Class I-Histone Deacetylase (HDAC) Inhibition is Superior to pan-HDAC Inhibition in Modulating Cisplatin Potency in High Grade Serous Ovarian Cancer Cell Lines

 ,
, 
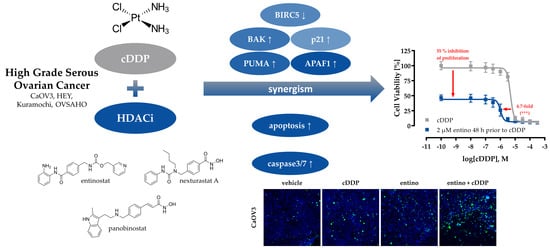
Abstract
:
1. Introduction
2. Results
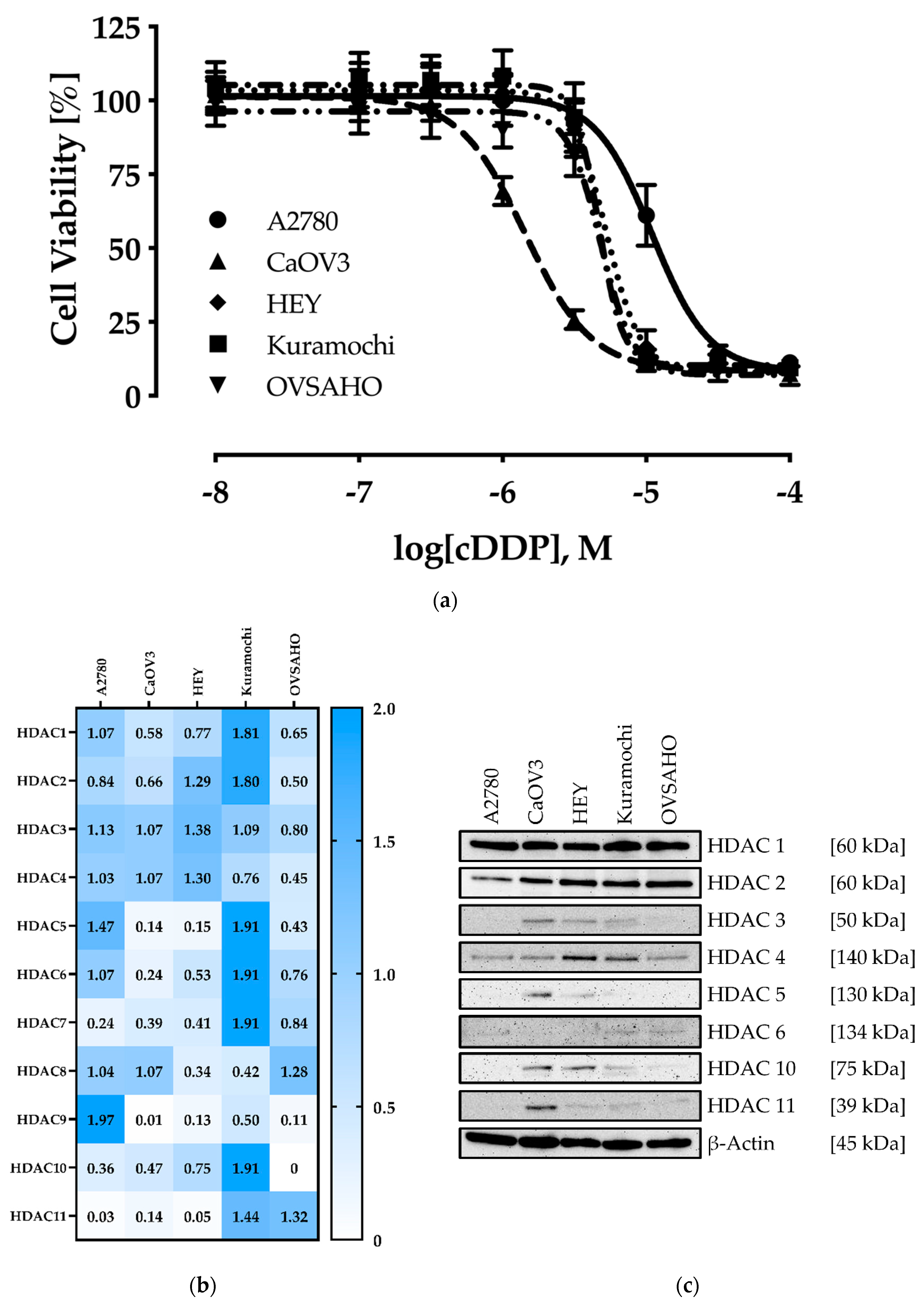
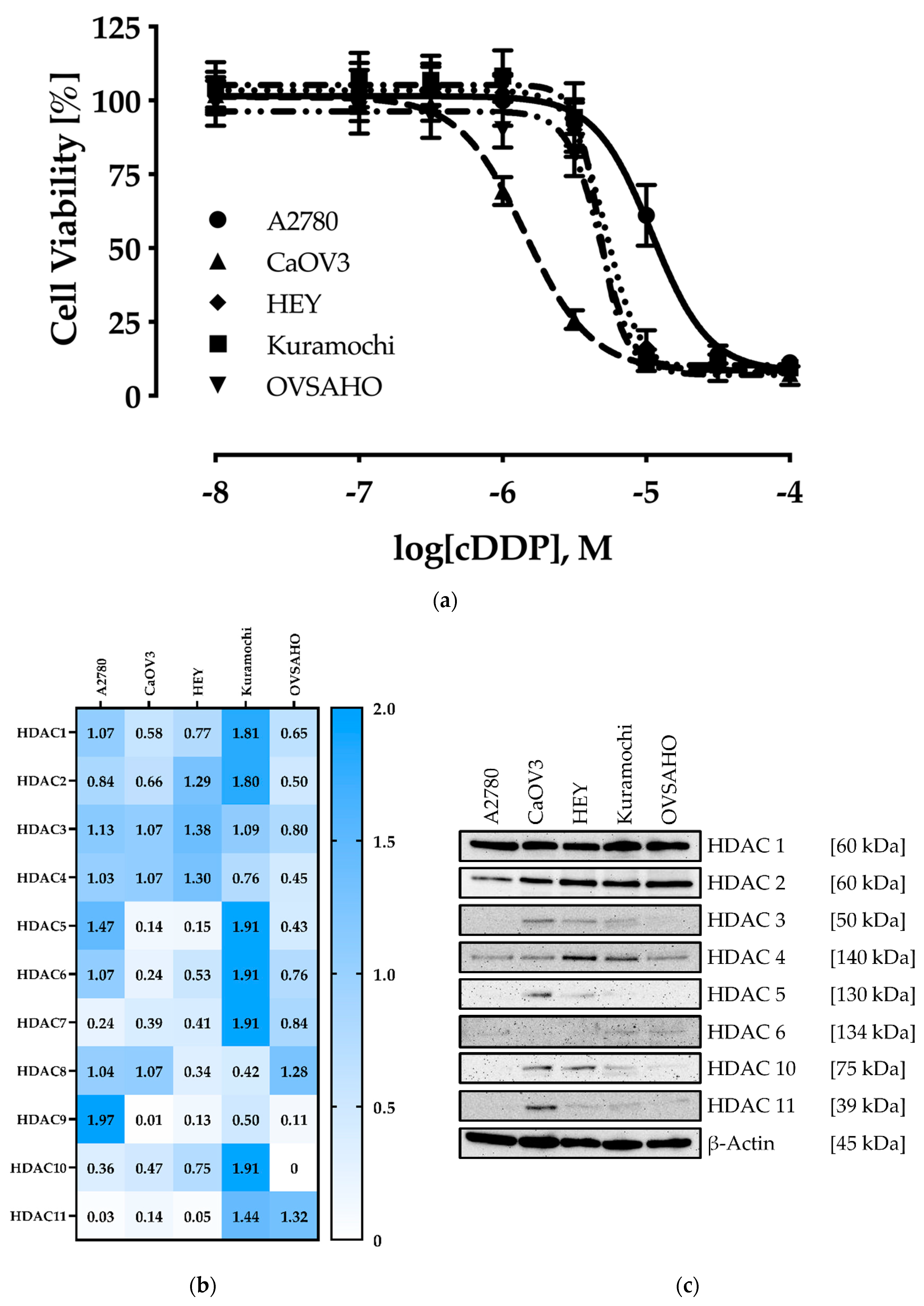
2.1. Characterization of the Human Ovarian Cancer Cell Lines
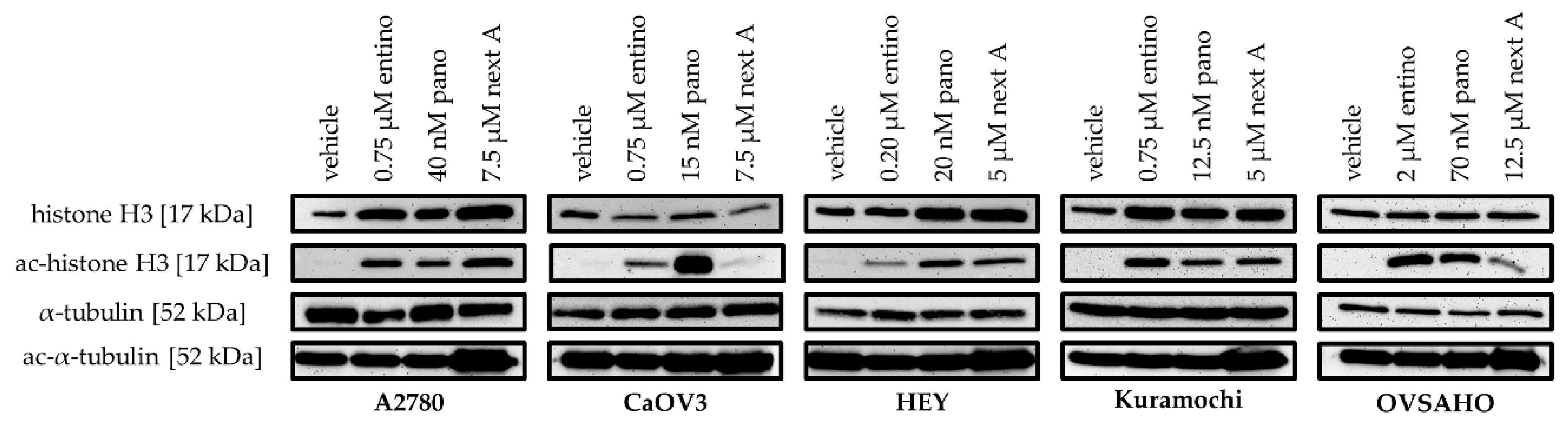
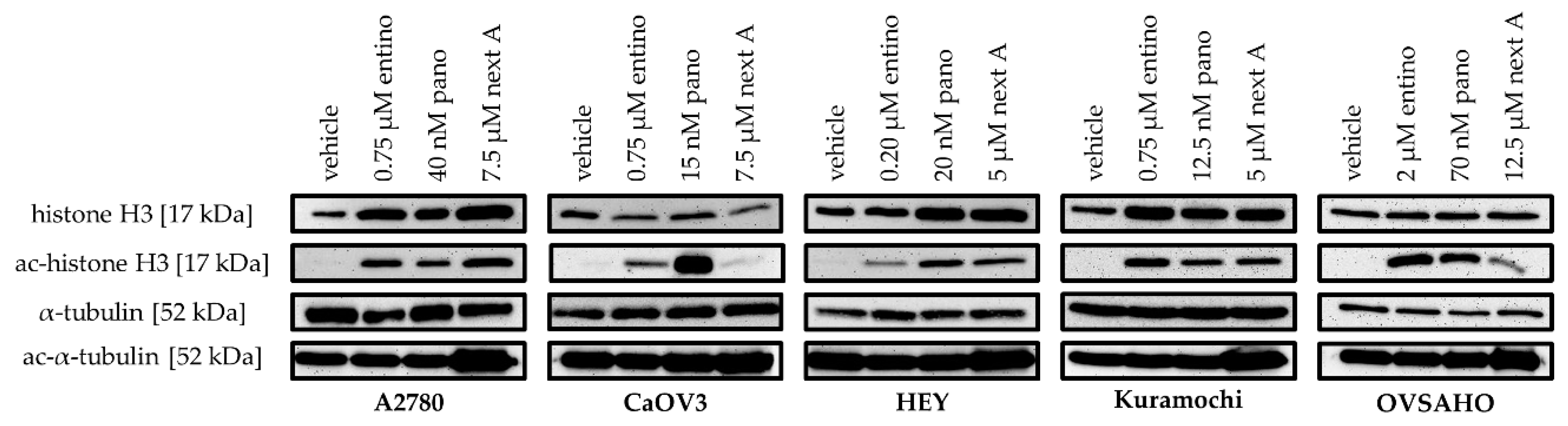
2.2. Cytotoxic and HDAC-Inhibitory Effects of Entinostat, Panobinostat, and Nexturastat A
2.3. Enhancement of Cisplatin-Induced Cytotoxicity
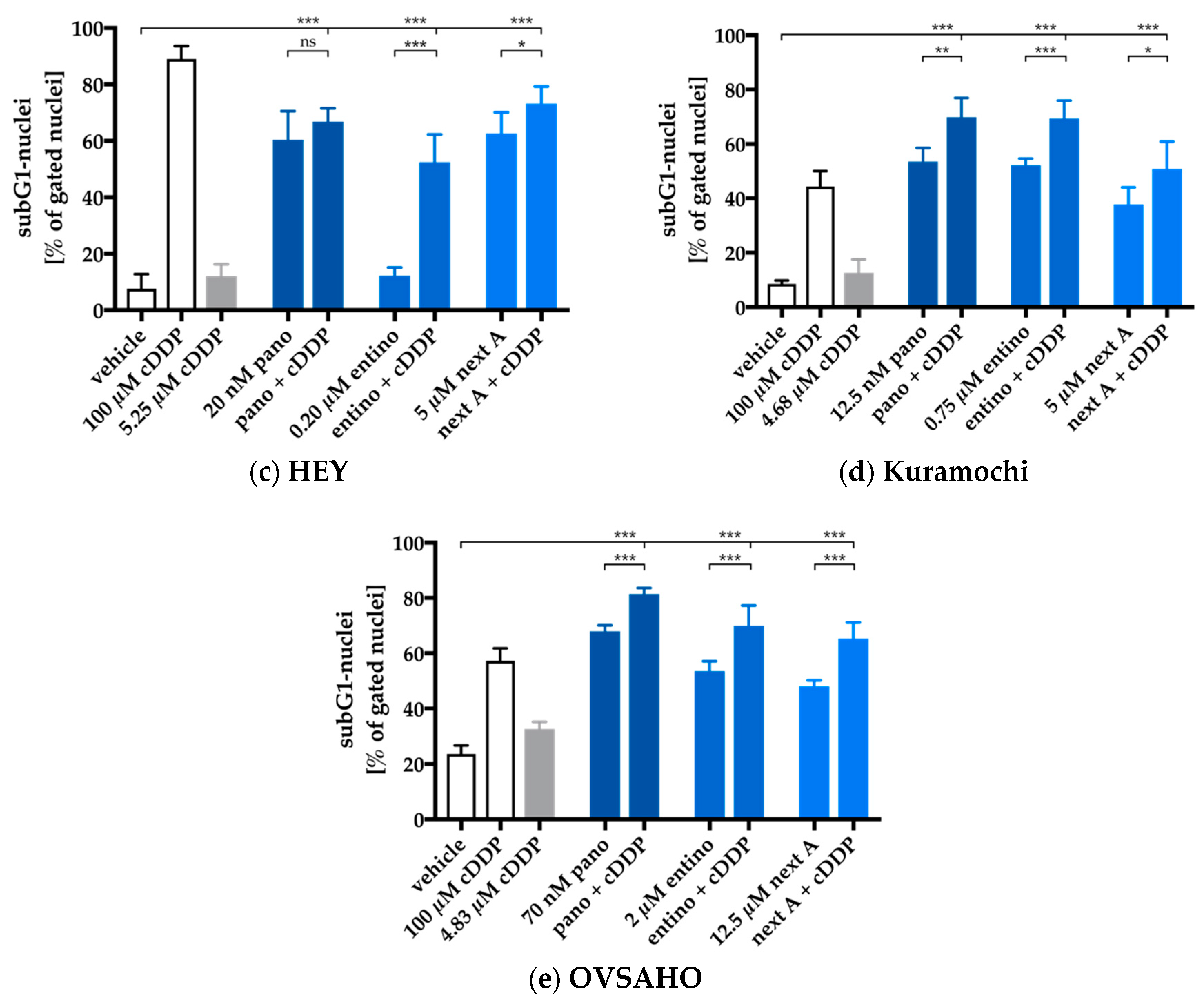
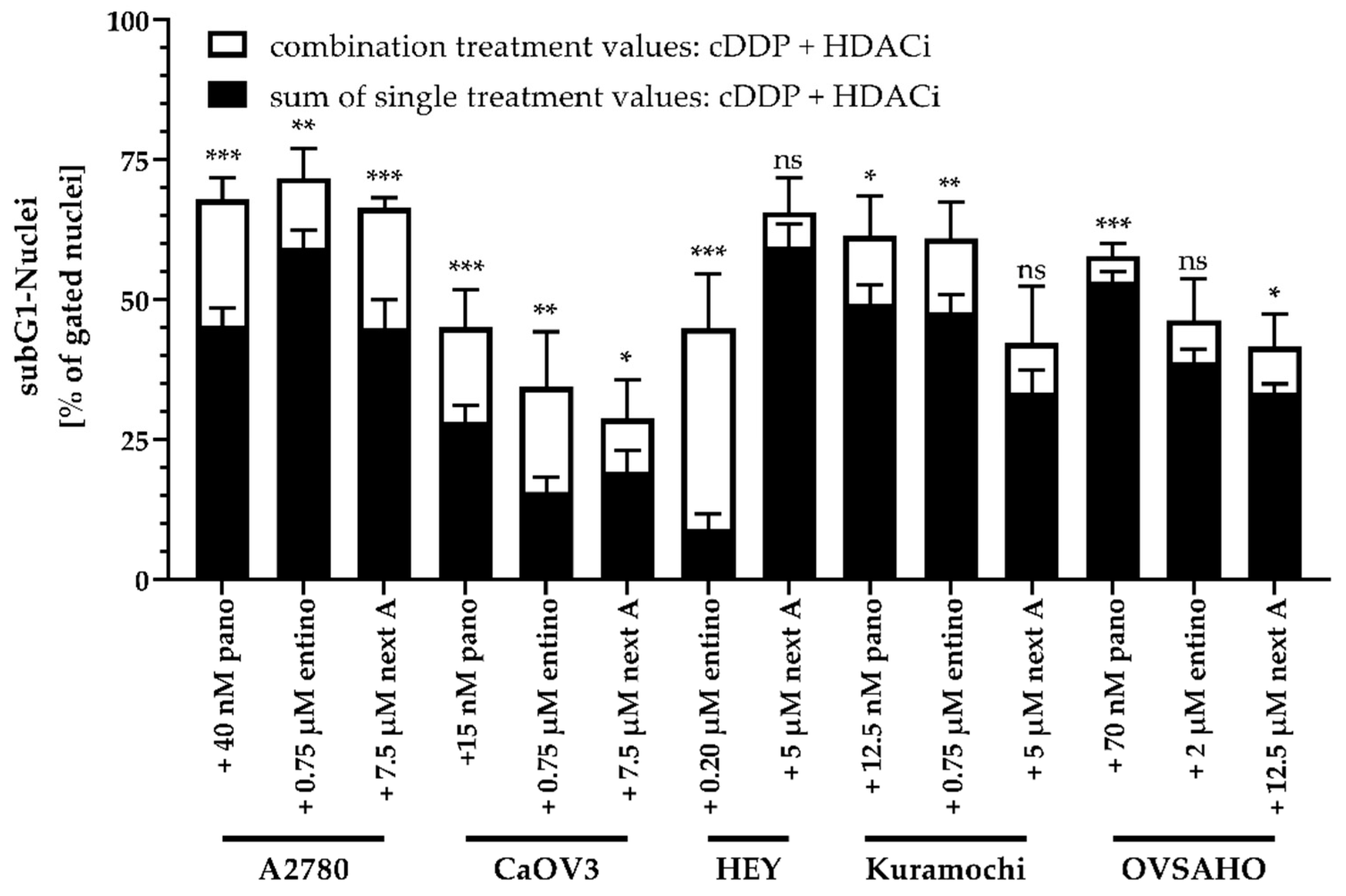
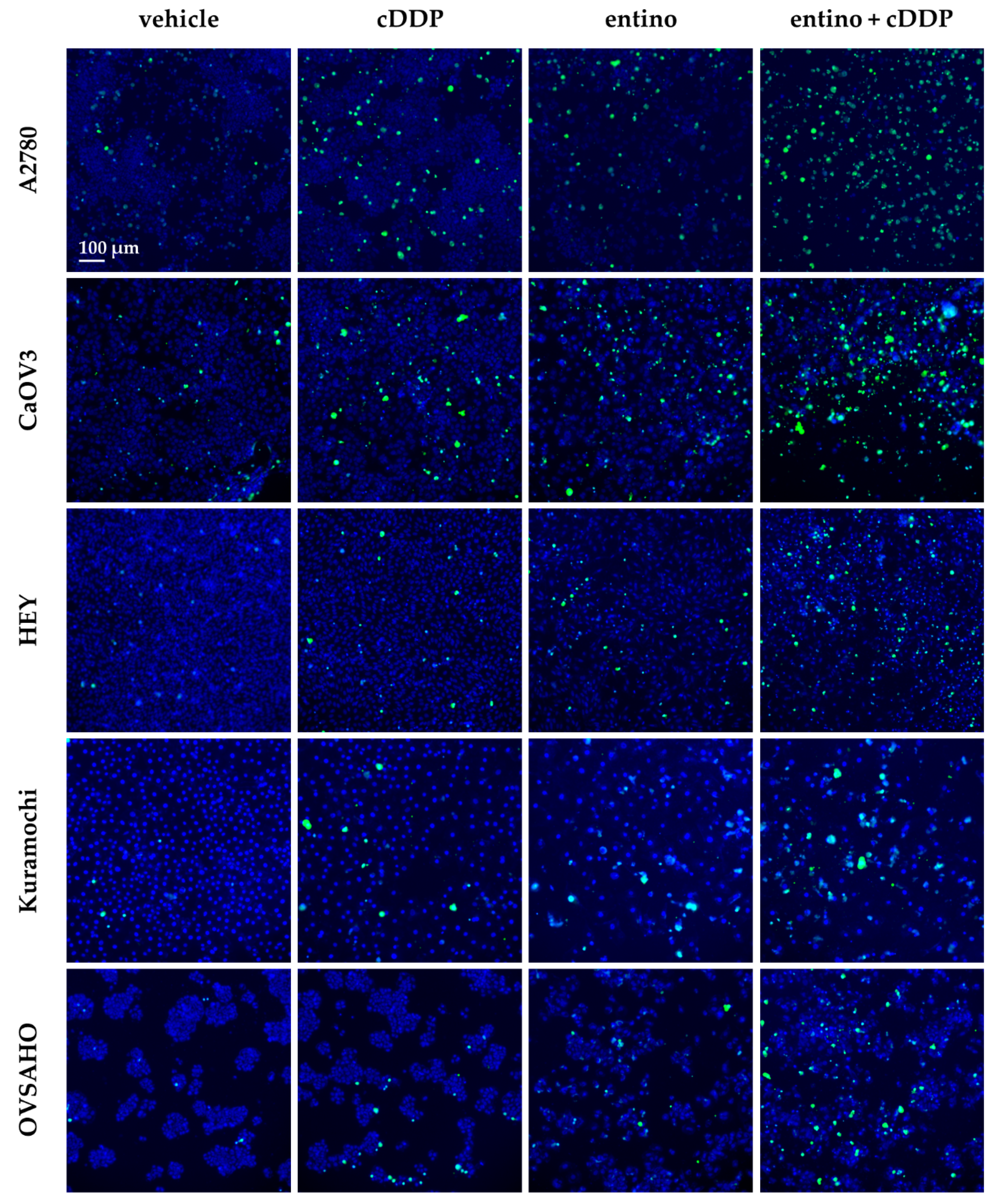
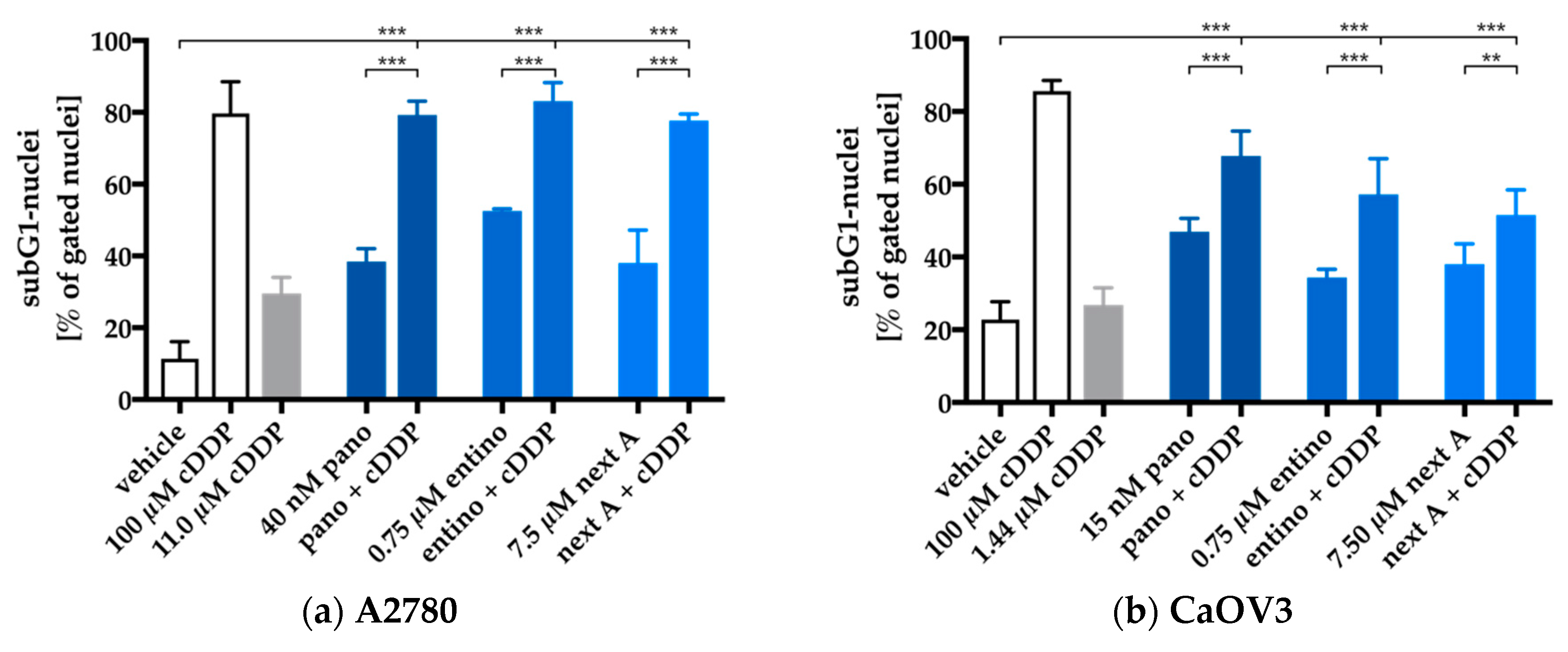
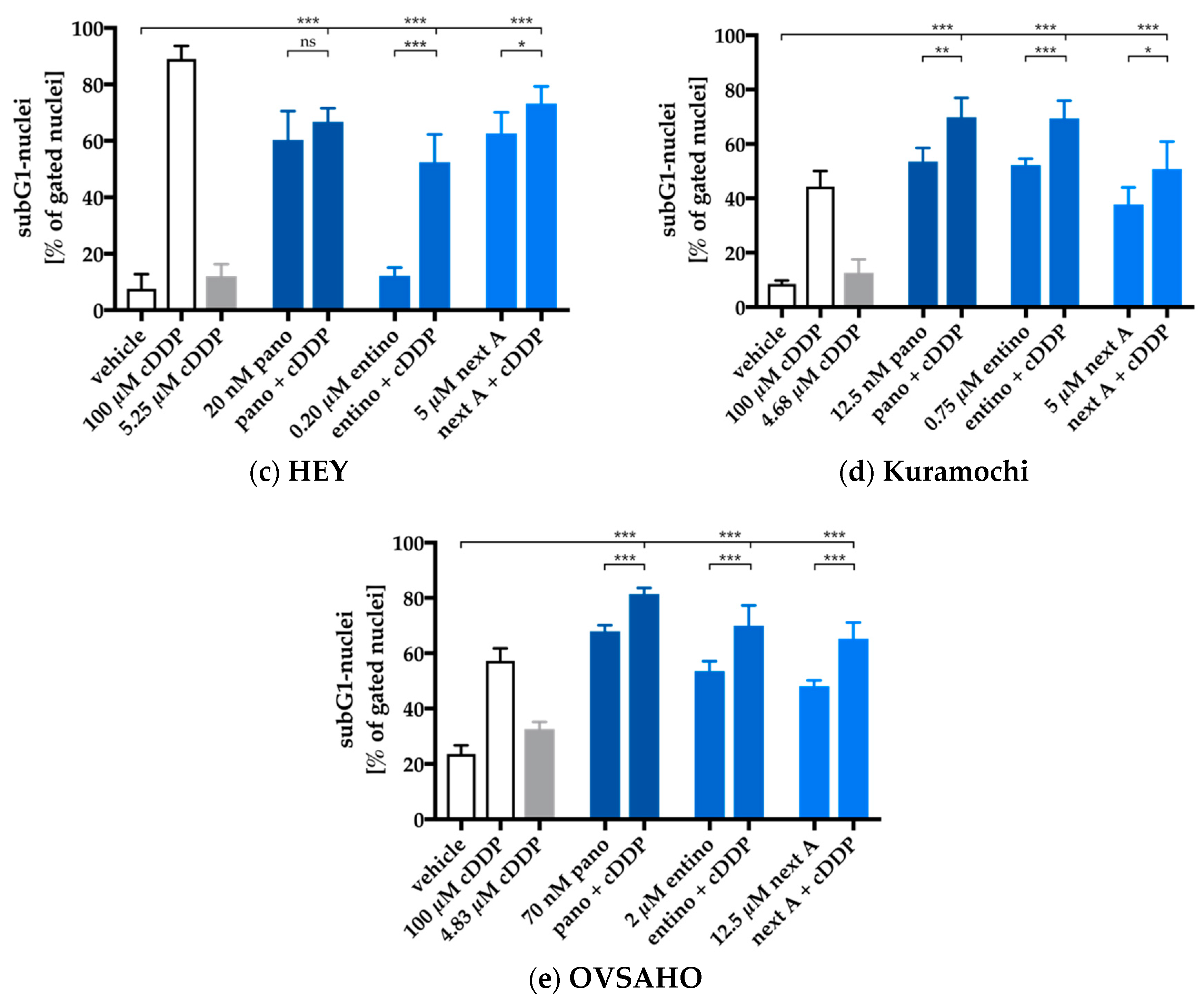
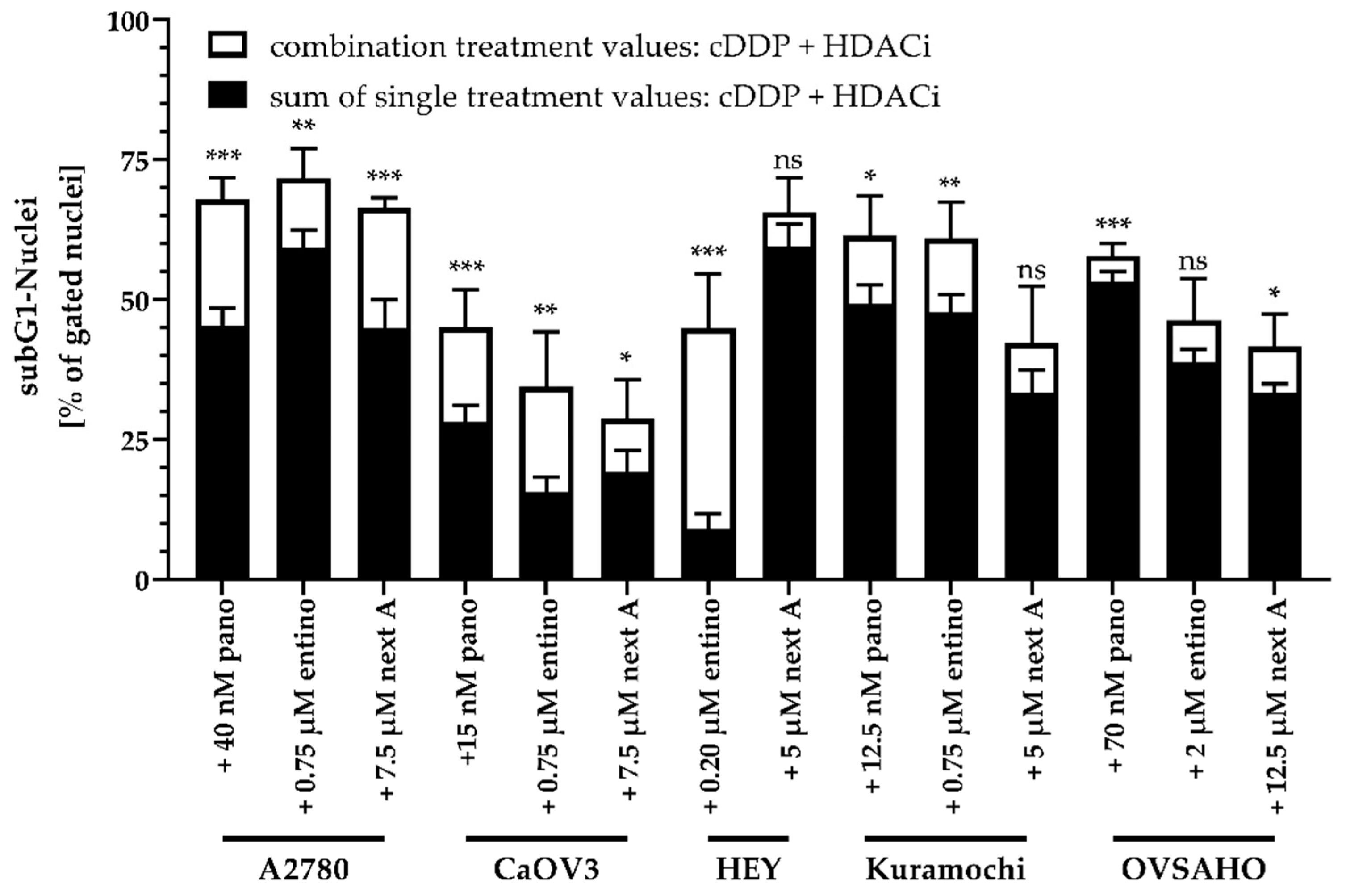
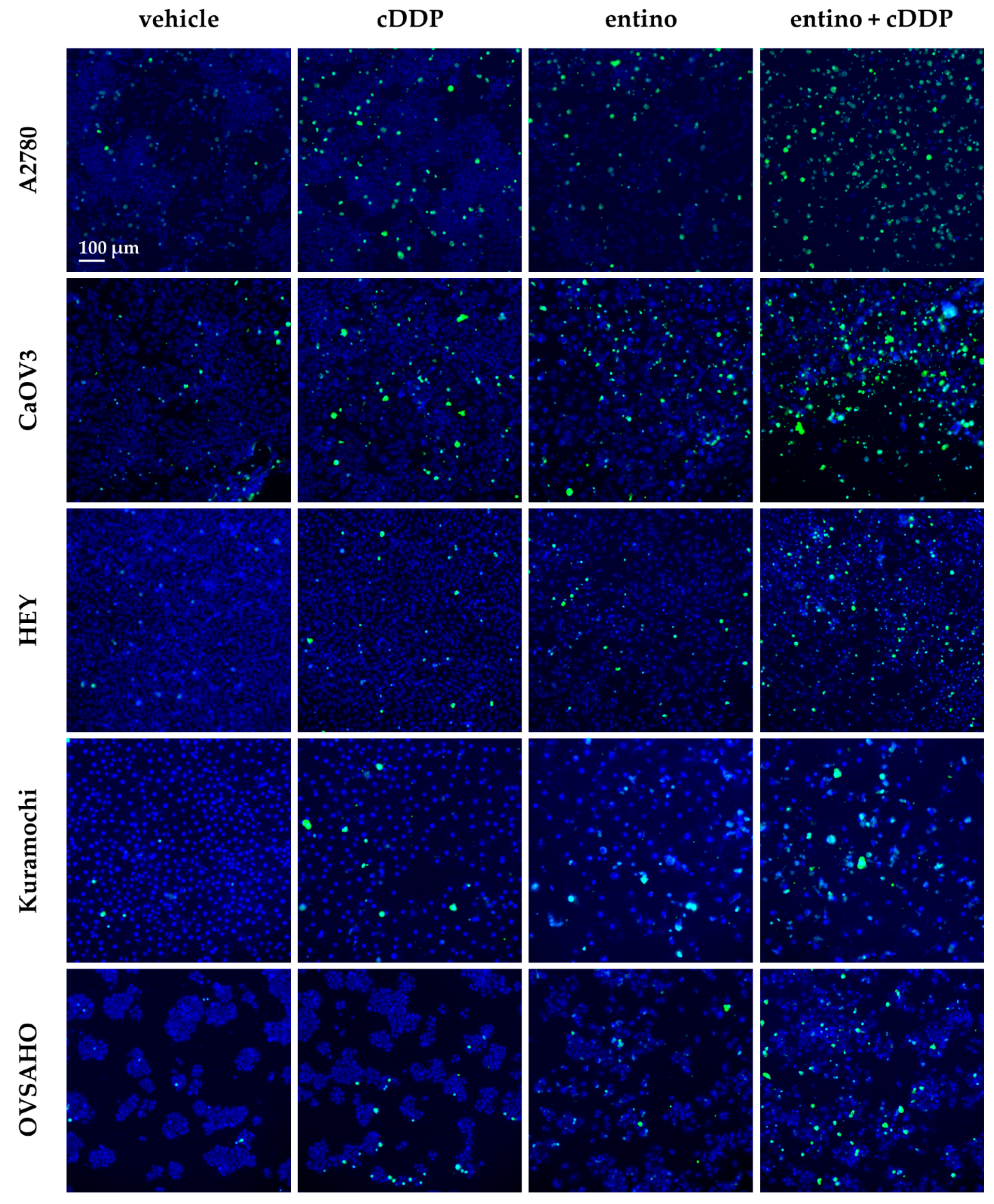
2.4. Enhancement of Cisplatin-Induced Cytotoxicity is Mediated via Apoptosis-Induction
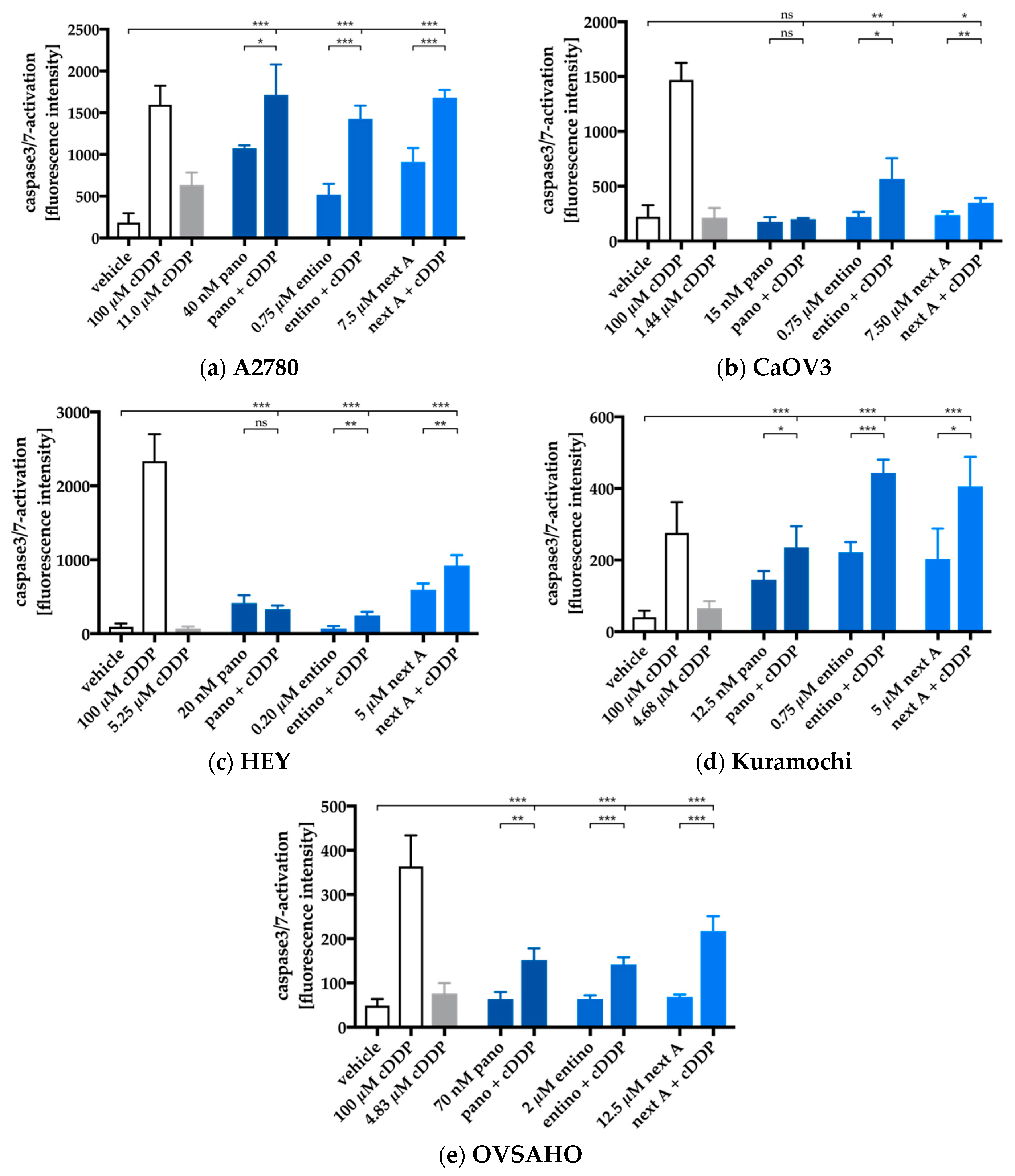
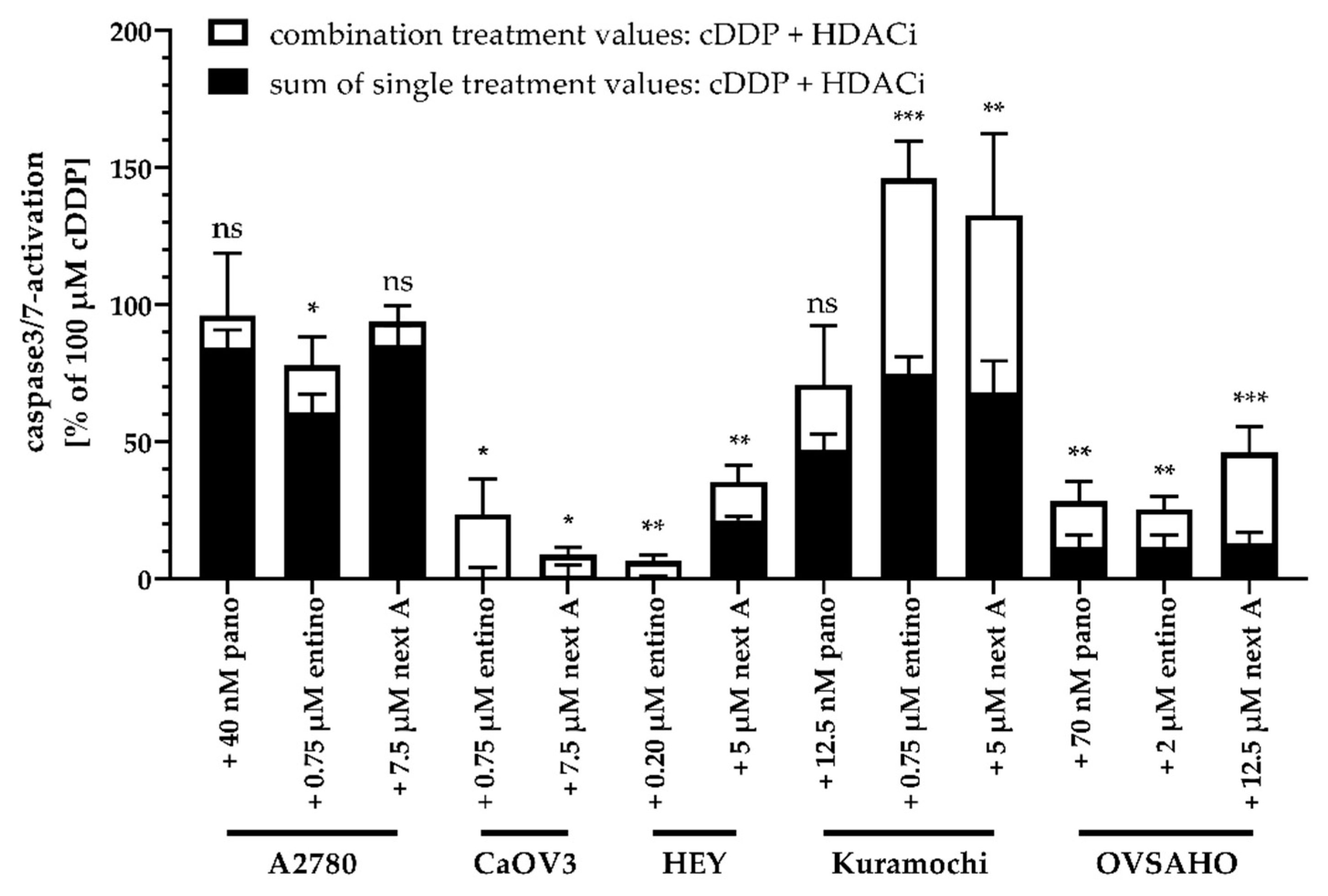
2.5. Apoptosis-Induction of the Combination Treatment is Caspase3/7-Driven
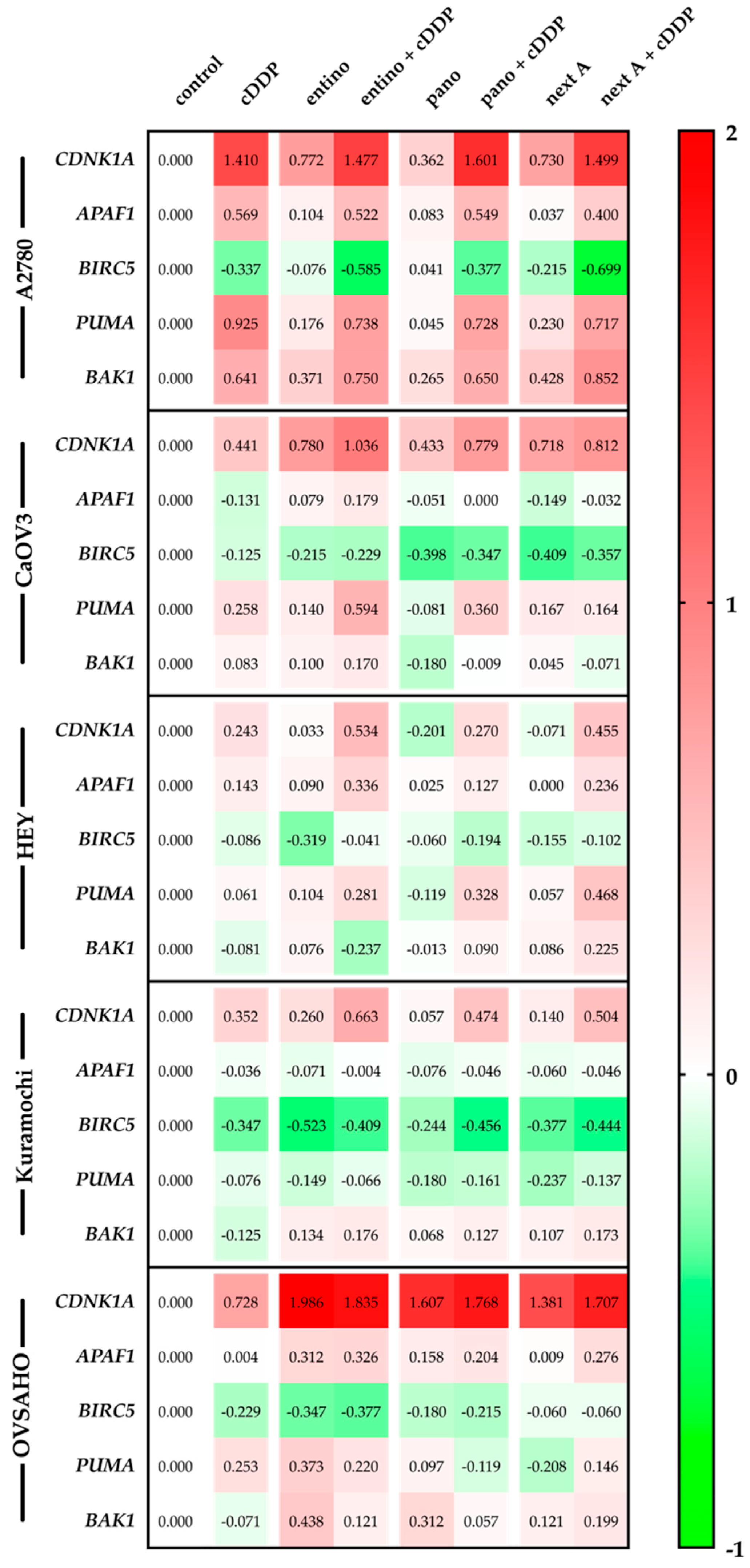
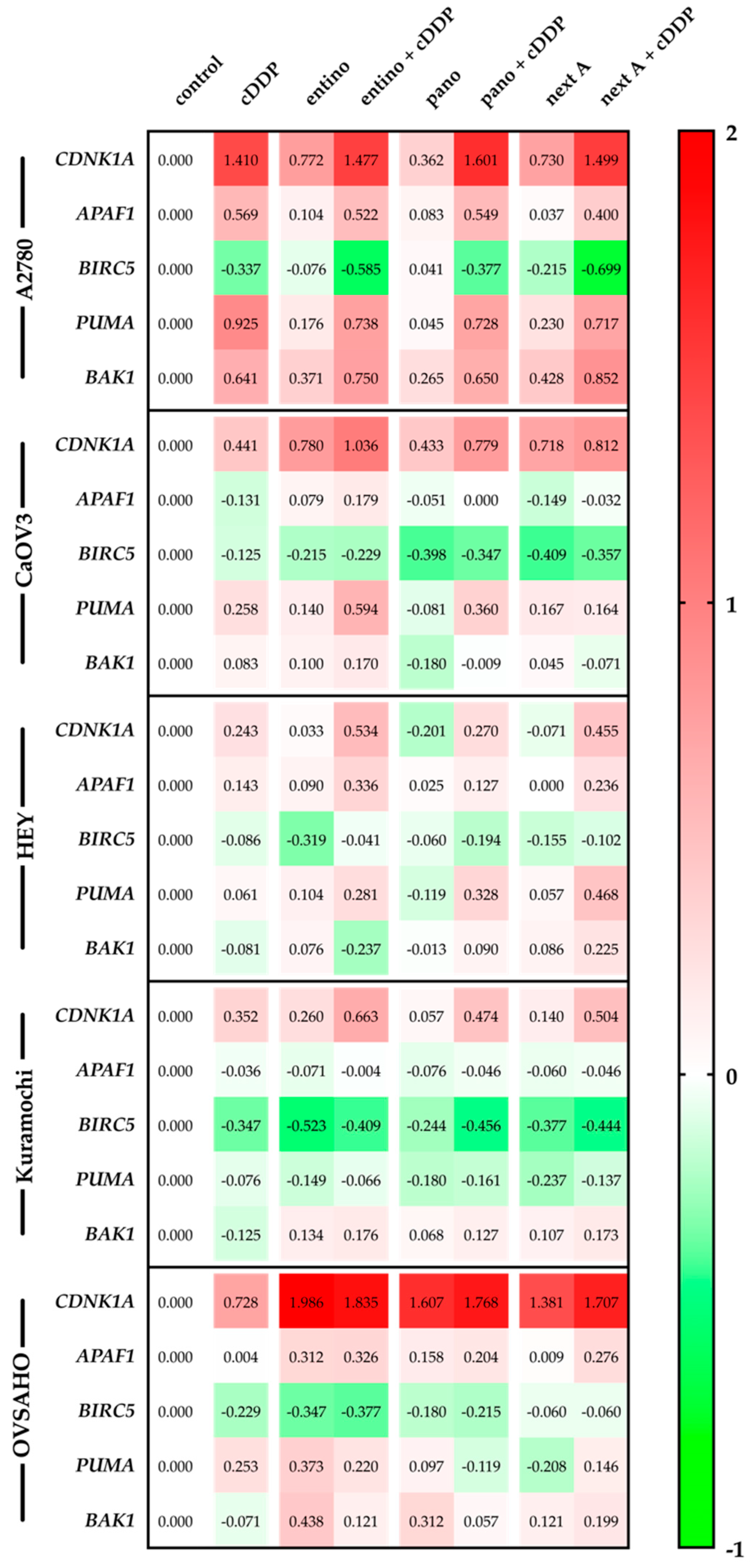
2.6. Alterations in Apoptosis-Related Gene Expression
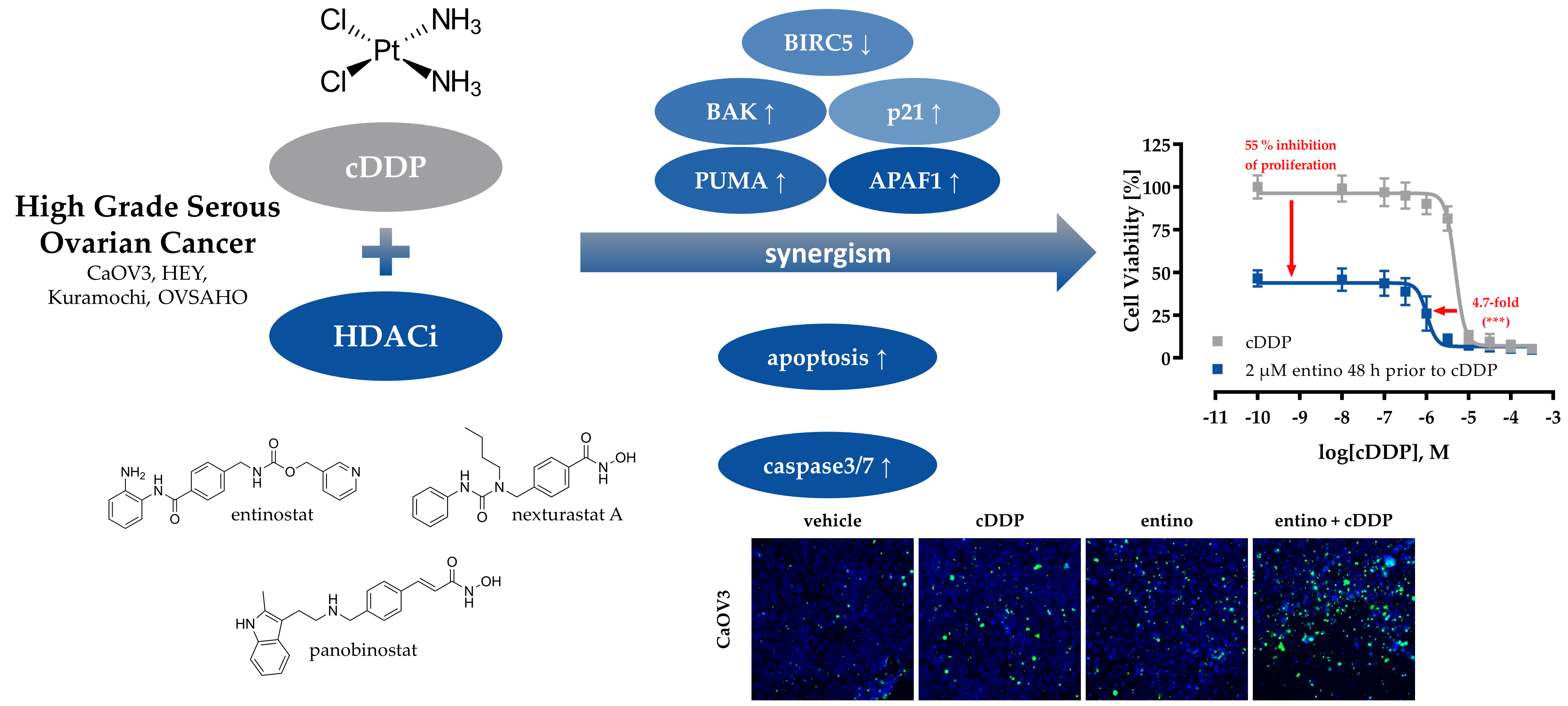
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cell Lines and Cell Culture
4.3. MTT Cell Viability Assay
4.4. Whole-Cell HDAC Inhibition Assay
4.5. Combination Experiments
4.6. Enzyme HDAC Inhibition Assay
4.7. Measurement of Apoptotic Nuclei
4.8. Caspase 3/7 Activation Assay
4.9. Immunoblotting
4.10. RT-PCR
4.11. Data Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| HDAC | histone deacetylase |
| HDACi | histone deacetylase inhibitor |
| entino | entinostat |
| cDDP | cis-diamminedichloridoplatinum(II) (cisplatin) |
| pano | panobinostat |
| next A | nexturastat A |
| BIRC5, survivin | baculoviral inhibitor of apoptosis repeat-containing 5 |
| PUMA | p53 upregulated modulator of apoptosis |
| BAK/Bak | Bcl-2 homologous antagonist killer |
| CDNK1A, p21 | cyclin-dependent kinase inhibitor 1 |
| APAF1 | apoptotic protease activating factor 1 |
| IAP | inhibitors of apoptosis |
| HPRT1 | hypoxanthine-guanine phosphoribosyltransferase |
| GUSB | beta-glucuronidase |
| TBP | TATA binding protein |
| HGSOC | high grade serous ovarian cancer |
| MTT | 3-(4,5-Dimethylthiazole-2-yl)-2,5-diphenyltetrazoliumbromide |
| CI | combination index |
| RT-PCR | reverse transcriptase polymerase chain reaction |
References
- Cancer of the Ovary - Cancer Stat Facts. Available online: https://seer.cancer.gov/statfacts/html/ovary.html (accessed on 24 April 2019).
- Cancer of the Cervix Uteri - Cancer Stat Facts. Available online: https://seer.cancer.gov/statfacts/html/cervix.html (accessed on 24 April 2019).
- Cancer of the Breast (Female) - Cancer Stat Facts. Available online: https://seer.cancer.gov/statfacts/html/breast.html (accessed on 24 April 2019).
- Cancer of the Vulva - Cancer Stat Facts. Available online: https://seer.cancer.gov/statfacts/html/vulva.html (accessed on 24 April 2019).
- Cancer of the Endometrium - Cancer Stat Facts. Available online: https://seer.cancer.gov/statfacts/html/corp.html (accessed on 24 April 2019).
- Krebs – Datenbankabfrage. Available online: https://www.krebsdaten.de/Krebs/SiteGlobals/Forms/Datenbankabfrage/datenbankabfrage_stufe2_form.html (accessed on 14 May 2019).
- Singh, N.; McCluggage, W.G.; Gilks, C.B. High-grade serous carcinoma of tubo-ovarian origin: recent developments. Histopathology 2017, 71, 339–356. [Google Scholar] [CrossRef] [PubMed]
- Köbel, M.; Kalloger, S.E.; Huntsman, D.G.; Santos, J.L.; Swenerton, K.D.; Seidman, J.D.; Gilks, C.B. Cheryl Brown Ovarian Cancer Outcomes Unit of the British Columbia Cancer Agency, Vancouver BC Differences in tumor type in low-stage versus high-stage ovarian carcinomas. Int. J. Gynecol. Pathol. Off. J. Int. Soc. Gynecol. Pathol. 2010, 29, 203–211. [Google Scholar]
- Kohn, E.C.; Ivy, S.P. Whence High-Grade Serous Ovarian Cancer. Am. Soc. Clin. Oncol. Educ. Book Am. Soc. Clin. Oncol. Annu. Meet. 2017, 37, 443–448. [Google Scholar] [CrossRef] [PubMed]
- Koshiyama, M.; Matsumura, N.; Konishi, I. Subtypes of Ovarian Cancer and Ovarian Cancer Screening. Diagnostics 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Eisenhauer, E.A. Real-world evidence in the treatment of ovarian cancer. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2017, 28, viii61–viii65. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S. Cisplatin: The first metal based anticancer drug. Bioorganic Chem. 2019, 88, 102925. [Google Scholar] [CrossRef] [PubMed]
- Ohmichi, M.; Hayakawa, J.; Tasaka, K.; Kurachi, H.; Murata, Y. Mechanisms of platinum drug resistance. Trends Pharmacol. Sci. 2005, 26, 113–116. [Google Scholar] [CrossRef]
- Jain, A.; Jahagirdar, D.; Nilendu, P.; Sharma, N.K. Molecular approaches to potentiate cisplatin responsiveness in carcinoma therapeutics. Expert Rev. Anticancer Ther. 2017, 17, 815–825. [Google Scholar] [CrossRef]
- Eckschlager, T.; Plch, J.; Stiborova, M.; Hrabeta, J. Histone Deacetylase Inhibitors as Anticancer Drugs. Int. J. Mol. Sci. 2017, 18, 1414. [Google Scholar] [CrossRef]
- Kim, H.-J.; Bae, S.-C. Histone deacetylase inhibitors: molecular mechanisms of action and clinical trials as anti-cancer drugs. Am. J. Transl. Res. 2011, 3, 166–179. [Google Scholar]
- Gregoretti, I.; Lee, Y.-M.; Goodson, H.V. Molecular Evolution of the Histone Deacetylase Family: Functional Implications of Phylogenetic Analysis. J. Mol. Biol. 2004, 338, 17–31. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Yang, Y.; Zhou, N.; Tang, K.; Lau, W.B.; Lau, B.; Wang, W.; Xu, L.; Yang, Z.; Huang, S.; et al. Epigenetics in ovarian cancer: premise, properties, and perspectives. Mol. Cancer 2018, 17, 109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yano, M.; Yasuda, M.; Sakaki, M.; Nagata, K.; Fujino, T.; Arai, E.; Hasebe, T.; Miyazawa, M.; Miyazawa, M.; Ogane, N.; et al. Association of histone deacetylase expression with histology and prognosis of ovarian cancer. Oncol. Lett. 2018, 15, 3524–3531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marek, L.; Hamacher, A.; Hansen, F.K.; Kuna, K.; Gohlke, H.; Kassack, M.U.; Kurz, T. Histone deacetylase (HDAC) inhibitors with a novel connecting unit linker region reveal a selectivity profile for HDAC4 and HDAC5 with improved activity against chemoresistant cancer cells. J. Med. Chem. 2013, 56, 427–436. [Google Scholar] [CrossRef] [PubMed]
- Stenzel, K.; Hamacher, A.; Hansen, F.K.; Gertzen, C.G.W.; Senger, J.; Marquardt, V.; Marek, L.; Marek, M.; Romier, C.; Remke, M.; et al. Alkoxyurea-Based Histone Deacetylase Inhibitors Increase Cisplatin Potency in Chemoresistant Cancer Cell Lines. J. Med. Chem. 2017, 60, 5334–5348. [Google Scholar] [CrossRef] [PubMed]
- Krieger, V.; Hamacher, A.; Gertzen, C.G.W.; Senger, J.; Zwinderman, M.R.H.; Marek, M.; Romier, C.; Dekker, F.J.; Kurz, T.; Jung, M.; et al. Design, Multicomponent Synthesis, and Anticancer Activity of a Focused Histone Deacetylase (HDAC) Inhibitor Library with Peptoid-Based Cap Groups. J. Med. Chem. 2017, 60, 5493–5506. [Google Scholar] [CrossRef] [PubMed]
- Cellosaurus Cell Line Caov-3 (CVCL_0201). Available online: https://web.expasy.org/cellosaurus/CVCL_0201 (accessed on 17 May 2019).
- Cellosaurus Cell Line HEY (CVCL_0297). Available online: https://web.expasy.org/cellosaurus/CVCL_0297 (accessed on 17 May 2019).
- Cellosaurus Cell Line Kuramochi (CVCL_1345). Available online: https://web.expasy.org/cellosaurus/CVCL_1345 (accessed on 17 May 2019).
- Cellosaurus Cell Line OVSAHO (CVCL_3114). Available online: https://web.expasy.org/cellosaurus/CVCL_3114 (accessed on 17 May 2019).
- Domcke, S.; Sinha, R.; Levine, D.A.; Sander, C.; Schultz, N. Evaluating cell lines as tumour models by comparison of genomic profiles. Nat. Commun. 2013, 4, 2126. [Google Scholar] [CrossRef]
- Cellosaurus Cell Line A2780 (CVCL_0134). Available online: https://web.expasy.org/cellosaurus/CVCL_0134 (accessed on 17 May 2019).
- Beaufort, C.M.; Helmijr, J.C.A.; Piskorz, A.M.; Hoogstraat, M.; Ruigrok-Ritstier, K.; Besselink, N.; Murtaza, M.; van IJcken, W.F.J.; Heine, A.A.J.; Smid, M.; et al. Ovarian Cancer Cell Line Panel (OCCP): Clinical Importance of In Vitro Morphological Subtypes. PLoS ONE 2014, 9, e103988. [Google Scholar] [CrossRef]
- Panteix, G.; Beaujard, A.; Garbit, F.; Chaduiron-Faye, C.; Guillaumont, M.; Gilly, F.; Baltassat, P.; Bressolle, F. Population pharmacokinetics of cisplatin in patients with advanced ovarian cancer during intraperitoneal hyperthermia chemotherapy. Anticancer Res. 2002, 22, 1329–1336. [Google Scholar]
- Engelke, L.H.; Hamacher, A.; Proksch, P.; Kassack, M.U. Ellagic Acid and Resveratrol Prevent the Development of Cisplatin Resistance in the Epithelial Ovarian Cancer Cell Line A2780. J. Cancer 2016, 7, 353–363. [Google Scholar] [CrossRef] [Green Version]
- Chou, T.-C. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010, 70, 440–446. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.-C.; Talalay, P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv. Enzyme Regul. 1984, 22, 27–55. [Google Scholar] [CrossRef]
- Waldman, T.; Kinzler, K.W.; Vogelstein, B. p21 Is Necessary for the p53-mediated G1 Arrest in Human Cancer Cells. Cancer Res. 1995, 55, 5187–5190. [Google Scholar] [PubMed]
- Pop, C.; Timmer, J.; Sperandio, S.; Salvesen, G.S. The Apoptosome Activates Caspase-9 by Dimerization. Mol. Cell 2006, 22, 269–275. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Flemington, C.; Houghton, A.B.; Gu, Z.; Zambetti, G.P.; Lutz, R.J.; Zhu, L.; Chittenden, T. Expression of bbc3, a pro-apoptotic BH3-only gene, is regulated by diverse cell death and survival signals. Proc. Natl. Acad. Sci. 2001, 98, 11318–11323. [Google Scholar] [CrossRef] [PubMed]
- Nakano, K.; Vousden, K.H. PUMA, a Novel Proapoptotic Gene, Is Induced by p53. Mol. Cell 2001, 7, 683–694. [Google Scholar] [CrossRef]
- McArthur, K.; Whitehead, L.W.; Heddleston, J.M.; Li, L.; Padman, B.S.; Oorschot, V.; Geoghegan, N.D.; Chappaz, S.; Davidson, S.; Chin, H.S.; et al. BAK/BAX macropores facilitate mitochondrial herniation and mtDNA efflux during apoptosis. Science 2018, 359, eaao6047. [Google Scholar] [CrossRef] [Green Version]
- Shin, S.; Sung, B.-J.; Cho, Y.-S.; Kim, H.-J.; Ha, N.-C.; Hwang, J.-I.; Chung, C.-W.; Jung, Y.-K.; Oh, B.-H. An Anti-apoptotic Protein Human Survivin Is a Direct Inhibitor of Caspase-3 and -7. Biochemistry 2001, 40, 1117–1123. [Google Scholar] [CrossRef]
- Vandesompele, J.; De Preter, K.; Pattyn, F.; Poppe, B.; Van Roy, N.; De Paepe, A.; Speleman, F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002, 3, RESEARCH0034. [Google Scholar] [CrossRef]
- Benton, C.B.; Fiskus, W.; Bhalla, K.N. Targeting Histone Acetylation: Readers and Writers in Leukemia and Cancer. Cancer J. Sudbury Mass 2017, 23, 286–291. [Google Scholar] [CrossRef]
- Pchejetski, D.; Alfraidi, A.; Sacco, K.; Alshaker, H.; Muhammad, A.; Monzon, L. Histone deacetylases as new therapy targets for platinum-resistant epithelial ovarian cancer. J. Cancer Res. Clin. Oncol. 2016, 142, 1659–1671. [Google Scholar] [CrossRef] [PubMed]
- Spiegel, S.; Milstien, S.; Grant, S. Endogenous modulators and pharmacological inhibitors of histone deacetylases in cancer therapy. Oncogene 2012, 31, 537–551. [Google Scholar] [CrossRef] [PubMed]
- Ozaki, K.; Kishikawa, F.; Tanaka, M.; Sakamoto, T.; Tanimura, S.; Kohno, M. Histone deacetylase inhibitors enhance the chemosensitivity of tumor cells with cross-resistance to a wide range of DNA-damaging drugs. Cancer Sci. 2008, 99, 376–384. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhao, Y.; Gou, W.; Zhao, S.; Takano, Y.; Zheng, H. The Anti-Tumor Effects and Molecular Mechanisms of Suberoylanilide Hydroxamic Acid (SAHA) on the Aggressive Phenotypes of Ovarian Carcinoma Cells. PLoS ONE 2013, 8, e79781. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.-Y.; Lin, H.; Moh, J.-S.; Chen, K.-D.; Wang, I.-W.; Ou, Y.-C.; You, Y.-S.; Lung, C.-C. Low-dose LBH589 increases the sensitivity of cisplatin to cisplatin-resistant ovarian cancer cells. Taiwan. J. Obstet. Gynecol. 2011, 50, 165–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, R.R. Safety and Tolerability of Histone Deacetylase (HDAC) Inhibitors in Oncology. Drug Saf. 2019, 42, 235–245. [Google Scholar] [CrossRef] [PubMed]
- Van Veggel, M.; Westerman, E.; Hamberg, P. Clinical Pharmacokinetics and Pharmacodynamics of Panobinostat. Clin. Pharmacokinet. 2018, 57, 21–29. [Google Scholar] [CrossRef]
- Tzogani, K.; van Hennik, P.; Walsh, I.; De Graeff, P.; Folin, A.; Sjöberg, J.; Salmonson, T.; Bergh, J.; Laane, E.; Ludwig, H.; et al. EMA Review of Panobinostat (Farydak) for the Treatment of Adult Patients with Relapsed and/or Refractory Multiple Myeloma. The Oncologist 2018, 23, 631–636. [Google Scholar] [CrossRef]
- Takai, N.; Narahara, H.; Narahara, T. Human Endometrial and Ovarian Cancer Cells: Histone Deacetylase Inhibitors Exhibit Antiproliferative Activity, Potently Induce Cell Cycle Arrest, and Stimulate Apoptosis. Curr. Med. Chem. 2007, 14, 2548–2553. [Google Scholar] [CrossRef]
- Buurman, R.; Sandbothe, M.; Schlegelberger, B.; Skawran, B. HDAC inhibition activates the apoptosome via Apaf1 upregulation in hepatocellular carcinoma. Eur. J. Med. Res. 2016, 21, 26. [Google Scholar] [CrossRef] [Green Version]
- Jaiswal, P.K.; Goel, A.; Mittal, R.D. Survivin: A molecular biomarker in cancer. Indian J. Med. Res. 2015, 141, 389–397. [Google Scholar] [PubMed]
- Tong, Q.-S.; Zheng, L.-D.; Wang, L.; Liu, J.; Qian, W. BAK overexpression mediates p53-independent apoptosis inducing effects on human gastric cancer cells. BMC Cancer 2004, 4, 33. [Google Scholar] [CrossRef] [PubMed]
- Rose, S.L.; Goodheart, M.J.; DeYoung, B.R.; Smith, B.J.; Buller, R.E. p21 expression predicts outcome in p53-null ovarian carcinoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2003, 9, 1028–1032. [Google Scholar]
- Yu, J.; Zhang, L. The transcriptional targets of p53 in apoptosis control. Biochem. Biophys. Res. Commun. 2005, 331, 851–858. [Google Scholar] [CrossRef] [PubMed]
- Sanders, Y.Y.; Hagood, J.S.; Liu, H.; Zhang, W.; Ambalavanan, N.; Thannickal, V.J. Histone deacetylase inhibition promotes fibroblast apoptosis and ameliorates pulmonary fibrosis in mice. Eur. Respir. J. 2014, 43, 1448–1458. [Google Scholar] [CrossRef] [PubMed]
- Glozak, M.A.; Seto, E. Histone deacetylases and cancer. Oncogene 2007, 26, 5420–5432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heltweg, B.; Jung, M. A Microplate Reader-Based Nonisotopic Histone Deacetylase Activity Assay. Anal. Biochem. 2002, 302, 175–183. [Google Scholar] [CrossRef]
- Ciossek, T.; Julius, H.; Wieland, H.; Maier, T.; Beckers, T. A homogeneous cellular histone deacetylase assay suitable for compound profiling and robotic screening. Anal. Biochem. 2008, 372, 72–81. [Google Scholar] [CrossRef]
- Bonfils, C.; Kalita, A.; Dubay, M.; Siu, L.L.; Carducci, M.A.; Reid, G.; Martell, R.E.; Besterman, J.M.; Li, Z. Evaluation of the pharmacodynamic effects of MGCD0103 from preclinical models to human using a novel HDAC enzyme assay. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2008, 14, 3441–3449. [Google Scholar] [CrossRef]
- Hoffmann, K.; Brosch, G.; Loidl, P.; Jung, M. A non-isotopic assay for histone deacetylase activity. Nucleic Acids Res. 1999, 27, 2057–2058. [Google Scholar] [CrossRef]
- Ye, J.; Coulouris, G.; Zaretskaya, I.; Cutcutache, I.; Rozen, S.; Madden, T.L. Primer-BLAST: A tool to design target-specific primers for polymerase chain reaction. BMC Bioinform. 2012, 13, 134. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | IC50 [µM] | pIC50 ± SEM |
|---|---|---|
| A2780 | 11.0 | 4.96 ± 0.01 |
| CaOV3 | 1.44 | 5.84 ± 0.01 |
| HEY | 5.25 | 5.28 ± 0.01 |
| Kuramochi | 4.68 | 5.33 ± 0.02 |
| OVSAHO | 4.83 | 5.32 ± 0.01 |
| Cell Line | HDACi | |||||
|---|---|---|---|---|---|---|
| Entinostat | Panobinostat | Nexturastat A | ||||
| IC50 [nM] | pIC50 ± SEM | IC50 [nM] | pIC50 ± SEM | IC50 [nM] | pIC50 ± SEM | |
| A2780 | 606 | 6.22 ± 0.03 | 15.3 | 7.82 ± 0.02 | 7778 | 5.11 ± 0.09 |
| CaOV3 | 1146 | 5.94 ± 0.03 | 7.62 | 8.12 ± 0.03 | 5291 | 5.28 ± 0.03 |
| HEY | 251 | 6.60 ± 0.02 | 2.68 | 8.57 ± 0.02 | 1724 | 5.76 ± 0.03 |
| Kuramochi | 485 | 6.31 ± 0.03 | 11.2 | 7.95 ± 0.02 | 5302 | 5.28 ± 0.02 |
| OVSAHO | 1828 | 5.74 ± 0.02 | 42.4 | 7.37 ± 0.05 | 16,218 | 4.79 ± 0.02 |
| Cell Line | HDACi | |||||
|---|---|---|---|---|---|---|
| Entinostat | Panobinostat | Nexturastat A | ||||
| IC50 [nM] | pIC50 ± SEM | IC50 [nM] | pIC50 ± SEM | IC50 [nM] | pIC50 ± SEM | |
| A2780 | 313 | 6.50 ± 0.04 | 12.1 | 7.91 ± 0.06 | 3633 | 5.44 ± 0.04 |
| CaOV3 | 333 | 6.48 ± 0.03 | 7.88 | 8.10 ± 0.05 | 3500 | 5.46 ± 0.03 |
| HEY | 219 | 6.60 ± 0.04 | 13.6 | 7.87 ± 0.03 | 3874 | 5.41 ± 0.03 |
| Kuramochi | 339 | 6.47 ± 0.04 | 9.87 | 8.01 ± 0.07 | 3733 | 5.43 ± 0.03 |
| OVSAHO | 326 | 6.49 ± 0.02 | 23.1 | 7.64 ± 0.04 | 5249 | 5.28 ± 0.02 |
| Cell Line | Entinostat [nM] | Panobinostat [nM] | Nexturastat A [nM] |
|---|---|---|---|
| A2780 | 750 | 40.0 | 7500 |
| CaOV3 | 750 | 15.0 | 7500 |
| HEY | 200 | 20.0 | 5000 |
| Kuramochi | 750 | 12.5 | 5000 |
| OVSAHO | 2000 | 70.0 | 12,500 |
| HDAC2 | HDAC4 | HDAC6 | HDAC8 | |||||
|---|---|---|---|---|---|---|---|---|
| Compd. | IC50 ± SD [µM] | Ki [µM] | IC50 ± SD [µM] | Ki [µM] | IC50 ± SD [µM] | Ki [µM] | IC50 ± SD [µM] | Ki [µM] |
| Nexturastat A | 1.99 ± 0.25 | 1.25 | 12.0 ± 1.69 | 7.57 | 0.05 ± 0.01 | 0.03 | 22.6 ± 2.55 | 12.8 |
| Cell Line | Cisplatin | + 48 h HDACi Pretreatment | |||||
|---|---|---|---|---|---|---|---|
| Entinostat | Panobinostat | Nexturastat A | |||||
| IC50 | IC50 | SF | IC50 | SF | IC50 | SF | |
| A2780 | 11.0 | 4.99 | 2.2 (***) | 2.58 | 5.6 (***) | 1.44 | 7.6 (***) |
| CaOV3 | 1.44 | 0.72 | 2.0 (*) | 0.74 | 2.0 (ns) | 1.33 | 1.1 (ns) |
| HEY | 5.25 | 1.39 | 3.8 (***) | 2.75 | 1.9 (*) | 1.28 | 4.1 (***) |
| Kuramochi | 4.68 | 3.28 | 1.4 (*) | 5.14 | < 1 (ns) | 3.22 | 1.5 (**) |
| OVSAHO | 4.83 | 1.02 | 4.7 (***) | 3.01 | 1.6 (**) | 1.43 | 3.4 (***) |
| Cisplatin [µM] | Entinostat [µM] | Panobinostat [nM] | Nexturastat A [µM] | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0.10 | 0.25 | 0.50 | 0.75 | 10 | 20 | 30 | 40 | 1.25 | 2.50 | 5.00 | 7.50 | ||
| A2780 | 0.50 | * | 0.40 | 0.36 | 0.38 | 0.21 | 0.10 | 0.15 | 0.15 | * | 0.32 | 0.24 | 0.28 |
| 1.00 | 0.64 | 0.38 | 0.35 | 0.39 | 0.25 | 0.12 | 0.17 | 0.18 | * | 0.36 | 0.28 | 0.32 | |
| 2.00 | 0.83 | 0.43 | 0.34 | 0.40 | 0.35 | 0.16 | 0.22 | 0.21 | 0.97 | 0.47 | 0.30 | 0.33 | |
| 4.00 | 0.73 | 0.44 | 0.34 | 0.38 | 0.35 | 0.21 | 0.27 | 0.25 | >1.1 | 0.57 | 0.38 | 0.39 | |
| 6.00 | 0.59 | 0.35 | 0.32 | 0.37 | 0.29 | 0.23 | 0.26 | 0.26 | 0.83 | 0.52 | 0.37 | 0.40 | |
| 0.25 | 0.50 | 0.75 | 1.00 | - | - | - | - | - | - | - | - | ||
| CaOV3 | 0.20 | * | 0.66 | 0.71 | 0.42 | ◦ | ◦ | ◦ | ◦ | ◦ | ◦ | ◦ | ◦ |
| 0.40 | * | 0.56 | 0.59 | 0.33 | ◦ | ◦ | ◦ | ◦ | ◦ | ◦ | ◦ | ◦ | |
| 0.60 | 0.56 | 0.41 | 0.49 | 0.28 | ◦ | ◦ | ◦ | ◦ | ◦ | ◦ | ◦ | ◦ | |
| 0.80 | 0.33 | 0.30 | 0.31 | 0.23 | ◦ | ◦ | ◦ | ◦ | ◦ | ◦ | ◦ | ◦ | |
| 1.00 | 0.21 | 0.20 | 0.21 | 0.16 | ◦ | ◦ | ◦ | ◦ | ◦ | ◦ | ◦ | ◦ | |
| 0.10 | 0.15 | 0.20 | 0.25 | 5.0 | 10.0 | 15.0 | 20.0 | 1.25 | 2.50 | 3.75 | 5.00 | ||
| HEY | 0.32 | * | * | * | 0.27 | * | * | 0.19 | 0.14 | * | * | 0.32 | 0.27 |
| 0.50 | * | * | * | 0.26 | * | * | 0.21 | 0.15 | * | * | 0.33 | 0.29 | |
| 1.00 | * | * | 0.31 | 0.25 | * | * | 0.25 | 0.16 | * | * | 0.36 | 0.27 | |
| 2.00 | * | 0.37 | 0.29 | 0.24 | * | 0.41 | 0.25 | 0.17 | * | 0.63 | 0.33 | 0.26 | |
| 4.00 | 0.35 | 0.28 | 0.24 | 0.23 | 0.51 | 0.33 | 0.28 | 0.20 | 0.62 | 0.42 | 0.29 | 0.27 | |
| 0.25 | 0.50 | 0.75 | 1.00 | - | - | - | - | 1.25 | 2.50 | 3.75 | 5.00 | ||
| Kuramochi | 1.00 | 0.43 | 0.42 | 0.51 | 0.62 | ◦ | ◦ | ◦ | ◦ | * | 0.73 | 0.66 | 0.66 |
| 2.00 | 0.42 | 0.43 | 0.49 | 0.60 | ◦ | ◦ | ◦ | ◦ | * | 0.78 | 0.74 | 0.72 | |
| 3.00 | 0.46 | 0.44 | 0.48 | 0.57 | ◦ | ◦ | ◦ | ◦ | * | 0.80 | 0.77 | 0.75 | |
| 4.00 | 0.44 | 0.42 | 0.44 | 0.53 | ◦ | ◦ | ◦ | ◦ | 0.84 | 0.79 | 0.77 | 0.74 | |
| 5.00 | 0.45 | 0.43 | 0.47 | 0.52 | ◦ | ◦ | ◦ | ◦ | 0.78 | 0.78 | 0.74 | 0.70 | |
| 0.50 | 1.00 | 1.50 | 2.00 | 40 | 50 | 60 | 70 | 5.00 | 7.50 | 10.0 | 12.5 | ||
| OVSAHO | 1.00 | 0.50 | 0.39 | 0.41 | 0.44 | 0.36 | 0.33 | 0.27 | 0.26 | * | 0.82 | 0.84 | 0.78 |
| 2.00 | 0.41 | 0.29 | 0.34 | 0.37 | 0.25 | 0.23 | 0.21 | 0.21 | 0.87 | 0.77 | 0.75 | 0.67 | |
| 3.00 | 0.42 | 0.28 | 0.32 | 0.36 | 0.24 | 0.22 | 0.21 | 0.21 | 0.80 | 0.76 | 0.65 | 0.60 | |
| 4.00 | 0.39 | 0.28 | 0.31 | 0.35 | 0.23 | 0.22 | 0.21 | 0.21 | 0.72 | 0.69 | 0.61 | 0.57 | |
| 5.00 | 0.39 | 0.29 | 0.33 | 0.36 | 0.24 | 0.23 | 0.22 | 0.22 | 0.64 | 0.59 | 0.53 | 0.53 | |
| Gene | Primer Forward | Primer Reverse | Efficacy [%] |
|---|---|---|---|
| HPRT1 | CCTGGCGTCGTGATTAGTGA | CGAGCAAGACGTTCAGTCCT | 93.6 |
| TBP | GTGACCCAGCATCACTGTTTC | GAGCATCTCCAGCACACTCT | 86.9 |
| GUSB | ACCTCCAAGTATCCCAAGGGT | GTCTTGCTCCACGCTGGT | 83.1 |
| HDAC1 | TGCAAAGAAGTCCGAGGCAT | ACCCTCTGGTGATACTTTAGCA | 84.9 |
| HDAC2 | AATGGAAATATATAGGCCCC | GTTATCTGGTCTTATTGACCG | 96.4 |
| HDAC3 | GGCAACTTCCACTACGGAGC | GCATATTGGTGGGGCTGACT | 97.2 |
| HDAC4 | TTGGATGTCACAGACTCCGC | CCTTCTCGTGCCACAAGTCT | 80.8 |
| HDAC5 | GGAGAGCTCAAGAATGGATTTGC | CTGCTGTAGGAGTTTTGCG | 97.2 |
| HDAC6 | CTGGCGGAGTGGAAGAACC | TCTGCCTACTTCTTCGCTGC | 104 |
| HDAC7 | TCTCGTGAGCTAAAGAATGG | CTGTTGAATGATCTGCATGG | 96.5 |
| HDAC8 | CCACCTTCCACACTGATGCT | GCTGGGCAGTCATAACCTAGC | 97.7 |
| HDAC9 | TGTAGCTGGTGGAGTTCCCT | CTCTGAGGCAAAGGTGCAGA | 103 |
| HDAC10 | TGGCCTTTGAGTTTGACCCT | CCGATGGCTGAGTCAAATCCT | 97.3 |
| HDAC11 | CGGAAAATGGGGCAAAGTGA | CAACAGCAAAGGACCACTTG | 100 |
| CDNK1A (p21) | TGCCGAAGTCAGTTCCTTGT | GTTCTGACATGGCGCCTCC | 94.7 |
| BIRC5 (Survivin) | TGAGAACGAGCCAGACTTGG | TGTTCCTCTATGGGGTCGTCA | 108 |
| APAF1 | AGTGGAATAACTTCGTATGTAAGGA | AAACAACTGGCCTCTGTGGT | 98.7 |
| BAK1 | TCATCGGGGACGACATCAAC | CAAACAGGCTGGTGGCAATC | 111 |
| PUMA | GAGCGGCGGAGACAAGAG | TAAGGGCAGGAGTCCCATGA | 94.7 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bandolik, J.J.; Hamacher, A.; Schrenk, C.; Weishaupt, R.; Kassack, M.U. Class I-Histone Deacetylase (HDAC) Inhibition is Superior to pan-HDAC Inhibition in Modulating Cisplatin Potency in High Grade Serous Ovarian Cancer Cell Lines. Int. J. Mol. Sci. 2019, 20, 3052. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20123052
Bandolik JJ, Hamacher A, Schrenk C, Weishaupt R, Kassack MU. Class I-Histone Deacetylase (HDAC) Inhibition is Superior to pan-HDAC Inhibition in Modulating Cisplatin Potency in High Grade Serous Ovarian Cancer Cell Lines. International Journal of Molecular Sciences. 2019; 20(12):3052. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20123052
Chicago/Turabian StyleBandolik, Jan J., Alexandra Hamacher, Christian Schrenk, Robin Weishaupt, and Matthias U. Kassack. 2019. "Class I-Histone Deacetylase (HDAC) Inhibition is Superior to pan-HDAC Inhibition in Modulating Cisplatin Potency in High Grade Serous Ovarian Cancer Cell Lines" International Journal of Molecular Sciences 20, no. 12: 3052. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20123052