Cerebrovascular and Neurological Disorders: Protective Role of NRF2


1
Department of Pharmaceutical Sciences, Texas Tech University Health Sciences Center, Amarillo, TX 79106, USA
2
Center for Blood Brain Barrier Research, Texas Tech University Health Sciences Center, Amarillo, TX 79106, USA
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2019, 20(14), 3433; https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20143433
Submission received: 13 June 2019
/
Revised: 5 July 2019
/
Accepted: 6 July 2019
/
Published: 12 July 2019
(This article belongs to the Special Issue The Nrf2 Pathway: Regulation, Functions, and Potential Applications)
Abstract
:Cellular defense mechanisms, intracellular signaling, and physiological functions are regulated by electrophiles and reactive oxygen species (ROS). Recent works strongly considered imbalanced ROS and electrophile overabundance as the leading cause of cellular and tissue damage, whereas oxidative stress (OS) plays a crucial role for the onset and progression of major cerebrovascular and neurodegenerative pathologies. These include Alzheimer’s disease (AD), Parkinson’s disease (PD), amyotrophic lateral sclerosis (ALS), Huntington’s disease (HD), stroke, and aging. Nuclear factor erythroid 2-related factor (NRF2) is the major modulator of the xenobiotic-activated receptor (XAR) and is accountable for activating the antioxidative response elements (ARE)-pathway modulating the detoxification and antioxidative responses of the cells. NRF2 activity, however, is also implicated in carcinogenesis protection, stem cells regulation, anti-inflammation, anti-aging, and so forth. Herein, we briefly describe the NRF2–ARE pathway and provide a review analysis of its functioning and system integration as well as its role in major CNS disorders. We also discuss NRF2-based therapeutic approaches for the treatment of neurodegenerative and cerebrovascular disorders.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
Oxidative stress (OS), one of the main indications of various pathologic processes, results from the production of ROS including hydrogen peroxide, superoxide, and hydroxyl free radicals. In turn, these highly reactive compounds promote lipid peroxidation, protein backbone fragmentation, genotoxicity, mitochondrial depolarization, and apoptosis that subsequently cause serious damage to tissues and organs including the brain [1,2,3]. The majority of this excessive reactive oxidative species is expected to be produced by oxidative phosphorylation responses in the mitochondria [4,5]. Cells have developed several defense mechanisms equipped with the capacity to upregulate the expression levels of cytoprotective enzyme genes in order to scavenge free radicals and reduce the risk for the cellular damaging effect of ROS [6,7]. Nuclear factor erythroid 2-related factor (NRF2) is one of the most important transcription factors that enhance the expression of antioxidant reaction to oxidant stress [5].
NRF2 (a member of the Cap-n-Collar family of basic leucine zipper proteins) as a ubiquitously expressed redox-sensitive transcription factor, primarily modulates the activation of biological systems encompassing anti-inflammatory molecules, antioxidants (such as thioredoxin, glutathione, and others), phase I and II drug metabolizing enzymes (such as cytochrome P450s), phase III enzymes (efflux transporters), and free radical scavengers [6,8,9,10,11,12]. Consequently, cell vulnerability to the detrimental effects of ROS and OS damage to mitochondrial function (leading to cell apoptosis) are enhanced by downregulation or suppression of NRF2 activity [13,14,15,16]. Cellular OS initiates a sequence of biological responses so that NRF2 (residing in the cytoplasm at a low basal level) translocates into the nucleus [17] where it forms a heterodimer with small Maf proteins (MafG, MafK, MafF). Coupling with Mafs endows NRF2 with a DNA-linking capacity to bind to the ARE sequence and initiate the transcription of ROS detoxification genes [18].
Due to the fact that NRF2 is likely to control, modulate, and sustain the expression of detoxification and antioxidative response elements and other types of protective elements (which include the ubiquitary iron exporter ferroportin 1, anti-apoptotic B-cell lymphoma 2, brain-derived neurotrophic factors, the peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α), the mitochondrial-nuclear respiratory factor 1 (Nrf1), and the autophagic protein p62) [19,20,21,22,23,24,25], its activation plays a considerable role in counteracting acute injuries, effects of xenobiotics, inflammation, and many other stimuli that are promoted by OS [17,26]. With regard to OS being implicated in several pathologies, current research is focused on pathogenic mechanisms that lead to mitochondrial dysfunction and redox imbalance [26]. In this review, we provide a detailed analysis of the current understanding of the NRF2–ARE system and its role in major CNS disorders. We also include NRF2-focused therapeutic approaches for the treatment of cerebrovascular and neurodegenerative diseases.
2. NRF2 Regulation and Response to Oxidative Stress
Domain analysis by high-resolution crystal structure and nuclear magnetic resonance spectroscopy has shown that the molecular structure of NRF2 includes seven functional domains (Neh1–Neh7) that regulate its transcriptional activity and stability [27]. The first conserved domain, Neh1, containing basic bZIP motif binds, to the ARE sequence exposing a nuclear localization signal required for translocation of released NRF2 from Kelch-like ECH-associated protein 1 (KEAP1) into the nucleus [28,29,30]. Neh1 and Neh2 play differing roles with respect to NRF2 regulation. While Neh1 modulates NRF2 protein stability through interaction with the E2 ubiquitin-conjugating enzyme, Neh2, a negative regulatory domain located in the N-terminal region, promotes NRF2 ubiquitination followed by proteasomal degradation, which is a result of increased KEAP1–NRF2 binding [31]. While Neh3 (which is located in the carboxyl-terminal region of the protein) modulates the transcriptional activation of the ARE genes [27,32], the Neh4 and Neh5 domains play a cooperative role in facilitating NRF2 transcription by binding to a transcriptional co-activator [33] and also increases NRF2–ARE gene expression by interfacing with the nuclear cofactor RAC3/AIB1/SRC-3 [33,34]. The Neh6 and Neh7 domains control KEAP1-independent degradation of NRF2 and regulate the activity of NRF2 so that KEAP1-alternative pathway of NRF2 degradation arises based on the recognition of phosphorylated Neh6 by the E3 ligase adapter beta-TrCP [35,36,37] and Neh7 inhibits NRF2 via interaction with retinoid X receptor α [38]. The main step in detoxification is the nuclear and cytoplasmic disposition of NRF2 so that under basal conditions, NRF2 is rapidly polyubiquitinated while cellular redox homeostasis is sustained by the accumulation of the NRF2 in the nucleus to mediate the normal expression of ARE-dependent genes [26,39].
KEAP1, the main intracellular regulator of NRF2, is composed of three main domains (totaling 624 amino acids) including the Broad-complex (1), Tramtrack (2), and the Bric-a-Brac (BTB) domain (3) which includes a cysteine-rich region and a double glycine repeat -DGR- binding site between KEAP1 and NRF2. Several cysteine residues within the BTB domain act as OS sensors and/or inducer ligands within the cell’s environment [27]. The activity of the NRF2–ARE signaling pathway is controlled by degradation and sequestration of NRF2 in the cytoplasm through binding with KEAP1 [5,26]. Other factors including post-transcription changes, gene polymorphisms in the promoter region, and protein–protein interactions are also influenced by NRF2 basal activity [39,40]. It is noteworthy that in response to mitochondrial oxidative stressors NRF2 provides direct interaction with the mitochondrial membrane [41].
Under basal conditions, KEAP1 binds to NRF2 in the cytoplasm and enhances the ubiquitination and proteasomal degradation of NRF2, whereas in response to oxidative stress condition, the NRF2 DLG motif is released from the DGR domain in KEAP1, which then undergoes conformational changes and dissociate NRF2 from itself to shift into the nucleus freely [39]. Independently from KEAP1 activity, not only phosphorylation of NRF2’s serine enhance separation of NRF2 from KEAP1 [26], but also glycogen synthase kinase-3β (GSK-3β), synthesis of specific microRNAs, methylation of CpG islands, histone phosphorylation, and acetylation modulate the expression and activation of NRF2 [6,42,43,44]. This cytoprotective pathway encompasses detoxification systems such as oxidation/reduction factors (Phase I), conjugation enzymes (Phase II), and drug efflux transporters (Phase III) [39,44]. Phase I includes more than 500 products encompassing genes encoding proteins for redox balancing factors, stress response proteins, detoxifying enzymes, and metabolic enzymes such as NAD(P)H:quinone oxidoreductase 1 (NQO1), heme oxygenase-1 (HO-1), superoxide dismutase (SOD), glutathione S-transferase (GST), glutathione reductase (GSR), glutathione peroxidase (GSH-Px), carbonyl reductase (CR), and glutamate-cysteine ligase (GCL) [45,46]. Metabolites then undergo Phase II metabolism where they are conjugated with large polar groups (such as glucuronic acid, glutathione, sulfate, or glycine) to increase their water solubility and facilitate their excretion. Thus, it seems that NRF2 plays a major role in modulating/promoting the process of detoxification [5,30]. NRF2 also controls/enhances the expression of active efflux transporters that remove or keep out potentially detrimental endogenous or xenobiotics of the cell [39] as well as tight junction (TJ) expression and BBB integrity at the neurovascular unit [47]. NRF2 nuclear accumulation can also have harmful effects [29] which accounts for the necessity of autoregulating the cellular levels of NRF2 [32,48]. For this end, the cullin3/ring box 1 (Cul3/Rbx1) E3 ubiquitin ligase complex promotes polyubiquitination of NRF2 coupled with KEAP 1 followed by NRF2 proteasomal degradation. This mechanism controls the “switching off” of NRF2-activated gene expression in the nucleus [32].
3. Crosstalk between NRF2 and Other Regulatory Signaling Pathways
According to numerous studies, there is a crosstalk between NRF2, KEAP1, and other signaling pathways. In the KEAP1-independent degradation pathway mediated by the cullin3/ring box 1 (Cul3/Rbx1) E3 ubiquitin ligase complex, NRF2 acts as a regulator for the complex. This occurs through NRF2 binding to the KEAP1 region via the Neh2 domain, which enhances NRF2 polyubiquitination and then degradation by the 26S proteasome [1,26,35]. Moreover, this Cul3/Rbx1-mediated degradation of NRF2 is controlled by phosphorylation of the Neh6 domain of NRF2 which is regulated by GSK-3 [37]. Furthermore, deletion of PTEN (phosphatase and tensin homolog) deleted from chromosome10 promotes NRF2 accumulation in KEAP1-deficient liver, suggesting that the inactivation of GSK-3 and the activation of the PI3K-Akt pathway by the loss of PTEN lead to the inhibition of NRF2 degradation via the Cul1/Rbx1 pathway [7,49].
Another essential crosstalk occurs between the KEAP1–NRF2 system and the cellular autophagy pathway. Autophagy-adaptor protein p62 competes with KEAP1 for binding with NRF2 using the STGE motif so that p62 promotes the stabilization of NRF2 and the upregulation of NRF2 activity through this competition [24]. Interestingly, phosphorylation of the STGE motif in p62 causes enhanced binding affinity to KEAP1 [50]. A considerable finding is that NRF2 is predominantly degraded through the proteasome pathway, whereas KEAP1 is degraded through the autophagy pathway maintaining the integrity and homeostasis of the KEAP1–NRF2 system by governing KEAP1 turnover [51]. Thus, the KEAP1–NRF2 system is regulated through the two significant cellular proteolysis pathways.
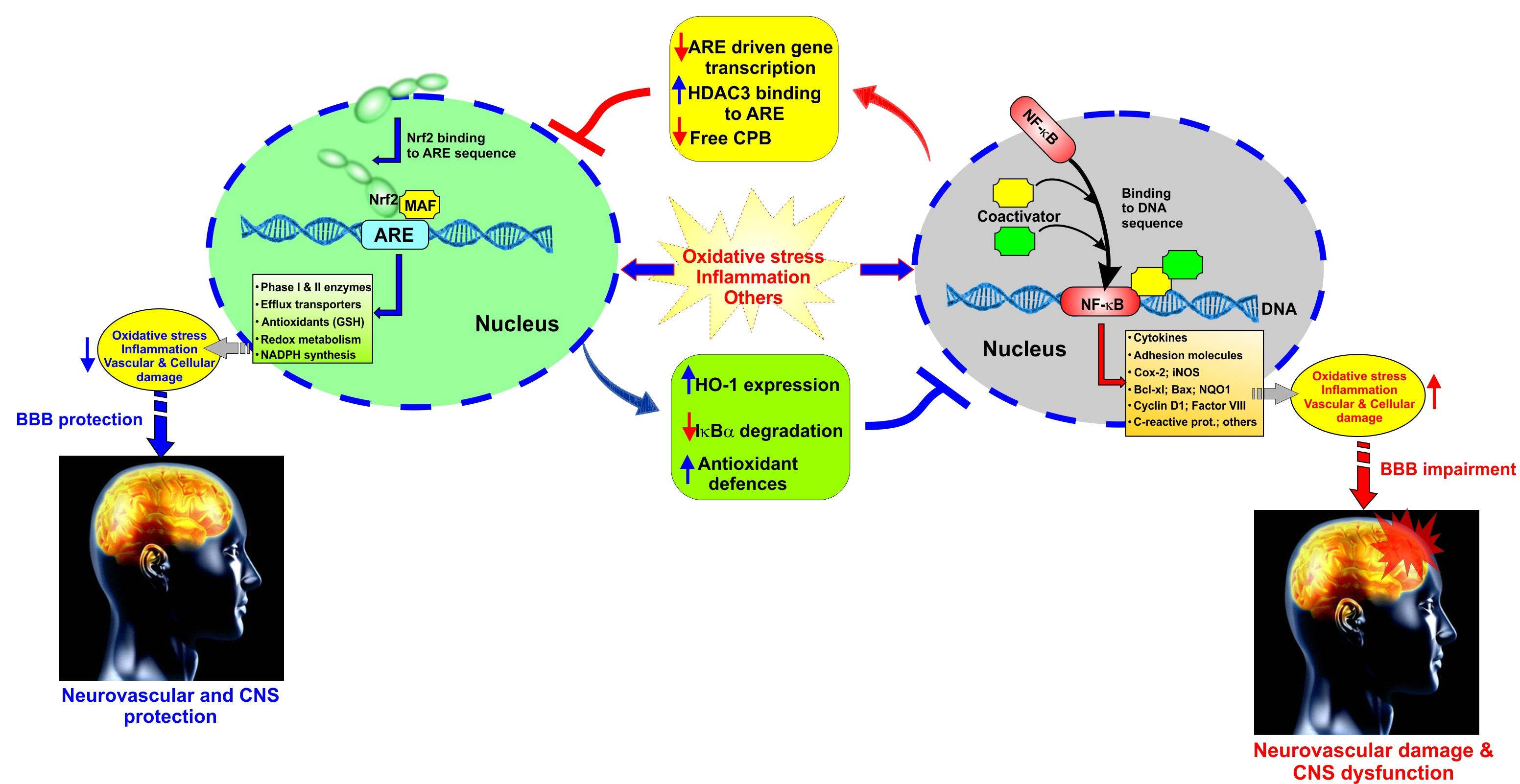
NF-κB is a rapid response factor to harmful cellular stimuli which is associated with Inhibitor of κB (IκB) proteins (which include IκBα, IκBβ, IκBε, and Bcl-3 although IκBα is the major IκB protein). In response to noxious stimuli (such as OS and pro-inflammatory cytokines like TNF-α), the IκB proteins (which under normal conditions mask the nuclear localization signals—NLS—of NF-κB and keep this factor sequestered in an inactive state in the cytoplasm) undergo phosphorylation by the IκB kinase complex, followed by ubiquitination, and proteasomal degradation [5]. Degradation of IκB frees NF-κB can then translocate into the nucleus, and alter (promote) the transcription of genes responsible for both the innate and adaptive immune responses [52]. Recent findings have confirmed the crosstalk between NRF2 and NF-κB signaling pathways under OS conditions as well as a variety of pathophysiological conditions [53] in us much that pro-inflammatory and immune responses promoted by the activity of NF-κB are contrasted by upregulation and activation of NRF2 [5]. By contrast, enhanced inflammation could be associated with downregulation of NRF2 activity (see Figure 1).
In a study on NRF2 knock out mice, hypersensitiveness to the inflammation induced by LPS was observed while these harmful effects were repealed following treatment with sulforaphane as a NRF2-dependent gene expression enhancer [54]. Although several studies on other cell types reported similar observations, the interconnection of NRF2 and NF-κB to coordinate anti-oxidative and inflammatory responses is not completely understood [55,56,57,58,59,60]. In a study by Cuadrado et al., the investigators observed that upregulation of RAC1 (a pleiotropic a small G-protein of the Rho family and a modulator of NF-κB transcription) activated the inflammatory pathway through an interplay between NF-κB and NRF2 [61] so that RAC1 participated in NADPH oxidase-dependent production of superoxide, whereas activation of NF-κB was mediated through RAC1 dependent ROS formation [62]. In fact, NRF2 deletion promoted the activation of NF-κB dependent inflammatory markers, while NRF2 overexpression inhibited RAC1-dependent activation of NF-κB pathway, suggesting that RAC1 affects NRF2 and NF-κB.
4. Role of NRF2 in Aging and Traumatic Brain Injury
Non-effective antioxidative responses to excessive ROS production and changes in redox signaling playing are one of the major reasons for advanced aging, whereas the inability to properly counteract OS leads to the progressive accumulation of OS-induced cellular damage [63,64]. Recent studies have, in fact, shown that age-related OS damages are dependent upon decreased antioxidant responses, as well as proteasome reduction, and reduced efficiency of mitochondrial proteases. The resulting effect of this impaired antioxidative response and inability to effectively maintain the redox balance is an accumulation of intracellular and intramitochondrial aggregates of oxidized proteins [65]. Similarly, the decreased level NADPH and GSH with aging could likely be due to a decreased cellular antioxidative capacity, as well as reduced intake of dietary antioxidants. Although there is a lot of dispute over the effect of age on the basal expression levels of antioxidative response factors, it seems that the primary underlying cause is a reduced NRF2 activity [66,67,68].
In respect to traumatic brain injury (TBI), disruption in the normal brain function following TBI is one of the foremost causes of death as well as severe emotional, physical, and cognitive impairments [69,70,71]. In spite of the pathogenic role of the primary brain injury immediate to TBI, the post-traumatic secondary injury derived from OS, inflammation, excitotoxicity, enhanced vascular permeability, and BBB impairment can significantly worsen post-traumatic brain damage as well as clinical outcomes [72,73]. Excessive ROS generation following cell damage, neuronal cell death, and brain dysfunction are the results of several secondary biochemical and metabolic changes in the cells [74]. According to recent studies, NRF2 plays a neuroprotective role in TBI so that NRF2 activation counteracts TBI-induced OS, loss of BBB integrity, etc. Unsurprisingly, impairments of the NRF2–ARE pathway leading to reduced activity of this protective system can result in more extensive post-TBI tissue damage, thus aggravating the secondary injury and worsening outcome. Accordingly, promoting upregulation of NRF2 activity could be exploited to reduce post-traumatic brain injuries, improve clinical outcomes, and reduce the risk of additional neurological disorders [75,76].
5. Ischemic Stroke and Protective Role of NRF2
Stroke, the fifth leading cause of death in the United States and a major cause of permanent disability, is defined by a bursting or blockage of blood vessels resulting in the sudden interruption of the local blood supply and the initiation of an anoxic and hypoglycemic state in the affected brain tissue [4]. Moreover, neuronal cell membrane depolarization causes the release of the neurotransmitter glutamate, which is the activator of the ionotropic glutamate receptor N-methyl-d-aspartate (NMDA) [5]. The resulting opening of these non-selective cation channels leads to calcium overload and neuronal cell death [77]. These events are associated with excessive ROS production by the mitochondria which overwhelm the antioxidant defenses, leading to post-ischemic inflammation and enhanced brain tissue damage [78,79,80,81]. Furthermore, degradation of the structural proteins of the vascular wall and loss of BBB integrity also occur as a result of blood flow restoration, which suddenly enhances tissue oxygenation further exacerbating ROS production, inflammatory responses, and OS damage [5]. Adhesion of leukocytes across the blood vessels and transmigration into the brain parenchyma is facilitated by the concurrent expression of vascular adhesion molecules on the luminal surface of the vascular walls. Based on these premises, it is evident that control of ROS levels and OS prevention could be a potential therapeutic strategy to address post-ischemic secondary brain injury and improve stroke outcome [5,82,83,84]. Recent studies demonstrated that the protective effect of interactions between p62 and the NRF2–EpRE signaling pathway inhibited OS damage during cerebral ischemia/ reperfusion in rat undergoing transient middle artery occlusion (tMCAO) and also promoted NRF2 activity to lower the infarct volume and post-ischemic neurocognitive impairments [85,86]. NRF2 activity is also crucial to protect the brain against injury. In fact, NRF2 activation through the use of pharmacological enhancers improved neuronal cell viability, decreased BBB permeability, and promoted the transcription of cytoprotective genes [87,88]. Furthermore, enhanced infarct size, inflammatory damages, and neurological deficits were reported in NRF2 KO mice when compared to controls (wild-type mice). By contrast to controls, the use of NRF2 enhancer in knock out mice did not elicit any beneficial effect [89]. Most recently, other studies have shown that NRF2 downregulated the activity of the NOD-like receptor protein 3 (NLRP3) inflammasome by acting on thioredoxin-1 (Trx1)/thioredoxin interacting protein (TXNIP) complex [82]. The NLRP3 inflammasome plays a key role in inflammation damage in cerebral ischemia-reperfusion injury by promoting the activity of interleukin-23/interleukin-17 axis which contributes to the ischemic reperfusion damage at the CNS [90]. The activation of NLRP3 is dependent upon the interaction with TXNIP, which dissociates from the Trx1/TXNIP complex under OS. Thus, it is clear how targeting NRF2 represent a viable target for the treatment of ischemia and reperfusion injury.
6. Role of NRF2 in Neurodegenerative Diseases
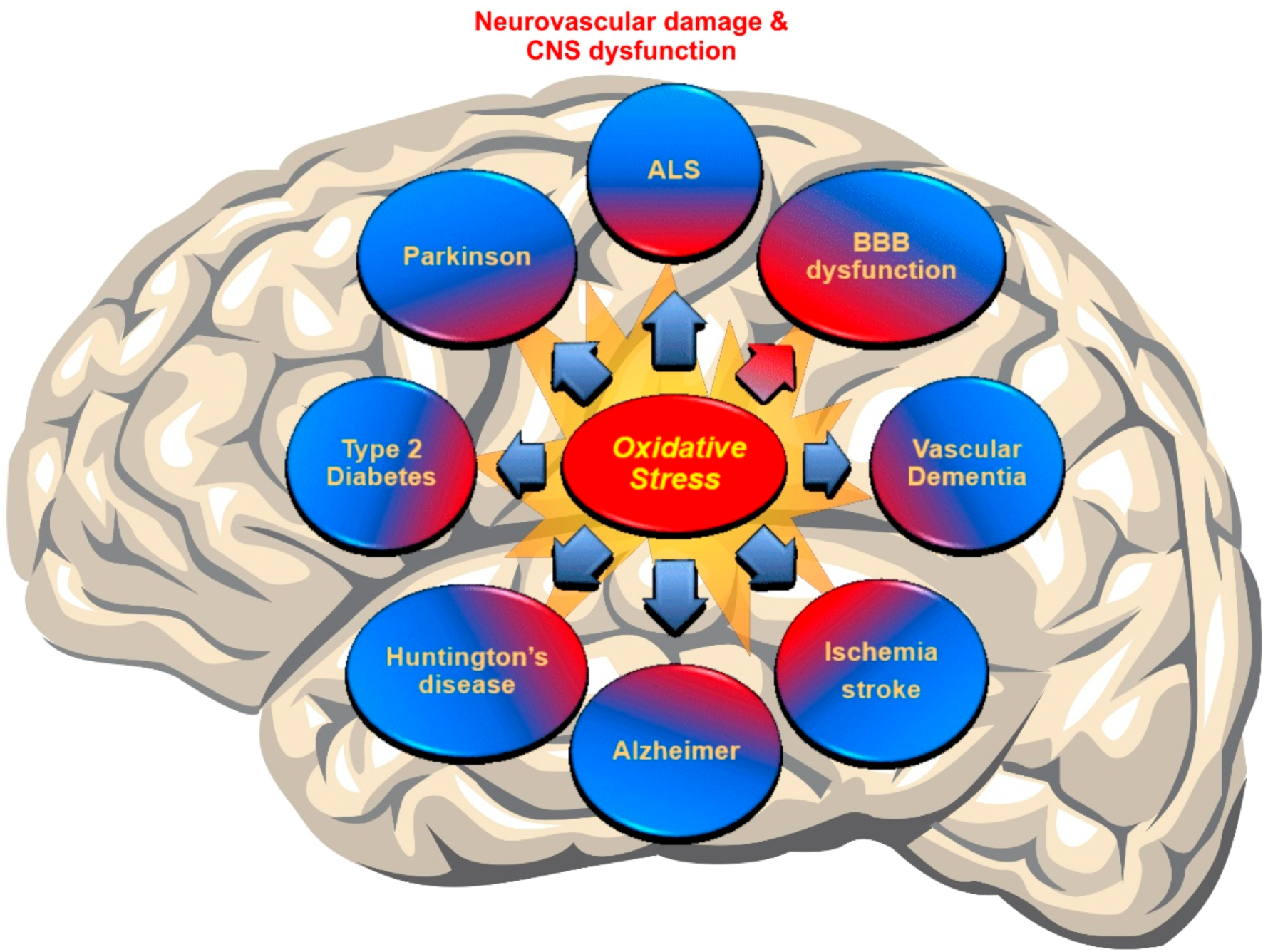
Recent discoveries have mentioned OS as a major pathogenesis of neurodegenerative disorders (NDDs) due to the accumulation of ROS [4,91]. In fact, a failure in maintaining the proper balance between ROS generation and their neutralization causes a disruption of brain homeostasis leading to neurodegenerative disorders [92,93] (see Figure 2).
As commonly observed in neurodegenerative disorders, including Alzheimer’s disease (AD), Parkinson’s disease (PD), amyotrophic lateral sclerosis (ALS) and Huntington’s disease (HD), ROS production overwhelms the antioxidative response system causing cellular damage so that elevated levels of oxidative markers and damaged cell components have been diagnosed in patients with neurodegenerative diseases [94]. In spite of the poor understanding of the underlying mechanisms linking NRF2 with the onset/progression of these disorders, proteinopathies are the pathogenic hallmark shared between these disorders [6]. The impairment of mitochondrial function is another common feature of these neurodegenerative disorders and also promotes ROS generation, ATP depletion, and inflammation [6,51,95,96,97,98]. Thus, this concept could provide opportunities for interventions focused on restoring/normalizing the cellular antioxidative response and decreasing inflammation and following the reduced progression of these diseases.
Alzheimer’s disease: A neuropathological hallmark of AD is the formation of intracellular neurofibrillary tangles (NFTs) and extracellular senile plaques (SPs) composed of small Aβ peptides [99]. Several studies propose that OS is an early prodromal event for progressive neurodegenerative disorders [100]. According to several studies on AD, NRF2 was able to provide a neuroprotective effect by decreasing ROS generation and ROS-induced toxicity mediated by Aβ [101,102]. Supporting data have shown that NRF2 activators, such as sulforaphane (SFN) lower toxin-induced Aβ1-42 secretion, while enhancing cell viability and improving cognitive function [103,104]. These beneficial effects may be due to the formation of Aβ aggregates or the inhibition of the release of monomer/oligomeric Aβ from dead cells [105]. A recent study also outlined the role of NRF2 in facilitating autophagy as well as altering β-Amyloid precursor proteins (APP) and Aβ processing whereas NRF2 knockout APP/PS1 mice showed increased accumulation of insoluble APP fragments and Aβ as well as mammalian targets of rapamycin (mTOR) activity [5,106]. The investigators also found that overexpression of mitochondria catalase in APP transgenic mice (Tg2576), decreases the formation of full-length APPs and lowers soluble and insoluble Aβ levels. From a clinical perspective, this may translate into extending the lifespan of the patient while improving working memory [5]. In a recent study, Rojo et al. demonstrated the protective effect of NRF2 against exacerbation of astrogliosis and microgliosis using transgenic mouse models [105]. Specifically, the investigators have shown a reduction in homeostatic responses with aging along with NRF2 activity resulting in reduced protection against proteotoxic, inflammatory and oxidative stress stimuli [5,107].
Parkinson’s disease: PD is a progressive neurodegenerative disorder characterized by lowered dopamine levels in the striatum due to the loss of dopaminergic neurons located in the substantia nigra affecting movement [94]. The initial symptoms in PD patients sometimes are tremors affecting one hand or slowing of movement. With the progression of the disease controls over movement is completely compromised and the effects are extended to neurocognitive functions dementia [5]. The certain diagnosis of PD in both familial and sporadic PD patients is the presence of Lewy bodies (LBs) as abnormal protein aggregates developing inside nerve cells. The main constituent of LBs is Alpha-synuclein (αSyn) which is a small protein with 140 amino acids. αSyn is abundant in presynaptic nerve terminals playing a role in synaptic transmission and dopamine levels adjustment [5]. Recent studies strongly postulate the association between PD with abnormal ROS production promoted by the dopamine metabolism, excitatory amino acids and iron content [100]. Moreover, it is emphasized that this increased OS plays a pivotal role in αSyn proteostasis, whereas NRF2 activity can counteract αSyn production and the associated cellular damage [101,108,109,110,111]. Recently, NRF2 overexpression has not only confirmed the reduction of the generation of αSyn aggregates in the CNS [112], but also the activation of NRF2 has appeared to prevent the loss of dopaminergic neurons mediated by αSyn and the consequent impairment of motor functions [5,113]. NRF2 deficiency and promoted expression of αSyn experienced enhanced loss of dopaminergic neuron and increased neuroinflammation and protein aggregation, whereas the enhanced expression level of NRF2 in a mutant αSyn transgenic mouse model, provided neuroprotective effects [5,57,111].
Huntington’s disease: HD as an inherited neurodegenerative disease is characterized by the loss of GABAergic inhibitory spiny projection neurons in the striatum [94] due to abnormally elongated poly-glutamine (polyQ) stretch encoded by the atypical expansion of adenine, cytosine, and guanine (CAG) trinucleotide repeats at the huntingtin protein (Htt). According to several in vitro studies, NRF2 activation can play a protective role in the reduction of mHtt-induced toxicity, while in HD patients the initiation of the NRF2–ARE system in striatal cells in response to OS failed because of the concurrent activation of the autophagy pathway [114,115]. Moreover, additional data have confirmed that Htt aggregation directly enhanced ROS generation promoting cell toxicity [116]. Furthermore, co-transfection of NRF2 with mHtt in primary striatal neurons, reduction of the mean lifetime of mHtt N-terminal fragments, and, subsequently, improvement of cell viability suggest that NRF2 is more likely to decrease mHtt -toxicity by negatively affecting its aggregation [117].
Amyotrophic lateral sclerosis: ALS is a progressive neurodegenerative disease characterized by the loss of motor neurons in the ventral horn of the spinal cord and in the motor cortex. The disease leads to progressive motor weakness and loss of controls of voluntary movements [5,94]. Although, for more than two decades, the mutation of Cu–Zn superoxide dismutase 1 (SOD1) was the only genetic aberration relevant to the initiation of familial ALS, recent studies have found more abnormalities associated with the onset of sporadic and non-SOD1 familial ALS, including a host of RNA/DNA-binding proteins such as the 43-kDa transactive response (TAR) DNA-binding protein (TDP-43) and the fused in sarcoma/translocated in liposarcoma (FUS/TLS) [5]. Several recent studies support that NRF2 activation plays a protective role against OS and cell death promoted by the SOD1 mutant protein so that glial NRF2 overexpression improves the survival of the spinal cord’s motor neurons and extends their viable lifespan [118,119]. Additional studies will be required to evaluate the impact of NRF2 on cellular proteostasis as well as other ALS-associated gene mutations and the effect of NRF2 stimulation on late-stage microglia activation to prevent OS.
7. Role of NRF2 in Blood-Brain Barrier (BBB) Integrity and Function
In the central nervous system, the vascular endothelium acquires a set of specific characteristics and functions that differ from other vascular beds [120]. This specialized endothelium, which forms the BBB, becomes a dynamic functional interface between the blood and the brain that strictly regulates the passage of substances, maintains the brain homeostasis, and protects the brain from pathogens as well as endogenous and xenobiotic substances [69]. According to numerous studies, there is a relationship between NRF2 and BBB relevant to cerebrovascular disorders, so that NRF2 signaling plays a neurovascular protective role in the conservation of the BBB and CNS [8,11,121,122]. With regard to BBB endothelium, it has been emphasized that NRF2 upregulates the expression of tight junctional proteins (TJ), promotes redox metabolic functions, and produces ATP with mitochondrial biogenesis [3,11,122,123]. In fact, recently published data from side by side experiments investigating the impact of electronic cigarettes (e-Cig) versus TS on mouse primary brain microvascular endothelial cells (BMVEC) clearly showed that NRF2 was strongly activated by the resulting OS and promoted upregulation of its downstream signaling molecule NQO-1 [122], whereas NQO-1 exerts acute detoxification and cytoprotective functions. However, chronic exposure to these pro-oxidative stimuli ended up compromising NRF2 activity and that of its downstream effector NQO-1. These resulted in an overall impairment of BBB integrity associated with increased permeability to paracellular markers and decreased trans-endothelial electrical resistance (TEER) [83,122]. In addition to the loss of BBB integrity, in vivo data also showed upregulation of inflammatory markers including vascular adhesion molecules and pro-inflammatory cytokines as well as blood hemostasis changes favoring blood coagulation and, therefore, risk of stroke. Recent preliminary data and work by others have also clearly demonstrated that NRF2 modulates mitochondrial biogenesis, redox metabolism, and antioxidant/detoxification functions, thus, strongly suggesting that impairment of NRF2 activity can negatively affect mitochondrial biogenesis and function [3]. Altogether, these studies have shown that NRF2 plays a major role in critical BBB cellular functions ranging from modulation of barrier integrity, inflammatory responses, redox metabolism, and antioxidative responses [4,5,6,44,66,88,105,121,122,124,125]. In fact, cerebrovascular and neurodegenerative disorders such as subarachnoid brain hemorrhage, MS, ALS, AD, PD, stroke, and type-2 diabetes mellitus (TD2M) have been tied to dysfunctions of NRF2 activity [6,11,51,121,126,127,128,129,130]. Unsurprisingly, the activation of the NRF2–ARE system can potentially prevent/reduce the BBB impairments and, consequently, decrease some types of brain injury [131]. Since vascular endothelial dysfunction and consequent CNS damages have been relevant to ROS [132,133,134] and OS-driven inflammation [135], NRF2 activation is likely to preserve the BBB by maintaining ROS homeostasis that ultimately leads to a decrease in the risk of cerebrovascular, neurodegenerative, and CNS disorders [85,131,136,137,138]. For instance, the well-known NRF2 promoter/activator Sulforaphane (SFN) has been shown to have neuroprotective characteristics that counteract oxidative stress by enhancing NRF2 activation [51,131,139,140,141,142,143,144,145,146] and regulating antioxidant reactions [18].
8. Role of NRF2 in Tobacco Smoke-Induced Cerebrovascular Disorders
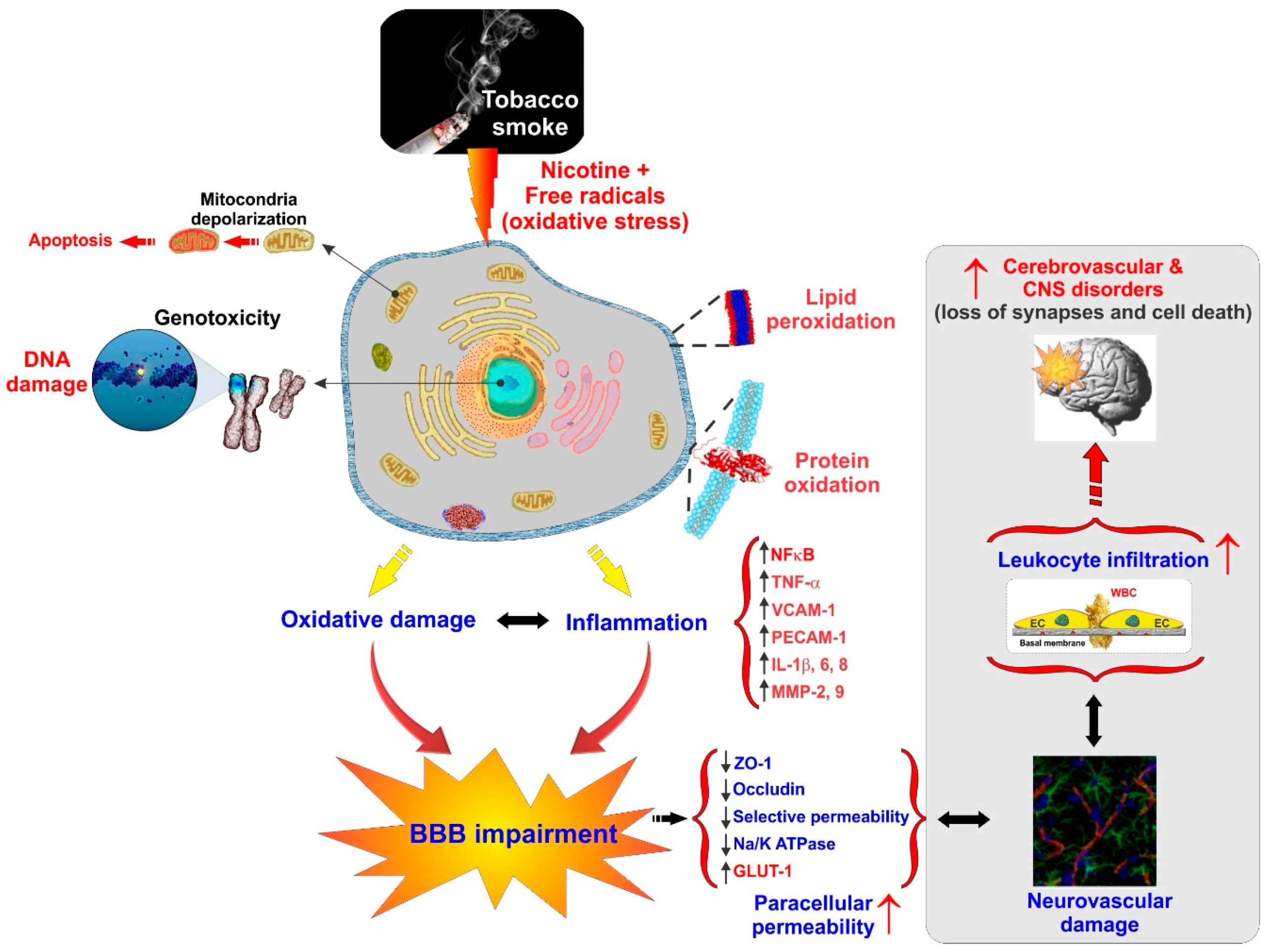
Cerebrovascular and BBB dysfunction promoted by tobacco smoke (TS) are also associated with the initiation of various neurovascular and neurodegenerative diseases linked to dysregulation of NRF2 activity such as stroke, vascular dementia, and previously noted neurogenerative disorders [39,85,122]. This is not surprising since TS contains over 7000 chemicals including nicotine and various ROS (e.g., H2O2, epoxides, nitrogen dioxide, peroxynitrite-ONOO−, etc.) that cross the lung alveolar wall and raise systemic OS. At the cerebrovascular level, this promotes oxidative damage and BBB breakdown via tight junction (TJ) modification and the activation of proinflammatory pathways [11,147]. Under normal conditions, ROS are scavenged by endogenous antioxidants involving vitamins such as ascorbic acid and α-tocopherol or intracellularly converted into less reactive molecules by superoxide dismutase (SOD), catalase (CAT), and glutathione peroxidase (GSH-Px). However, containing excessive pro-oxidant substances, chronic exposure to active and passive smoking can overwhelm these protective mechanisms. Furthermore, the protective nature of NRF2 may be altered in smokers by means of somatic mutation, epigenetic alteration, and accumulation of disruptor proteins, thereby promoting cell resistance and proliferation of cancerous cells as indicated by other studies [122,148,149].
According to several recent studies, NRF2 enhancers can counteract OS and possibly decrease the burden of neuropathologies including ischemic and cerebral stroke [5,85,150,151,152,153]. Recently Prasad et al. have confirmed the upregulation of NRF2 upon acute TS/e-Cig exposure and its impact on mitochondria biogenesis and bioenergetic functions at the BBB. Their results strongly confirmed the positive role of NRF2 in regulating the redox metabolic interplay that triggers the expression of antioxidative active elements and ultimately the protection of the BBB against OS damage [122]. NRF2 nuclear translocation and increased transcription of detoxifying enzymes and antioxidants effectively protect against chronic TS exposure [5]. However, they demonstrated impairment of NRF2 activity by chronic TS exposure, resulting in a suboptimal antioxidative response and consequent cellular OS damage, while acute exposure to TS and vapors from an electronic cigarette (e-Cig) initially enhances NRF2 expression and activation [11,85,122,123,154,155]. This facet of chronic TS and e-cig exposure and their effect on the NRF2–ARE system needs to be considered due to early-stage former smokers remaining at a high risk of developing cerebrovascular disorders for years after quitting [39]. Similar results have been observed in our recent work confirming TS/e-Cig induced cerebrovascular dysfunction and possibly other detrimental xenobiotics affects the BBB via oxidative stress [156] (see Figure 3).
9. Role of NRF2 in Hyperglycemia
Hyperglycemia or high blood sugar is described as an abnormally elevated rate of blood sugar (fasting levels of glucose > 130 mg/dL) and a sign of type 1 and type 2 diabetes [5]. Recent studies on the impact of hyperglycemia on NRF2 expression have revealed that while hyperglycemia does not directly affect NRF2 expression, it promotes its nuclear translocation [5,11]. It appears that both acute TS exposure and hyperglycemia significantly increase the nuclear/cytoplasmic NRF2 ratio and subsequent activation of antioxidant mechanisms. Indirectly, these data also suggest that TD2M-promoted and TS promote OS damage through similar mechanisms [8]. This is part of an acute response system activated in response to oxidative stress. In fact, recent studies have shown that inhibition of NLRP3 by NRF2 can improve diabetes-mediated cognitive and cerebrovascular impairments [157]. However, chronic persistence of hyperglycemia and/or exposure to TS impairs NRF2 activity leading to cerebrovascular damage and heighten the risk of neurological disorders.
In spite of the observed facts such as enhanced TS-induced NRF2 activation by hyperglycemia (accounting for the existence of a cooperative effect) and enhanced-activation of endothelial pro-inflammatory responses by hyperglycemia, additional studies will be necessary to validate the underlying mechanisms and determine the corresponding pathophysiological implications relevant to the cerebrovascular system.
10. NRF2 Enhancers for the Treatment of Cerebrovascular Disorders: Repurposing of Antidiabetic Drugs
Recent studies have provided several dietary and therapeutic agents currently approved for the treatment of non-vascular and non-neurodegenerative pathologies that, on the contrary, do possess protective effects against the initiation/progression of neurodegenerative [1] and cerebrovascular diseases [5,122]. Metformin (MF), an oral anti-hyperglycemic agent, not only can enhance neurogenesis for the injured or degenerating neurovasculature [158], but it is also likely to diminish BBB disruption and decrease/inhibit ischemic injury upon stroke and impairment in neurodegenerative disorders [159,160,161,162,163]. MF has been shown to activate counteractive mechanisms which drastically reduce OS toxicity at the cerebrovascular and BBB levels such as those promoted by chronic TS exposure [83,122]. These beneficial effects are seemingly mediated by MF‘s activation of NRF2 [159] and include the suppression of TJ proteins downregulation and loss of BBB integrity by TS, the reduction of inflammation and oxidative stress, the renormalization of the expression levels of the major BBB glucose transporter Glut-1 and that of the anticoagulant factor thrombomodulin. Both AMPK-dependent and independent mechanisms are known to be involved in the mechanism of action of Metformin [164,165]. According to a study by Montalvo et al., treatment with Metformin resulted in an increased lifespan of aging mice brought about by the increase in antioxidant property and AMPK activation [166]. In addition, pre-treatment with MF has been discovered in distinct research to avoid ischemia-induced brain injury by activating the AMPK and NRF2 pathways and promoting rearrangement with ZO-1, occludin, and claudin-5 [159,160]. Recently our team showed that pretreatment with MF in vitro prevented downregulation of tight junction protein (ZO-1 and occludin) following chronic exposure to soluble cigarette smoke extract (sCSE). This effect was dependent upon activation of the NRF2 and AMPK pathways. Our study also revealed that that NRF2 activation (upregulation and translocation to the nucleus) is not exclusively dependent upon the activation of AMPK [122].
MF also has shown a neuroprotective effect on TS-induced cerebrovascular/BBB impairments to diminish the cerebrovascular toxicity accounting for a functional role of NRF2 and NRF2–ARE signaling pathways in protecting BBB integrity in chronically TS-exposed human BBB microvascular endothelial cells [11,85,122,123,159]. A recent study also demonstrated that treatment with MF in TS exposed mice restored levels of NRF2 and NQO1 to control levels in a dose-dependent manner. Along with these effects, levels of TJ proteins ZO-1 and Occludin were also restored [83]. In supporting the neuroprotective effect of anti-diabetic drug against oxidative stress in another study, Rosiglitazone (RSG) is a thiazolidinedione compound used for the treatment of TD2M that is well known to improve insulin resistance by regulating adiponectin gene expression [167]. Rosiglitazone is also considered as a transcription factor peroxisome proliferator-activated receptor (PPARγ) agonist [168,169]. Although the exact mechanism of action of rosiglitazone is not fully understood, recent studies have shown that this drug also possesses antioxidative features and can protect against OS damage and inhibit the inflammatory cascade through signaling inactivation by p38, JNK, and NF-κB [170,171,172]. Recently, Ceolotto et al. demonstrated that RSG protects endothelial cells against glucose-induced OS with an AMPK-dependent mechanism [156,168]. AMPK has been shown to promote NRF2 activity via nuclear accumulation further implying that RSG-mediated upregulation of PPARγ can be also associated with increased NRF2 activity [156]. In another study, Kadam et al. observed upregulation of NRF2 and its downstream target HO-1 as well as downregulation of toll-like receptor 4 (Tlr4) following RSG administration [173]. Activation of Tlr4 promotes NF-κB activity followed by pro-inflammatory cytokine production and stimulation of the innate immune system. A very recent study by our group has shown that RSG can seemingly mediate NRF2 upregulation and activation through upregulation of PPARγ expression [156]. Our data strongly suggest the possibility for a PPARγ-mediated mechanism of NRF2 upregulation/activation leading to the repairing of BBB integrity, decreased endothelial inflammatory responses as well as upregulation of NRF2 downstream signaling molecule NQO-1 which exert acute detoxification and cytoprotective functions [122]. Along this line, it is conceivable that other drugs presenting similar NRF2 enhancing effects (including nutritional dietary phytochemicals) could be repurposed for the treatment of OS and pro-inflammatory-dependent cerebrovascular and neurological disorders. However, more in vitro and in vivo studies will be necessary to confirm the feasibility and the beneficial effects of each treatment.
11. Conclusions
NRF2 plays a pivotal role in regulating redox homeostasis as well as the activation and modulation of antioxidant, anti-inflammatory, drug metabolism, detoxification, and radical scavenging functions [5,44]. Indeed, the NRF2–ARE signaling is significant for cytoprotection of cell survival against oxidative stress and preservation of the proper redox balance in cells and tissues by promoting antioxidative defenses neutralizing ROS and also blocking transcription of pro-inflammatory genes [174,175]. Nowadays, there is a growing research interest in investigating the cerebrovascular and neurodegenerative protective effect of NRF2 on maintaining the functional integrity of the BBB, preventing harmful CNS disorders, and the initiation/progression of neuroinflammatory and also the identification of novel approaches targeting NRF2 to prevent and/or reduce brain injury [14,88,125].
Author Contributions
F.S. conceived the study and prepared the drafting of the manuscript. A.B. helped with the manuscript preparation. L.C. assisted with the drafting of the manuscript and preparation of the figures. L.C. also oversaw the entire project and provided funding support. All authors reviewed the manuscript.
Funding
This work was supported by the National Institutes of Health/National Institute on Drug Abuse 2R01-DA029121-01A1 and ARDF to Luca Cucullo.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
| αSyn | Alpha-synuclein |
| AD | Alzheimer’s disease |
| ALS | Amyotrophic lateral sclerosis |
| AMPK | AMP-activated protein kinase |
| ARE | Anti-oxidant response element |
| BBB | Blood–brain barrier |
| BMVEC | Brain microvascular endothelial cells |
| CAT | Catalase |
| CNC | Cap‘n’collar |
| COPD | Chronic obstructive pulmonary disease |
| EGCG | Epigallocatechin gallate |
| Glut 1 | Glucose transporter |
| GSH Px | Glutathione peroxidase |
| HD | Huntington’s disease |
| HO-1 | Heme oxygenase-1 |
| ICAM-1 | Intercellular adhesion molecule-1 |
| IKK | IκB kinase |
| KEAP1 | Kelc-like ECH-associated protein 1 |
| MF | Metformin |
| MS | Multiple sclerosis |
| NLRP3 | Nod-like receptor protein 3 |
| NQO-1 | NAD(P)H: quinone reductase I |
| NTF2 | Nuclear factor erythroid 2-related factor |
| PAMPs | Pathogen-associated molecular patterns |
| PD | Parkinson’s disease |
| PGC-1α | Proliferator-activated receptor gamma coactivator 1-alpha |
| PKC | Protein kinase C |
| PPARγ | Peroxisome proliferator-activated receptor gamma |
| PRR | Pattern recognition receptor |
| ROS | Reactive oxygen species |
| RSG | Rosiglitazone |
| SFN | Sulforaphane |
| SOD | Superoxide dismutase |
| TD2M | Type-2 diabetes mellitus |
| TBHQ | Terbutylhydroquinone |
| TBI | Traumatic brain injury |
| TEER | Trans-endothelial electrical resistance |
| TLRs | Toll-like receptors |
| TJ | Tight junction |
| TMCAO | Transient middle artery occlusion |
| TS | Tobacco smoking |
| Trx1 | Thioredoxin-1 |
| ZO-1 | Zonulae occludentes-1 |
References
- Huang, Y.; Li, W.J.; Su, Z.Y.; Kong, A.-N.T. The complexity of the Nrf2 pathway: beyond the antioxidant response. J. Nutr. Biochem. 2015, 26, 1401–1413. [Google Scholar] [CrossRef] [PubMed]
- Ding, K.; Wang, H.; Xu, J.; Li, T.; Zhang, L.; Ding, Y.; Zhu, L.; He, J.; Zhou, M. Melatonin stimulates antioxidant enzymes and reduces oxidative stress in experimental traumatic brain injury: the Nrf2–ARE signaling pathway as a potential mechanism. Free Radic. Biol. Med. 2014, 73, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Sajja, R.K.; Kaisar, M.A.; Vijay, V.; Desai, V.G.; Prasad, S.; Cucullo, L. In Vitro Modulation of Redox and Metabolism Interplay at the Brain Vascular Endothelium: Genomic and Proteomic Profiles of Sulforaphane Activity. Sci. Rep. 2018, 8, 12708. [Google Scholar] [CrossRef] [PubMed]
- Buendia, I.; Michalska, P.; Navarro, E.; Gameiro, I.; Egea, J.; León, R. Nrf2–ARE pathway: An emerging target against oxidative stress and neuroinflammation in neurodegenerative diseases. Pharmacol. Ther. 2016, 157, 84–104. [Google Scholar] [CrossRef] [PubMed]
- Sivandzade, F.; Prasad, S.; Bhalerao, A.; Cucullo, L. NRF2 and NF-κB interplay in cerebrovascular and neurodegenerative disorders: Molecular mechanisms and possible therapeutic approaches. Redox Biol. 2019, 21, 101059. [Google Scholar] [CrossRef] [PubMed]
- Sandberg, M.; Patil, J.; D’angelo, B.; Weber, S.G.; Mallard, C. NRF2-regulation in brain health and disease: implication of cerebral inflammation. Neuropharmacology 2014, 79, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Yamamoto, M. Molecular basis of the Keap1–Nrf2 system. Free Radic. Biol. Med. 2015, 88, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Sajja, R.K.; Prasad, S.; Tang, S.; Kaisar, M.A.; Cucullo, L. Blood-brain barrier disruption in diabetic mice is linked to Nrf2 signaling deficits: Role of ABCB10? Neurosci. Lett. 2017, 653, 152–158. [Google Scholar] [CrossRef] [PubMed]
- Kowluru, R.A.; Mishra, M. Epigenetic regulation of redox signaling in diabetic retinopathy: Role of Nrf2. Free Radic. Biol. Med. 2017, 103, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Dinkova-Kostova, A.T.; Abramov, A.Y. The emerging role of Nrf2 in mitochondrial function. Free Radic. Biol. Med. 2015, 88, 179–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prasad, S.; Sajja, R.K.; Park, J.H.; Naik, P.; Kaisar, M.A.; Cucullo, L. Impact of cigarette smoke extract and hyperglycemic conditions on blood–brain barrier endothelial cells. Fluids Barriers CNS 2015, 12, 18. [Google Scholar] [CrossRef] [PubMed]
- Tufekci, K.U.; Civi Bayin, E.; Genc, S.; Genc, K. The Nrf2/ARE pathway: A promising target to counteract mitochondrial dysfunction in Parkinson’s disease. Parkinson’s Dis. 2011, 2011. [Google Scholar] [CrossRef] [PubMed]
- Holmström, K.M.; Baird, L.; Zhang, Y.; Hargreaves, I.; Chalasani, A.; Land, J.M.; Stanyer, L.; Yamamoto, M.; Dinkova-Kostova, A.T.; Abramov, A.Y. Nrf2 impacts cellular bioenergetics by controlling substrate availability for mitochondrial respiration. Biol. Open 2013, 2, 761–770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, T.; Wang, H.; Ding, Y.; Zhou, M.; Zhou, X.; Zhang, X.; Ding, K.; He, J.; Lu, X.; Xu, J.; et al. Genetic elimination of Nrf2 aggravates secondary complications except for vasospasm after experimental subarachnoid hemorrhage in mice. Brain Res. 2014, 1558, 90–99. [Google Scholar] [CrossRef] [PubMed]
- Mitsuishi, Y.; Taguchi, K.; Kawatani, Y.; Shibata, T.; Nukiwa, T.; Aburatani, H.; Yamamoto, M.; Motohashi, H. Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming. Cancer Cell 2012, 22, 66–79. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.M.; Shih, A.Y.; Murphy, T.H.; Johnson, J.A. NF-E2-related factor-2 mediates neuroprotection against mitochondrial complex I inhibitors and increased concentrations of intracellular calcium in primary cortical neurons. J. Biol. Chem. 2003, 278, 37948–37956. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.; Krishnan, J.; Ruckmani, K. Cigarette smoke and related risk factors in neurological disorders: An update. Biomed. Pharmacother. 2017, 85, 79–86. [Google Scholar]
- Santín-Márquez, R.; Alarcón-Aguilar, A.; López-Diazguerrero, N.E.; Chondrogianni, N.; Königsberg, M. Sulfoaphane—Role in aging and neurodegeneration. GeroScience 2019, 1–16. [Google Scholar] [CrossRef]
- Pajares, M.; Rojo, A.I.; Cuadrad, A. P 087—NRF2 controls proteostasis through the transcriptional regulation of autophagy. In Proceedings of the OCC World Congress and Annual SFRR-E Conference 2017 Metabolic Stress and Redox Regulation, Berlin, Germany, 21–23 June 2017; p. S47. [Google Scholar]
- Pajares, M.; Rojo, A.I.; Arias, E.; Diaz-Carretero, A.; Cuervo, A.M.; Cuadrado, A. Transcription factor NFE2L2/NRF2 modulates chaperone-mediated autophagy through the regulation of LAMP2A. Autophagy 2018, 14, 1310–1322. [Google Scholar] [CrossRef]
- Niture, S.K.; Jaiswal, A.K. Nrf2 Protein Up-regulates Antiapoptotic Protein Bcl-2 and Prevents Cellular Apoptosis. J. Biol. Chem. 2012, 287, 9873–9886. [Google Scholar] [CrossRef]
- Harada, N.; Kanayama, M.; Maruyama, A.; Yoshida, A.; Tazumi, K.; Hosoya, T.; Mimura, J.; Toki, T.; Maher, J.M.; Yamamoto, M. Nrf2 regulates ferroportin 1-mediated iron efflux and counteracts lipopolysaccharide-induced ferroportin 1 mRNA suppression in macrophages. Arch. Biochem. Biophys. 2011, 508, 101–109. [Google Scholar] [CrossRef]
- Sakata, H.; Niizuma, K.; Yoshioka, H.; Kim, G.S.; Jung, J.E.; Katsu, M.; Narasimhan, P.; Maier, C.M.; Nishiyama, Y.; Chan, P.H. Minocycline-preconditioned neural stem cells enhance neuroprotection after ischemic stroke in rats. J. Neurosci. 2012, 32, 3462–3473. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, M.; Kurokawa, H.; Waguri, S.; Taguchi, K.; Kobayashi, A.; Ichimura, Y.; Sou, Y.-S.; Ueno, I.; Sakamoto, A.; Tong, K.I. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat. Cell Biol. 2010, 12, 213. [Google Scholar] [CrossRef] [PubMed]
- Piantadosi, C.A.; Withers, C.M.; Bartz, R.R.; MacGarvey, N.C.; Fu, P.; Sweeney, T.E.; Welty-Wolf, K.E.; Suliman, H.B. Heme oxygenase-1 couples activation of mitochondrial biogenesis to anti-inflammatory cytokine expression. J. Biol. Chem. 2011, 286, 16374–16385. [Google Scholar] [CrossRef] [PubMed]
- Vomhof-DeKrey, E.E.; Picklo Sr, M.J. The Nrf2-antioxidant response element pathway: a target for regulating energy metabolism. J. Nutr. Biochem. 2012, 23, 1201–1206. [Google Scholar] [CrossRef] [PubMed]
- Cardozo, L.F.; Pedruzzi, L.M.; Stenvinkel, P.; Stockler-Pinto, M.B.; Daleprane, J.B.; Leite Jr, M.; Mafra, D. Nutritional strategies to modulate inflammation and oxidative stress pathways via activation of the master antioxidant switch Nrf2. Biochimie 2013, 95, 1525–1533. [Google Scholar] [CrossRef]
- Baird, L.; Dinkova-Kostova, A.T. The cytoprotective role of the Keap1–Nrf2 pathway. Arch. Toxicol. 2011, 85, 241–272. [Google Scholar] [CrossRef] [PubMed]
- Cominacini, L.; Mozzini, C.; Garbin, U.; Pasini, A.; Stranieri, C.; Solani, E.; Vallerio, P.; Tinelli, I.A.; Pasini, A.F. Endoplasmic reticulum stress and Nrf2 signaling in cardiovascular diseases. Free Radic. Biol. Med. 2015, 88, 233–242. [Google Scholar] [CrossRef]
- Bellezza, I.; Giambanco, I.; Minelli, A.; Donato, R. Nrf2-Keap1 signaling in oxidative and reductive stress. Biochim. Biophys. Acta. Mol. Cell Res. 2018, 1865, 721–733. [Google Scholar] [CrossRef]
- Plafker, K.S.; Nguyen, L.; Barneche, M.; Mirza, S.; Crawford, D.F.; Plafker, S.M. The ubiquitin conjugating enzyme, UbcM2, can regulate the stability and activity of the anti-oxidant transcription factor, Nrf2. J. Biol. Chem. 2010, 285, 23064–23074. [Google Scholar] [CrossRef]
- Niture, S.K.; Khatri, R.; Jaiswal, A.K. Regulation of Nrf2—An update. Free Radic. Biol. Med. 2014, 66, 36–44. [Google Scholar] [CrossRef]
- Xiang, M.; Namani, A.; Wu, S.; Wang, X. Nrf2: bane or blessing in cancer? J. Cancer Res. Clin. Oncol. 2014, 140, 1251–1259. [Google Scholar] [CrossRef] [PubMed]
- Krajka-Kuźniak, V.; Paluszczak, J.; Baer-Dubowska, W. The Nrf2-ARE signaling pathway: an update on its regulation and possible role in cancer prevention and treatment. Pharmacol. Rep. 2017, 69, 393–402. [Google Scholar] [CrossRef] [PubMed]
- Rada, P.; Rojo, A.I.; Chowdhry, S.; McMahon, M.; Hayes, J.D.; Cuadrado, A. SCF/{beta}-TrCP promotes glycogen synthase kinase 3-dependent degradation of the Nrf2 transcription factor in a Keap1-independent manner. Mol. Cell Biol. 2011, 31, 1121–1133. [Google Scholar] [CrossRef] [PubMed]
- Rada, P.; Rojo, A.I.; Evrard-Todeschi, N.; Innamorato, N.G.; Cotte, A.; Jaworski, T.; Tobon-Velasco, J.C.; Devijver, H.; Garcia-Mayoral, M.F.; Van Leuven, F.; et al. Structural and functional characterization of Nrf2 degradation by the glycogen synthase kinase 3/beta-TrCP axis. Mol. Cell Biol. 2012, 32, 3486–3499. [Google Scholar] [CrossRef] [PubMed]
- Chowdhry, S.; Zhang, Y.; McMahon, M.; Sutherland, C.; Cuadrado, A.; Hayes, J.D. Nrf2 is controlled by two distinct beta-TrCP recognition motifs in its Neh6 domain, one of which can be modulated by GSK-3 activity. Oncogene 2013, 32, 3765–3781. [Google Scholar] [CrossRef] [PubMed]
- Bai, X.; Chen, Y.; Hou, X.; Huang, M.; Jin, J. Emerging role of NRF2 in chemoresistance by regulating drug-metabolizing enzymes and efflux transporters. Drug Metab. Rev. 2016, 48, 541–567. [Google Scholar] [CrossRef] [PubMed]
- Naik, P.; Cucullo, L. Pathobiology of tobacco smoking and neurovascular disorders: untied strings and alternative products. Fluids Barriers CNS 2015, 12, 25. [Google Scholar] [CrossRef] [PubMed]
- Bryan, H.K.; Olayanju, A.; Goldring, C.E.; Park, B.K. The Nrf2 cell defence pathway: Keap1-dependent and-independent mechanisms of regulation. Biochem. Pharmacol. 2013, 85, 705–717. [Google Scholar] [CrossRef] [PubMed]
- Strom, J.; Xu, B.; Tian, X.; Chen, Q.M. Nrf2 protects mitochondrial decay by oxidative stress. FASEB J. 2016, 30, 66–80. [Google Scholar] [CrossRef]
- Salazar, M.; Rojo, A.I.; Velasco, D.; de Sagarra, R.M.; Cuadrado, A. Glycogen synthase kinase-3beta inhibits the xenobiotic and antioxidant cell response by direct phosphorylation and nuclear exclusion of the transcription factor Nrf2. J. Biol. Chem. 2006, 281, 14841–14851. [Google Scholar] [CrossRef]
- Espada, S.; Rojo, A.I.; Salinas, M.; Cuadrado, A. The muscarinic M1 receptor activates Nrf2 through a signaling cascade that involves protein kinase C and inhibition of GSK-3beta: connecting neurotransmission with neuroprotection. J. Neurochem. 2009, 110, 1107–1119. [Google Scholar] [CrossRef] [PubMed]
- Hayes, J.D.; Dinkova-Kostova, A.T. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem. Sci. 2014, 39, 199–218. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Palliyaguru, D.L.; Kensler, T.W. Frugal chemoprevention: targeting Nrf2 with foods rich in sulforaphane. Semin. Oncol. 2016, 43, 146–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuse, Y.; Kobayashi, M. Conservation of the Keap1-Nrf2 system: An evolutionary journey through stressful space and time. Molecules 2017, 22, 436. [Google Scholar] [CrossRef] [PubMed]
- Sajja, R.K.; Green, K.N.; Cucullo, L. Altered Nrf2 signaling mediates hypoglycemia-induced blood-brain barrier endothelial dysfunction in vitro. PLoS ONE 2015, 10, e0122358. [Google Scholar] [CrossRef] [PubMed]
- Niture, S.K.; Jain, A.K.; Shelton, P.M.; Jaiswal, A.K. Src subfamily kinases regulate nuclear export and degradation of transcription factor Nrf2 to switch off Nrf2-mediated antioxidant activation of cytoprotective gene expression. J. Biol. Chem. 2017, 292, 2048. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, K.; Hirano, I.; Itoh, T.; Tanaka, M.; Miyajima, A.; Suzuki, A.; Motohashi, H.; Yamamoto, M. Nrf2 Enhances Cholangiocyte Expansion in Pten-Deficient Livers. Mol. Cell. Biol. 2014, 34, 900–913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ichimura, Y.; Waguri, S.; Sou, Y.-s.; Kageyama, S.; Hasegawa, J.; Ishimura, R.; Saito, T.; Yang, Y.; Kouno, T.; Fukutomi, T.; et al. Phosphorylation of p62 Activates the Keap1-Nrf2 Pathway during Selective Autophagy. Mol. Cell 2013, 51, 618–631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taguchi, K.; Fujikawa, N.; Komatsu, M.; Ishii, T.; Unno, M.; Akaike, T.; Motohashi, H.; Yamamoto, M. Keap1 degradation by autophagy for the maintenance of redox homeostasis. Proc. Natl. Acad. Sci. USA 2012, 109, 13561–13566. [Google Scholar] [CrossRef] [Green Version]
- Smith, E.M.; Gregg, M.; Hashemi, F.; Schott, L.; Hughes, T.K. Corticotropin Releasing Factor (CRF) activation of NF-κB-directed transcription in leukocytes. Cell Mol. Neurobiol. 2006, 26, 1021–1036. [Google Scholar] [CrossRef] [PubMed]
- Wardyn, J.D.; Ponsford, A.H.; Sanderson, C.M. Dissecting molecular cross-talk between Nrf2 and NF-κB response pathways. Biochem. Soc. Trans. 2015, 43, 621–626. [Google Scholar] [CrossRef] [PubMed]
- Innamorato, N.G.; Rojo, A.I.; García-Yagüe, Á.J.; Yamamoto, M.; de Ceballos, M.L.; Cuadrado, A. The Transcription Factor Nrf2 Is a Therapeutic Target against Brain Inflammation. J. Immunol. 2008, 181, 680–689. [Google Scholar] [CrossRef] [PubMed]
- Brandenburg, L.-O.; Kipp, M.; Lucius, R.; Pufe, T.; Wruck, C.J. Sulforaphane suppresses LPS-induced inflammation in primary rat microglia. Inflamm. Res. 2010, 59, 443–450. [Google Scholar] [CrossRef] [PubMed]
- Innamorato, N.G.; Lastres-Becker, I.; Cuadrado, A. Role of microglial redox balance in modulation of neuroinflammation. Curr. Opin. Neurol. 2009, 22, 308–314. [Google Scholar] [CrossRef] [PubMed]
- Innamorato, N.G.; Cuadrado, A.; Lastres-Becker, I.; Kirik, D.; Sahin, G.; Ulusoy, A.; Rábano, A. α-Synuclein expression and Nrf2 deficiency cooperate to aggravate protein aggregation, neuronal death and inflammation in early-stage Parkinson’s disease. Hum. Mol. Genet. 2012, 21, 3173–3192. [Google Scholar]
- Rojo, A.I.; Innamorato, N.G.; Martín-Moreno, A.M.; De Ceballos, M.L.; Yamamoto, M.; Cuadrado, A. Nrf2 regulates microglial dynamics and neuroinflammation in experimental Parkinson’s disease. Glia 2010, 58, 588–598. [Google Scholar] [CrossRef] [PubMed]
- Rushworth, S.A.; MacEwan, D.J.; O’Connell, M.A. Lipopolysaccharide-Induced Expression of NAD(P)H:Quinone Oxidoreductase 1 and Heme Oxygenase-1 Protects against Excessive Inflammatory Responses in Human Monocytes. J. Immunol. 2008, 181, 6730–6737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wakabayashi, N.; Slocum, S.L.; Skoko, J.J.; Shin, S.; Kensler, T.W. When NRF2 Talks, Who’s Listening? Antioxid. Redox Signal 2010, 13, 1649–1663. [Google Scholar] [CrossRef]
- Cuadrado, A.; Martín-Moldes, Z.; Ye, J.; Lastres-Becker, I. Transcription factors NRF2 and NF-κB are coordinated effectors of the Rho family, GTP-binding protein RAC1 during inflammation. J. Biol. Chem. 2014, 289, 15244–15258. [Google Scholar] [CrossRef]
- Sanlioglu, S.; Williams, C.M.; Samavati, L.; Butler, N.S.; Wang, G.; McCray, P.B.; Ritchie, T.C.; Hunninghake, G.W.; Zandi, E.; Engelhardt, J.F. Lipopolysaccharide induces Rac1-dependent reactive oxygen species formation and coordinates tumor necrosis factor-α secretion through IKK regulation of NF-κB. J. Biol. Chem. 2001, 276, 30188–30198. [Google Scholar] [CrossRef]
- Zhang, H.; Davies, K.J.; Forman, H.J. Oxidative stress response and Nrf2 signaling in aging. Free Radic. Biol. Med. 2015, 88, 314–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sohal, R.S.; Orr, W.C. The redox stress hypothesis of aging. Free Radic. Biol. Med. 2012, 52, 539–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ngo, J.K.; Pomatto, L.C.; Davies, K.J. Upregulation of the mitochondrial Lon Protease allows adaptation to acute oxidative stress but dysregulation is associated with chronic stress, disease, and aging. Redox Biol. 2013, 1, 258–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tarantini, S.; Valcarcel-Ares, M.N.; Yabluchanskiy, A.; Tucsek, Z.; Hertelendy, P.; Kiss, T.; Gautam, T.; Zhang, X.A.; Sonntag, W.E.; de Cabo, R.; et al. Nrf2 Deficiency Exacerbates Obesity-Induced Oxidative Stress, Neurovascular Dysfunction, Blood-Brain Barrier Disruption, Neuroinflammation, Amyloidogenic Gene Expression, and Cognitive Decline in Mice, Mimicking the Aging Phenotype. J. Gerontol. A Biol. Sci. Med. Sci. 2018, 73, 853–863. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Liu, H.; Davies, K.J.; Sioutas, C.; Finch, C.E.; Morgan, T.E.; Forman, H.J. Nrf2-regulated phase II enzymes are induced by chronic ambient nanoparticle exposure in young mice with age-related impairments. Free Radic. Biol. Med. 2012, 52, 2038–2046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva-Palacios, A.; Ostolga-Chavarria, M.; Zazueta, C.; Konigsberg, M. Nrf2: Molecular and epigenetic regulation during aging. Ageing Res. Rev. 2018, 47, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Sivandzade, F.; Cucullo, L. In-vitro blood–brain barrier modeling: A review of modern and fast-advancing technologies. J. Cereb. Blood Flow Metab. 2018, 0271678X18788769. [Google Scholar] [CrossRef]
- Hasan, A.; Deeb, G.; Rahal, R.; Atwi, K.; Mondello, S.; Marei, H.E.; Gali, A.; Sleiman, E. Mesenchymal stem cells in the treatment of traumatic brain injury. Front. Neurol. 2017, 8, 28. [Google Scholar] [CrossRef]
- Semple, B.D.; Zamani, A.; Rayner, G.; Shultz, S.R.; Jones, N.C. Affective, neurocognitive and psychosocial disorders associated with traumatic brain injury and post-traumatic epilepsy. Neurobiol. Dis. 2019, 123, 27–41. [Google Scholar] [CrossRef]
- Dong, W.; Yang, B.; Wang, L.; Li, B.; Guo, X.; Zhang, M.; Jiang, Z.; Fu, J.; Pi, J.; Guan, D. Curcumin plays neuroprotective roles against traumatic brain injury partly via Nrf2 signaling. Toxicol. Appl. Pharmacol. 2018, 346, 28–36. [Google Scholar] [CrossRef]
- Angeloni, C.; Prata, C.; Vieceli Dalla Sega, F.; Piperno, R.; Hrelia, S. Traumatic brain injury and NADPH oxidase: a deep relationship. Oxidative Med. Cell. Longev. 2015, 2015. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.A.; Park, S.; Krause, J.S.; Banik, N.L. Oxidative stress, DNA damage, and the telomeric complex as therapeutic targets in acute neurodegeneration. Neurochem. Int. 2013, 62, 764–775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, Y.; Yan, H.; Ni, H.; Liang, W.; Jin, W. Expression of nuclear factor erythroid 2-related factor 2 following traumatic brain injury in the human brain. Neuroreport 2019, 30, 344–349. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Tian, M.; Wang, H.D.; Gao, C.C.; Zhu, L.; Lin, Y.X.; Fang, J.; Ding, K. Activation of the Nrf2-ARE signal pathway after blast induced traumatic brain injury in mice. Int. J. Neurosci. 2019, 129, 801–807. [Google Scholar] [CrossRef] [PubMed]
- Suvanish Kumar, V.; Gopalakrishnan, A.; Naziroglu, M.; Rajanikant, G. Calcium ion–the key player in cerebral ischemia. Curr. Med. Chem. 2014, 21, 2065–2075. [Google Scholar] [CrossRef]
- Pradeep, H.; Diya, J.B.; Shashikumar, S.; Rajanikant, G.K. Oxidative stress–assassin behind the ischemic stroke. Folia Neuropathol. 2012, 50, 219–230. [Google Scholar] [CrossRef] [PubMed]
- Stephenson, D.; Yin, T.; Smalstig, E.B.; Hsu, M.A.; Panetta, J.; Little, S.; Clemens, J. Transcription factor nuclear factor-κB is activated in neurons after focal cerebral ischemia. J. Cereb. Blood Flow Metab. 2000, 20, 592–603. [Google Scholar] [CrossRef] [PubMed]
- Kunz, A.; Abe, T.; Hochrainer, K.; Shimamura, M.; Anrather, J.; Racchumi, G.; Zhou, P.; Iadecola, C. Nuclear factor-κB activation and postischemic inflammation are suppressed in CD36-null mice after middle cerebral artery occlusion. J. Neurosci. 2008, 28, 1649–1658. [Google Scholar] [CrossRef]
- Harari, O.A.; Liao, J.K. NF-κB and innate immunity in ischemic stroke. Ann. N Y Acad. Sci. 2010, 1207, 32–40. [Google Scholar] [CrossRef]
- Hou, Y.; Wang, Y.; He, Q.; Li, L.; Xie, H.; Zhao, Y.; Zhao, J. Nrf2 inhibits NLRP3 inflammasome activation through regulating Trx1/TXNIP complex in cerebral ischemia reperfusion injury. Behav. Brain Res. 2018, 336, 32–39. [Google Scholar] [CrossRef]
- Kaisar, M.A.; Villalba, H.; Prasad, S.; Liles, T.; Sifat, A.E.; Sajja, R.K.; Abbruscato, T.J.; Cucullo, L. Offsetting the impact of smoking and e-cigarette vaping on the cerebrovascular system and stroke injury: Is Metformin a viable countermeasure? Redox Biol. 2017, 13, 353–362. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Locascio, L.M.; Dore, S. Critical Role of Nrf2 in Experimental Ischemic Stroke. Front. Pharm. 2019, 10, 153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prasad, S.; Kaisar, M.A.; Cucullo, L. Unhealthy smokers: scopes for prophylactic intervention and clinical treatment. BMC Neurosci. 2017, 18, 70. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Kang, J.; Li, H.; Su, J.; Wu, J.; Xu, Y.; Yu, H.; Xiang, X.; Yi, H.; Lu, Y. Regulation of endoplasmic reticulum stress in rat cortex by p62/ZIP through the Keap1-Nrf2-ARE signalling pathway after transient focal cerebral ischaemia. Brain Inj. 2013, 27, 924–933. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Zhu, X.; Kim, Y.; Li, J.; Huang, S.; Saleem, S.; Li, R.C.; Xu, Y.; Dore, S.; Cao, W. Histone deacetylase inhibition activates transcription factor Nrf2 and protects against cerebral ischemic damage. Free Radic. Biol. Med. 2012, 52, 928–936. [Google Scholar] [CrossRef]
- Zhao, J.; Moore, A.N.; Redell, J.B.; Dash, P.K. Enhancing expression of Nrf2-driven genes protects the blood brain barrier after brain injury. J. Neurosci. 2007, 27, 10240–10248. [Google Scholar] [CrossRef]
- Li, L.; Zhang, X.; Cui, L.; Wang, L.; Liu, H.; Ji, H.; Du, Y. Ursolic acid promotes the neuroprotection by activating Nrf2 pathway after cerebral ischemia in mice. Brain Res. 2013, 1497, 32–39. [Google Scholar] [CrossRef]
- Wang, H.; Zhong, D.; Chen, H.; Jin, J.; Liu, Q.; Li, G. NLRP3 inflammasome activates interleukin-23/interleukin-17 axis during ischaemia-reperfusion injury in cerebral ischaemia in mice. Life Sci. 2019, 227, 101–113. [Google Scholar] [CrossRef]
- Kaisar, M.A.; Sivandzade, F.; Bhalerao, A.; Cucullo, L. Conventional and electronic cigarettes dysregulate the expression of iron transporters and detoxifying enzymes at the brain vascular endothelium: In vivo evidence of a gender-specific cellular response to chronic cigarette smoke exposure. Neurosci. Lett. 2018, 682, 1–9. [Google Scholar] [CrossRef]
- Carvalho, C.; Machado, N.; Mota, P.C.; Correia, S.C.; Cardoso, S.; Santos, R.X.; Santos, M.S.; Oliveira, C.R.; Moreira, P.I. Type 2 diabetic and Alzheimer’s disease mice present similar behavioral, cognitive, and vascular anomalies. J. Alzheimer’s Dis. 2013, 35, 623–635. [Google Scholar] [CrossRef]
- Wevers, N.R.; de Vries, H.E. Morphogens and blood-brain barrier function in health and disease. Tissue Barriers 2016, 4, e1090524. [Google Scholar] [CrossRef] [PubMed]
- Gan, L.; Johnson, J.A. Oxidative damage and the Nrf2-ARE pathway in neurodegenerative diseases. Biochim. Et Biophys. Acta (BBA)-Mol. Basis. Dis. 2014, 1842, 1208–1218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramsey, C.P.; Glass, C.A.; Montgomery, M.B.; Lindl, K.A.; Ritson, G.P.; Chia, L.A.; Hamilton, R.L.; Chu, C.T.; Jordan-Sciutto, K.L. Expression of Nrf2 in neurodegenerative diseases. J. Neuropathol. Exp. Neurol. 2007, 66, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Salminen, A.; Kaarniranta, K.; Haapasalo, A.; Hiltunen, M.; Soininen, H.; Alafuzoff, I. Emerging role of p62/sequestosome-1 in the pathogenesis of Alzheimer’s disease. Prog. Neurobiol. 2012, 96, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Sarlette, A.; Krampfl, K.; Grothe, C.; Neuhoff, N.; Dengler, R.; Petri, S. Nuclear erythroid 2-related factor 2-antioxidative response element signaling pathway in motor cortex and spinal cord in amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 2008, 67, 1055–1062. [Google Scholar] [CrossRef] [PubMed]
- Tanji, K.; Maruyama, A.; Odagiri, S.; Mori, F.; Itoh, K.; Kakita, A.; Takahashi, H.; Wakabayashi, K. Keap1 is localized in neuronal and glial cytoplasmic inclusions in various neurodegenerative diseases. J. Neuropathol. Exp. Neurol. 2013, 72, 18–28. [Google Scholar] [CrossRef] [PubMed]
- De Strooper, B. Proteases and proteolysis in Alzheimer disease: a multifactorial view on the disease process. Physiol. Rev. 2010, 90, 465–494. [Google Scholar] [CrossRef]
- Freeman, L.R.; Keller, J.N. Oxidative stress and cerebral endothelial cells: Regulation of the blood–brain-barrier and antioxidant based interventions. Biochim. Biophys. Acta Mol. Cell Res. 2012, 1822, 822–829. [Google Scholar] [CrossRef]
- Bae, E.-J.; Ho, D.-H.; Park, E.; Jung, J.W.; Cho, K.; Hong, J.H.; Lee, H.-J.; Kim, K.P.; Lee, S.-J. Lipid peroxidation product 4-hydroxy-2-nonenal promotes seeding-capable oligomer formation and cell-to-cell transfer of α-synuclein. Antioxid. Redox Signal 2013, 18, 770–783. [Google Scholar] [CrossRef]
- Li, X.-H.; Li, C.-Y.; Lu, J.-M.; Tian, R.-B.; Wei, J. Allicin ameliorates cognitive deficits ageing-induced learning and memory deficits through enhancing of Nrf2 antioxidant signaling pathways. Neurosci. Lett. 2012, 514, 46–50. [Google Scholar] [CrossRef]
- Eftekharzadeh, B.; Maghsoudi, N.; Khodagholi, F. Stabilization of transcription factor Nrf2 by tBHQ prevents oxidative stress-induced amyloid β formation in NT2N neurons. Biochimie 2010, 92, 245–253. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.V.; Kim, H.Y.; Ehrlich, H.Y.; Choi, S.Y.; Kim, D.J.; Kim, Y. Amelioration of Alzheimer’s disease by neuroprotective effect of sulforaphane in animal model. Amyloid 2013, 20, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Rojo, A.I.; Pajares, M.; Garcia-Yague, A.J.; Buendia, I.; Van Leuven, F.; Yamamoto, M.; Lopez, M.G.; Cuadrado, A. Deficiency in the transcription factor NRF2 worsens inflammatory parameters in a mouse model with combined tauopathy and amyloidopathy. Redox Biol. 2018, 18, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Mao, P.; Manczak, M.; Calkins, M.J.; Truong, Q.; Reddy, T.P.; Reddy, A.P.; Shirendeb, U.; Lo, H.-H.; Rabinovitch, P.S.; Reddy, P.H. Mitochondria-targeted catalase reduces abnormal APP processing, amyloid β production and BACE1 in a mouse model of Alzheimer’s disease: implications for neuroprotection and lifespan extension. Hum. Mol. Genet. 2012, 21, 2973–2990. [Google Scholar] [CrossRef] [PubMed]
- Rojo, A.I.; Pajares, M.; Rada, P.; Nunez, A.; Nevado-Holgado, A.J.; Killik, R.; Van Leuven, F.; Ribe, E.; Lovestone, S.; Yamamoto, M.; et al. NRF2 deficiency replicates transcriptomic changes in Alzheimer’s patients and worsens APP and TAU pathology. Redox Biol. 2017, 13, 444–451. [Google Scholar] [CrossRef] [PubMed]
- Näsström, T.; Fagerqvist, T.; Barbu, M.; Karlsson, M.; Nikolajeff, F.; Kasrayan, A.; Ekberg, M.; Lannfelt, L.; Ingelsson, M.; Bergström, J. The lipid peroxidation products 4-oxo-2-nonenal and 4-hydroxy-2-nonenal promote the formation of α-synuclein oligomers with distinct biochemical, morphological, and functional properties. Free Radic. Biol. Med. 2011, 50, 428–437. [Google Scholar] [CrossRef] [PubMed]
- Lastres-Becker, I.; Garcia-Yague, A.J.; Scannevin, R.H.; Casarejos, M.J.; Kugler, S.; Rabano, A.; Cuadrado, A. Repurposing the NRF2 Activator Dimethyl Fumarate as Therapy Against Synucleinopathy in Parkinson’s Disease. Antioxid Redox Signal 2016, 25, 61–77. [Google Scholar] [CrossRef] [PubMed]
- Woo, S.Y.; Kim, J.H.; Moon, M.K.; Han, S.-H.; Yeon, S.K.; Choi, J.W.; Jang, B.K.; Song, H.J.; Kang, Y.G.; Kim, J.W. Discovery of vinyl sulfones as a novel class of neuroprotective agents toward Parkinson’s disease therapy. J. Med. Chem. 2014, 57, 1473–1487. [Google Scholar] [CrossRef]
- Gan, L.; Vargas, M.R.; Johnson, D.A.; Johnson, J.A. Astrocyte-specific overexpression of Nrf2 delays motor pathology and synuclein aggregation throughout the CNS in the alpha-synuclein mutant (A53T) mouse model. J. Neurosci. 2012, 32, 17775–17787. [Google Scholar] [CrossRef]
- He, Q.; Song, N.; Jia, F.; Xu, H.; Yu, X.; Xie, J.; Jiang, H. Role of α-synuclein aggregation and the nuclear factor E2-related factor 2/heme oxygenase-1 pathway in iron-induced neurotoxicity. Int. J. Biochem. Cell Biol. 2013, 45, 1019–1030. [Google Scholar] [CrossRef]
- Barone, M.C.; Sykiotis, G.P.; Bohmann, D. Genetic activation of Nrf2 signaling is sufficient to ameliorate neurodegenerative phenotypes in a Drosophila model of Parkinson’s disease. Dis. Models Mech. 2011, 4, 701–707. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.N.; Yanxun, V.Y.; Gundemir, S.; Jo, C.; Cui, M.; Tieu, K.; Johnson, G.V. Impaired mitochondrial dynamics and Nrf2 signaling contribute to compromised responses to oxidative stress in striatal cells expressing full-length mutant huntingtin. PLoS ONE 2013, 8, e57932. [Google Scholar] [CrossRef] [PubMed]
- Stack, C.; Ho, D.; Wille, E.; Calingasan, N.Y.; Williams, C.; Liby, K.; Sporn, M.; Dumont, M.; Beal, M.F. Triterpenoids CDDO-ethyl amide and CDDO-trifluoroethyl amide improve the behavioral phenotype and brain pathology in a transgenic mouse model of Huntington’s disease. Free Radic. Biol. Med. 2010, 49, 147–158. [Google Scholar] [CrossRef] [PubMed]
- Hands, S.; Sajjad, M.U.; Newton, M.J.; Wyttenbach, A. In vitro and in vivo aggregation of a fragment of huntingtin protein directly causes free radical production. J. Biol. Chem. 2011, 286, 44512–44520. [Google Scholar] [CrossRef] [PubMed]
- Tsvetkov, A.S.; Arrasate, M.; Barmada, S.; Ando, D.M.; Sharma, P.; Shaby, B.A.; Finkbeiner, S. Proteostasis of polyglutamine varies among neurons and predicts neurodegeneration. Nat. Chem. Biol. 2013, 9, 586. [Google Scholar] [CrossRef] [PubMed]
- Kanno, T.; Tanaka, K.; Yanagisawa, Y.; Yasutake, K.; Hadano, S.; Yoshii, F.; Hirayama, N.; Ikeda, J.-E. A novel small molecule, N-(4-(2-pyridyl)(1,3-thiazol-2-yl))-2-(2,4,6-trimethylphenoxy) acetamide, selectively protects against oxidative stress-induced cell death by activating the Nrf2–ARE pathway: Therapeutic implications for ALS. Free Radic. Biol. Med. 2012, 53, 2028–2042. [Google Scholar] [CrossRef] [PubMed]
- Mead, R.J.; Higginbottom, A.; Allen, S.P.; Kirby, J.; Bennett, E.; Barber, S.C.; Heath, P.R.; Coluccia, A.; Patel, N.; Gardner, I. S [+] Apomorphine is a CNS penetrating activator of the Nrf2-ARE pathway with activity in mouse and patient fibroblast models of amyotrophic lateral sclerosis. Free Radic. Biol. Med. 2013, 61, 438–452. [Google Scholar] [CrossRef]
- Liebner, S.; Czupalla, C.J.; Wolburg, H. Current concepts of blood-brain barrier development. Int. J. Dev. Biol. 2011, 55, 467–476. [Google Scholar] [CrossRef]
- Alfieri, A.; Srivastava, S.; Siow, R.C.; Modo, M.; Fraser, P.A.; Mann, G.E. Targeting the Nrf2–Keap1 antioxidant defence pathway for neurovascular protection in stroke. J. Physiol. 2011, 589, 4125–4136. [Google Scholar] [CrossRef]
- Prasad, S.; Sajja, R.K.; Kaisar, M.A.; Park, J.H.; Villalba, H.; Liles, T.; Abbruscato, T.; Cucullo, L. Role of Nrf2 and protective effects of Metformin against tobacco smoke-induced cerebrovascular toxicity. Redox Biol. 2017, 12, 58–69. [Google Scholar] [CrossRef]
- Naik, P.; Sajja, R.K.; Prasad, S.; Cucullo, L. Effect of full flavor and denicotinized cigarettes exposure on the brain microvascular endothelium: a microarray-based gene expression study using a human immortalized BBB endothelial cell line. BMC Neurosci. 2015, 16, 38. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.-Y.; Wang, H.-D.; Xu, J.-G.; Ding, K.; Li, T. Deletion of Nrf2 exacerbates oxidative stress after traumatic brain injury in mice. Cell. Mol. Neurobiol. 2015, 35, 713–721. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Campos, C.R.; Peart, J.C.; Smith, L.K.; Boni, J.L.; Cannon, R.E.; Miller, D.S. Nrf2 upregulates ATP binding cassette transporter expression and activity at the blood–brain and blood–spinal cord barriers. J. Neurosci. 2014, 34, 8585–8593. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Fang, Q.; Zhang, J.; Zhou, D.; Wang, Z. Role of the Nrf2-ARE pathway in early brain injury after experimental subarachnoid hemorrhage. J. Neurosci. Res. 2011, 89, 515–523. [Google Scholar] [CrossRef] [PubMed]
- Petri, S.; Körner, S.; Kiaei, M. Nrf2/ARE signaling pathway: key mediator in oxidative stress and potential therapeutic target in ALS. Neurol. Res. Int. 2012, 2012. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.-H.; Gold, R.; Linker, R.A. Mechanisms of oxidative damage in multiple sclerosis and neurodegenerative diseases: therapeutic modulation via fumaric acid esters. Int. J. Mol. Sci. 2012, 13, 11783–11803. [Google Scholar] [CrossRef] [PubMed]
- Prasad, S.; Sajja, R.K.; Naik, P.; Cucullo, L. Diabetes mellitus and blood-brain barrier dysfunction: an overview. J. Pharmacovigil. 2014, 2, 125. [Google Scholar] [PubMed]
- Wang, W.; Wu, Y.; Zhang, G.; Fang, H.; Wang, H.; Zang, H.; Xie, T.; Wang, W. Activation of Nrf2-ARE signal pathway protects the brain from damage induced by epileptic seizure. Brain Res. 2014, 1544, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Alfieri, A.; Srivastava, S.; Siow, R.C.; Cash, D.; Modo, M.; Duchen, M.R.; Fraser, P.A.; Williams, S.C.; Mann, G.E. Sulforaphane preconditioning of the Nrf2/HO-1 defense pathway protects the cerebral vasculature against blood–brain barrier disruption and neurological deficits in stroke. Free Radic. Biol. Med. 2013, 65, 1012–1022. [Google Scholar] [CrossRef]
- Patel, M. Targeting Oxidative Stress in Central Nervous System Disorders. Trends Pharm. Sci. 2016, 37, 768–778. [Google Scholar] [CrossRef] [Green Version]
- Salim, S. Oxidative Stress and the Central Nervous System. J. Pharm. Exp. 2017, 360, 201–205. [Google Scholar] [CrossRef] [PubMed]
- Yaribeygi, H.; Panahi, Y.; Javadi, B.; Sahebkar, A. The Underlying Role of Oxidative Stress in Neurodegeneration: A Mechanistic Review. CNS Neurol. Disord. Drug Targets 2018, 17, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Solleiro-Villavicencio, H.; Rivas-Arancibia, S. Effect of Chronic Oxidative Stress on Neuroinflammatory Response Mediated by CD4(+)T Cells in Neurodegenerative Diseases. Front. Cell Neurosci. 2018, 12, 114. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Mao, X.; Liu, A.; Gao, X.; Chen, X.; Ye, M.; Ye, J.; Liu, P.; Xu, S.; Liu, J. Osthole, a natural coumarin improves cognitive impairments and BBB dysfunction after transient global brain ischemia in C57 BL/6J mice: involvement of Nrf2 pathway. Neurochem. Res. 2015, 40, 186–194. [Google Scholar] [CrossRef] [PubMed]
- Imai, T.; Takagi, T.; Kitashoji, A.; Yamauchi, K.; Shimazawa, M.; Hara, H. Nrf2 activator ameliorates hemorrhagic transformation in focal cerebral ischemia under warfarin anticoagulation. Neurobiol. Dis. 2016, 89, 136–146. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Suwanwela, N.C.; Patumraj, S. Curcumin by down-regulating NF-kB and elevating Nrf2, reduces brain edema and neurological dysfunction after cerebral I/R. Microvasc. Res. 2016, 106, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Tarozzi, A.; Angeloni, C.; Malaguti, M.; Morroni, F.; Hrelia, S.; Hrelia, P. Sulforaphane as a potential protective phytochemical against neurodegenerative diseases. Oxidative Med. Cell. Longev. 2013, 2013, 415078. [Google Scholar] [CrossRef] [PubMed]
- Mao, L.; Yang, T.; Li, X.; Lei, X.; Sun, Y.; Zhao, Y.; Zhang, W.; Gao, Y.; Sun, B.; Zhang, F. Protective effects of sulforaphane in experimental vascular cognitive impairment: Contribution of the Nrf2 pathway. J. Cereb. Blood Flow Metab. 2018, 0271678X18764083. [Google Scholar] [CrossRef] [PubMed]
- Soane, L.; Li Dai, W.; Fiskum, G.; Bambrick, L.L. Sulforaphane protects immature hippocampal neurons against death caused by exposure to hemin or to oxygen and glucose deprivation. J. Neurosci. Res. 2010, 88, 1355–1363. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; He, Q.; Zheng, J.; Li, L.Y.; Hou, Y.H.; Song, F.Z. Sulforaphane improves outcomes and slows cerebral ischemic/reperfusion injury via inhibition of NLRP3 inflammasome activation in rats. Int. Immunopharmacol. 2017, 45, 74–78. [Google Scholar] [CrossRef] [PubMed]
- Holloway, P.M.; Gillespie, S.; Becker, F.; Vital, S.A.; Nguyen, V.; Alexander, J.S.; Evans, P.C.; Gavins, F.N. Sulforaphane induces neurovascular protection against a systemic inflammatory challenge via both Nrf2-dependent and independent pathways. Vasc. Pharmacol. 2016, 85, 29–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dinkova-Kostova, A.T.; Kostov, R.V. Glucosinolates and isothiocyanates in health and disease. Trends Mol. Med. 2012, 18, 337–347. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Wen, L.; Dong, M.; Lu, X. Sulforaphane activates the cerebral vascular Nrf2–ARE pathway and suppresses inflammation to attenuate cerebral vasospasm in rat with subarachnoid hemorrhage. Brain Res. 2016, 1653, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Takaya, K.; Suzuki, T.; Motohashi, H.; Onodera, K.; Satomi, S.; Kensler, T.W.; Yamamoto, M. Validation of the multiple sensor mechanism of the Keap1-Nrf2 system. Free Radic. Biol. Med. 2012, 53, 817–827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Noel, J.; Kaplan, H.J.; Dean, D.C. Circulating reactive oxidant causes apoptosis of retinal pigment epithelium and cone photoreceptors in the mouse central retina. Ophthalmol. Eye Dis. 2011, 3, 45–54. [Google Scholar] [CrossRef]
- Muller, T.; Hengstermann, A. Nrf2: friend and foe in preventing cigarette smoking-dependent lung disease. Chem. Res. Toxicol. 2012, 25, 1805–1824. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.; He, X. Molecular basis of electrophilic and oxidative defense: promises and perils of Nrf2. Pharm. Rev. 2012, 64, 1055–1081. [Google Scholar] [CrossRef]
- Kaisar, M.A.; Prasad, S.; Cucullo, L. Protecting the BBB endothelium against cigarette smoke-induced oxidative stress using popular antioxidants: Are they really beneficial? Brain Res. 2015, 1627, 90–100. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.-Y.; Kuan, Y.-H.; Li, J.-R.; Chen, W.-Y.; Ou, Y.-C.; Pan, H.-C.; Liao, S.-L.; Raung, S.-L.; Chang, C.-J.; Chen, C.-J. Docosahexaenoic acid reduces cellular inflammatory response following permanent focal cerebral ischemia in rats. J. Nutr. Biochem. 2013, 24, 2127–2137. [Google Scholar] [CrossRef]
- Ren, J.; Fan, C.; Chen, N.; Huang, J.; Yang, Q. Resveratrol pretreatment attenuates cerebral ischemic injury by upregulating expression of transcription factor Nrf2 and HO-1 in rats. Neurochem. Res. 2011, 36, 2352. [Google Scholar] [CrossRef]
- Zhang, J.; Fu, B.; Zhang, X.; Zhang, L.; Bai, X.; Zhao, X.; Chen, L.; Cui, L.; Zhu, C.; Wang, L. Bicyclol upregulates transcription factor Nrf2, HO-1 expression and protects rat brains against focal ischemia. Brain Res. Bull. 2014, 100, 38–43. [Google Scholar] [CrossRef] [PubMed]
- Cantin, A.M. Cellular response to cigarette smoke and oxidants: adapting to survive. Proc. Am. Thorac. Soc. 2010, 7, 368–375. [Google Scholar] [CrossRef] [PubMed]
- Tuder, R.M.; Petrache, I. Pathogenesis of chronic obstructive pulmonary disease. J. Clin. Investig. 2012, 122, 2749–2755. [Google Scholar] [CrossRef] [PubMed]
- Sivandzade, F.; Cucullo, L. Assessing the protective effect of rosiglitazone against electronic cigarette/tobacco smoke-induced blood–brain barrier impairment. BMC Neurosci. 2019, 20, 15. [Google Scholar] [CrossRef] [PubMed]
- Ward, R.; Li, W.; Abdul, Y.; Jackson, L.; Dong, G.; Jamil, S.; Filosa, J.; Fagan, S.C.; Ergul, A. NLRP3 inflammasome inhibition with MCC950 improves diabetes-mediated cognitive impairment and vasoneuronal remodeling after ischemia. Pharm. Res. 2019, 142, 237–250. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Ma, C.; Meng, C.J.; Zhu, G.Q.; Sun, X.B.; Huo, L.; Zhang, J.; Liu, H.X.; He, W.C.; Shen, X.M. Melatonin activates the Nrf2-ARE pathway when it protects against early brain injury in a subarachnoid hemorrhage model. J. Pineal Res. 2012, 53, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Ashabi, G.; Khalaj, L.; Khodagholi, F.; Goudarzvand, M.; Sarkaki, A. Pre-treatment with metformin activates Nrf2 antioxidant pathways and inhibits inflammatory responses through induction of AMPK after transient global cerebral ischemia. Metab. Brain Dis. 2015, 30, 747–754. [Google Scholar] [CrossRef]
- Liu, Y.; Tang, G.; Li, Y.; Wang, Y.; Chen, X.; Gu, X.; Zhang, Z.; Wang, Y.; Yang, G.Y. Metformin attenuates blood-brain barrier disruption in mice following middle cerebral artery occlusion. J. Neuroinflammation 2014, 11, 177. [Google Scholar] [CrossRef]
- Jin, Q.; Cheng, J.; Liu, Y.; Wu, J.; Wang, X.; Wei, S.; Zhou, X.; Qin, Z.; Jia, J.; Zhen, X. Improvement of functional recovery by chronic metformin treatment is associated with enhanced alternative activation of microglia/macrophages and increased angiogenesis and neurogenesis following experimental stroke. Brain Behav. Immun. 2014, 40, 131–142. [Google Scholar] [CrossRef]
- Ou, Z.; Kong, X.; Sun, X.; He, X.; Zhang, L.; Gong, Z.; Huang, J.; Xu, B.; Long, D.; Li, J.; et al. Metformin treatment prevents amyloid plaque deposition and memory impairment in APP/PS1 mice. Brain Behav. Immun. 2018, 69, 351–363. [Google Scholar] [CrossRef]
- Tanokashira, D.; Kurata, E.; Fukuokaya, W.; Kawabe, K.; Kashiwada, M.; Takeuchi, H.; Nakazato, M.; Taguchi, A. Metformin treatment ameliorates diabetes-associated decline in hippocampal neurogenesis and memory via phosphorylation of insulin receptor substrate 1. FEBS Open Biol. 2018, 8, 1104–1118. [Google Scholar] [CrossRef] [PubMed]
- Pryor, R.; Cabreiro, F. Repurposing metformin: an old drug with new tricks in its binding pockets. Biochem. J. 2015, 471, 307–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ming, M.; Sinnett-Smith, J.; Wang, J.; Soares, H.P.; Young, S.H.; Eibl, G.; Rozengurt, E. Dose-dependent AMPK-dependent and independent mechanisms of berberine and metformin inhibition of mTORC1, ERK, DNA synthesis and proliferation in pancreatic cancer cells. PLoS ONE 2014, 9, e114573. [Google Scholar] [CrossRef] [PubMed]
- Martin-Montalvo, A.; Mercken, E.M.; Mitchell, S.J.; Palacios, H.H.; Mote, P.L.; Scheibye-Knudsen, M.; Gomes, A.P.; Ward, T.M.; Minor, R.K.; Blouin, M.J.; et al. Metformin improves healthspan and lifespan in mice. Nat. Commun. 2013, 4, 2192. [Google Scholar] [CrossRef] [PubMed]
- Malinowski, J.M.; Bolesta, S. Rosiglitazone in the treatment of type 2 diabetes mellitus: A critical review. Clin 2000, 22, 1151–1168, discussion 1149–1150. [Google Scholar] [CrossRef]
- Ceolotto, G.; Gallo, A.; Papparella, I.; Franco, L.; Murphy, E.; Iori, E.; Pagnin, E.; Fadini, G.P.; Albiero, M.; Semplicini, A. Rosiglitazone reduces glucose-induced oxidative stress mediated by NAD (P) H oxidase via AMPK-dependent mechanism. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 2627–2633. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, Z.; Liu, J.Z.; Hu, J.X.; Chen, H.L.; Li, W.L.; Hai, C.X. Double antioxidant activities of rosiglitazone against high glucose-induced oxidative stress in hepatocyte. Toxicol. Vitr. 2011, 25, 839–847. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.E.; Park, J.H.; Jang, S.J.; Koh, H.C. Rosiglitazone inhibits chlorpyrifos-induced apoptosis via modulation of the oxidative stress and inflammatory response in SH-SY5Y cells. Toxicol. Appl. Pharmacol. 2014, 278, 159–171. [Google Scholar] [CrossRef]
- Markowicz-Piasecka, M.; Sikora, J.; Szydłowska, A.; Skupień, A.; Mikiciuk-Olasik, E.; Huttunen, K.M. Metformin–a future therapy for neurodegenerative diseases. Pharm. Res. 2017, 34, 2614–2627. [Google Scholar] [CrossRef]
- Sayan-Ozacmak, H.; Ozacmak, V.H.; Barut, F.; Jakubowska-Dogru, E. Rosiglitazone treatment reduces hippocampal neuronal damage possibly through alleviating oxidative stress in chronic cerebral hypoperfusion. Neurochem. Int. 2012, 61, 287–290. [Google Scholar] [CrossRef]
- Kadam, L.; Gomez-Lopez, N.; Mial, T.N.; Kohan-Ghadr, H.R.; Drewlo, S. Rosiglitazone Regulates TLR4 and Rescues HO-1 and NRF2 Expression in Myometrial and Decidual Macrophages in Inflammation-Induced Preterm Birth. Reprod. Sci. 2017, 24, 1590–1599. [Google Scholar] [CrossRef] [PubMed]
- Soares, M.P.; Seldon, M.P.; Gregoire, I.P.; Vassilevskaia, T.; Berberat, P.O.; Yu, J.; Tsui, T.Y.; Bach, F.H. Heme oxygenase-1 modulates the expression of adhesion molecules associated with endothelial cell activation. J. Immunol. 2004, 172, 3553–3563. [Google Scholar] [CrossRef] [PubMed]
- Kansanen, E.; Kuosmanen, S.M.; Leinonen, H.; Levonen, A.L. The Keap1-Nrf2 pathway: Mechanisms of activation and dysregulation in cancer. Redox Biol. 2013, 1, 45–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Figure 1.
Schematic representation of the NRF2–NF-kB cross talk with respect to oxidative stress and inflammatory stimuli. Note that an “up arrow” indicates upregulation while a “down arrow” indicates downregulation.
Figure 1.
Schematic representation of the NRF2–NF-kB cross talk with respect to oxidative stress and inflammatory stimuli. Note that an “up arrow” indicates upregulation while a “down arrow” indicates downregulation.

Figure 2.
Schematic representation of the Cerebrovascular and neurodegenerative diseases associated with impaired redox metabolism and oxidative stress.
Figure 2.
Schematic representation of the Cerebrovascular and neurodegenerative diseases associated with impaired redox metabolism and oxidative stress.

Figure 3.
Schematic illustration of the impact of smoking on BBB impairment and the onset of cerebrovascular and CNS disorders. Note that an “up arrow” indicates upregulation while a “down arrow” indicates downregulation.
Figure 3.
Schematic illustration of the impact of smoking on BBB impairment and the onset of cerebrovascular and CNS disorders. Note that an “up arrow” indicates upregulation while a “down arrow” indicates downregulation.

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Sivandzade, F.; Bhalerao, A.; Cucullo, L. Cerebrovascular and Neurological Disorders: Protective Role of NRF2. Int. J. Mol. Sci. 2019, 20, 3433. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20143433
AMA Style
Sivandzade F, Bhalerao A, Cucullo L. Cerebrovascular and Neurological Disorders: Protective Role of NRF2. International Journal of Molecular Sciences. 2019; 20(14):3433. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20143433
Chicago/Turabian StyleSivandzade, Farzane, Aditya Bhalerao, and Luca Cucullo. 2019. "Cerebrovascular and Neurological Disorders: Protective Role of NRF2" International Journal of Molecular Sciences 20, no. 14: 3433. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20143433
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.