
- Cordoba-Chacon, J.; Majumdar, N.; List, E.O.; Diaz-Ruiz, A.; Frank, S.J.; Manzano, A.; Bartrons, R.; Puchowicz, M.; Kopchick, J.J.; Kineman, R.D. Growth Hormone Inhibits Hepatic De Novo Lipogenesis in Adult Mice. Diabetes 2015, 64, 3093–3103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sos, B.C.; Harris, C.; Nordstrom, S.M.; Tran, J.L.; Balázs, M.; Caplazi, P.; Febbraio, M.; Applegate, M.A.; Wagner, K.U.; Weiss, E.J. Abrogation of growth hormone secretion rescues fatty liver in mice with hepatocyte-specific deletion of JAK2. J. Clin. Invest. 2011, 121, 1412–1423. [Google Scholar] [CrossRef] [PubMed]
- Barclay, J.L.; Nelson, C.N.; Ishikawa, M.; Murray, L.A.; Kerr, L.M.; McPhee, T.R.; Powell, E.E.; Waters, M.J. GH-dependent STAT5 signaling plays an important role in hepatic lipid metabolism. Endocrinology 2011, 152, 181–192. [Google Scholar] [CrossRef]
- Nishizawa, H.; Takahashi, M.; Fukuoka, H.; Iguchi, G.; Kitazawa, R.; Takahashi, Y. GH-independent IGF-1 action is essential to prevent the development of nonalcoholic steatohepatitis in a GH-deficient rat model. Biochem. Biophys. Res. Commun. 2012, 423, 295–300. [Google Scholar] [CrossRef] [PubMed]
- Puche, J.E.; García-Fernández, M.; Muntané, J.; Rioja, J.; González-Barón, S.; Castilla Cortazar, I. Low doses of insulin-like growth factor-I induce mitochondrial protection in aging rats. Endocrinology 2008, 149, 2620–2627. [Google Scholar] [CrossRef] [PubMed]
- Karamitri, A.; Jockers, R. Melatonin in type 2 diabetes mellitus and obesity. Nat. Rev. Endocrinol. 2019, 15, 105–125. [Google Scholar] [CrossRef]
- Zhou, H.; Du, W.; Li, Y.; Shi, C.; Hu, N.; Ma, S.; Wang, W.; Ren, J. Effects of melatonin on fatty liver disease: The role of NR4A1/DNA-PKcs/p53 pathway, mitochondrial fission, and mitophagy. J. Pineal Res. 2018, 64, e12450. [Google Scholar] [CrossRef]
- Takahashi, Y.; Iida, K.; Takahashi, K.; Yoshioka, S.; Fukuoka, H.; Takeno, R.; Imanaka, M.; Nishizawa, H.; Takahashi, M.; Seo, Y.; et al. Growth hormone reverses nonalcoholic steatohepatitis in a patient with adult growth hormone deficiency. Gastroenterology 2007, 132, 938–943. [Google Scholar] [CrossRef]
- Nishizawa, H.; Iguchi, G.; Fukuoka, H.; Takahashi, M.; Suda, K.; Bando, H.; Matsumoto, R.; Yoshida, K.; Odake, Y.; Ogawa, W.; et al. IGF-I induces senescence of hepatic stellate cells and limits fibrosis in a p53-dependent manner. Sci. Rep. 2016, 6, 34605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Attanasio, A.F.; Bates, P.C.; Ho, K.K.; Webb, S.M.; Ross, R.J.; Strasburger, C.J.; Bouillon, R.; Crowe, B.; Selander, K.; Valle, D.; et al. Hypoptiuitary Control and Complications Study International Advisory Board. Human growth hormone replacement in adult hypopituitary patients: long-term effects on body composition and lipid status--3-year results from the Hypo CCS Database. J. Clin. Endocrinol. Metab. 2002, 87, 1600–1606. [Google Scholar]
- Molitch, M.E.; Clemmons, D.R.; Malozowski, S.; Merriam, G.R.; Vance, M.L. Endocrine Society. Evaluation and treatment of adult growth hormone deficiency: An Endocrine Society clinical practice guideline. J. Clin. Endocrinol. Metab. 2011, 96, 1587–1609. [Google Scholar] [CrossRef]
- Takano, S.; Kanzaki, S.; Sato, M.; Kubo, T.; Seino, Y. Effect of growth hormone on fatty liver in panhypopituitarism. Arch. Dis. Child. 1997, 76, 537–538. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, R.; Fukuoka, H.; Iguchi, G.; Nishizawa, H.; Bando, H.; Suda, K.; Takahashi, M.; Takahashi, Y. Long-term effects of growth hormone replacement therapy on liver function in adult patients with growth hormone deficiency. Growth Horm. IGF Res. 2014, 24, 174–179. [Google Scholar] [CrossRef] [PubMed]
- Sesmilo, G.; Biller, B.M.; Llevadot, J.; Hayden, D.; Hanson, G.; Rifai, N.; Klibanski, A. Effects of growth hormone administration on inflammatory and other cardiovascular risk markers in men with growth hormone deficiency. A randomized, controlled clinical trial. Ann. Intern Med. 2000, 33, 111–122. [Google Scholar] [CrossRef]
- Fukuda, I.; Hizuka, N.; Yasumoto, K.; Morita, J.; Kurimoto, M.; Takano, K. Metabolic co-morbidities revealed in patients with childhood-onset adult GH deficiency after cessation of GH replacement therapy for short stature. Endocr. J. 2008, 55, 977–984. [Google Scholar] [CrossRef]
- Zhang, H.; Liu, Y.; Wang, L.; Li, Z.; Zhang, H.; Wu, J.; Rahman, N.; Guo, Y.; Li, D.; Li, N.; et al. Differential effects of estrogen/androgen on the prevention of nonalcoholic fatty liver disease in the male rat. J. Lipid Res. 2013, 54, 345–357. [Google Scholar] [CrossRef] [Green Version]
- Grossmann, M.; Wierman, M.E.; Angus, P.; Handelsman, D.J. Reproductive Endocrinology of Nonalcoholic Fatty Liver Disease. Endocr. Rev. 2019, 40, 417–446. [Google Scholar] [CrossRef]
- Li, J.; Su, J.M.; Zeng, X.J. Is primary biliary cirrhosis another example of an immune-mediated complication of klinefelter syndrome? J. Clin. Rheumatol. 2004, 10, 286–287. [Google Scholar] [CrossRef]
- Tian, Y.; Wang, C.; Liu, J.X.; Wang, H.H. Primary Biliary Cirrhosis-Related Autoimmune Hemolytic Anemia: Three Case Reports and Review of the Literature. Case Rep. Gastroenterol. 2009, 3, 240–247. [Google Scholar] [CrossRef] [PubMed]
- Machlab, S.; Miquel, M.; Voltà, T.; Escoda, M.R.; Vergara, M. Turner syndrome as a cause of liver cirrhosis. Gastroenterol. Hepatol. 2018, 41, 308–309. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Ref. | Study Population | Thyroid Function Tests—Definition of Hypothyroidism | NAFLD Diagnosis; Prevalence of NAFLD | Adjustments | Main Findings |
|---|---|---|---|---|---|
| Observational studies | |||||
| [53] | Case-control study: 174 US patients with NASH and 442 age-, sex-, race-, and BMI-matched controls | Hypothyroidism was defined by the self-reported use of levothyroxine | Biopsy | Diabetes, dyslipidemia, hypertension | Hypothyroidism was independently associated with an increased risk of NASH |
| [54] | Cross-sectional study: 878 Chinese elderly individuals | TSH, FT4, and FT3 | USG; 26% with NAFLD | Age, BMI, waist circumference, triglyceride, fasting glucose, uric acid, creatinine | FT4 levels were independently associated with an increased risk of NAFLD |
| [55] | Cross-sectional study: 1322 Chinese individuals | Hypothyroidism was defined as TSH levels >2.5 mIU/L | USG; 25% with NAFLD | Age, sex, BMI, body fat, triglycerides, systolic blood pressure, diastolic blood pressure, fasting glucose | Hypothyroidism was not independently associated with an increased risk of NAFLD |
| [56] | Population-based study (Study of Health in Pomerania): 3661 German individuals | Subclinical hypothyroidism was defined as TSH >3 mIU/L and normal FT4; overt hypo thyroidism was defined as TSH >3 mIU/L and FT4 <7 pmol/L | USG; 16% with NAFLD | Age, waist circumference, physical activity, alcohol intake, food intake pattern | Hypothyroidism was not independently associated with an increased risk of NAFLD. Contrariwise, serum FT4 levels were inversely associated with NAFLD |
| [57] | Case-control study: 246 US patients with biopsy-proven NAFLD and 430 age-, sex-, race-, and BMI-matched controls | Hypothyroidism was defined by the self-reported use of levothyroxine | Biopsy | Alcohol use | Hypothyroidism was independently associated with an increased risk of NAFLD |
| [58] | Cross-sectional study: 4648 South Korean adults | Subclinical hypothyroidism was defined as TSH ≥4.1 mIU/L and normal FT4; overt hypothyroidism was defined as FT4 <0.7 ng/dL | USG; 25% with NAFLD | Age, sex, BMI, waist circumference, lipids hypertension, diabetes | Subclinical and overt hypothyroidism were independently associated with an increased risk of NAFLD |
| [59] | Prospective case-control study: 327 Chinese patients with subclinical hypothyroidism and 327 age-, sex-, and BMI-matched euthyroid controls | Subclinical hypothyroidism was defined as TSH ≥4.5 mIU/L and normal FT4 levels | USG; 15% of individuals developed NAFLD over a median follow-up of ~5 years | Waist circumference, systolic blood pressure, diastolic blood pressure, lipids, fasting glucose | Subclinical hypothyroidism was independently associated with an increased risk of NAFLD during the follow-up |
| [60] | Cross-sectional study: 69 Italian euthyroid patients with biopsy-proven NAFLD | TSH | Biopsy; 44 patients had NASH, whereas 25 patients had only steatosis | Age, sex, BMI, HOMA | TSH levels were independently and positively associated with an increased risk of NASH |
| [61] | Cross-sectional study: 832 Iranian healthy individuals | Subclinical hypothyroidism was defined as TSH >5.2 mIU/L and normal FT4 levels; overt hypothyroidism was defined as TSH >5.2 mIU/L and FT4 <11.5 pmol/L | USG; 15% with NAFLD | None | Subclinical and overt hypothyroidism were not independently associated with an increased risk of NAFLD |
| [62] | Cross-sectional study: 753 Mexican adults from the Genetics of Atherosclerotic Disease study | Subclinical hypothyroidism was defined as TSH >4.5 mIU/L and normal FT4 levels | Computed tomography; 31% with NAFLD | None | Subclinical hypothyroidism was not independently associated with an increased risk of NAFLD |
| [63] | Cross-sectional study: 739 Chinese euthyroid individuals | TSH, FT4 | USG; 26% with NAFLD | Age, sex, BMI, smoking status, systolic blood pressure, diastolic blood pressure, lipids, FT3 | TSH and FT4 levels were independently associated with an increased risk of NAFLD |
| [64] | Cross-sectional study: 2576 euthyroid South Koreans | TSH, FT4, FT3 | USG; 38% with NAFLD | Age, sex, smoking status, hypertension, fasting glucose, lipids, creatinine, uric acid | FT3 levels, but not TSH and FT4 levels, were independently associated with an increased risk of NAFLD |
| [65] | Cross-sectional study: 1154 Chinese with chronic hepatitis B | Subclinical hypothyroidism was defined as serum TSH >5.3 mUI/L and normal FT4 levels, whereas overt hypothyroidism was defined as serum FT4 level <7.9 pmol/L and TSH >5.3 mIU/L | Biopsy; 23% with NAFLD | Age, sex | TSH levels were independently and positively associated with an increased risk of NAFLD |
| [66] | Case-control study: 500 biopsy-proven NAFLD Indians and 300 age-, sex-, and BMI-matched controls | Hypothyroidism was defined by the self-reported use of levothyroxine | Biopsy | Age, sex, BMI, transaminases | Hypothyroidism was independently associated with an increased risk of NAFLD |
| [67] | Population-based study: 1276 Germans | Subclinical hypothyroidism was defined as TSH ≥3.4 mIU/L and normal FT4 levels; overt hypothyroidism was defined as total T4 levels <12.8 pmol/L and TSH levels ≥3.4 mIU/L | USG; 25% with NAFLD | Age, sex, BMI, waist circumference | Subclinical and overt hypothyroidism were not independently associated with an increased risk of NAFLD |
| [68] | Longitudinal study: 18,544 healthy South Koreans NAFLD-free at baseline | Subclinical hypothyroidism was defined as TSH >4.2 mIU/L normal FT4 levels; overt hypothyroidism was defined as TSH >4.2 mIU/L and FT4 <0.9 ng/dL | USG; 2348 individuals developed incident NAFLD during a mean follow-up of 4 years | Age, sex, BMI, metabolic syndrome | Subclinical and overt hypothyroidism were not independently associated with an increased risk of NAFLD |
| [69] | Cross-sectional study: 232 USA euthyroid patients with T2D | FT4 | MRS; liver biopsy was performed in patients with a diagnosis of NAFLD; 63% with NAFLD | Age, BMI, hemoglobin A1c | FT4 levels were significantly associated with hepatic triglyceride. However, no associations between thyroid function tests and liver histological parameters (such as inflammation, hepatocyte ballooning, and advanced fibrosis) were observed |
| [70] | A population-based, prospective cohort study (the Rotterdam study): 9419 Dutch euthyroid individuals | Subclinical hypothyroidism was defined as serum TSH levels >4.0 mIU/L and normal FT4; overt hypothyroidism was defined as serum TSH >4.0 mIU/L and FT4 levels <0.8 ng/dL | Fatty liver index at baseline; USG and Fibroscan® at follow-up; 13% of participants developed incident NAFLD over a median follow-up of 10 years | Age, sex, BMI, alcohol intake, smoking status, follow-up time, use of lipid-lowering agents, lipids, hypertension, diabetes | Compared to euthyroidism, any form of hypothyroidism was associated with an increased risk of NAFLD. Moreover, subclinical and overt hypothyroidism were associated with an increased risk of liver fibrosis as detected by Fibroscan® |
| [71] | Cross-sectional study: 115 Turkish individuals | FT3/FT4 ratio | USG; 60% with NAFLD | Waist circumference, lipids, uric acid, HOMA | FT3/FT4 ratio was independently associated with an increased risk of NAFLD |
| [72] | Population-based study (Lifelines Cohort Study): 20,289 Dutch euthyroid individuals | TSH, FT4, FT3 | FLI (≥60); 21% with NAFLD | Age, sex | FLI ≥60 was independently associated with higher FT3 and lower FT4 levels |
| [73] | Cross-sectional study: 580 Filipino adults | TSH >4.5 mIU/L without previous history of thyroid disease | USG; 48% with NAFLD | None | NAFLD was not independently associated with TSH levels |
| [74] | Case-control study 100 Indian adult non-obese hypothyroid patients and 100 age-, sex-, and BMI-matched euthyroid controls | Subclinical hypothyroidism was defined as serum TSH levels ≥4.1 mIU/L and normal FT4 levels, whereas overt hypothyroidism was defined as serum TSH levels ≥4.1 mIU/L and FT4 levels <0.7 ng/dL | USG; 42% with NAFLD | None | NAFLD was diagnosed in 30 patients with hypothyroid and in 12 controls |
| [75] | Cross-sectional study: 425 South Koreans with biopsy-proven NAFLD | Subclinical hypothyroidism was defined as serum TSH >4.5 mIU/L and normal FT4 | Biopsy | Age, sex, BMI, smoking status, hypertension, diabetes, lipids, visceral adipose tissue area, HOMA-IR | Subclinical hypothyroidism was independently associated with an increased risk of NASH and advanced fibrosis |
| [76] | Population-based study: 3452 Koreans from Korea National Health and Nutrition Examination Survey 2013 to 2015 | Subclinical hypothyroidism was defined as a serum TSH levels >6.7 mIU/L with serum FT4 within a normal range | Hepatic steatosis index (≥36) | Age, smoking status, physical activity, income, MetS, waist circumference, lipids, systolic blood pressure, diastolic blood pressure, fasting glucose, urine iodine, thyroid peroxidase antibodies | Subclinical hypothyroidism was independently associated with an increased risk of NAFLD in males, but not in females |
| Meta-analyses | |||||
| [77] | 14 observational studies for a total of 42,143 individuals | Self-reported history of hypothyroidism with use of levothyroxine replacement therapy or by presence of abnormal thyroid function tests | Imaging and biopsy | Multiple demographic and clinical variables | NAFLD was not independently associated with hypothyroidism |
| [78] | 13 observational studies for a total of 37,194 individuals | Self-reported history of hypothyroidism with use of levothyroxine replacement therapy or by presence of abnormal thyroid function tests | Liver enzymes, imaging, and biopsy | Multiple demographic and clinical variables | Hypothyroidism was independently associated with an increased risk of NAFLD |
| [79] | 12 cross-sectional and 3 longitudinal studies for a total of 44,140 individuals | Self-reported history of hypothyroidism with use of levothyroxine replacement therapy or by presence of abnormal thyroid function tests | Imaging and biopsy | Multiple demographic and clinical variables | Hypothyroidism was independently associated with an increased risk of prevalent NAFLD. Meta-analysis of data from 3 longitudinal studies documented that subclinical hypothyroidism was not independently associated with an increased risk of incident over a median follow-up of 5 years |
| [80] | 26 observational studies for a total of 61,548 individuals | Self-reported history of hypothyroidism with use of levothyroxine replacement therapy or by presence of abnormal thyroid function tests | Imaging, FLI, and biopsy | Multiple demographic and clinical variables | NAFLD patients had significantly higher TSH levels compared to controls. In addition, hypothyroidism was significantly associated with the risk of NAFLD |
| Primary (hypergonadotropic) |
| Congenital anatomical abnormalities of the gonads |
| Castration |
| Specific syndromes (e.g., Turner’s syndrome; Alstrom’s syndrome; Kallmann syndrome) |
| Drugs (e.g., antiandrogens, antiestrogens, chemotherapy) |
| Radiation |
| Hemochromatosis |
| Autoimmune |
| Secondary to pituitary/hypothalamic failure (hypogonadotropic) |
| Congenital |
| Tumor |
| Trauma |
| Radiation |
| Functional |
| Ref. | Study Characteristics | Diagnosis of NAFLD | Main Findings |
|---|---|---|---|
| Hypogonadism in men | |||
| [88] | A cohort of 117 hypogonadic men (34–69 years), who were treated with HRT (testosterone undecanoate for 1 year) | ALT, AST, and CRP | HRT was associated with a significant reduction in adiposity measures, lipid profile, and rate of individuals who met the criteria of the MetS. Steatosis was strongly associated with all components of the MetS and RLE, which were associated with higher plasma CRP concentrations, significantly decreased following 1-year HRT |
| [89] | 1944 non-drinking men submitted to repeated liver ultrasonography over a median 4.2-year period, with available baseline serum TT level were evaluated | Ultrasonography | Baseline levels of TT were significantly lower in NAFLD individuals in cross-sectional analyses. However, TT level was not independently associated with either development or regression of NAFLD during follow-up |
| [90] | Retrospective study comparing 75 Chinese IHH men (mean age 21.4 ± 3.8 years, range 17–30 years) to 135 age- and sex-matched healthy controls | Ultrasonography | Compared to healthy controls, IHH men had higher serum ACTH levels, lower cortisol levels, deranged glycolipidic profile and a higher prevalence of NAFLD. NAFLD was independently associated with ACTH levels |
| [91] | 55 consecutive men with chronic SCI admitted to a rehabilitation program were submitted to clinical/biochemical evaluations and liver ultrasonography | Ultrasonography | NAFLD was diagnosed in 49% of cases. TT and FT levels were independently associated with NAFLD; the risk of NAFLD increased by 1% for each decrement of 1 ng/dL of TT and of 3% for each decrement of 1 pg/mL of FT |
| [92] | Out of 380,669 men with histologically proven prostate cancer, 31,117 elderly men who received ADT were identified by using a representative cancer registry. Individuals with metastatic disease, pre-existent MetS, diabetes, preexisting liver disease, or a history of alcoholism/ alcohol related disorders were excluded | Diagnostic and procedural codes from physician office or inpatient visits | Elderly men submitted to ADT were more likely to be diagnosed with NAFLD, cirrhosis, liver necrosis, and any liver disease |
| Hypogonadism in women | |||
| [93] | Retrospective analysis of serum liver enzymes in 80 women with TS, followed by a prospective study in 20 women with TS following 3 months on-and-off HRT | GGT, AST, and ALP | 44% of women with TS had RLE. HRT resulted in a significant reduction in serum liver enzymes without improving serum protein concentrations |
| [94] | Prospective, double-blind, RCT vs. placebo recruiting 5408 women who underwent hysterectomies and enrolled into the multicentric Italian tamoxifen chemoprevention trial | NAFLD was suspected based on de novo incident chronic unexplained hyper-transaminasemia (× 1.5 n.v.) over a 6-month period, in the absence of competing etiologies and confirmed with ultrasonography. NAFLD women defined as above were offered confirmative liver biopsy | 52 out of 64 women who met the predefined criteria in the course of follow-up developed suspected US-confirmed NAFLD: 34 on tamoxifen vs. 18 on placebo, HR = 2.0 (95% CI 1.1–3.5; p = 0.04). Further to tamoxifen, overweight (HR 2.4, 95% CI 1.2–4.8), obesity (HR 3.6, 95%CI 1.7–7.6), hypercholesterolemia (HR 3.4, 9%% CI 1.4–7.8), and hypertension (HR 2.0, 95% CI 1.0–3.8) were associated with increased risk of incident NAFLD. Out of 20 women submitted to LB, 15 had mild-to-moderate NASH (12 tamoxifen vs. 3 placebo), and 5 had simple steatosis (1 tamoxifen vs. 4 placebo). None developed cirrhosis over a mean 8.7-year follow-up |
| [95] | 50 women with T2D entered a double-blind, RCT of HRT vs. placebo | AST, ALT, GGT, and ALP | Compared to those randomized to placebo, women randomized and compliant to HRT (n = 19) had significant reductions in serum liver enzymes |
| [96] | Serum liver enzymes were assessed in 169 women (14 with TS and 11 controls with hypogonadism) on HRT with oral E2 | GGT, ALT, ALP, albumin, and bilirubin | The prevalence and incidence rates of RLE among women with TS were 91% and 2.1% per year, respectively. RLE were associated with total cholesterol and BMI, and reversed by increasing doses of HRT |
| [97] | 218 women with TS (mean age 33 ± 13, range 16–71 years) from outpatient clinics at Swedish university hospitals. | AST, ALT, ALP, GGT, serology for viral hepatitis and liver-specific auto-antibodies | 36% of TS women had one or more RLE; the most prevalent was serum GGT level, which was independently associated with total cholesterol both at baseline and at 5 years. Liver histology findings in 6 TS women submitted to LB included cholangitis (n = 1), hepatitis C (n = 1), steatosis (n = 2), and normal (n = 2). Withdrawal of estrogen substitution did not influence serum liver enzymes |
| [98] | This Mexican cross-sectional study compared anthropometric, metabolic, hormonal (serum estradiol and cortisol concentrations), and biochemical variables in 93 women with NAFLD and as many NAFLD-free controls | Ultrasonography and transient elastography; APRI, NFS | The prevalence of NAFLD in premenopausal, post-menopausal, and PCOS patients was 32.2, 57.9, and 62%, respectively. Age, adiposity measures, fasting glucose, HOMA-IR, and insulin were significantly higher in NAFLD patients. Compared to NAFLD women, those NAFLD-free women had significantly higher levels of serum E2 |
| [99] | 541 individuals with biopsy-proven NASH were recruited. LRA was used to determine the association among sex, menopause, and severity of hepatic fibrosis | Liver biopsy | Compared to pre-menopausal women, men and post-menopausal women had a higher risk of advanced liver fibrosis suggesting that estrogens may protect from fibrosis |
| [100] | Survey of questionnaires referring to 492 patients with TS (age 17.1–42.5 years) administered by attending physicians throughout Japan. For comparison purposes, data from the National Health and Nutrition Survey were used | Liver dysfunction was defined as AST ≥41 U/L, ALT ≥36 U/L, or GGT ≥60 U/L | Women with TS tend to become obese from young ages (15–39 years). Compared to the Japanese general female population, women with TS had higher prevalence rates of diabetes, hypertension, dyslipidemia, and liver dysfunction which were associated with increasing BMI rather than karyotypes |
| [101] | Retrospective analysis of incident fatty liver and/or RLE during SERM treatment in 1061 women who were treated for breast cancer | Imaging and/or ALT | SERM treatment was independently associated with an increased the risk of incident raised serum ALT levels and/or fatty liver. Consistently, SERM discontinuation was associated with normalization of raised ALT levels in virtually all cases. No cases of liver-related death/ progression to cirrhosis were registered |
| [102] | Analysis of data from 488 post-menopausal women with histologically proven NAFLD and self-reported information on age at menopause | Liver histology | In post-menopausal women with NAFLD, the duration of estrogen deficiency affected risk of liver fibrosis. |
| Glucose Metabolism | Lipid Metabolism | |||
|---|---|---|---|---|
| Estrogen | ↑ Insulin clearance | ↑ Glycogen storage | ↑ Lipolysis | ↑ Cholesterol removal |
| ↓Gluconeogenesis | ↓Lipogenesis ↓Lipid uptake | ↓Cholesterol synthesis | ||
| Androgen | ↑ Insulin receptor | ↑ Glycogen synthesis | ↑ Cholesterol uptake | ↑ Cholesterol synthesis |
| ↓Glucose uptake | ↓Lipogenesis | ↓Cholesterol removal | ||
| Ref. | Method | Findings | Comment |
|---|---|---|---|
| [115] | Overall, 439 patients with biopsy-proven mild/advanced NAFLD were enrolled in the USA Simple steatosis and NASH with initial fibrosis (i.e., stages 0–2) were categorized together as “mild NAFLD” NASH with fibrosis stage 3–4 was defined as “advanced NAFLD” DHEA-S was measured with ELISA. | Compared to patients with mild disease, individuals with advanced NAFLD exhibited lower serum concentrations of DHEA-S Decreasing DHEA-S paralleled increasing stages of fibrosis All patients who had advanced NAFLD exhibited as low DHEA-S levels which were compatible with adrenal insufficiency in the majority of cases | NASH with advanced stages of fibrosis is strongly associated with low serum concentrations of DHEA-S |
| [116] | Data from 133 Japanese patients with biopsy-proven NAFLD (90 with NASH: 73 patients had fibrosis stage 0–2, and 17 had advanced disease as defined by fibrosis stage 3 or 4) were compared to 399 sex- and age-matched healthy controls DHEA-S was measured by CEI | DHEA-S serum concentrations were not statistically different from those found in the controls Lower DHEA-S paralleled with increasing stages of fibrosis and this association remained after adjustment for confounding factors (age, sex, and IR) | Low circulating DHEA-S could have a causal role in the progression of NAFLD to fibrosing NASH |
| [117] | 69 Japanese men who had NAFLD diagnosed with ultrasonography were extracted from a larger sample of 158 Japanese men who had neither viral liver diseases nor an alcoholic intake >20 g/day DHEA-S was measured by RIA | At multivariate regression analysis, serum ALT was positively correlated with serum DHEA-S, serum triglyceride, and BMI | DHEAS levels are increased in patients with NAFLD with elevated ALT levels |
| [118] | 160 individuals with morbid obesity were submitted to liver biopsy which showed SS in 72, NASH with fibrosis stage 0–1 in 60, and NASH with fibrosis stage ≥2 in 12 patients. 16 had normal liver histology DHEA-S was measured by ELISA | With one exception, all patients with NASH and fibrosis stage 2–3 had low serum DHEA concentrations, i.e., <123 μg/dL. | Low serum levels of DHEA are very common among morbidly obese individuals with NASH and advanced fibrosis |
| [119] | This study enrolled (a) a training cohort of 44 patients with biopsy proven NAFLD and (b) a validation cohort comprising 105 patients with biopsy-proven NAFLD and 26 with biopsy-proven PBC patients. Moreover, 48 age-matched healthy controls who had normal liver tests and no infection with hepatitis viruses were evaluated Metabolites with LMW were identified with capillary electrophoresis and liquid chromatography with mass spectrometry | In the training cohort, increasing severity of hepatic fibrosis was associated with a decrease of DHEA-S and etiocholanolone-S and an increase of 16-OH-DHEA-S. In the validation cohort, the 16-OH-DHEA-S/DHEA-S ratio and 16-OH-DHEA-S/etiocholanolone-S ratio were also strongly associated with the stage of fibrosis. Conversely, DHEA-S, etiocholanolone-S, and the two ratios were not associated with the stage of fibrosis in patients with PBC | Disturbances in the hormonal profile are a specific feature of NAFLD, which could be exploited for therapeutic purposes |
| [90] | Retrospective case-control study enrolling 75 Chinese men with IHH and 135 age- and sex-matched healthy controls. The diagnosis of presence/absence of NAFLD was based on ultrasonography to which only 63 individuals with IHH were submitted DHEA-S was measured by chemiluminescence | DHEA-S serum concentrations were significantly more elevated among those 22 patients with IHH and NAFLD than among those 41 with IHH without NAFLD | There is a complex interaction among the HPA axis, testosterone deficiency, and perturbed metabolic health in men with IHH |
| Author, Ref. | Method, Cohort, and Diagnostic Criteria | Findings |
|---|---|---|
| [143] | Longitudinal cohort study. 879 patients with hypothalamic or pituitary dysfunction. NAFLD diagnosis based on with imaging and liver enzyme alteration. Liver biopsy was performed in a subgroup of 10 NAFLD patients | NAFLD was found in 21 patients with metabolic syndrome-like phenotype (prevalence of 2.3%). The majority of biopsy-proven NAFLD patients exhibited advanced forms, i.e., cirrhosis and NASH with fibrosis |
| [144] | Cross-sectional retrospective study. 66 adults with hypopituitarism and GHD compared to 83 age-, gender-, and BMI-matched healthy controls. 19 patients received GH replacement therapy according to clinical recommendations. GHD diagnosis based on insulin tolerance test or GH releasing peptide-2 test. NAFLD diagnosis based on ultrasound. Liver biopsy was performed in 16 patients | NAFLD prevalence was significantly higher of 6.4-fold in GHD group compared to healthy controls, independently of obesity. Overweight and insulin resistance were more prevalent in GHD group with NAFLD. Histological NASH was found in 14 out of 16. GHRT was associated with reduction of liver enzyme and improvement of steatosis and fibrosis |
| [145] | Cross-sectional study. 18 adult patients with hypopituitarism, of these 13 with GHD. NAFLD diagnosis based on computer tomography (CT) (liver/spleen CT value <0.9) | Prevalence of hepatic steatosis higher in GHD subjects compared to hypopituitaric subjects without GHD, independently of BMI and triglycerides levels |
| [146] | Cross-sectional observational study. 34 Korean patients with hypopituitarism and 40 age- and sex-matched lean healthy controls. GHD defined as peak GH level of <3 ng/mL. NAFLD diagnosis based on ultrasound | Prevalence of fatty liver was significantly higher in men with hypopituitarism than controls. Among NAFLD patients with hypopituitarism, GH levels were lower and negatively correlated with degree of steatosis |
| [147] | Cross-sectional study. 28 patients with GHD and 24 age- and BMI-matched controls. 12 GHD patients evaluated longitudinally before and 6 months after start of GHRT. GHD diagnosis based on GH response <3 mg/L after glucagon stimulation. NAFLD defined as MRI-assessed intrahepatic lipid content (IHLC) > 5.6% | Although GHD patients exhibited higher visceral fat, the 2 groups presented similar liver enzyme levels and IHLC. GHRT was associated with reduction of subcutaneous and visceral adipose tissue and, in those with baseline high liver fat, with a positive trend in reduction of IHLC |
| [148] | Cross-sectional study. 22 adult patients with GHD and 44 age-, gender-, and BMI-matched healthy controls. 9 GHD patients received GHRT. GHD diagnosis based on GH levels <7.8 mU/L after glucagon stimulation test. NAFLD diagnosis based on IHLC >5.56% assessed with proton magnetic resonance spectroscopy (1H-MRS) | No significant difference in IHLC and prevalence of hepatic steatosis was observed in the two groups. In GHD patients receiving GHRT, no changes in IHLC were observed |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lonardo, A.; Mantovani, A.; Lugari, S.; Targher, G. NAFLD in Some Common Endocrine Diseases: Prevalence, Pathophysiology, and Principles of Diagnosis and Management. Int. J. Mol. Sci. 2019, 20, 2841. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20112841
Lonardo A, Mantovani A, Lugari S, Targher G. NAFLD in Some Common Endocrine Diseases: Prevalence, Pathophysiology, and Principles of Diagnosis and Management. International Journal of Molecular Sciences. 2019; 20(11):2841. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20112841
Chicago/Turabian StyleLonardo, Amedeo, Alessandro Mantovani, Simonetta Lugari, and Giovanni Targher. 2019. "NAFLD in Some Common Endocrine Diseases: Prevalence, Pathophysiology, and Principles of Diagnosis and Management" International Journal of Molecular Sciences 20, no. 11: 2841. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20112841