High Functioning Autism with Missense Mutations in Synaptotagmin-Like Protein 4 (SYTL4) and Transmembrane Protein 187 (TMEM187) Genes: SYTL4- Protein Modeling, Protein-Protein Interaction, Expression Profiling and MicroRNA Studies

 , ,
, ,
Abstract
:1. Introduction
1.1. Synaptotagmin-Like Protein 4 (SYTL4) Gene
1.2. Transmembrane Protein 187 (TEM187) Gene
2. Results
2.1. Genomic Study
2.2. Synaptotagmin-Like 4 (SYTL4) Gene
2.2.1. Deleterious and Damaging Nature of the SYTL4- Variant
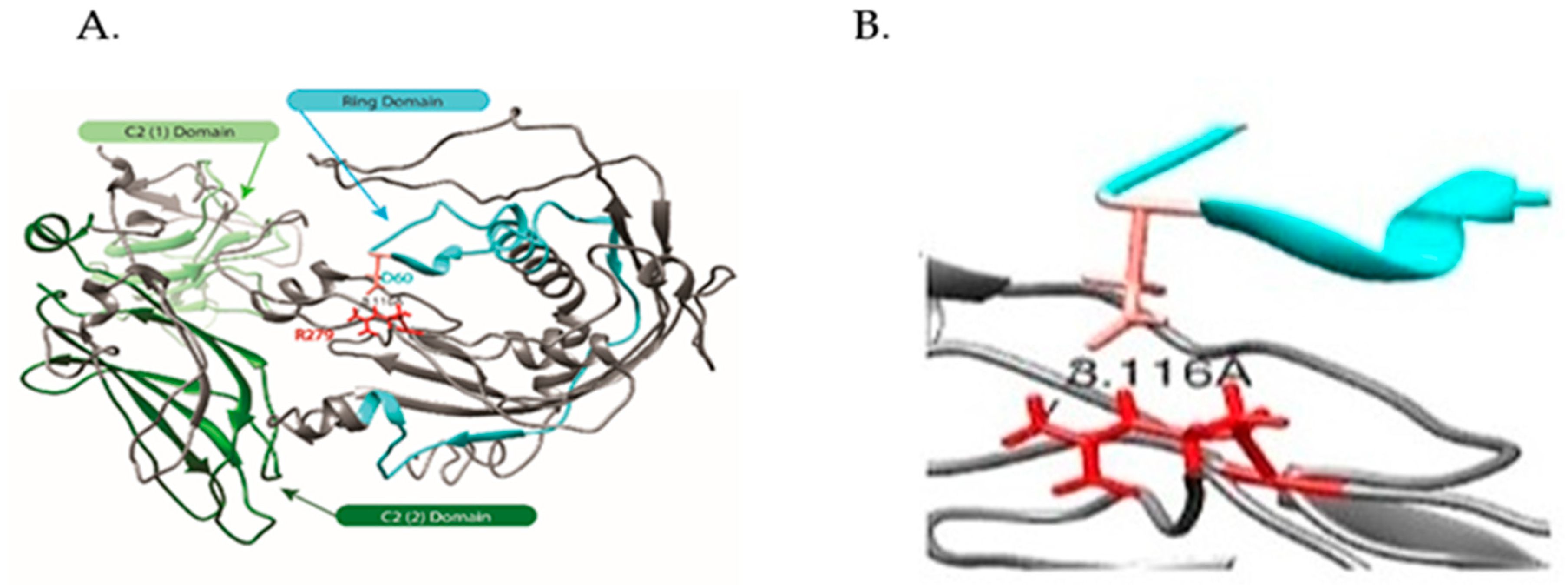
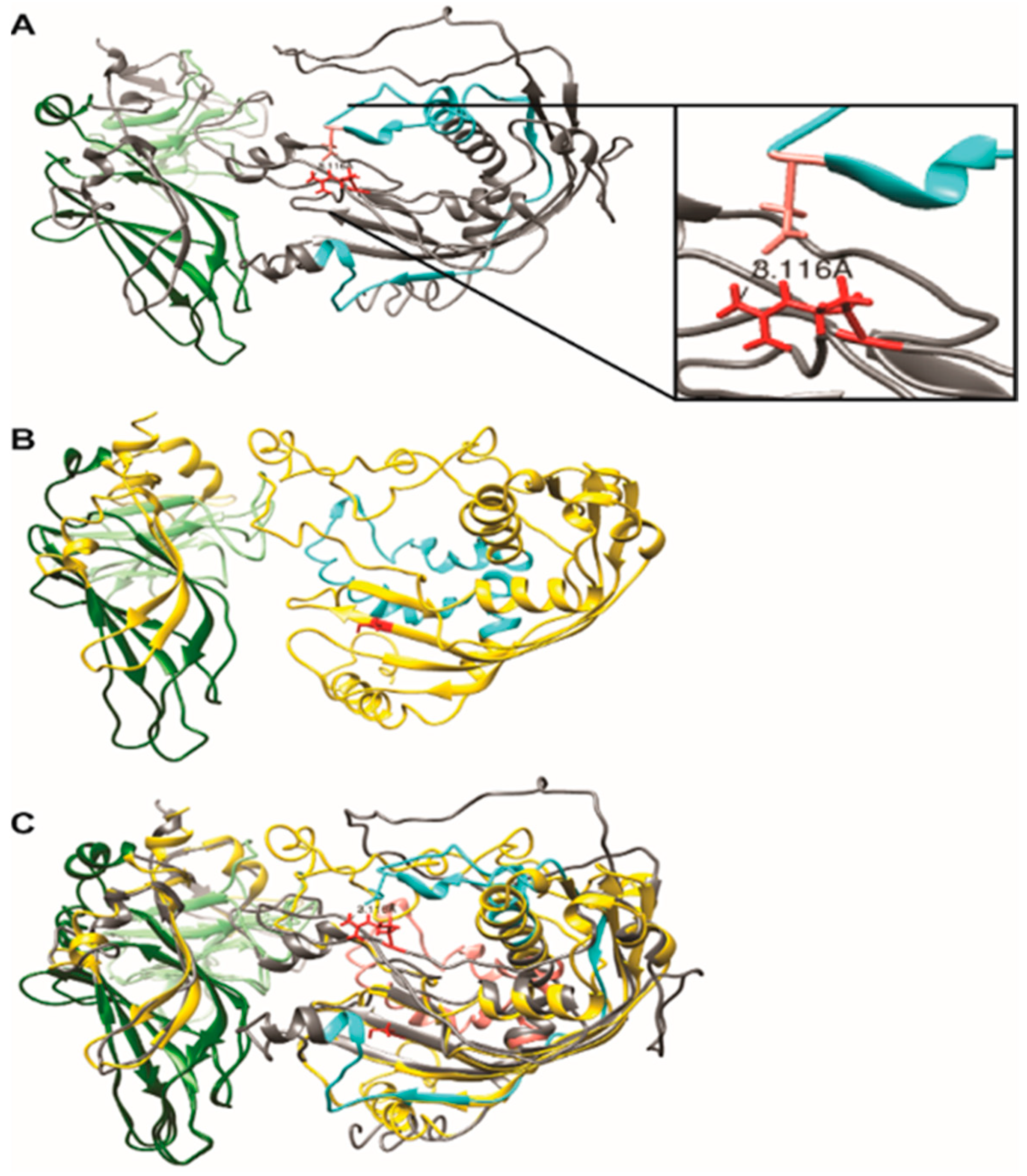
2.2.2. Modeling of Native and R279C Mutant for SYTL4 Gene
2.2.3. Hierarchical Protein Structural Modeling Study of Both Native and R (279) C SYTL4
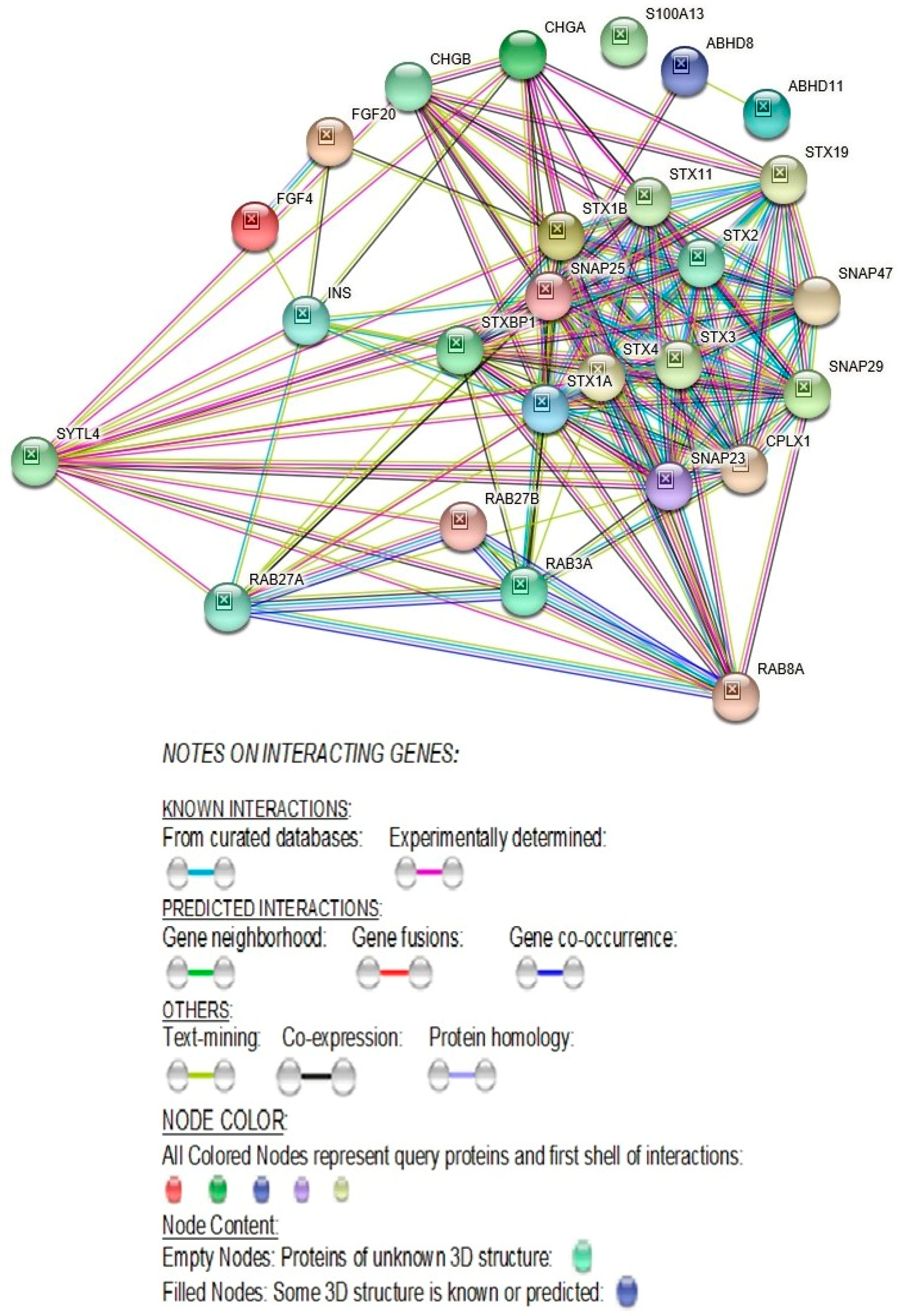
2.2.4. STRING- Protein–Protein Interaction Network Study Reveals Direct Interaction of SYTL4 with Other Known Autism Genes
2.2.5. SYTL4- Molecular Pathways and Associated Diseases
2.2.6. SYTL4- Networks of Biological Processes
2.2.7. SYTL4- Molecular Functions
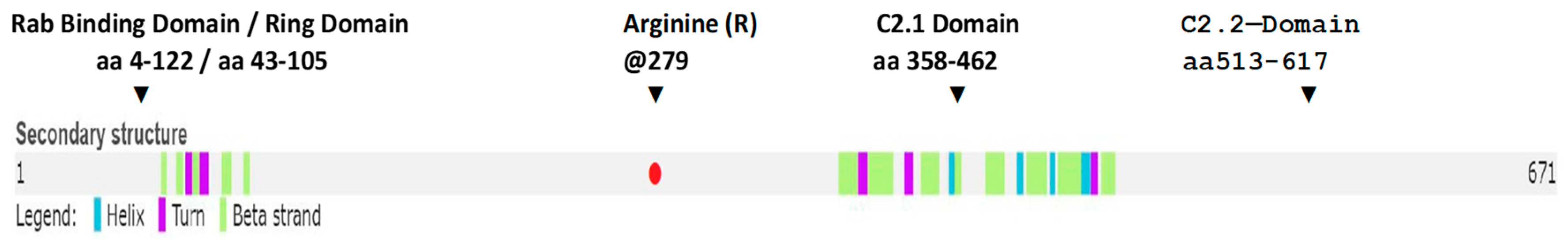
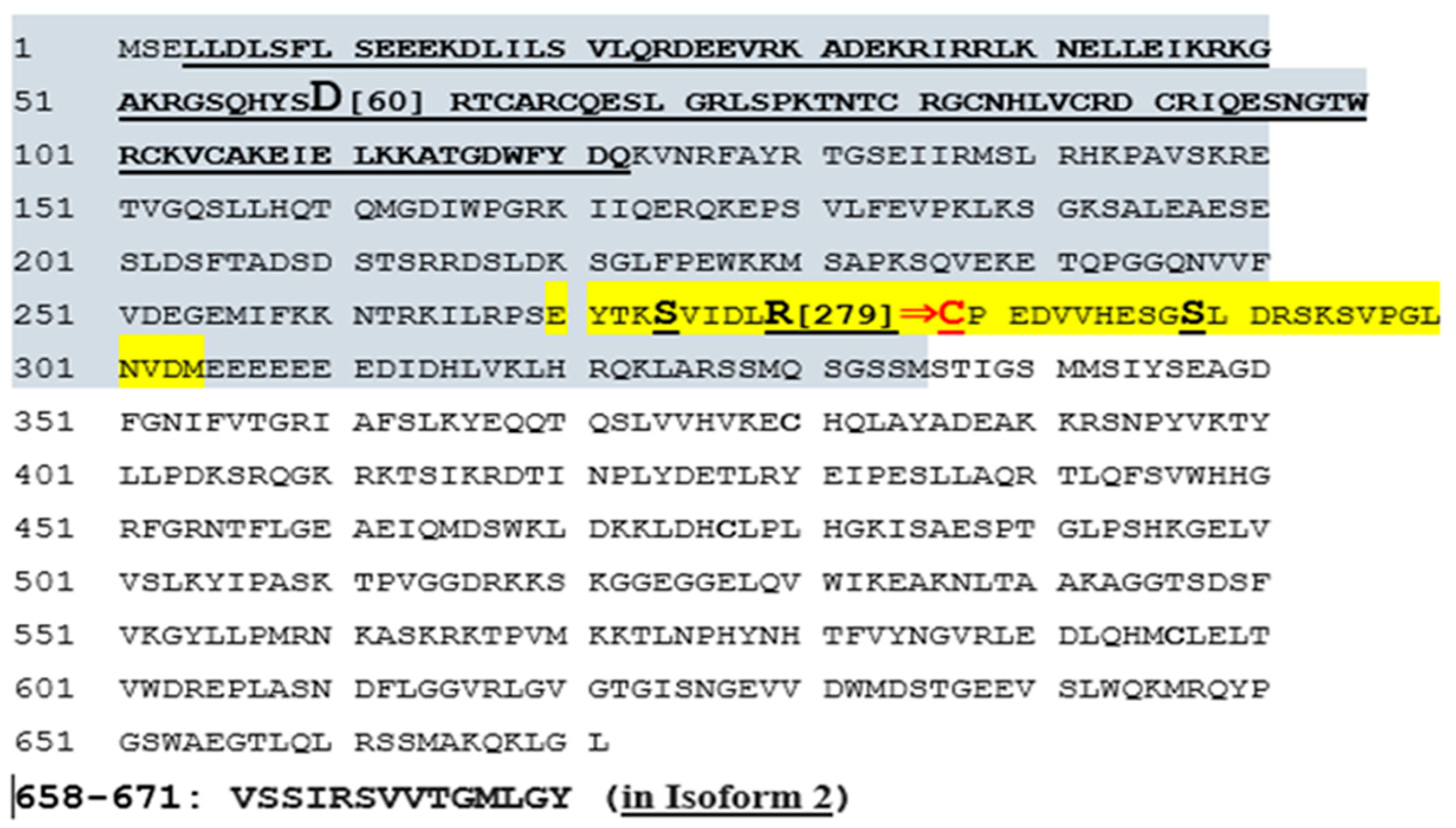
2.2.8. Missense Mutation Causing R (279) C Amino Acid Change Affects the Structures of Canonical SYTL4 Gene as Well as Its Shorter Isoform
2.2.9. Autism Predictive Human Serum MicroRNAs with Predicted Interaction with Mouse Sytl4 Gene
2.3. Transmembrane Protein 187 (TEM187) Gene
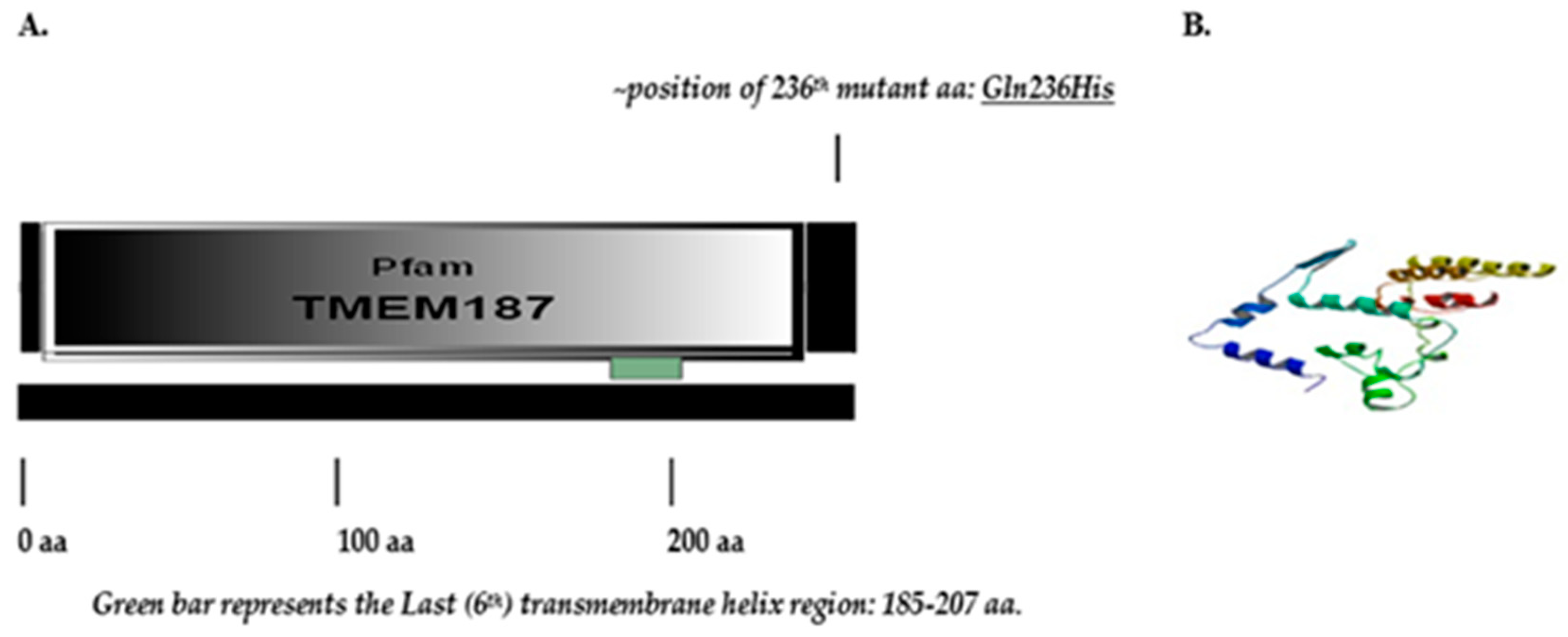
2.3.1. Structure of TMEM187 Gene, Expression and Location of the Novel Variant
2.3.2. Deleterious and Damaging Nature of the Novel TMEM187 Gene Variant
2.3.3. TMEM187 Gene Is Expressed in the Brain
2.3.4. Latest STRING- Gene Interaction Network Study Reveals Direct Protein–Protein Interactions of TMEM187 with Several Other Known Autism Genes
3. Discussion
3.1. Synaptotagmin-Like 4 (SYTL4) Gene
3.1.1. Protein Structure Altering Rare Variants Have Been Observed to Be More Frequent in Individuals with Autism
3.1.2. Deleterious and Damaging Nature of the SYTL4 Gene Variant
3.1.3. Randomness of X Chromosome Inactivation Could Render the Mother Asymptomatic
3.1.4. Our Modeling Results Show Large Conformational Changes Proximal to the R (279) C Amino Acid Variation
3.1.5. Missense Mutations Change the Size or Properties of Amino Acids Preventing the Function of Proteins
3.1.6. SYTL4 Amino Acid Change R (279) C in Exon 9: RAB-Binding Domain
3.1.7. Effect of the R[Arg]⇒C[Cys] Amino Acid Change at 279 on the functionality of the RAB-Binding Domain
3.1.8. Potentially Deleterious R (279) C Amino Acid Change “Likely” To Affect Its Neighboring Active Phosphorylation Sites
3.1.9. Role of Arginine (R279) in SYTL4 Protein Structure and Function
3.1.10. Arginine Disfavors Cysteine for Substitution
3.1.11. Dysfunction of Evolutionarily Conserved RAB-Binding GTPases Play a Role in Autism and Neuronal Disorders
3.1.12. Significance of Defect in RAB- Protein Binding Region of N-Terminal Half of SYTL4 Protein due to R (279) C Amino Acid Variant
3.1.13. SYTL4-Protein-Protein Interactions with RAB27A and with Other RAB-Family of Genes
3.1.14. Upregulation of RAB27A Protein Associated with Mild Cognitive Impairment and Alzheimer Disease
3.1.15. Dysfunction of Conserved RAB-Binding GTPases Play a Role in X-Linked Mental Retardation with Autism
3.1.16. SYTL4 Gene Is Relevant to Neuronal System Function and Disorders
3.1.17. SYTL4 Protein Is Abundantly Expressed in the Bed Nucleus of Stria Terminalis and Is Upregulated in Male Brain
3.1.18. Targeted Knockout Mutant Mammalian Phenotypes for Sytl4 Includes Abnormal Behavior and Abnormal Neurological Phenotype
3.1.19. Targeted Sytl4 Knock Out Mouse Model Studies Affirm That Defective SYTL4 Protein Function is Likely to Effectuate Neurological and Phenotypic Defects
3.1.20. SYTL4 Protein Directly Interacts with Proteins Known to Cause Autism
3.1.21. SYTL4 Gene Sequence Shows Similarity to a Known Autism Gene: SYT1
3.1.22. SYTL4 Gene Sequence Alignment Shows Similarity to SYT1(Synaptotagmin 1) Gene Which is a Known ASD Gene
3.1.23. Direct Protein-Protein STRING Interactions of the SYTL4 Gene with Other ASD Genes
3.1.24. SYTL4- Molecular Pathways, Biological Processes and Molecular Functions
3.1.25. Synaptic Dysfunction in Neurodevelopmental Disorders Is Associated with Autism and Intellectual Disabilities
3.1.26. ASD-Predictive MicroRNAs among Mouse Sytl4- Interacting MicroRNAs
3.1.27. Dysregulation of miR-320—Most Predictive for ASD in Serum and Brain Tissues
3.1.28. SYTL4 Interacting miR181b-1- Being Predictive of ASD
3.1.29. SYTL4 Interacting miR130a-Being Predictive of ASD
3.1.30. SYTL4-Interacting miR106b and miR328 Dysregulated in ASD Cerebellar Cortex and Altered among Schizophrenics
3.1.31. SYTL Interacting miR63, miR103, 5nd miR132 Are Dysregulated in Superior Temporal Gyrus of ASD
3.2. Transmembrane Protein 187 (TMEM187) Gene
3.2.1. TMEM187 Gene Belongs to a Group of Genes Which Host MicroRNA Genes in Their Introns or Exons
3.2.2. Novel TMEM187 Missense Variant c.708G>T: Glutamine(Q)236 Histidine(H)
3.2.3. Deleterious and Damaging Nature of the Novel TMEM187- Variant
3.2.4. TMEM187 Protein Is Expressed in Brain
3.2.5. STRING–Gene Interaction Network Study Reveals Direct Protein–Protein Interactions of the TMEM187 Gene with Several Other Known Autism Genes
3.2.6. Significance of TMEM187 Protein-Protein Interacting Autism Genes
3.2.7. Significance of Other Protein-Protein Interactions of TMEM187
3.2.8. Other TMEM Proteins Gene Family Members Are Known Autism, Bipolar and Panic Disorder Genes
3.2.9. X-chromosome Harbors Disproportionately Higher Number of TMEM187-Interacting Autism and Nervous System Disorder Genes: Implications for Boys vs Girls Ratio
4. Materials and Methods
4.1. Clinical Report
4.2. Genomic Investigations
4.3. Modeling of Native and R279C Variant for SYTL4 Gene
4.4. MicroRNAs
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Shashi, V.; McConkie-Rosell, A.; Rosell, B.; Schoch, K.; Vellore, K.; McDonald, M.; Jiang, Y.H.; Xie, P.; Need, A.; Goldstein, D.B. The utility of the traditional medical genetic diagnostic evaluation in the context of next-generation sequencing for undiagnosed genetic disorders. Genet. Med. 2014, 16, 176–182. [Google Scholar] [CrossRef] [PubMed]
- LePichon, J.B.; Saunders, C.; Soden, S.E. The future of next-generation sequencing in neurology. JAMA Neurol. 2015, 72, 971–972. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.; Ankala, A.; Wilcox, W.R.; Hegde, M.R. Solving the molecular diagnostic testing conundrum for Mendelian disorders in the era of next-generation sequencing: Single-gene, gene panel, or exome/genome sequencing. Genet. Med. 2015, 17, 444–451. [Google Scholar] [CrossRef] [PubMed]
- O’Roak, B.J.; Deriziotis, P.; Lee, C.; Vives, L.; Schwartz, J.J.; Girirajan, S.; Karakoc, E.; Mackenzie, A.P.; Ng, S.B.; Baker, C.; et al. Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nat. Genet. 2011, 43, 585–589. [Google Scholar] [CrossRef] [PubMed]
- Dixon-Salazar, T.J.; Silhavy, J.L.; Udpa, N.; Schroth, J.; Bielas, S.; Schaffer, A.E.; Olvera, J.; Bafna, V.; Zaki, M.S.; Abdel-Salam, G.H.; et al. Exome sequencing can improve diagnosis and alter patient management. Sci. Transl. Med. 2012, 4, 138ra78. [Google Scholar] [CrossRef] [PubMed]
- Sanders, S.J.; Murtha, M.T.; Gupta, A.R.; Murdoch, J.D.; Raubeson, M.J.; Willsey, A.J.; Ercan-Sencicek, A.G.; DiLullo, N.M.; Parikshak, N.N.; Stein, J.L.; et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature 2012, 485, 237–241. [Google Scholar] [CrossRef]
- Yu, T.W.; Chahrour, M.H.; Coulter, M.E.; Jiralerspong, S.; Okamura-Ikeda, K.; Ataman, B.; Schmitz-Abe, K.; Harmin, D.A.; Adli, M.; Malik, A.N.; et al. Using whole-exome sequencing to identify inherited causes of autism. Neuron 2013, 77, 259–273. [Google Scholar] [CrossRef]
- Vasieva, O.; Cetiner, S.; Savage, A.; Schumann, G.G.; Bubb, J.V.; Quinn, J.P. Primate specific retrotransposons, SVAs, in the evolution of networks that alter brain function in Neurons and Cognition. arXiv 2016, arXiv:1602.07642v2. [Google Scholar]
- Xu, X.; Coats, J.K.; Yang, C.F.; Wang, A.; Ahmed, O.M.; Alvarado, M.; Izumi, T.; Shah, N.M. Modular genetic control of sexually dimorphic behaviors. Cell 2012, 48, 596–607. [Google Scholar] [CrossRef]
- Lebow, M.; Chen, A. Overshadowed by the amygdala: The bed nucleus of the stria terminalis emerges as key to psychiatric disorders. Mol. Psychiatry 2016, 21, 450–463. [Google Scholar] [CrossRef]
- Kerman, I.A.; Bernard, R.; Bunney, W.E.; Jones, E.G.; Schatzberg, A.F.; Myers, R.M.; Barchas, J.D.; Akil, H.; Watson, S.J.; Thompson, R.C. Evidence for transcriptional factor dysregulation in the dorsal raphe nucleus of patients with major depressive disorder. Front. Neurosci. 2012, 6, 135. [Google Scholar] [CrossRef] [PubMed]
- Szego, E.M.; Janáky, T.; Szabó, Z.; Csorba, A.; Kompagne, H.; Müller, G.; Lévay, G.; Simor, A.; Juhász, G.; Kékesi, K.A. A mouse model of anxiety molecularly characterized by altered protein networks in the brain proteome. Eur. Neuropsychopharmacol. 2010, 20, 96–111. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Ishizaki, R.; Xu, J.; Kasai, K.; Kobayashi, E.; Gomi, H.; Izumi, T. The Rab27a effector exophilin7 promotes fusion of secretory granules that have not been docked to the plasma membrane. Mol. Biol. Cell 2013, 24, 319–330. [Google Scholar] [CrossRef] [PubMed]
- Xu, X. Modular genetic control of innate behaviors. Bioessays 2013, 35, 421–424. [Google Scholar] [CrossRef]
- Butler, M.G.; Rafi, S.K.; Hossain, W.; Stephan, D.A.; Manzardo, A.M. Whole exome sequencing in females with autism implicates novel and candidate genes. Int. J. Mol. Sci. 2015, 16, 1312–1335. [Google Scholar] [CrossRef] [PubMed]
- Gomi, H.; Mori, K.; Itohara, S.; Izumi, T. Rab27b is expressed in a wide range of exocytic cells and involved in the delivery of secretory granules near the plasma membrane. Mol. Biol. Cell 2007, 18, 4377–4386. [Google Scholar] [CrossRef] [PubMed]
- Boivin, V.; Deschamps-Francoeur, G.; Scott, M.S. Protein coding genes as hosts for noncoding RNA expression. Semin. Cell Dev. Biol. 2018, 75, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Bonanno, J.B.; Rutter, M.; Bain, K.T.; Miller, S.; Romero, R.; Wasserman, S.; Sauder, J.M.; Burley, S.K.; Almo, S.C. Protein Data Bank in Europe (PDBe) ID: 3fdw: Crystal Structure of a C2 Domain from Human Synaptotagmin-Like Protein 4; New York SGX Research Center for Structural Genomics (NYSGXRC): New York, NY, USA, 2008. [Google Scholar]
- Miyamoto, K.; Sato, M.; Koshiba, S.; Inoue, M.; Kigawa, T.; Yokoyama, S. Protein Data Bank (PDB) ID: 2CSZ: Solution Structure of the RING Domain of the Synaptotagmin-Like Protein 4. RIKEN Structural Genomics/Proteomics Initiative (RSGI), 2005. Available online: https://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/Structure/pdb/2CSZ (accessed on 8 July 2018).
- Zhang, Y. I-TASSER server for protein 3D structure prediction. BMC Bioinform. 2008, 9, 40. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Fuson, K.L.; Montes, M.; Robert, J.J.; Sutton, R.B. Structure of human synaptotagmin 1 C2AB in the absence of Ca2+ reveals a novel domain association. Biochemistry 2007, 46, 13041–13048. [Google Scholar] [CrossRef]
- Schauder, C.M.; Wu, X.; Saheki, Y.; Narayanaswamy, P.; Torta, F.; Wenk, M.R.; De Camilli, P.; Reinisch, K.M. Structure of a lipid-bound extended synaptotagmin indicates a role in lipid transfer. Nature 2014, 510, 552–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donald, J.E.; Kulp, D.W.; DeGrado, W.F. Salt bridges: Geometrically specific, designable interactions. Proteins 2011, 79, 898–915. [Google Scholar] [CrossRef]
- Gregoret, L.M.; Rade, S.D.; Fletterick, R.J.; Cohen, F.E. Hydrogen bonds involving sulfur atoms in proteins. Proteins 1991, 9, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Franceschini, A.; Wyder, S.; Forslund, K.; Heller, D.; Huerta-Cepas, J.; Simonovic, M.; Roth, A.; Santos, A.; Tsafou, K.P.; et al. STRING v10: Protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015, 43, D447–D452. [Google Scholar] [CrossRef] [PubMed]
- Okada, M.; Itoh, M.; Haraguchi, M.; Okajima, T.; Inoue, M.; Oishi, H.; Matsuda, Y.; Iwamoto, T.; Kawano, T.; Fukumoto, S.; et al. b-series Ganglioside deficiency exhibits no definite changes in the neurogenesis and the sensitivity to Fas-mediated apoptosis but impairs regeneration of the lesioned hypoglossal nerve. J. Biol. Chem. 2002, 277, 1633–1636. [Google Scholar] [CrossRef] [PubMed]
- Stamberger, H.; Nikanorova, M.; Willemsen, M.H.; Accorsi, P.; Angriman, M.; Baier, H.; Benkel-Herrenbrueck, I.; Benoit, V.; Budetta, M.; Caliebe, A.; et al. STXBP1 Encephalopathy: A neurodevelopmental disorder including epilepsy. Neurology 2016, 86, 954–962. [Google Scholar] [CrossRef]
- Söllner, T.; Bennett, M.K.; Whiteheart, S.W.; Scheller, R.H.; Rothman, J.E. A protein assembly-disassembly pathway in vitro that may correspond to sequential steps of synaptic vesicle docking, activation, and fusion. Cell 1993, 75, 409–430. [Google Scholar] [CrossRef]
- Neves, S.R.; Ram, P.T.; Iyengar, R. G protein pathways. Science 2002, 296, 1636–1639. [Google Scholar] [CrossRef]
- Rapoport, J.; Chavez, A.; Greenstein, D.; Addington, A.; Gogtay, N. Autism spectrum disorders and childhood-onset schizophrenia: Clinical and biological contributions to a relation revisited. J. Am. Acad. Child Adolesc. Psychiatry 2009, 48, 10–18. [Google Scholar] [CrossRef]
- Beveridge, N.J.; Cairns, M.J. MicroRNA dysregulation in schizophrenia. Neurobiol. Dis. 2012, 46, 263–271. [Google Scholar] [CrossRef] [Green Version]
- Shi, W.; Du, J.; Qi, Y.; Liang, G.; Wang, T.; Li, S.; Xie, S.; Zeshan, B.; Xiao, Z. Aberrant expression of serum miRNAs in schizophrenia. J. Psychiatr. Res. 2012, 46, 198–204. [Google Scholar] [CrossRef] [PubMed]
- Vasu, M.; Anitha, A.; Thanseem, I.; Suzuki, K.; Yamada, K.; Takahashi, T.; Wakuda, T.; Iwata, K.; Tsujii, M.; Sugiyama, T.; et al. Serum microRNA profiles in children with autism. Mol. Autism 2014, 5, 40. [Google Scholar] [CrossRef] [PubMed]
- Abu-Elneel, K.; Liu, T.; Gazzaniga, F.S.; Nishimura, Y.; Wall, D.P.; Geschwind, D.H.; Lao, K.; Kosik, K.S. Heterogeneous dysregulation of microRNAs across the autism spectrum. Neurogenetics 2008, 9, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Ghahramani Seno, M.M.; Hu, P.; Gwadry, F.G.; Pinto, D.; Marshall, C.R.; Casallo, G.; Scherer, S.W. Gene and miRNA expression profiles in autism spectrum disorders. Brain Res. 2011, 1380, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Stamova, B.; Ander, B.P.; Barger, N.; Sharp, F.R.; Schumann, C.M. Specific regional and age-related small noncoding RNA expression patterns within superior temporal gyrus of typical human brains are less distinct in autism brains. J. Child Neurol. 2015, 30, 1930–1946. [Google Scholar] [CrossRef] [PubMed]
- Anney, R.; Klei, L.; Pinto, D.; Almeida, J.; Bacchelli, E.; Baird, G.; Bolshakova, N.; Bölte, S.; Bolton, P.F.; Bourgeron, T.; et al. Individual common variants exert weak effects on the risk for autism spectrum disorders. Hum. Mol. Genet. 2012, 21, 4781–4792. [Google Scholar] [CrossRef] [PubMed]
- Klei, L.; Sanders, S.J.; Murtha, M.T.; Hus, V.; Lowe, J.K.; Willsey, A.J.; Moreno-De-Luca, D.; Yu, T.W.; Fombonne, E.; Geschwind, D.; et al. Common genetic variants, acting additively, are a major source of risk for autism. Mol. Autism 2012, 3, 9. [Google Scholar] [CrossRef]
- Stein, J.L.; Parikshak, N.N.; Geschwind, D.H. Rare inherited variation in autism: Beginning to see the forest. Neuron 2013, 77, 209–211. [Google Scholar] [CrossRef]
- Crestani, C.C.; Alves, F.H.; Gomes, F.V.; Resstel, L.B.; Correa, F.M.; Herman, J.P. Mechanisms in the bed nucleus of the stria terminalis involved in control of autonomic and neuroendocrine functions: A review. Curr. Neuropharmacol. 2013, 11, 141–159. [Google Scholar] [CrossRef]
- Lopes, A.M.; Burgoyne, P.S.; Ojarikre, A.; Bauer, J.; Sargent, C.A.; Amorim, A.; Affara, N.A. Transcriptional changes in response to X chromosome dosage in the mouse: Implications for X inactivation and the molecular basis of Turner Syndrome. BMC Genom. 2010, 11, 82. [Google Scholar] [CrossRef]
- Vitkup, D.; Sander, C.; Church, G.M. The amino-acid mutational spectrum of human genetic disease. Genome Biol. 2003, 4, R72. [Google Scholar] [CrossRef] [PubMed]
- Betts, M.J.; Russell, R.B. Amino acid properties and consequences of substitutions. In Bioinformatics for Geneticists; Barnes, M.R., Gray, I.C., Eds.; John Wiley & Sons: New York, NY, USA, 2003. [Google Scholar]
- Khan, S.; Vihinen, M. Spectrum of disease-causing mutations in protein secondary structures. BMC Struct. Biol. 2007, 7, 56. [Google Scholar] [CrossRef] [PubMed]
- Poussu, E.; Vihinen, M.; Paulin, L.; Savilahti, H. Probing the alpha-complementing domain of E. coli beta-galactosidase with use of an insertional pentapeptide mutagenesis strategy based on Mu in vitro DNA transposition. Proteins 2004, 54, 681–692. [Google Scholar] [CrossRef]
- Vihinen, M.; Vetri, D.; Maniar, H.S.; Ochs, H.D.; Zhu, Q.; Vorechovský, I.; Webster, A.D.; Notarangelo, L.D.; Nilsson, L.; Sowadski, J.M.; et al. Structural basis for chromosome X-linked agammaglobulinemia: A tyrosine kinase disease. Proc. Natl. Acad. Sci. USA 1994, 91, 12803–12807. [Google Scholar] [CrossRef] [PubMed]
- Yue, P.; Li, Z.; Moult, J. Loss of protein structure stability as a major causative factor in monogenic disease. J. Mol. Biol. 2005, 353, 459–473. [Google Scholar] [CrossRef] [PubMed]
- Rong, S.B.; Vihinen, M. Structural basis of Wiskott-Aldrich syndrome causing mutations in the WH1 domain. J. Mol. Med. (Berl.) 2000, 78, 530–537. [Google Scholar] [CrossRef] [PubMed]
- Lappalainen, I.; Vihinen, M. Structural basis of ICF-causing mutations in the methyltransferase domain of DNMT3B. Protein Eng. 2002, 15, 1005–1014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strom, M.; Hume, A.N.; Tarafder, A.K.; Barkagianni, E.; Seabra, M.C. A family of Rab27-binding proteins. Melanophilin links Rab27a and myosin Va function in melanosome transport. J. Biol. Chem. 2002, 277, 25423–25430. [Google Scholar] [CrossRef] [PubMed]
- Mufson, E.J.; Counts, S.E.; Ginsberg, S.D. Gene expression profiles of cholinergic nucleus basalis neurons in Alzheimer’s disease. Neurochem. Res. 2002, 27, 1035–1048. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Aravind, L. Identification of novel families and classification of the C2 domain superfamily elucidate the origin and evolution of membrane targeting activities in eukaryotes. Gene 2010, 469, 18–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyakhova, T.A.; Knight, J.D. The C2 domains of granuphilin are high-affinity sensors for plasma membrane lipids. Chem. Phys. Lipids 2014, 182, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Dephoure, N.; Zhou, C.; Villén, J.; Beausoleil, S.; Bakalarski, C.E.; Elledge, S.J.; Gygi, S.P. A quantitative atlas of mitotic phosphorylation. Proc. Natl. Acad. Sci. USA 2008, 105, 10762–10767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bian, Y.; Song, C.; Cheng, K.; Dong, M.; Wang, F.; Huang, J.; Sun, D.; Wang, L.; Ye, M.; Zou, H. An enzyme assisted RP-RPLC approach for in-depth analysis of human liver phosphoproteome. J. Proteom. 2014, 96, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Sharma, K.; D’Souza, R.C.; Tyanova, S.; Schaab, C.; Wiśniewski, J.R.; Cox, J.; Mann, M. Ultradeep human phosphoproteome reveals a distinct regulatory nature of Tyr and Ser/Thr-based signaling. Cell Rep. 2014, 8, 1583–1594. [Google Scholar] [CrossRef] [PubMed]
- Musafia, B.; Buchner, V.; Arad, D. Complex salt bridges in proteins: Statistical analysis of structure and function. J. Mol. Biol. 1995, 254, 761–770. [Google Scholar] [CrossRef] [PubMed]
- Mitra, S.; Cheng, K.W.; Mills, G.B. Rab GTPases implicated in inherited and acquired disorders. Semin. Cell Dev. Biol. 2010, 22, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Zhen, Y.; Stenmark, H. Cellular functions of Rab GTPases at a glance. J. Cell Sci. 2015, 128, 3171–3176. [Google Scholar] [CrossRef] [PubMed]
- Zerial, M.; McBride, H. Rab proteins as membrane organizers. Nat. Rev. Mol. Cell Biol. 2001, 2, 107–117. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, M.; Kanno, E. Analysis of the role of Rab27 effector Slp4-a/Granuphilin-a in dense-core vesicle exocytosis. Methods Enzymol. 2005, 403, 445–457. [Google Scholar]
- Izumi, T.; Gomi, H.; Torii, S. Functional analysis of Rab27a effector granuphilin in insulin exocytosis. Methods Enzymol. 2005, 403, 216–229. [Google Scholar]
- Chavas, L.M.; Ihara, K.; Kawasaki, M.; Torii, S.; Uejima, T.; Kato, R.; Izumi, T.; Wakatsuki, S. Elucidation of Rab27 recruitment by its effectors: Structure of Rab27a bound to Exophilin4/Slp2-a. Structure 2008, 16, 1468–1477. [Google Scholar] [CrossRef] [PubMed]
- Krzewski, K.; Cullinane, A.R. Evidence for defective Rab GTPase-dependent cargo traffic in immune disorders. Exp. Cell Res. 2013, 319, 2360–2367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giannandrea, M.; Bianchi, V.; Mignogna, M.L.; Sirri, A.; Carrabino, S.; D’Elia, E.; Vecellio, M.; Russo, S.; Cogliati, F.; Larizza, L.; et al. Mutations in the small GTPase gene RAB39B are responsible for X-linked mental retardation associated with autism, epilepsy, and macrocephaly. Am. J. Hum. Genet. 2010, 86, 185–195. [Google Scholar] [CrossRef] [PubMed]
- Goitre, L.; Trapani, E.; Trabalzini, L.; Retta, S.F. The Ras superfamily of small GTPases: The unlocked secrets. Methods Mol. Biol. 2014, 1120, 1–18. [Google Scholar] [PubMed]
- Yang, C.F.; Shah, N.M. Representing sex in the brain, one module at a time. Neuron 2014, 8, 261–278. [Google Scholar] [CrossRef] [PubMed]
- Palmer, N.; Beam, A.; Agniel, D.; Eran, A.; Manrai, A.; Spettell, C.; Steinberg, G.; Mandl, K.; Fox, K.; Nelson, S.F.; et al. Association of Sex with Recurrence of Autism Spectrum Disorder among Siblings. JAMA Pediatr. 2017, 171, 1107–1112. [Google Scholar] [CrossRef] [PubMed]
- Durdiakova, J.; Warrier, V.; Banerjee-Basu, S.; Baron-Cohen, S.; Chakrabarti, B. STX1A and Asperger syndrome: A replication study. Mol. Autism 2014, 5, 14. [Google Scholar] [CrossRef] [PubMed]
- Halladay, A.K.; Bishop, S.; Constantino, J.M.; Daniels, A.M.; Koenig, K.; Palmer, K.; Messinger, D.; Pelphrey, K.; Sanders, S.J.; Singer, A.T.; et al. Sex and gender differences in autism spectrum disorder: Summarizing evidence gaps and identifying emerging areas of priority. Mol. Autism 2015, 6, 36. [Google Scholar] [CrossRef] [PubMed]
- Gomi, H.; Mizutani, S.; Kasai, K.; Itohara, S.; Izumi, T. Granuphilin molecularly docks insulin granules to the fusion machinery. J. Cell Biol. 2005, 171, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Budnik, V.; Ruiz-Cañada, C.; Wendler, F. Extracellular vesicles round off communication in the nervous system. Nat. Rev. 2016, 17, 160–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Breevoort, D.; Snijders, A.P.; Hellen, N.; Weckhuysen, S.; van Hooren, K.W.; Eikenboom, J.; Valentijn, K.; Fernandez-Borja, M.; Ceulemans, B.; De Jonghe, P.; et al. STXBP1 promotes Weibel-Palade body exocytosis through its interaction with the Rab27A effector Slp4-a. Blood 2014, 123, 3185–3194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuen, R.K.; Thiruvahindrapuram, B.; Merico, D.; Walker, S.; Tammimies, K.; Hoang, N.; Chrysler, C.; Nalpathamkalam, T.; Pellecchia, G.; Liu, Y.; et al. Whole-genome sequencing of quartet families with autism spectrum disorder. Nat. Med. 2015, 21, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Franceschini, A.; Lin, J.; von Mering, C.; Jensen, L.J. SVD-phy: Improved prediction of protein functional associations through singular value decomposition of phylogenetic profiles. Bioinformatics 2016, 32, 1085–1087. [Google Scholar] [CrossRef] [PubMed]
- Braida, D.; Guerini, F.R.; Ponzoni, L.; Corradini, I.; De Astis, S.; Pattini, L.; Bolognesi, E.; Benfante, R.; Fornasari, D.; Chiappedi, M.; et al. Association between SNAP-25 gene polymorphisms and cognition in autism: Functional consequences and potential therapeutic strategies. Transl. Psychiatry 2015, 5, e500. [Google Scholar] [CrossRef] [PubMed]
- Hong, B.S.; Cho, J.H.; Kim, H.; Choi, E.J.; Rho, S.; Kim, J.; Kim, J.H.; Choi, D.S.; Kim, Y.K.; Hwang, D.; et al. Colorectal cancer cell-derived microvesicles are enriched in cell cycle-related mRNAs that promote proliferation of endothelial cells. BMC Genom. 2009, 10, 556. [Google Scholar] [CrossRef] [PubMed]
- Waites, C.L.; Garner, C.C. Presynaptic function in health and disease. Trends Neurosci. 2011, 34, 326–337. [Google Scholar] [CrossRef]
- Baker, K.; Gordon, S.L.; Grozeva, D.; van Kogelenberg, M.; Roberts, N.Y.; Pike, M.; Blair, E.; Hurles, M.E.; Chong, W.K.; Baldeweg, T.; et al. Identification of a human synaptotagmin-1 mutation that perturbs synaptic vesicle cycling. J. Clin. Investig. 2015, 125, 1670–1678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geppert, M.; Goda, Y.; Hammer, R.E.; Li, C.; Rosahl, T.W.; Stevens, C.F.; Südhof, T.C. Synaptotagmin I: A major Ca2+ sensor for transmitter release at a central synapse. Cell 1994, 79, 717–727. [Google Scholar] [CrossRef]
- Bai, J.; Wang, P.; Chapman, E.R. C2A activates a cryptic Ca (2+)-triggered membrane penetration activity within the C2B domain of synaptotagmin I. Proc. Natl. Acad. Sci. USA 2002, 99, 1665–1670. [Google Scholar] [CrossRef]
- Zoghbi, H.Y.; Bear, M.F. Synaptic dysfunction in neurodevelopmental disorders associated with autism and intellectual disabilities. Cold Spring Harb. Perspect. Biol. 2012, 4, a009886. [Google Scholar] [CrossRef]
- Bacaj, T.; Wu, D.; Yang, X.; Morishita, W.; Zhou, P.; Xu, W.; Malenka, R.C.; Südhof, T.C. Synaptotagmin-1 and synaptotagmin-7 trigger synchronous and asynchronous phases of neurotransmitter release. Neuron 2013, 80, 947–959. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Plaisance, V.; Abderrahmani, A.; Perret-Menoud, V.; Jacquemin, P.; Lemaigre, F.; Regazzi, R. MicroRNA-9 controls the expression of Granuphilin/Slp4 and the secretory response of insulin-producing cells. J. Biol. Chem. 2006, 281, 26932–26942. [Google Scholar] [CrossRef] [PubMed]
- Rolland, T.; Taşan, M.; Charloteaux, B.; Pevzner, S.J.; Zhong, Q.; Sahni, N.; Yi, S.; Lemmens, I.; Fontanillo, C.; Mosca, R.; et al. A proteome-scale map of the human interactome network. Cell 2014, 159, 1212–1226. [Google Scholar] [CrossRef] [PubMed]
- Eppig, J.T.; Blake, J.A.; Bult, C.J.; Kadin, J.A.; Richardson, J.E. Mouse Genome Database Group. The mouse genome database (MGD): Facilitating mouse as a model for human biology and disease. Nucleic Acids Res. 2015, 43, D726–D736. [Google Scholar] [CrossRef] [PubMed]
- Feng, R.; Sang, Q.; Zhu, Y.; Fu, W.; Liu, M.; Xu, Y.; Shi, H.; Xu, Y.; Qu, R.; Chai, R.; et al. MiRNA-320 in the human follicular fluid-development in vitro. Sci. Rep. 2015, 5, 8689. [Google Scholar] [CrossRef] [PubMed]
- Fatima, M.; Prajapati, B.; Saleem, K.; Kumari, R.; Mohindar Singh Singal, C.; Seth, P. Novel insights into role of miR-320a-VDAC1 axis in astrocyte-mediated neuronal damage in neuroAIDS. Glia 2017, 65, 250–263. [Google Scholar] [CrossRef] [PubMed]
- Song, H.T.; Sun, X.Y.; Zhang, L.; Zhao, L.; Guo, Z.M.; Fan, H.M.; Zhong, A.F.; Niu, W.; Dai, Y.H.; Zhang, L.Y.; et al. A preliminary analysis of association between the downregulation of microRNA-181b expression and symptomatology improvement in schizophrenia patients before and after antipsychotic treatment. J. Psychiatr. Res. 2014, 54, 134–140. [Google Scholar] [CrossRef] [PubMed]
- Khanzada, N.S.; Butler, M.G.; Manzardo, A.M. GeneAnalytics pathway analysis and genetic overlap among autism spectrum disorder, bipolar disorder and schizophrenia. Int. J. Mol. Sci. 2017, 18, 527. [Google Scholar] [CrossRef] [PubMed]
- Sundararajan, T.; Manzardo, A.M.; Butler, M.G. Functional analysis of schizophrenia genes using GeneAnalytics program and integrated databases. Gene 2018, 641, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Mor, M.; Nardone, S.; Sams, D.S.; Elliott, E. Hypomethylation of miR-142 promoter and upregulation of microRNAs that target the oxytocin receptor gene in the autism prefrontal cortex. Mol. Autism 2015, 6, 46. [Google Scholar] [CrossRef] [PubMed]
- Frattini, S.A.; Zucchi, I.; Patrosso, C.P. Characterization sequence/EST database screening. Genomics 1996, 34, 323–327. [Google Scholar]
- Joyner, A.H.; Cooper Roddey, J.; Bloss, C.S.; Bakken, T.E.; Rimol, L.M.; Melle, I.; Agartz, I.; Djurovic, S.; Topol, E.J.; Schork, N.J.; et al. A common MECP2 haplotype associates with reduced cortical surface area in humans in two independent populations. Proc. Natl. Acad. Sci. USA 2009, 106, 15483–15488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sklar, P.; Smoller, J.W.; Fan, J.; Ferreira, M.A.; Perlis, R.H.; Chambert, K.; Nimgaonkar, V.L.; McQueen, M.B.; Faraone, S.V.; Kirby, A.; et al. Whole-genome association study of bipolar disorder. Mol. Psychiatry 2008, 13, 558–569. [Google Scholar] [CrossRef] [PubMed]
- Erhardt, A.; Akula, N.; Schumacher, J.; Czamara, D.; Karbalai, N.; Müller-Myhsok, B.; Mors, O.; Borglum, A.; Kristensen, A.S.; Woldbye, D.P.; et al. Replication and meta-analysis of TMEM132D gene variants in panic disorder. Transl. Psychiatry 2012, 2, e156. [Google Scholar] [CrossRef] [PubMed]
- Butler, M.G.; Rafi, S.K.; Manzardo, A.M. High-resolution chromosome ideogram representation of currently recognized genes for autism spectrum disorders. Int. J. Mol. Sci. 2015, 16, 6464–6495. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.; Kucukural, A.; Zhang, Y. I-TASSER: A unified platform for automated protein structure and function prediction. Nat. Protoc. 2010, 5, 725–738. [Google Scholar] [CrossRef]
- Yang, J.; Yan, R.; Roy, A.; Xu, D.; Poisson, J.; Zhang, Y. The I-TASSER Suite: Protein structure and function prediction. Nat. Methods 2015, 12, 7–8. [Google Scholar] [CrossRef]
- Wu S, Zhang Y: LOMETS: A local meta-threading-server for protein structure prediction. Nucleic Acids Res. 2007, 35, 3375–3382. [CrossRef]
- Rual, J.F.; Venkatesan, K.; Hao, T.; Hirozane-Kishikawa, T.; Dricot, A.; Li, N.; Berriz, G.F.; Gibbons, F.D.; Dreze, M.; Ayivi-Guedehoussou, N.; et al. Towards a proteome-scale map of the human protein-protein interaction network. Nature 2005, 437, 1173–1178. [Google Scholar] [CrossRef]
- Smith, C.M.; Finger, J.H.; Hayamizu, T.F.; McCright, I.J.; Xu, J.; Berghout, J.; Campbell, J.; Corbani, L.E.; Forthofer, K.L.; Frost, P.J. The mouse gene expression database (GXD): 2014 update. Nucleic Acids Res. 2014, 42, D818–D824. [Google Scholar] [CrossRef] [PubMed]
- Gandal, M.J.; Haney, J.R.; Parikshak, N.N.; Leppa, V.; Ramaswami, G.; Hartl, C.; Schork, A.J.; Appadurai, V.; Buil, A.; Werge, T.M.; et al. Shared molecular neuropathology across major psychiatric disorders parallels polygenic overlap. Science 2018, 359, 693–697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaaf, C.P.; Sabo, A.; Sakai, Y.; Crosby, J.; Muzny, D.; Hawes, A.; Lewis, L.; Akbar, H.; Varghese, R.; Boerwinkle, E.; et al. Oligogenic heterozygosity in individuals with high-functioning autism spectrum disorders. Hum. Mol. Genet. 2011, 20, 3366–3375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, H.; Wang, T.; Wu, H.; Long, M.; Coe, B.P.; Li, H.; Xia, K. Inherited and multiple de novo mutations in autism/developmental delay risk genes suggest a multifactorial model. Mol. Autism 2018, 9, 64. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pathway ID | Pathway Description | Count in Gene Set | False Discovery Rate | Functional Description |
|---|---|---|---|---|
| 4721 | Synaptic vesicle cycle | 3 | 0.0161 | Communication between neurons is mediated by the release of neurotransmitter from synaptic vesicles (SVs). At the nerve terminal, SVs cycle through repetitive episodes of exocytosis and endocytosis. SVs are filled with neurotransmitters by active transport. DISEASES: Early infantile epileptic encephalopathy; Centronuclear myopathy; Episodic ataxias; Familial or sporadic hemiplegic migraine |
| 4911 | Insulin secretion | 3 | 0.0161 | Insulin secretion is regulated by several hormones and neurotransmitters. Peptide hormones, such as glucagon-like peptide 1 (GLP-1), increase cAMP levels and thereby potentiate insulin secretion via the combined action of PKA and Epac2. Acetylcholine (Ach), a major parasympathetic neurotransmitter. DISEASES: Type II diabetes mellitus; Defects in the degradation of ganglioside. |
| 4152 | AMPK signaling pathway | 3 | 0.0325 | AMP-activated protein kinase (AMPK) is a serine threonine kinase that is highly conserved through evolution. AMPK system acts as a sensor of cellular energy status. |
| 4130 | SNARE interactions | 2 | 0.0432 | SNARE proteins (an acronym derived from “SNAP (Soluble NSF Attachment Protein) Receptor”). The primary role of SNARE proteins is to mediate vesicle fusion, that is, the fusion of vesicles with their target membrane-bound compartments. The best studied SNAREs are those that mediate docking of synaptic vesicles with the presynaptic membrane in neurons. DISEASES: Pseudohypoparathyroidism and Cerebral dysgenesis, neuropathy, ichthyosis, and palmoplantar keratoderma syndrome; CEDNIK syndrome. |
| Pathway ID | Pathway Description | Count in Gene Set | False Discovery Rate |
|---|---|---|---|
| GO:0048489 | synaptic vesicle transport | 8 | 1.14 × 10−9 |
| GO:0097479 | synaptic vesicle localization | 8 | 1.14 × 10−9 |
| GO:0016079 | synaptic vesicle exocytosis | 7 | 1.79 × 10−9 |
| GO:0016082 | synaptic vesicle priming | 4 | 1.14 × 10−7 |
| GO:0031629 | synaptic vesicle fusion to presynaptic membrane | 4 | 5.45 × 10−7 |
| GO:0007269 | neurotransmitter secretion | 6 | 3.06 × 10−6 |
| GO:0048167 | regulation of synaptic plasticity | 5 | 0.000171 |
| GO:0032482 | RAB protein signal transduction | 4 | 0.000701 |
| GO:0031630 | regulation of synaptic vesicle fusion to presynaptic membrane | 2 | 0.00156 |
| GO:0014047 | glutamate secretion | 3 | 0.00165 |
| GO:0007268 | synaptic transmission | 6 | 0.0115 |
| GO:0023051 | regulation of signaling | 1 | 0.0211 |
| GO:0050803 | regulation of synapse structure or activity | 4 | 0.0249 |
| GO:0050804 | modulation of synaptic transmission | 4 | 0.0356 |
| GO:0007274 | neuromuscular synaptic transmission | 2 | 0.0385 |
| GO:0065008 | regulation of biological quality | 1 | 0.0389 |
| GO:0007409 | axonogenesis | 5 | 0.0393 |
| Pathway ID | Pathway Description | Count in Gene Set | False Discovery Rate | Functional Description and Associated Diseases |
|---|---|---|---|---|
| GO:0019905; GO:0017075 | Syntaxin binding | 6 | 5.46 × 10−7 | Syntaxin binding is essential for neurotransmission: syntaxin is a component of the synaptic vesicle fusion machinery. Mutations in Syntaxin binding protein 1 (STXBP1) have been associated with infantile-epileptic encephalopathy-4 [28]. |
| GO:0005484 | SNAP receptor activity | 4 | 0.000161 | SNAPRE activity also regulates neurotransmitter release to ensure vesicle-to-target specificity (SNAP receptors implicated in vesicle targeting and fusion [29]. |
| GO:0019003 | GDP binding | 3 | 0.019 | The trimeric-G-protein (GTP binding proteins) play a pivotal role in the signal transduction pathways for numerous hormones and neurotransmitters [30]. |
| Sytl4-miR=ASD-miR | Mouse Sytl4- miRs: Predicted Interactions with ASD & Schizophrenia- Associated miRs | Validation | Reference |
|---|---|---|---|
| miR93 | Sytl4 | predicted | MGI:1351606c |
| miR93 | ASD | Dysregulated in superior temporal gyrus of ASD | Stomova, et al., 2015 [37] |
| miR103-1; miR103-2 | Sytl4 | Predicted | MGI:1351606c |
| miR103 | ASD | Dysregulated in superior temporal gyrus of ASD | Stomova, et al., 2015 [37] |
| miR106b | Sytl4 | Predicted | MGI:1351606c |
| miR106b-5p (miR106b) * | ASD | Upregulated in ASD-serum; differentially expressed in ASD cerebellar cortex | Vasu, et al., 2014 [34] and Abu-Elneel K, et al., 2008 [35] |
| miR106b | Schizophrenia | Altered expression (serum/cortical) | Vasu, et al., 2014 [34] and Shi, et al., 2012 [33], Beveridge and Cairns, 2012 [32]. |
| miR130a | Sytl4 | Predicted | MGI:1351606c |
| miR130a-3p (miR130a) ** | ASD | Good predictive power for ASD- serum | Vasu, et al., 2014 [34] |
| miR-130a | Schizophrenia | Altered expression (serum/cortical) | Vasu, et al., 2014 [34]; Shi, et al., 2012 [33], Beveridge and Cairns, 2012 [32] |
| miR132 | Sytl4 | Predicted | MGI:1351606c |
| miR132 | ASD | Dysregulated in superior temporal gyrus of ASD | Stomova, et al., 2015 [37] |
| miR181b-1 (miR181b) *** | Sytl4 | Predicted | MGI:1351606c |
| miR181b-2 | Sytl4 | Predicted | MGI:1351606c |
| miR181b-5p (miR181b/b1) *** | ASD | Good predictive power for ASD in serum; differentially expressed in ASD cerebellar cortex | Vasu, et al. 2014 [34]. Abu-Elneel K, et al. 2008 [35]. Ghahramani Seno, et al., 2011 [36]. |
| miR181b | Schizophrenia | Altered expression (serum/cortical) | Vasu, et al., 2014 [34]; Shi, et al., 2012 [33], Beveridge and Cairns, 2012 [32]. |
| miR320 | Sytl4 | Predicted | MGI:1351606c |
| miR320a (miR320) **** | ASD | Good predictive power for ASD in serum | Vasu, et al., 2014 [34] |
| miR320 | ASD | Dysregulated in superior temporal gyrus of ASD | Stomova, et al., 2015 [37] |
| miR320 | ASD | Differentially expressed in ASD cerebellar cortex | Abu-Elneel K, et al. 2008 [35] |
| miR328 | Sytl4 | Predicted | MGI:1351606c |
| miR328 | ASD | Down regulated in serum; differentially expressed in ASD cerebellar cortex | Vasu, et al., 2014 [34] and Abu-Elneel K, et al., 2008 [35] |
| miR328 | Schizophrenia | Altered expression (serum/cortical) | Vasu, et al., 2014 [34] and Shi, et al., 2012 [33]; Beveridge and Cairns, 2012 [32] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rafi, S.K.; Fernández-Jaén, A.; Álvarez, S.; Nadeau, O.W.; Butler, M.G. High Functioning Autism with Missense Mutations in Synaptotagmin-Like Protein 4 (SYTL4) and Transmembrane Protein 187 (TMEM187) Genes: SYTL4- Protein Modeling, Protein-Protein Interaction, Expression Profiling and MicroRNA Studies. Int. J. Mol. Sci. 2019, 20, 3358. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20133358
Rafi SK, Fernández-Jaén A, Álvarez S, Nadeau OW, Butler MG. High Functioning Autism with Missense Mutations in Synaptotagmin-Like Protein 4 (SYTL4) and Transmembrane Protein 187 (TMEM187) Genes: SYTL4- Protein Modeling, Protein-Protein Interaction, Expression Profiling and MicroRNA Studies. International Journal of Molecular Sciences. 2019; 20(13):3358. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20133358
Chicago/Turabian StyleRafi, Syed K., Alberto Fernández-Jaén, Sara Álvarez, Owen W. Nadeau, and Merlin G. Butler. 2019. "High Functioning Autism with Missense Mutations in Synaptotagmin-Like Protein 4 (SYTL4) and Transmembrane Protein 187 (TMEM187) Genes: SYTL4- Protein Modeling, Protein-Protein Interaction, Expression Profiling and MicroRNA Studies" International Journal of Molecular Sciences 20, no. 13: 3358. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20133358