Early Response of Radish to Heat Stress by Strand-Specific Transcriptome and miRNA Analysis

 , and
, and
Abstract
:1. Introduction
2. Results
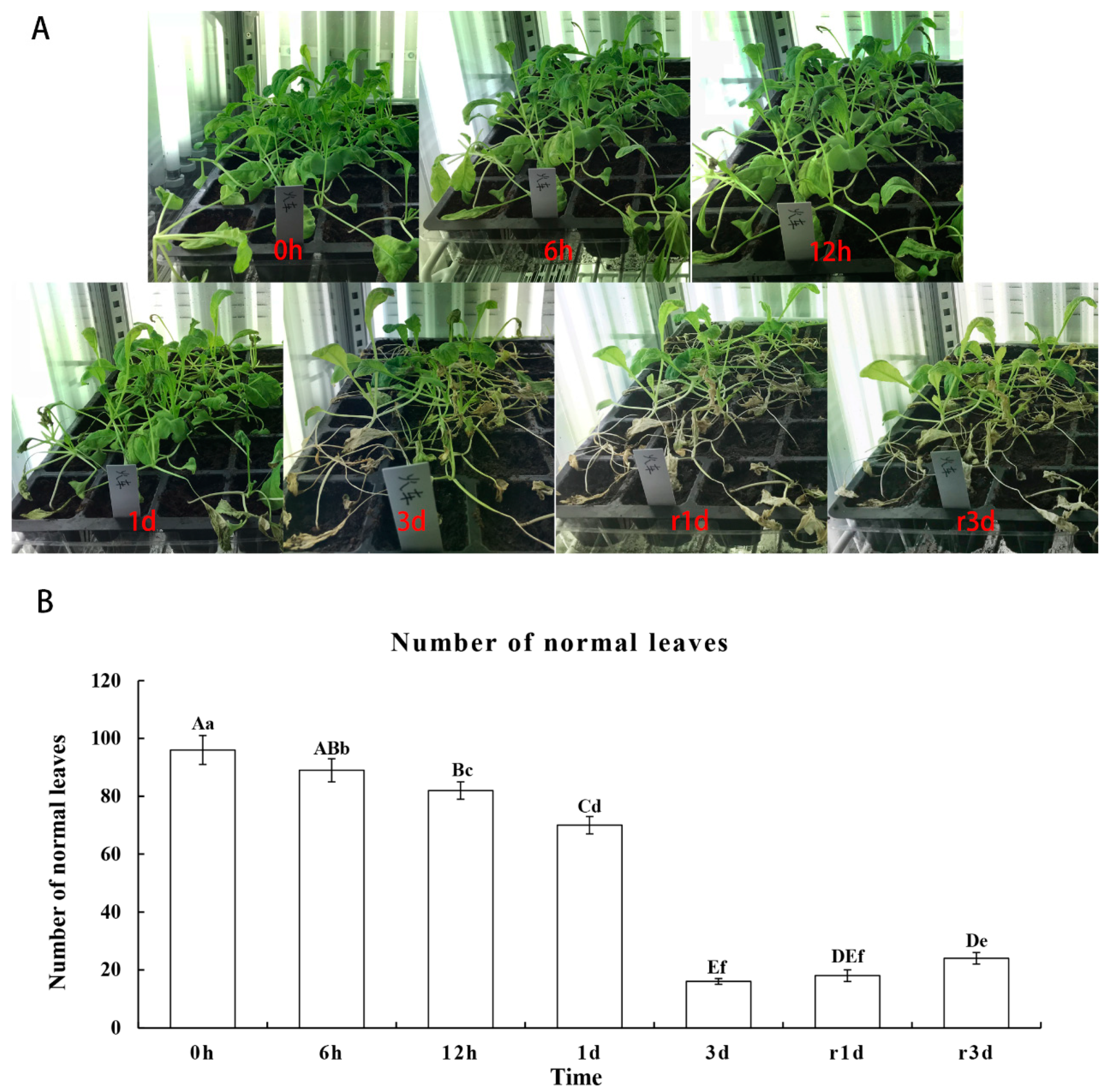
2.1. Morphological Changes Under Heat Stress
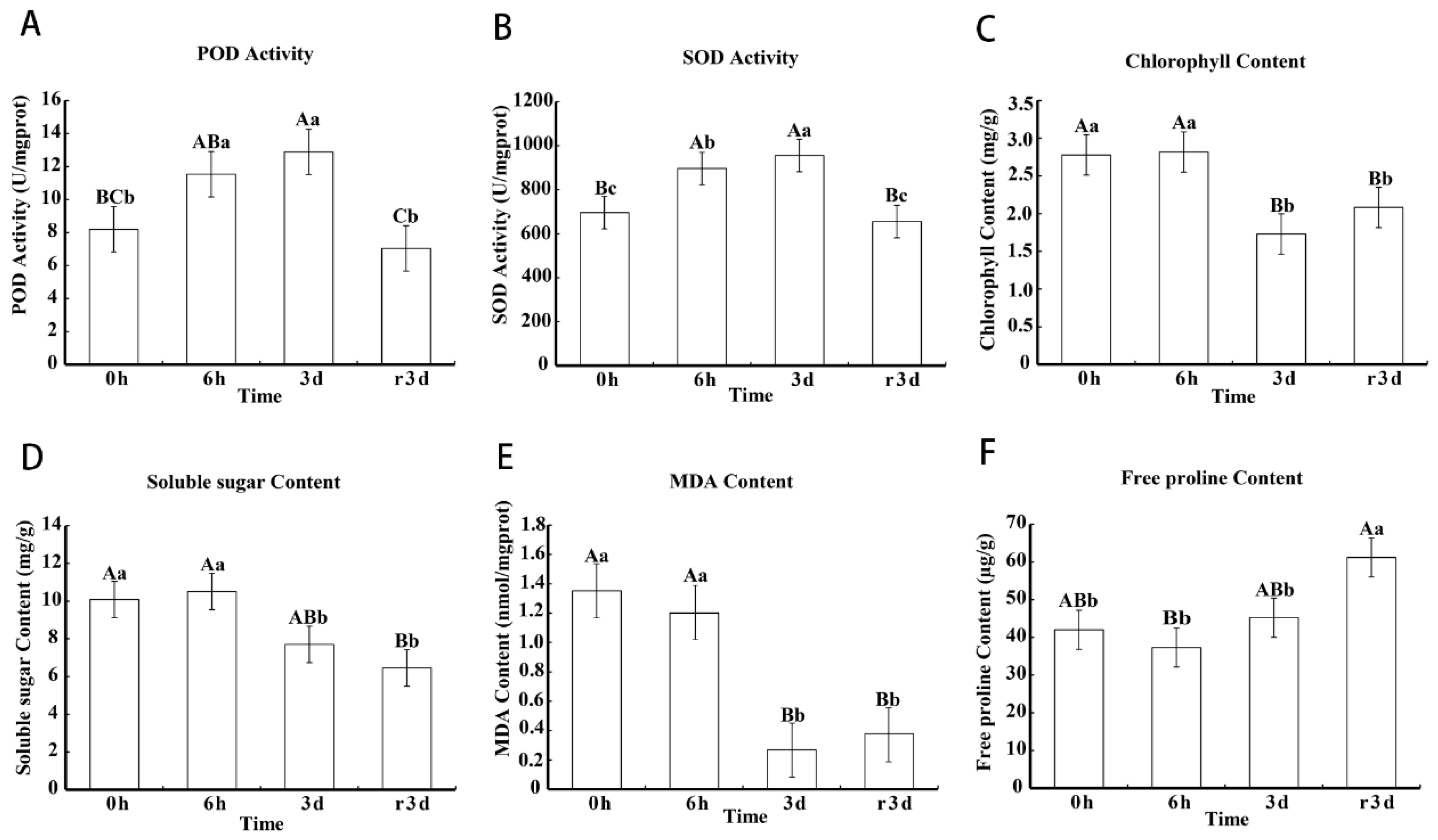
2.2. Physiological Response to Heat Stress in Leaves Of The Cultivar “Huoche”
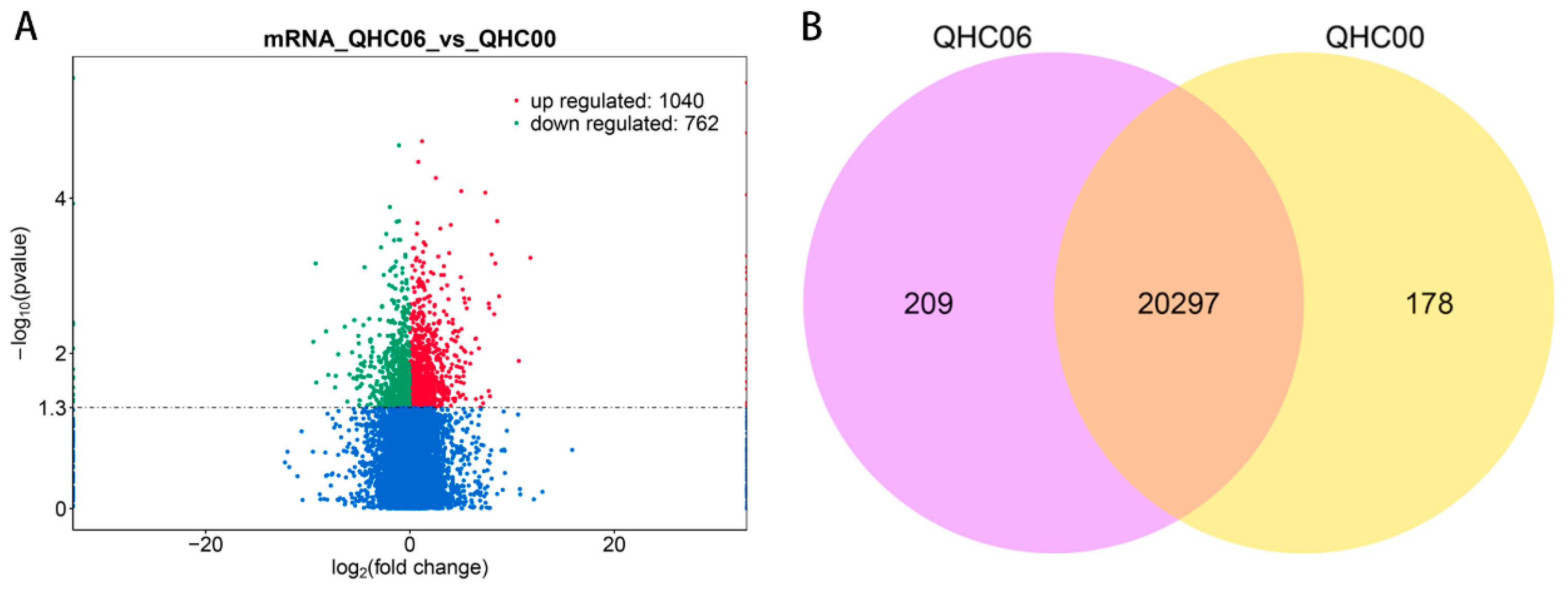
2.3. Mapping and Quantitative Assessment of Illumina Sequence
2.4. Analysis of DE mRNAs
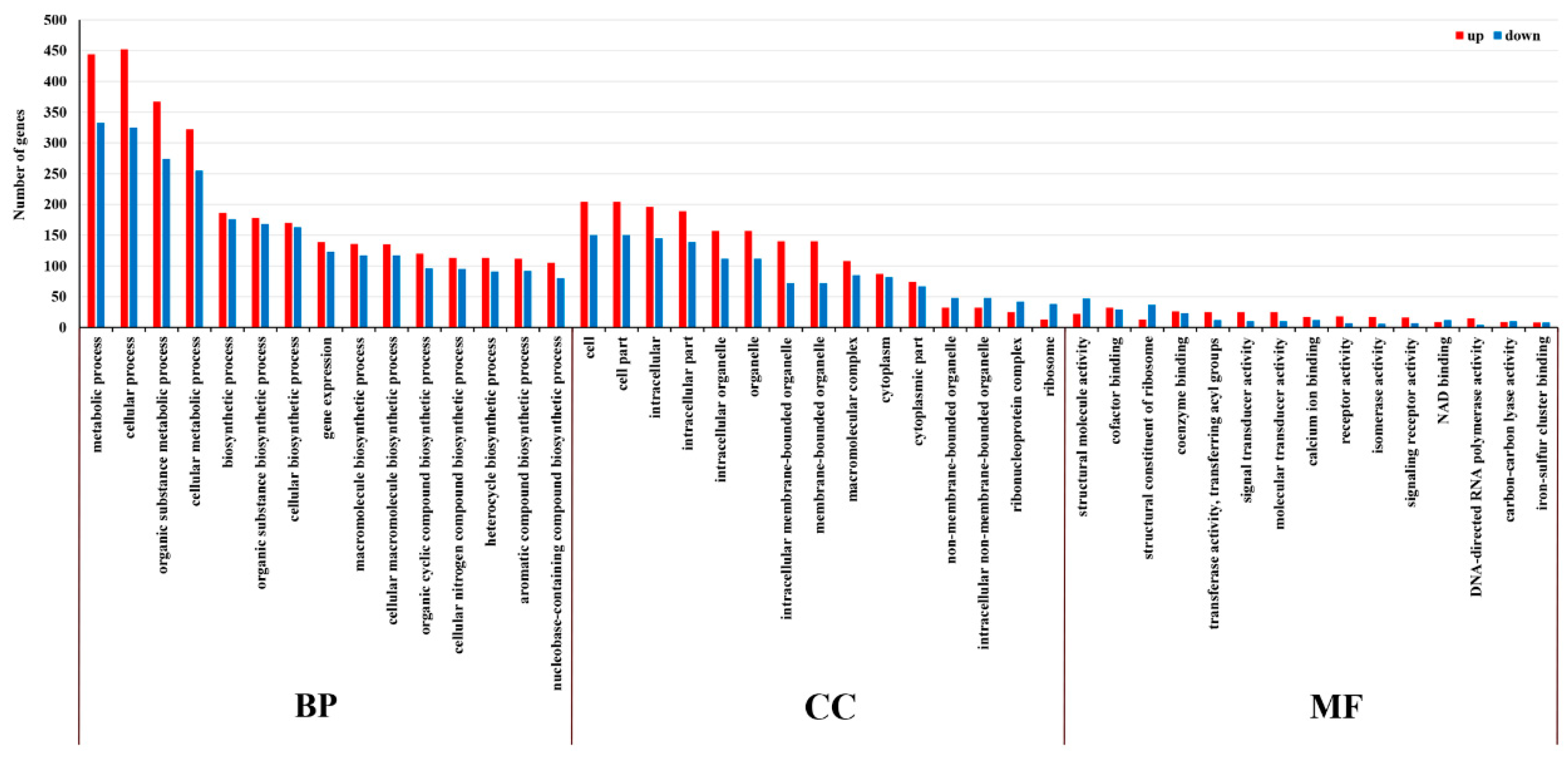
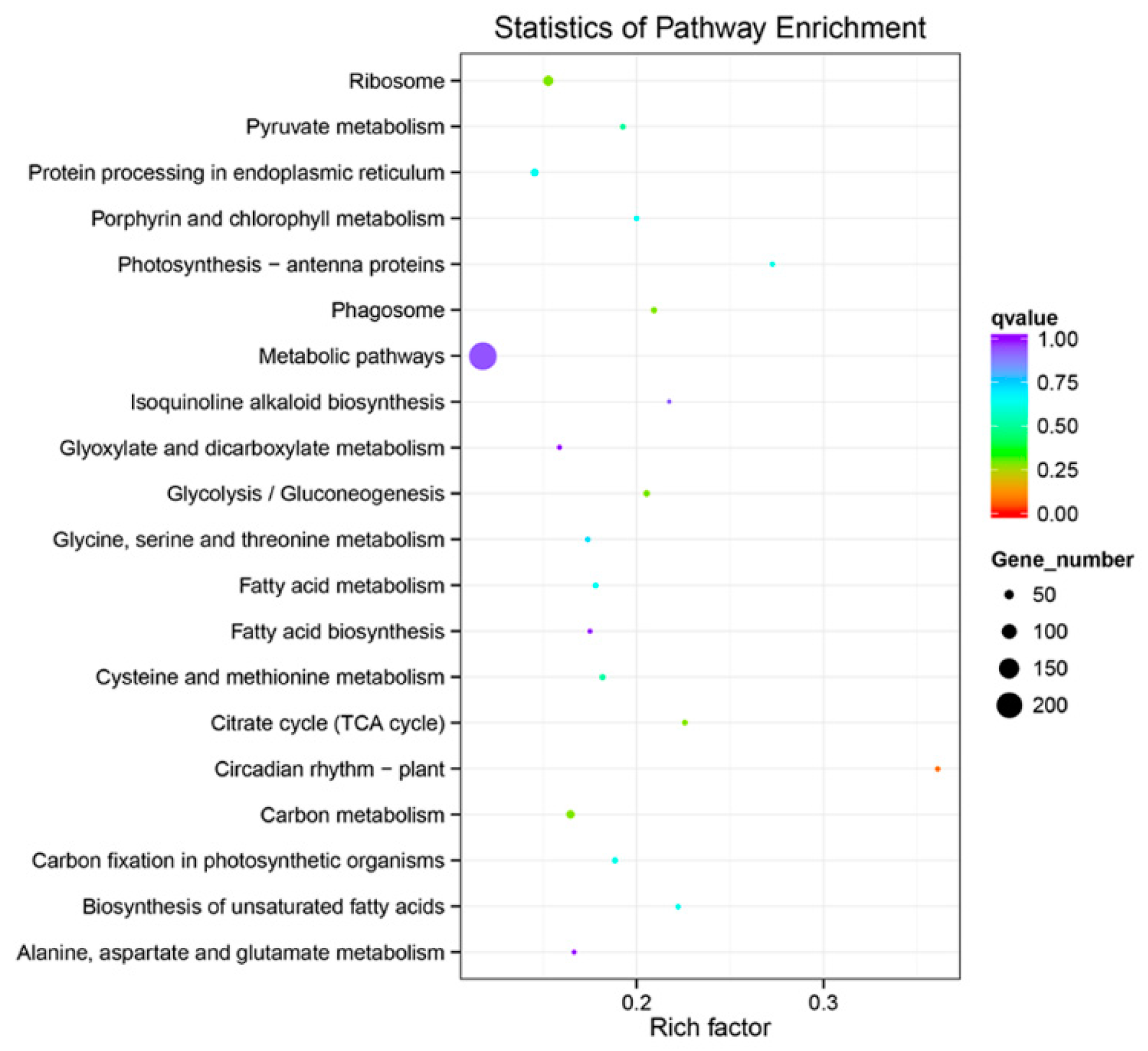
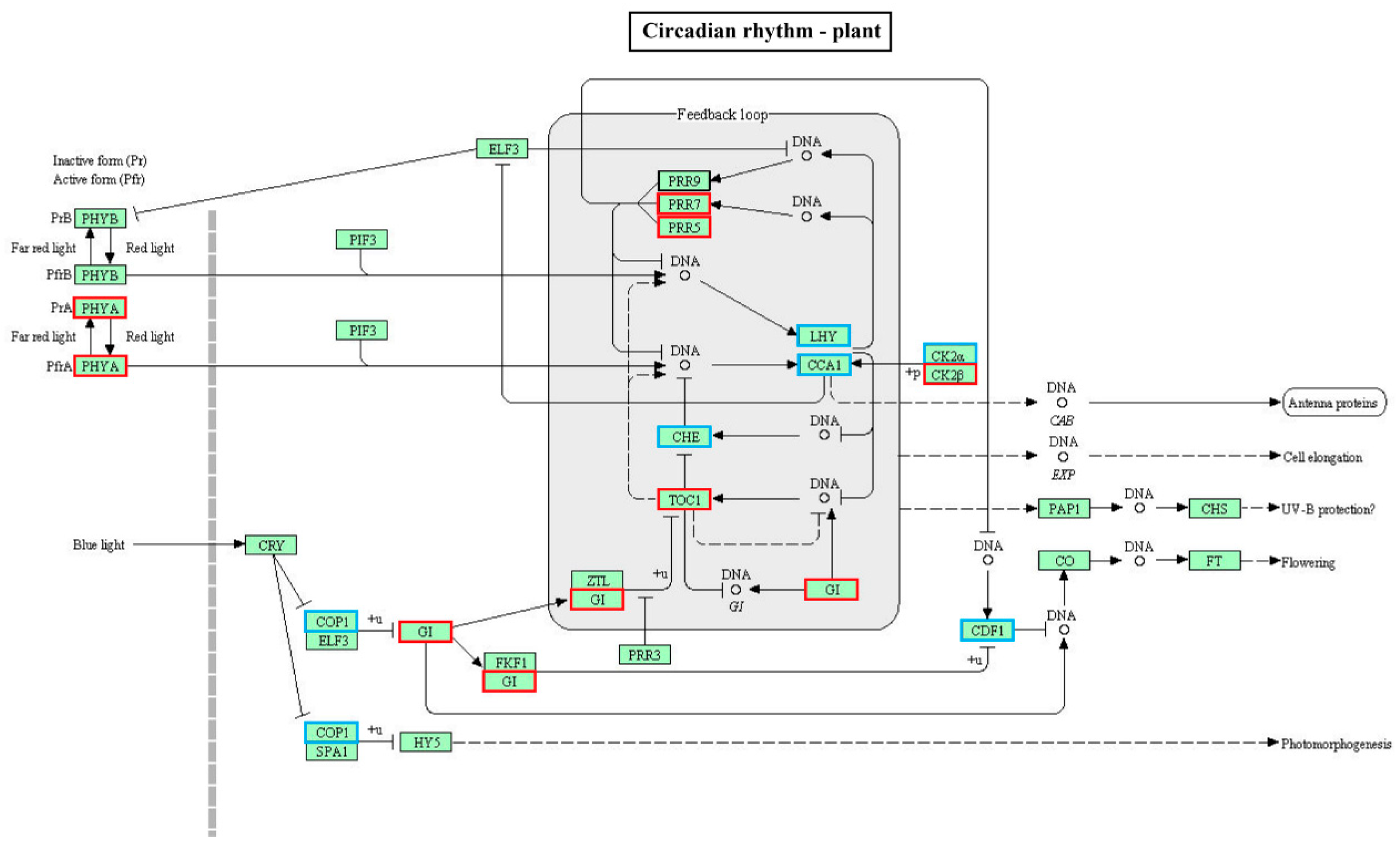
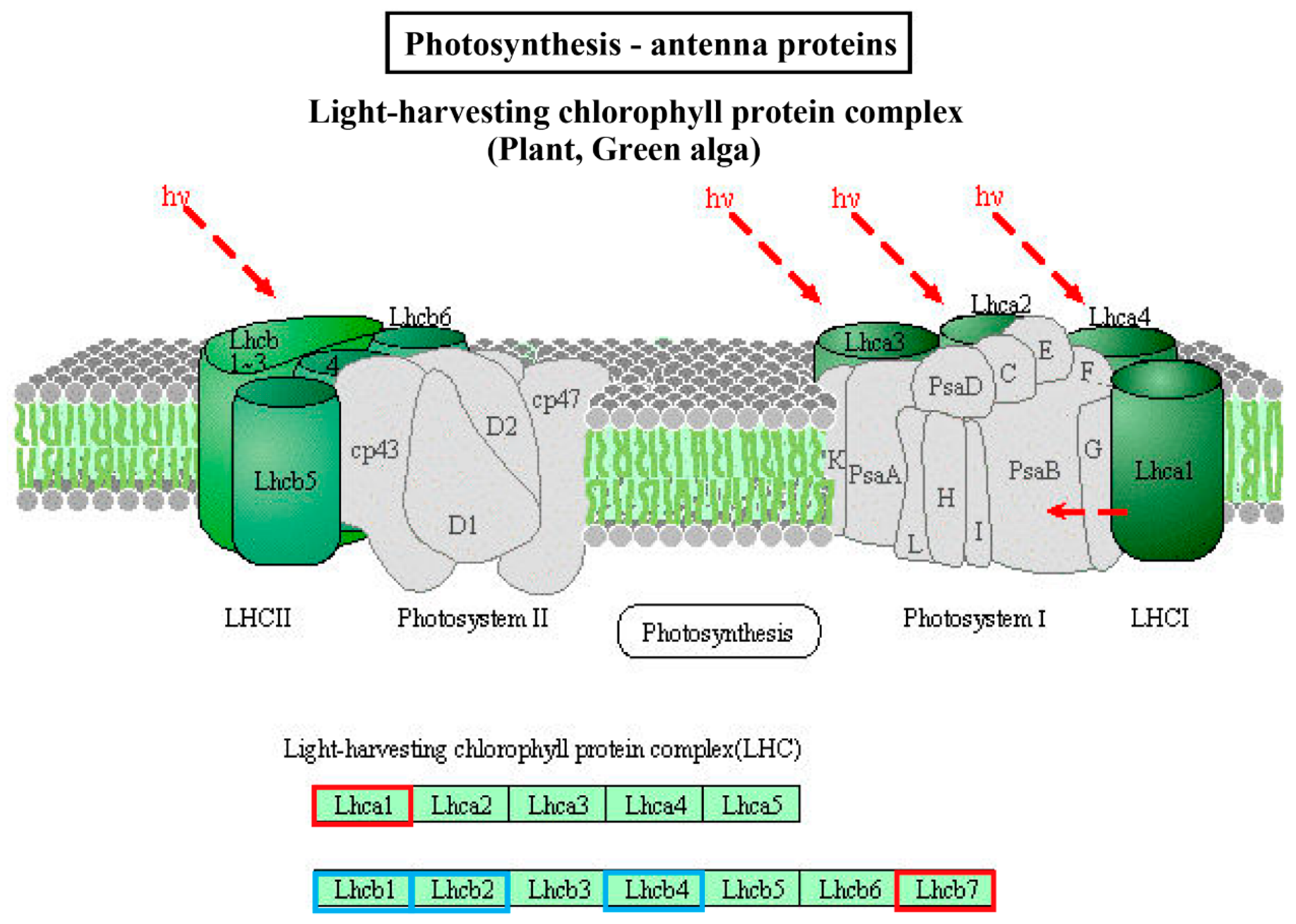
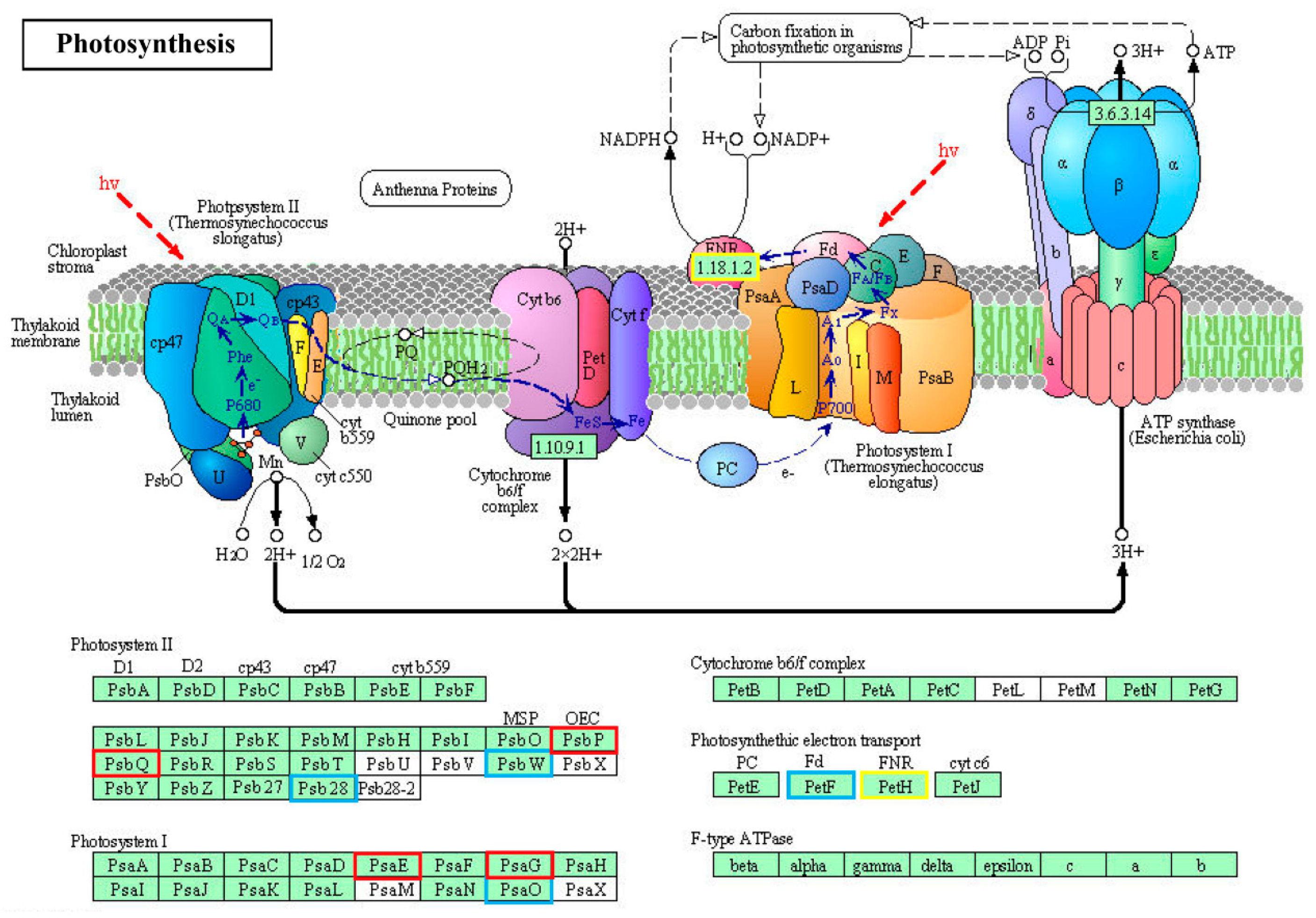
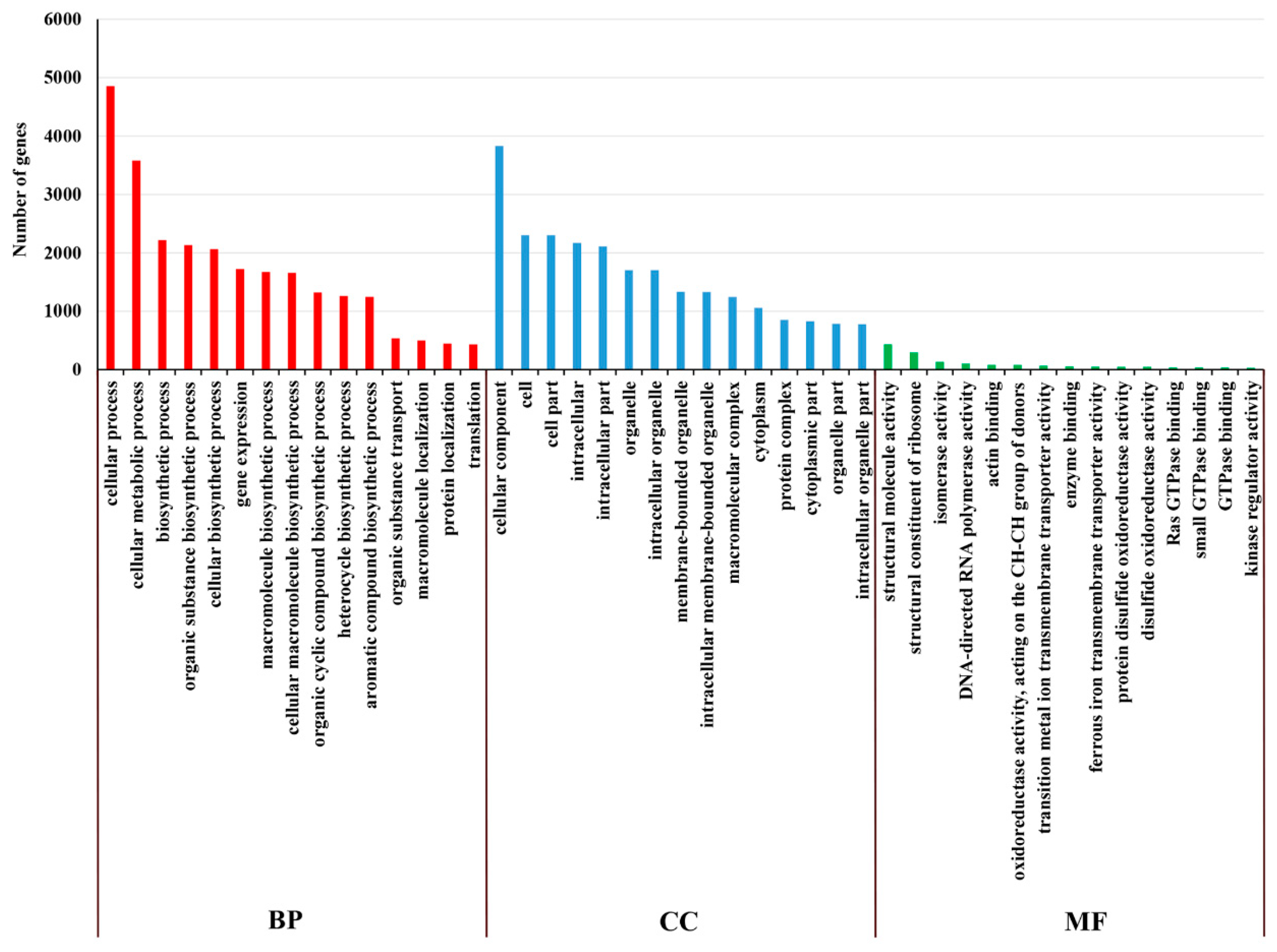
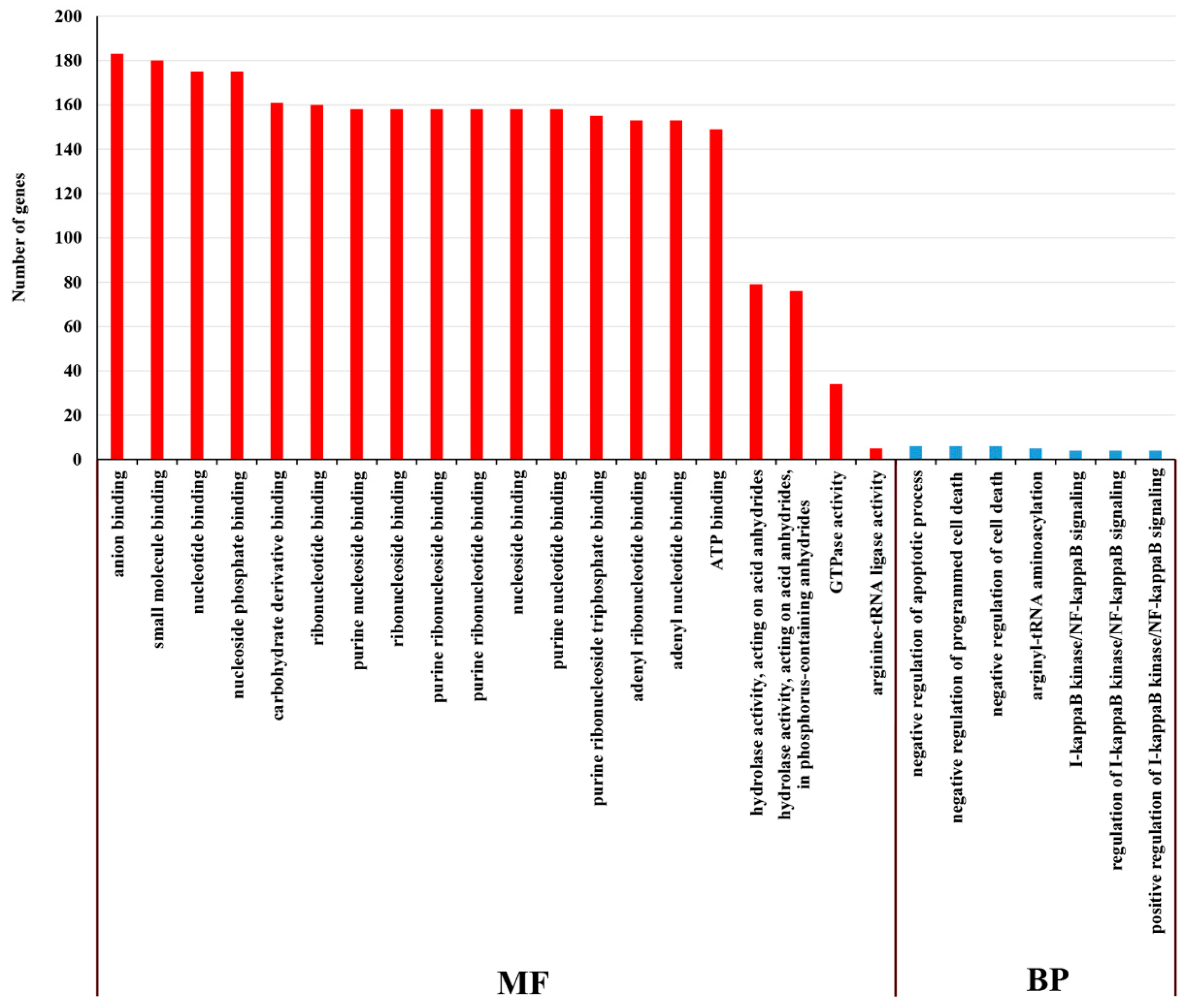
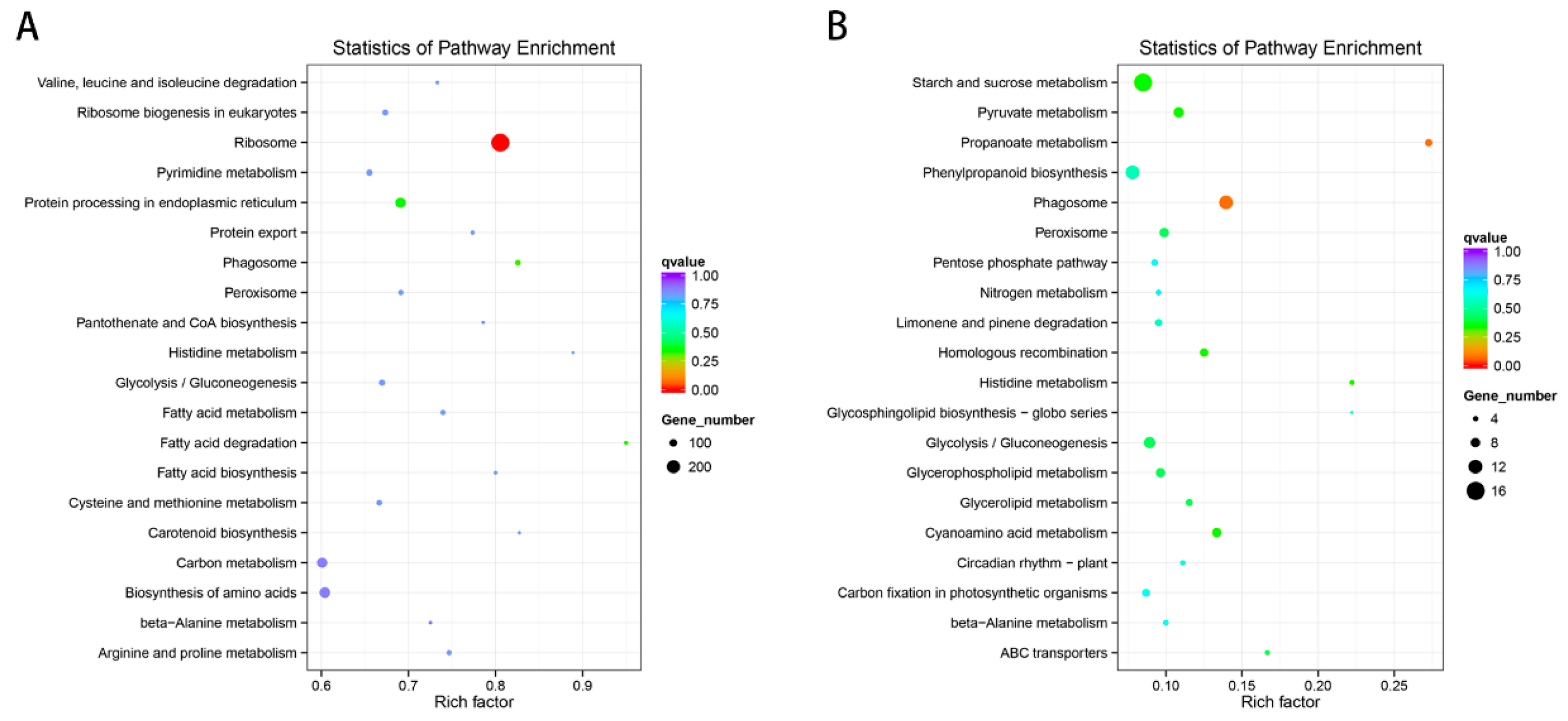
2.5. GO and KEGG Enrichment Analysis of DE mRNA Corresponding Genes under HS
2.6. DE mRNAs Encoding Transcription Factor
2.7. Identification of DE mRNA Encoding Heat Shock Protein (HSP) and Heat Shock Factor (HSF)
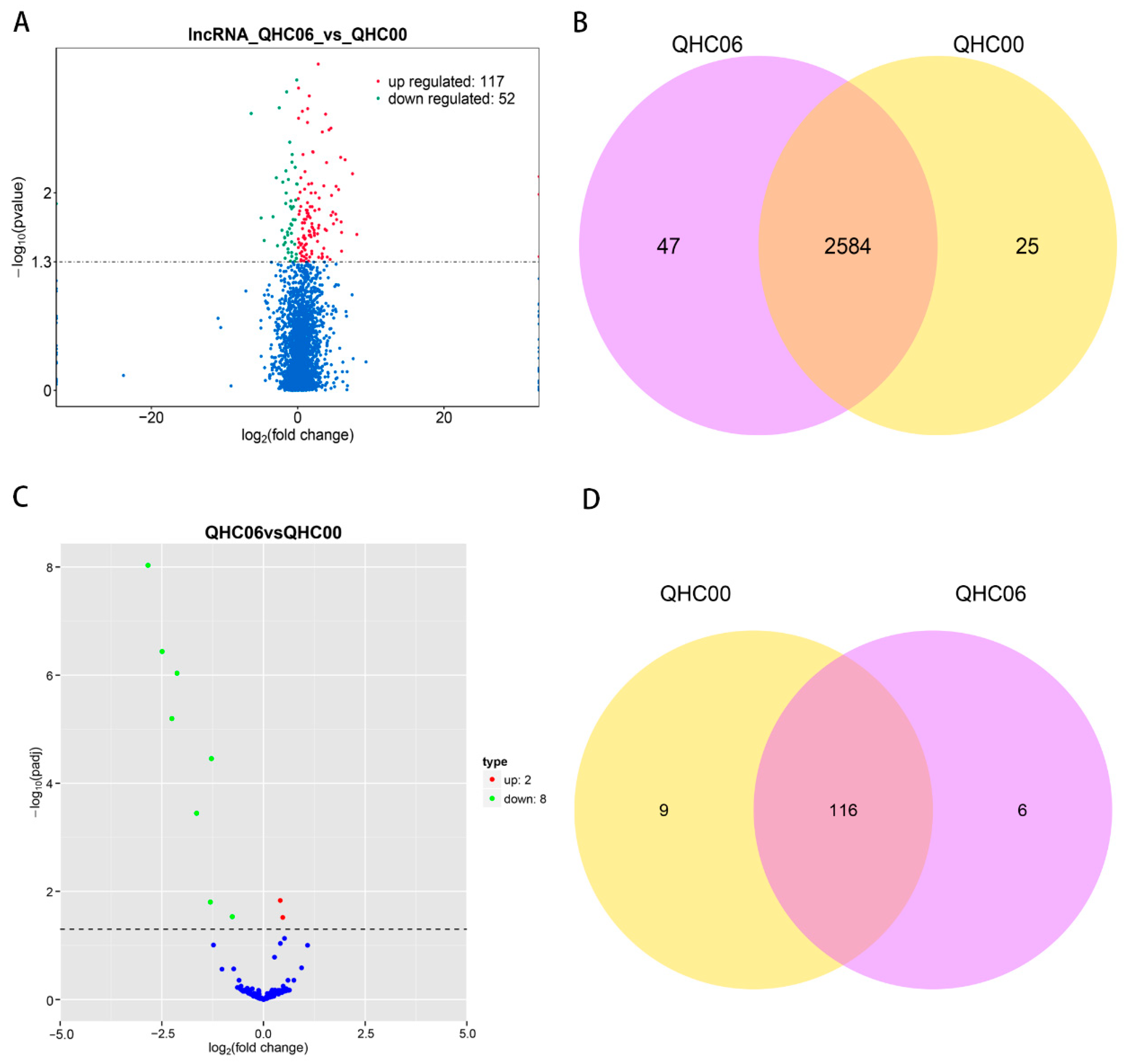
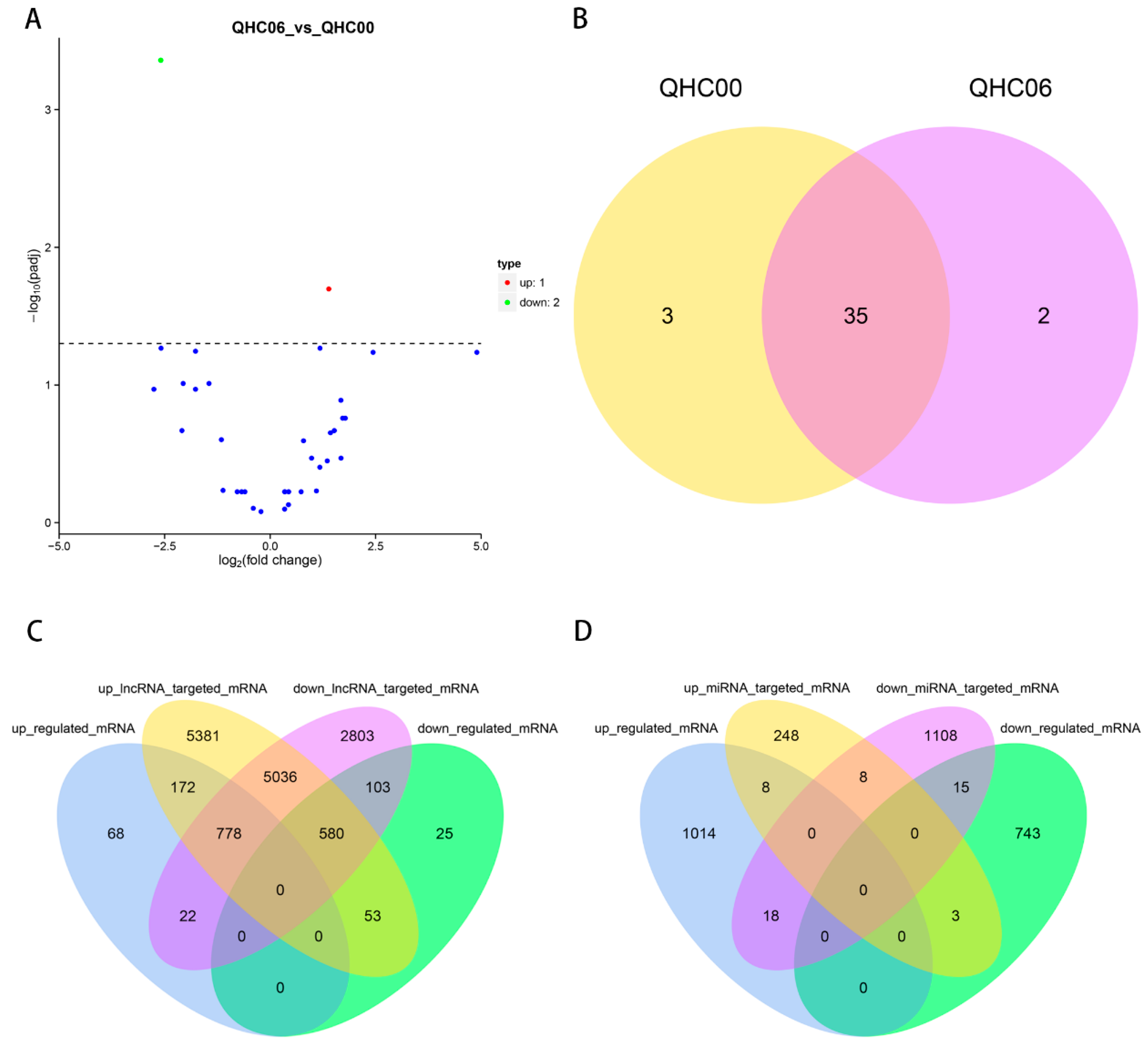
2.8. Analysis of DE lncRNAs, DE miRNAs and DE circRNAs
2.9. Functional Prediction of DE lncRNA and DE miRNA in the Cultivar “Huoche”
2.10. Regulatory Network in Response to HS
3. Discussion
3.1. TFs Response to HS
3.2. HS-Responsive HSPs and HSFs
3.3. HS-Induced miRNAs
4. Materials and Methods
4.1. Plant Materials and HS Treatment
4.2. Morphological and Physiological Analysis
4.3. RNA Isolation and RNA-Seq
4.4. Differential Expression Analysis
4.5. GO and KEGG Enrichment Analysis
4.6. Analysis of Transcription Factors
4.7. Construction of the Regulation Network of Competing Endogenous RNA
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ROS | Reactive oxygen species |
| SOD | Superoxide dismutase |
| POD | Peroxidases |
| HS | Heat stress |
| MDA | Malondialdehyde |
| RNA-Seq | RNA sequencing |
| DEGs | Differentially expressed genes |
| ncRNAs | Non-coding RNAs |
| r1d | Recovery treatment for 1 d |
| r3d | Recovery treatment for 3 d |
| DE | Differentially expressed |
| GO | Gene Ontology |
| BP | Biological processes |
| CC | Cellular components |
| MF | Molecular functions |
| KEGG | Kyoto Encyclopedia of Genes and Genome |
| PHYA | Phytochrome A |
| PRR5 | Pseudo-response regulator 5 |
| PRR7 | Pseudo-response regulator 7 |
| TOC1 | Pseudo-response regulator 1 |
| CK2β | Casein kinase II subunit beta |
| GI | Gigantea |
| COP1 | E3 ubiquitin-protein ligase RFWD2 |
| CHE | Transcription factor TCP21 (protein CCA1 HIKING EXPEDITION) |
| CCA1 | Circadian clock associated 1 |
| LHY | MYB-related transcription factor LHY |
| CDF1 | Dof zinc finger protein DOF5.5 |
| CK2α | Casein kinase II subunit alpha |
| KO | KEGG orthology |
| Lhca1 | Light-harvesting complex I chlorophyll a/b binding protein 1 |
| Lhcb1 | Light-harvesting complex II chlorophyll a/b binding protein 1 |
| Lhcb2 | Light-harvesting complex II chlorophyll a/b binding protein 2 |
| Lhcb4 | Light-harvesting complex II chlorophyll a/b binding protein 4 |
| Lhcb7 | Light-harvesting complex II chlorophyll a/b binding protein 7 |
| PsbP | Photosystem II oxygen-evolving enhancer protein 2 |
| PsbQ | Photosystem II oxygen-evolving enhancer protein 3 |
| PsbW | Photosystem II PsbW protein |
| Psb28 | Photosystem II 13kDa protein |
| PsaE | Photosystem I subunit IV |
| PsaG | Photosystem I subunit V |
| PsaO | Photosystem I subunit PsaO |
| PetF | ferredoxin |
| TFs | Transcription factors |
| ceRNA | Competing endogenous RNA |
| HSP | Heat shock protein |
| HSF | Heat shock factor |
| PIFs | Phytochrome interacting factors |
| C3H | Cinnamate 3-hydroxylase |
| NAC | NAM, ATAF, and CUC |
| sHSPs | Small HSPs |
| CuZn-SOD | Copper-zinc superoxide dismutase |
| MRE | MicroRNA response element |
| Ndufs4 | NADH dehydrogenase (ubiquinone) Fe-S protein 4 |
| Ndufs5 | NADH dehydrogenase (ubiquinone) Fe-S protein 5 |
| Ndufs6 | NADH dehydrogenase (ubiquinone) Fe-S protein 6 |
| Ndufs7 | NADH dehydrogenase (ubiquinone) Fe-S protein 7 |
| Ndufv1 | NADH dehydrogenase (ubiquinone) flavoprotein 1 |
| Ndufa2 | NADH dehydrogenase (ubiquinone) 1 alpha subcomplex subunit 2 |
| Ndufa5 | NADH dehydrogenase (ubiquinone) 1 alpha subcomplex subunit 5 |
| Ndufa6 | NADH dehydrogenase (ubiquinone) 1 alpha subcomplex subunit 6 |
| Ndufa8 | NADH dehydrogenase (ubiquinone) 1 alpha subcomplex subunit 8 |
| COX10 | Heme o synthase |
| COX17 | Cytochrome c oxidase assembly protein subunit 17 |
| PXMP2 | Peroxisomal membrane protein 2 |
| MPV17 | Protein Mpv17 |
| ACAA1 | Acetyl-CoA acyltransferase 1 |
| FAR | Alcohol-forming fatty acyl-CoA reductase |
| AGT | Alanine-glyoxylate transaminase/serine-glyoxylate transaminase/serine-pyruvate Transaminase |
| AUX1 | Auxin influx carrier (AUX1 LAX family) |
| ARF | Auxin response factor |
| GH3 | Auxin responsive GH3 gene family |
| SAUR | SAUR family protein |
| A-ARR | Two-component response regulator ARR-A family |
| PYR/PYL | Abscisic acid receptor PYR/PYL family |
| PP2C | Protein phosphatase 2C |
| ABF | ABA responsive element binding factor |
| EBF1/2 | EIN3-binding F-box protein |
| EIN3 | Ethylene-insensitive protein 3 |
| BSK | BR-signaling kinase |
| CYCD3 | Cyclin D3, plant |
| JAZ | Jasmonate ZIM domain-containing protein |
References
- Song, L.; Chow, W.S.; Sun, L.; Li, C.; Peng, C. Acclimation of photosystem II to high temperature in two Wedelia species from different geographical origins: Implications for biological invasions upon global warming. J. Exp. Bot. 2010, 61, 4087–4096. [Google Scholar] [CrossRef] [PubMed]
- Miller, G.A.D.; Mittler, R.O.N. Could heat shock transcription factors function as hydrogen peroxide sensors in plants? Ann. Bot. 2006, 98, 279–288. [Google Scholar] [CrossRef] [PubMed]
- Kotak, S.; Larkindale, J.; Lee, U.; von Koskull-Döring, P.; Vierling, E.; Scharf, K.D. Complexity of the heat stress response in plants. Curr. Opin. Plant Biol. 2007, 10, 310–316. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Zou, Z.; Li, Q.; Xin, H.; Zhu, X.; Chen, X.; Li, X. Heterologous expression of three Camellia sinensis small heat shock protein genes confers temperature stress tolerance in yeast and Arabidopsis thaliana. Plant Cell Rep. 2017, 36, 1125–1135. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yang, P.; Kang, J.; Gan, Y.; Yu, J.; Calderón-Urrea, A.; Lyu, J.; Zhang, G.; Feng, Z.; Xie, J. Transcriptome analysis of pepper (Capsicum annuum) revealed a role of 24-epibrassinolide in response to chilling. Front. Plant Sci. 2016, 7, 1281. [Google Scholar] [CrossRef]
- Gill, S.S.; Tuteja, N. Reactive oxygen species and antioxidant machinery in abiotic stress tolerance in crop plants. Plant Physiol. Biochem. 2010, 48, 909–930. [Google Scholar] [CrossRef] [PubMed]
- Anwar, A.; Yan, Y.; Liu, Y.; Li, Y.; Yu, X. 5-aminolevulinic acid improves nutrient uptake and endogenous hormone accumulation, enhancing low-temperature stress tolerance in cucumbers. Int. J. Mol. Sci. 2018, 19, 3379. [Google Scholar] [CrossRef]
- Mittler, R. Oxidative stress, antioxidants and stress tolerance. Trends Plant Sci. 2002, 7, 405–410. [Google Scholar] [CrossRef]
- Meloni, D.A.; Oliva, M.A.; Martinez, C.A.; Cambraia, J. Photosynthesis and activity of superoxide dismutase, peroxidase and glutathione reductase in cotton under salt stress. Environ. Exp. Bot. 2003, 49, 69–76. [Google Scholar] [CrossRef]
- Chaitanya, K.V.; Sundar, D.; Reddy, A.R. Mulberry leaf metabolism under high temperature stress. Biol. Plant 2001, 44, 379–384. [Google Scholar] [CrossRef]
- Marioni, J.C.; Mason, C.E.; Mane, S.M.; Stephens, M.; Gilad, Y. RNA-seq: An assessment of technical reproducibility and comparison with gene expression arrays. Genome Res. 2008, 18, 1509–1517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57. [Google Scholar] [CrossRef] [PubMed]
- Fu, C.; Wang, F.; Liu, W.; Liu, D.; Li, J.; Zhu, M.; Liao, Y.; Liu, Z.; Huang, H.; Zeng, X.; et al. Transcriptomic analysis reveals new insights into high-temperature-dependent glume-unclosing in an elite rice male sterile line. Front. Plant Sci. 2017, 8, 112. [Google Scholar] [CrossRef] [PubMed]
- Kitashiba, H.; Li, F.; Hirakawa, H.; Kawanabe, T.; Zou, Z.; Hasegawa, Y.; Tonosaki, K.; Shirasawa, S.; Fukushima, A.; Yokoi, S.; et al. Draft sequences of the radish (Raphanus sativus L.) genome. DNA Res. 2014, 21, 481–490. [Google Scholar] [CrossRef] [PubMed]
- Mitsui, Y.; Shimomura, M.; Komatsu, K.; Namiki, N.; Shibata-Hatta, M.; Imai, M.; Katayose, Y.; Mukai, Y.; Kanamori, H.; Kurita, K.; et al. The radish genome and comprehensive gene expression profile of tuberous root formation and development. Sci. Rep. 2015, 5, 10835. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Mei, Y.; Xu, L.; Zhu, X.; Wang, Y.; Guo, J.; Liu, L. Genome-wide characterization of differentially expressed genes provides insights into regulatory network of heat stress response in radish (Raphanus sativus L.). Funct. Integr. Genom. 2018, 18, 225–239. [Google Scholar] [CrossRef] [PubMed]
- Karanja, B.K.; Xu, L.; Wang, Y.; Muleke, E.M.m.; Jabir, B.M.; Xie, Y.; Zhu, X.; Cheng, W.; Liu, L. Genome-wide characterization and expression profiling of NAC transcription factor genes under abiotic stresses in radish (Raphanus sativus L.). PeerJ 2017, 5, 4172. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wu, Z.; Raitskin, O.; Sun, Q.; Dean, C. Antisense-mediated FLC transcriptional repression requires the P-TEFb transcription elongation factor. Proc. Natl. Acad. Sci. USA 2014, 111, 7468–7473. [Google Scholar] [CrossRef] [PubMed]
- Contreras-Cubas, C.; Palomar, M.; Arteaga-Vázquez, M.; Reyes, J.L.; Covarrubias, A.A. Non-coding RNAs in the plant response to abiotic stress. Planta 2012, 236, 943–958. [Google Scholar] [CrossRef]
- Nejat, N.; Mantri, N. Emerging roles of long non-coding RNAs in plant response to biotic and abiotic stresses. Crit. Rev. Biotechnol. 2018, 38, 93–105. [Google Scholar] [CrossRef]
- Li, B.; Gao, K.; Ren, H.; Tang, W. Molecular mechanisms governing plant responses to high temperatures. J. Integr. Plant Biol. 2018, 60, 757–779. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, K.; Yamaguchi-Shinozaki, K.; Shinozaki, K. The transcriptional regulatory network in the drought response and its crosstalk in abiotic stress responses including drought, cold, and heat. Front. Plant Sci. 2014, 5, 170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Essebier, A.; Lamprecht, M.; Piper, M.; Bodén, M. Bioinformatics approaches to predict target genes from transcription factor binding data. Methods 2017, 131, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.L.; Petty, J.; Hoyle, D.C.; Hayes, A.; Oliver, S.G.; Riba-Garcia, I.; Gaskell, S.J.; Stateva, L. Genome-wide analysis of the effects of heat shock on a Saccharomyces cerevisiae mutant with a constitutively activated cAMP-dependent pathway. Comp. Funct. Genom. 2004, 5, 419–431. [Google Scholar] [CrossRef] [PubMed]
- Lim, C.J.; Yang, K.A.; Hong, J.K.; Choi, J.S.; Yun, D.J.; Hong, J.C.; Chung, W.S.; Lee, S.Y.; Cho, M.J.; Lim, C.O. Gene expression profiles during heat acclimation in Arabidopsis thaliana suspension-culture cells. J. Plant Res. 2006, 119, 373–383. [Google Scholar] [CrossRef]
- Riechmann, J.L.; Meyerowitz, J.L. The AP2/EREBP family of plant transcription factors. Biol. Chem. 1998, 379, 633. [Google Scholar] [CrossRef] [PubMed]
- Tang, M.; Liu, X.; Deng, H.; Shen, S. Over-expression of JcDREB, a putative AP2/EREBP domain-containing transcription factor gene in woody biodiesel plant Jatropha curcas, enhances salt and freezing tolerance in transgenic Arabidopsis thaliana. Plant Sci. 2011, 181, 623–631. [Google Scholar] [CrossRef]
- Zhao, J.; Zhang, X.; Wollenweber, B.; Jiang, D.; Liu, F. Water deficits and heat shock effects on photosynthesis of a transgenic Arabidopsis thaliana constitutively expressing ABP9, a bZIP transcription factor. J. Exp. Bot. 2008, 59, 839–848. [Google Scholar] [CrossRef]
- Bita, C.; Gerats, T. Plant tolerance to high temperature in a changing environment: Scientific fundamentals and production of heat stress-tolerant crops. Front. Plant Sci. 2013, 4, 273. [Google Scholar] [CrossRef]
- Jain, M.; Tyagi, A.K.; Khurana, J.P. Genome-wide identification, classification, evolutionary expansion and expression analyses of homeobox genes in rice. FEBS J. 2008, 275, 2845–2861. [Google Scholar] [CrossRef]
- Feng, Z.J.; Xu, S.C.; Liu, N.; Zhang, G.W.; Hu, Q.Z.; Gong, Y.M. Soybean TCP transcription factors: Evolution, classification, protein interaction and stress and hormone responsiveness. Plant Physiol. Biochem. 2018, 127, 129–142. [Google Scholar] [CrossRef] [PubMed]
- Francis, A.; Dhaka, N.; Bakshi, M.; Jung, K.H.; Sharma, M.K.; Sharma, R. Comparative phylogenomic analysis provides insights into TCP gene functions in Sorghum. Sci. Rep. 2016, 6, 38488. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhang, B.; Li, M.J.; Yin, X.M.; Huang, L.F.; Cui, Y.C.; Wang, M.L.; Xia, X. OsMSR15 encoding a rice C2H2-type zinc finger protein confers enhanced drought tolerance in transgenic Arabidopsis. J. Plant Biol. 2016, 59, 271–281. [Google Scholar] [CrossRef]
- Martinez, V.; Mestre, T.C.; Rubio, F.; Girones-Vilaplana, A.; Moreno, D.A.; Mittler, R.; Rivero, R.M. Accumulation of flavonols over hydroxycinnamic acids favors oxidative damage protection under abiotic stress. Front. Plant Sci. 2016, 7, 838. [Google Scholar] [CrossRef] [PubMed]
- Shao, H.; Wang, H.; Tang, X. NAC transcription factors in plant multiple abiotic stress responses: Progress and prospects. Front. Plant Sci. 2015, 6, 902. [Google Scholar] [CrossRef]
- Shahnejat-Bushehri, S.; Mueller-Roeber, B.; Balazadeh, S. Arabidopsis NAC transcription factor JUNGBRUNNEN1 affects thermomemory-associated genes and enhances heat stress tolerance in primed and unprimed conditions. Plant Signal. Behav. 2012, 7, 1518–1521. [Google Scholar] [CrossRef] [PubMed]
- Wu, A.; Allu, A.D.; Garapati, P.; Siddiqui, H.; Dortay, H.; Zanor, M.-I.; Asensi-Fabado, M.A.; Munné-Bosch, S.; Antonio, C.; Tohge, T.; et al. JUNGBRUNNEN1, a reactive oxygen species–responsive NAC transcription factor, regulates longevity in Arabidopsis. Plant Cell 2012, 24, 482–506. [Google Scholar] [CrossRef]
- Chen, C.; Begcy, K.; Liu, K.; Folsom, J.J.; Wang, Z.; Zhang, C.; Walia, H. Heat stress yields a unique MADS box transcription factor in determining seed size and thermal sensitivity. Plant Physiol. 2016, 171, 606–622. [Google Scholar] [CrossRef] [Green Version]
- Duan, W.; Song, X.; Liu, T.; Huang, Z.; Ren, J.; Hou, X.; Li, Y. Genome-wide analysis of the MADS-box gene family in Brassica rapa (Chinese cabbage). Mol. Genet. Genom. 2015, 290, 239–255. [Google Scholar] [CrossRef]
- Chauhan, H.; Khurana, N.; Agarwal, P.; Khurana, P. Heat shock factors in rice (Oryza sativa L.): Genome-wide expression analysis during reproductive development and abiotic stress. Mol. Genet. Genom. 2011, 286, 171. [Google Scholar] [CrossRef]
- Wang, W.; Vinocur, B.; Shoseyov, O.; Altman, A. Role of plant heat-shock proteins and molecular chaperones in the abiotic stress response. Trends Plant Sci. 2004, 9, 244–252. [Google Scholar] [CrossRef] [PubMed]
- Al-Whaibi, M.H. Plant heat-shock proteins: A mini review. J. King Saud Univ. Sci. 2011, 23, 139–150. [Google Scholar] [CrossRef] [Green Version]
- Qu, A.L.; Ding, Y.F.; Jiang, Q.; Zhu, C. Molecular mechanisms of the plant heat stress response. Biochem. Biophys. Res. Commun. 2013, 432, 203–207. [Google Scholar] [CrossRef] [PubMed]
- Guo, M.; Liu, J.H.; Ma, X.; Luo, D.X.; Gong, Z.H.; Lu, M.H. The plant heat stress transcription factors (hsfs): Structure, regulation, and function in response to abiotic stresses. Front. Plant Sci. 2016, 7, 114. [Google Scholar] [CrossRef] [PubMed]
- Mishra, S.K.; Tripp, J.; Winkelhaus, S.; Tschiersch, B.; Theres, K.; Nover, L.; Scharf, K.D. In the complex family of heat stress transcription factors, HsfA1 has a unique role as master regulator of thermotolerance in tomato. Genes Dev. 2002, 16, 1555–1567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schramm, F.; Larkindale, J.; Kiehlmann, E.; Ganguli, A.; Englich, G.; Vierling, E.; Von Koskull-Döring, P. A cascade of transcription factor DREB2A and heat stress transcription factor HsfA3 regulates the heat stress response of Arabidopsis. Plant J. 2008, 53, 264–274. [Google Scholar] [CrossRef]
- Xue, G.-P.; Drenth, J.; McIntyre, C.L. TaHsfA6f is a transcriptional activator that regulates a suite of heat stress protection genes in wheat (Triticum aestivum L.) including previously unknown Hsf targets. J. Exp. Bot. 2015, 66, 1025–1039. [Google Scholar] [CrossRef]
- Zhao, J.; He, Q.; Chen, G.; Wang, L.; Jin, B. Regulation of non-coding RNAs in heat stress responses of plants. Front. Plant Sci. 2016, 7, 1213. [Google Scholar] [CrossRef]
- Rogers, K.; Chen, X. Biogenesis, turnover, and mode of action of plant microRNAs. Plant Cell 2013, 25, 2383–2399. [Google Scholar] [CrossRef]
- Xin, M.; Wang, Y.; Yao, Y.; Xie, C.; Peng, H.; Ni, Z.; Sun, Q. Diverse set of microRNAs are responsive to powdery mildew infection and heat stress in wheat (Triticum aestivum L.). BMC Plant Biol. 2010, 10, 123. [Google Scholar] [CrossRef]
- Hivrale, V.; Zheng, Y.; Puli, C.O.R.; Jagadeeswaran, G.; Gowdu, K.; Kakani, V.G.; Barakat, A.; Sunkar, R. Characterization of drought- and heat-responsive microRNAs in switchgrass. Plant Sci. 2016, 242, 214–223. [Google Scholar] [CrossRef] [PubMed]
- Reyes, J.L.; Chua, N.H. ABA induction of miR159 controls transcript levels of two MYB factors during Arabidopsis seed germination. Plant J. 2007, 49, 592–606. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Sun, Y.H.; Chiang, V.L. Stress-responsive microRNAs in Populus. Plant J. 2008, 55, 131–151. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Ren, Y.; Zhang, Y.; Xu, J.; Sun, F.; Zhang, Z.; Wang, Y. Genome-wide identification and expression analysis of heat-responsive and novel microRNAs in Populus tomentosa. Gene 2012, 504, 160–165. [Google Scholar] [CrossRef] [PubMed]
- Mahale, B.M.; Fakrudin, B.; Ghosh, S.; Krishnaraj, P.U. LNA mediated in situ hybridization of miR171 and miR397a in leaf and ambient root tissues revealed expressional homogeneity in response to shoot heat shock in Arabidopsis thaliana. J. Plant Biochem. Biotechnol. 2014, 23, 93–103. [Google Scholar] [CrossRef]
- Guan, Q.; Lu, X.; Zeng, H.; Zhang, Y.; Zhu, J. Heat stress induction of miR398 triggers a regulatory loop that is critical for thermotolerance in Arabidopsis. Plant J. 2013, 74, 840–851. [Google Scholar] [CrossRef] [PubMed]
- Sunkar, R.; Zhu, J.K. Novel and stress-regulated microRNAs and other small RNAs from arabidopsis. Plant Cell 2004, 16, 2001–2019. [Google Scholar] [CrossRef]
- Beauclair, L.; Yu, A.; Bouché, N. microRNA-directed cleavage and translational repression of the copper chaperone for superoxide dismutase mRNA in Arabidopsis. Plant J. 2010, 62, 454–462. [Google Scholar] [CrossRef]
- Sunkar, R.; Kapoor, A.; Zhu, J.K. Posttranscriptional induction of two Cu/Zn superoxide dismutase genes in Arabidopsis is mediated by downregulation of miR398 and important for oxidative stress tolerance. Plant Cell 2006, 18, 2051–2065. [Google Scholar] [CrossRef]
- Yu, X.; Wang, H.; Lu, Y.; de Ruiter, M.; Cariaso, M.; Prins, M.; van Tunen, A.; He, Y. Identification of conserved and novel microRNAs that are responsive to heat stress in Brassica rapa. J. Exp. Bot. 2012, 63, 1025–1038. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhao, G.; Li, Y.; Mo, N.; Zhang, J.; Liang, Y. Transcriptomic analysis implies that GA regulates sex expression via ethylene-dependent and ethylene-independent pathways in cucumber (Cucumis sativus L.). Front. Plant Sci. 2017, 8, 10. [Google Scholar] [CrossRef]
- Zhou, J.; Xiong, Q.; Chen, H.; Yang, C.; Fan, Y. Identification of the spinal expression profile of non-coding RNAs involved in neuropathic pain following spared nerve injury by sequence analysis. Front. Mol. Neurosci. 2017, 10, 91. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Zhu, D.; Li, H.; Li, H.; Feng, C.; Zhang, W. Characterization of circRNA-associated-ceRNA networks in a senescence-accelerated mouse prone 8 brain. Mol. Ther. 2017, 25, 2053–2061. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357. [Google Scholar] [CrossRef] [PubMed]
- Frazee, A.C.; Pertea, G.; Jaffe, A.E.; Langmead, B.; Salzberg, S.L.; Leek, J.T. Ballgown bridges the gap between transcriptome assembly and expression analysis. Nat. Biotechnol. 2015, 33, 243. [Google Scholar] [CrossRef]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef]
- Kanehisa, M.; Araki, M.; Goto, S.; Hattori, M.; Hirakawa, M.; Itoh, M.; Katayama, T.; Kawashima, S.; Okuda, S.; Tokimatsu, T.; et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2008, 36, D480–D484. [Google Scholar] [CrossRef]
- Mao, X.; Cai, T.; Olyarchuk, J.G.; Wei, L. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 2005, 21, 3787–3793. [Google Scholar] [CrossRef]
- Pérez-Rodríguez, P.; Riaño-Pachón, D.M.; Corrêa, L.G.G.; Rensing, S.A.; Kersten, B.; Mueller-Roeber, B. PlnTFDB: Updated content and new features of the plant transcription factor database. Nucleic Acids Res. 2010, 38, D822–D827. [Google Scholar] [CrossRef]
- Salmena, L.; Poliseno, L.; Tay, Y.; Kats, L.; Pandolfi, P.P. A ceRNA Hypothesis: The rosetta stone of a hidden rna language? Cell 2011, 146, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Tay, Y.; Rinn, J.; Pandolfi, P.P. The multilayered complexity of ceRNA crosstalk and competition. Nature 2014, 505, 344. [Google Scholar] [CrossRef] [PubMed]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| mRNA ID | Gene ID | Regulation | Gene Description |
|---|---|---|---|
| XM_018589933.1 | 108817282 | up | HEAT repeat-containing protein 5B%2C |
| XM_018607917.1 | 108834589 | up | heat stress transcription factor A-8-like |
| XM_018632926.1 | 108859078 | up | hsp70 nucleotide exchange factor fes1 |
| XM_018579151.1 | 108806935 | down | 28 kDa heat- and acid-stable phosphoprotein |
| XM_018607274.1 | 108833879 | down | heat shock factor-binding protein 1-like |
| XM_018631289.1 | 108857317 | down | hsp70 nucleotide exchange factor fes1-like |
| miRNA ID | QHC06 Read Count | QHC00 Read Count | log2 Fold Change | p-Value | q-Value | Regulation |
|---|---|---|---|---|---|---|
| ath-miR159b-3p | 22,398.8097 | 16761.99 | 0.41194 | 0.00079 | 0.014687 | up |
| ath-miR159c | 6837.4573 | 4883.9258 | 0.4737 | 0.00233 | 0.030311 | up |
| ath-miR398b-3p | 284.819304 | 4018.3814 | −2.8401 | 7.15E−11 | 9.30E−09 | down |
| ath-miR398a-3p | 16.1500394 | 158.0362 | −2.4915 | 5.62E−09 | 3.65E−07 | down |
| ath-miR165a-5p | 18.7396158 | 111.39067 | −2.1263 | 2.12E−08 | 9.20E−07 | down |
| ath-miR169g-3p | 2.63494616 | 22.542957 | −2.2541 | 1.95E−07 | 6.35E−06 | down |
| novel_86 | 28.5158186 | 74.079789 | −1.2807 | 1.34E−06 | 3.49E−05 | down |
| novel_107 | 6.19793394 | 24.899317 | −1.6446 | 1.64E−05 | 0.000356 | down |
| novel_21 | 778.011 | 2374.9094 | −1.3037 | 0.00097 | 0.01577 | down |
| ath-miR171b-3p | 129.729447 | 229.73667 | −0.76804 | 0.00204 | 0.029436 | down |
| circRNA ID | QHC06 Read Count | QHC00 Read Count | log2 Fold Change | p-Value | q-Value | Regulation |
|---|---|---|---|---|---|---|
| novel_circ_0000265 | 202.3891602 | 76.29972516 | 1.3908 | 0.0015051 | 0.020068 | up |
| novel_circ_0000325 | 7.966953075 | 46.82442697 | −2.5937 | 1.09E−05 | 0.0004377 | down |
| novel_circ_0000315 | 0 | 9.680244794 | −6.3484 | 0.000834 | 0.016679 | down |
| DE miRNA ID | miRNA Regulation | DE Targeted mRNA ID | mRNA Regulation |
|---|---|---|---|
| ath-miR171b-3p | down | XM_018577681.1 | up |
| ath-miR165a-5p | down | XM_018579231.1 | up |
| novel_21 | down | XM_018579451.1 | up |
| ath-miR165a-5p | down | XM_018585227.1 | up |
| ath-miR398b-3p, ath-miR398a-3p | down | XM_018585419.1 | up |
| ath-miR165a-5p | down | XM_018587204.1 | up |
| ath-miR169g-3p | down | XM_018589104.1 | up |
| novel_107 | down | XM_018590932.1 | up |
| ath-miR169g-3p | down | XM_018596526.1 | up |
| novel_86 | down | XM_018598856.1 | up |
| ath-miR171b-3p | down | XM_018599972.1 | up |
| ath-miR398a-3p, ath-miR398b-3p | down | XM_018604024.1 | up |
| novel_86 | down | XM_018618012.1 | up |
| novel_107 | down | XM_018620431.1 | up |
| ath-miR171b-3p | down | XM_018622399.1 | up |
| ath-miR398a-3p, ath-miR398b-3p | down | XM_018622487.1 | up |
| novel_107 | down | XM_018628114.1 | up |
| novel_86 | down | XM_018635962.1 | up |
| ath-miR159c | up | XM_018609293.1 | down |
| ath-miR159b-3p | up | XM_018617329.1 | down |
| ath-miR159b-3p, ath-miR159c | up | XM_018634874.1 | down |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, Z.; Li, W.; Su, X.; Ge, P.; Zhou, Y.; Hao, Y.; Shu, H.; Gao, C.; Cheng, S.; Zhu, G.; et al. Early Response of Radish to Heat Stress by Strand-Specific Transcriptome and miRNA Analysis. Int. J. Mol. Sci. 2019, 20, 3321. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20133321
Yang Z, Li W, Su X, Ge P, Zhou Y, Hao Y, Shu H, Gao C, Cheng S, Zhu G, et al. Early Response of Radish to Heat Stress by Strand-Specific Transcriptome and miRNA Analysis. International Journal of Molecular Sciences. 2019; 20(13):3321. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20133321
Chicago/Turabian StyleYang, Zhuang, Wen Li, Xiao Su, Pingfei Ge, Yan Zhou, Yuanyuan Hao, Huangying Shu, Chonglun Gao, Shanhan Cheng, Guopeng Zhu, and et al. 2019. "Early Response of Radish to Heat Stress by Strand-Specific Transcriptome and miRNA Analysis" International Journal of Molecular Sciences 20, no. 13: 3321. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20133321