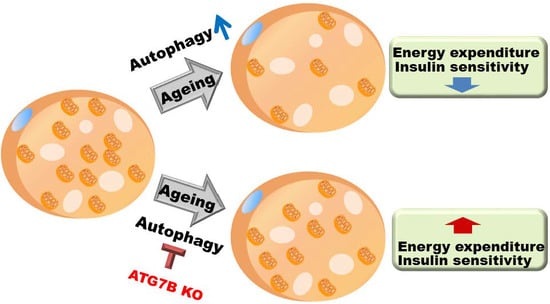
Suppression of Brown Adipocyte Autophagy Improves Energy Metabolism by Regulating Mitochondrial Turnover

 , ,
, ,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
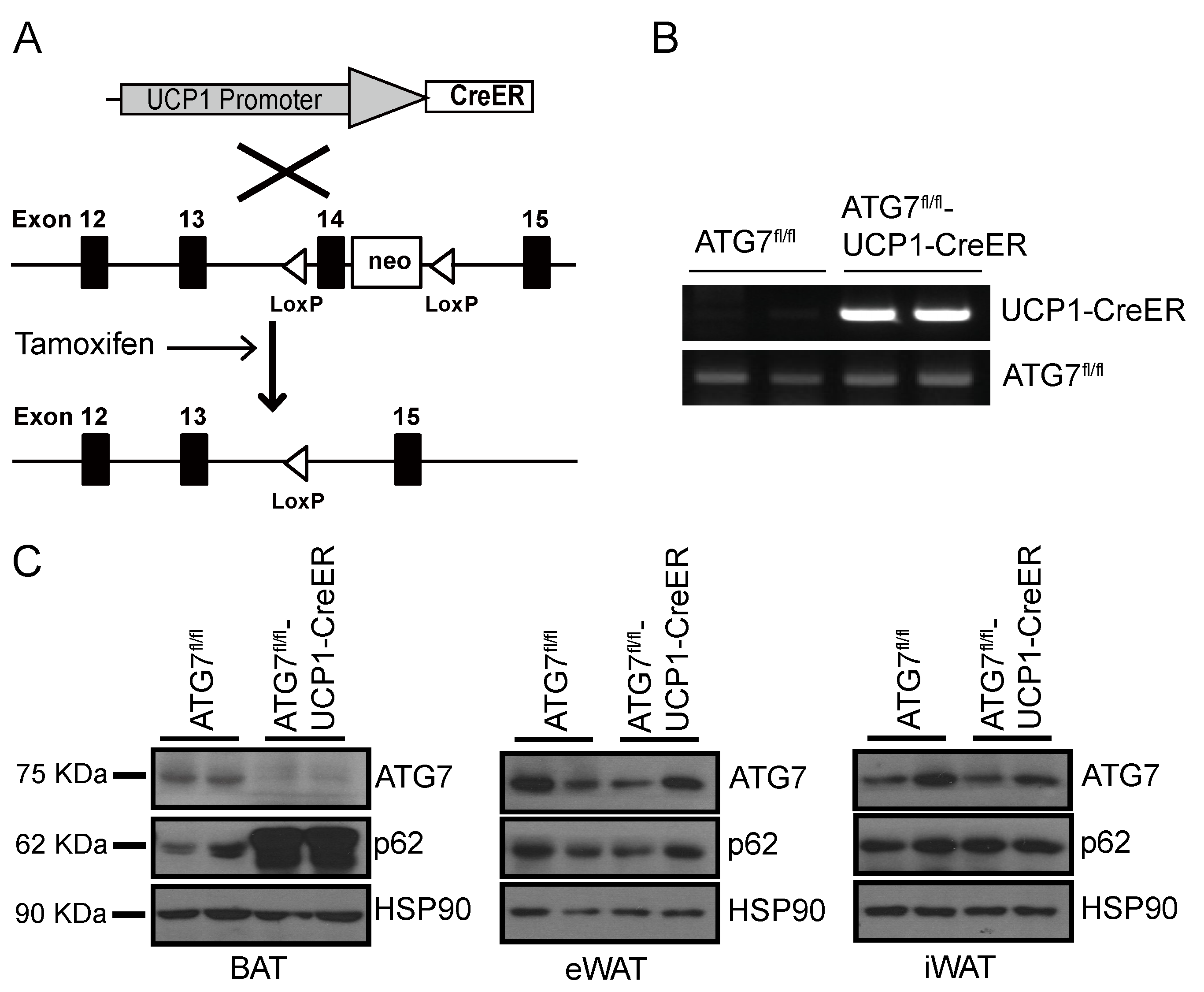
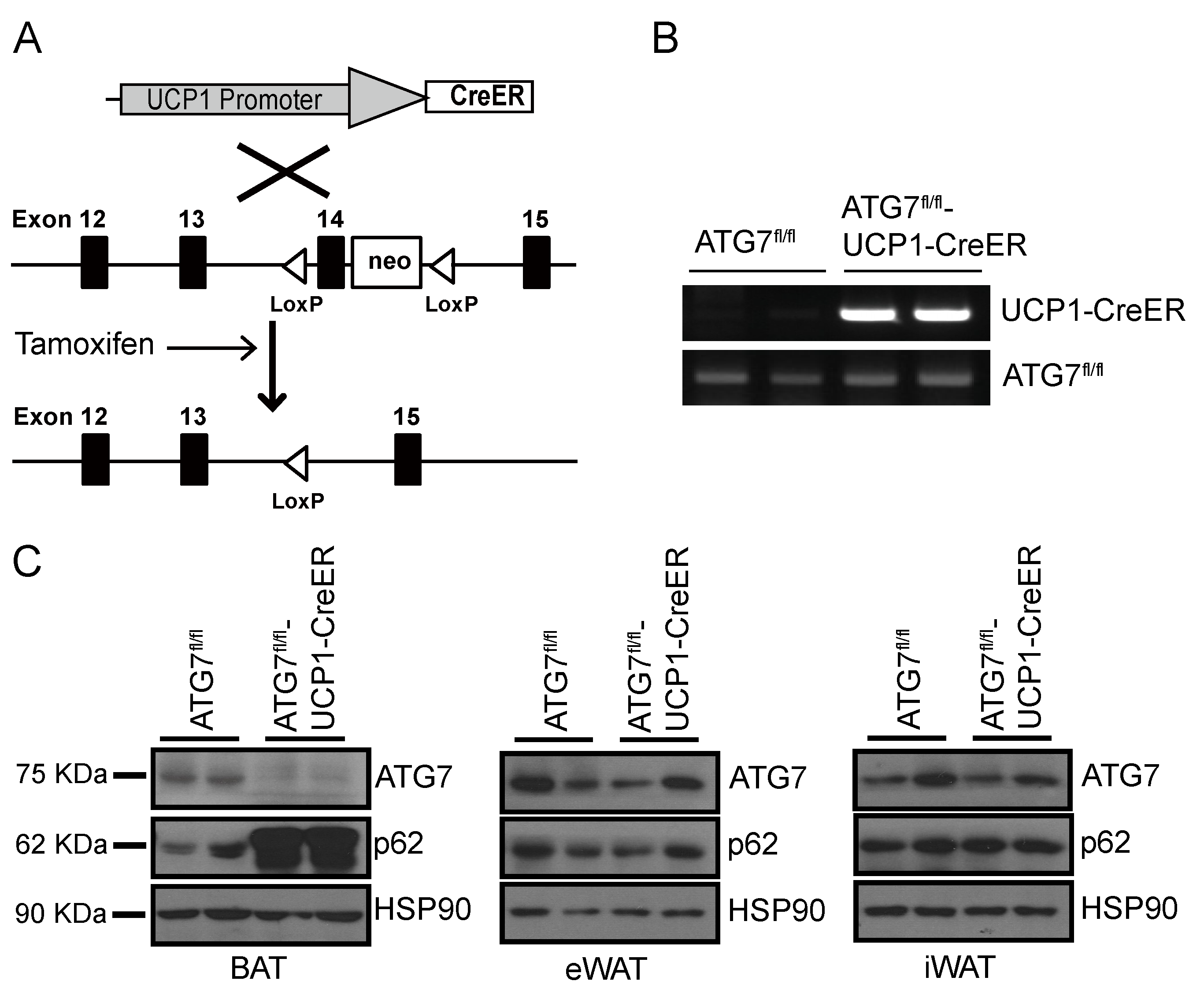
2.1. Generation of Brown Adipocyte-Specific Atg7 Knockout Mice
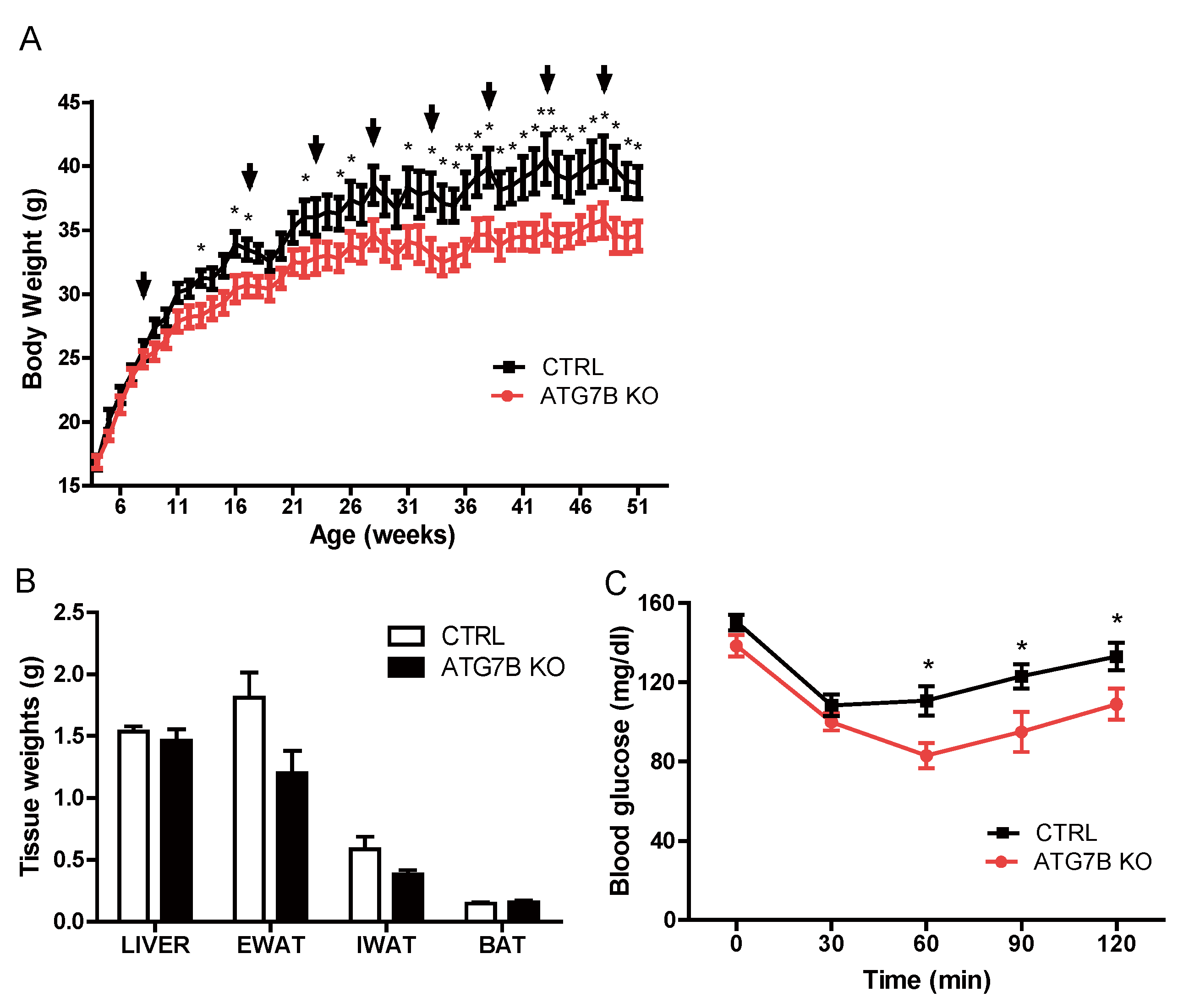
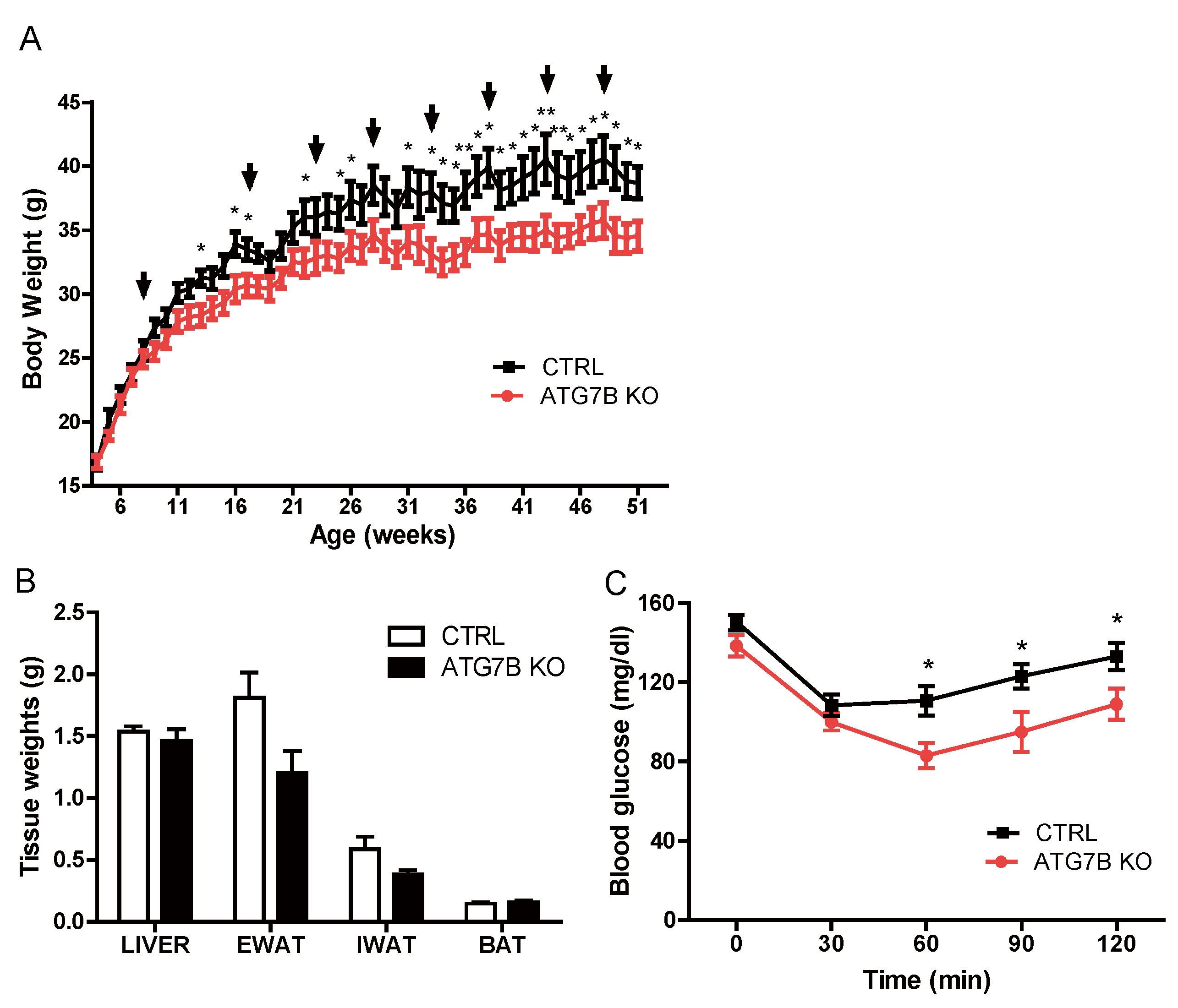
2.2. Suppression of BAT Autophagy Reduces Body Weight and Improves Glucose Metabolism
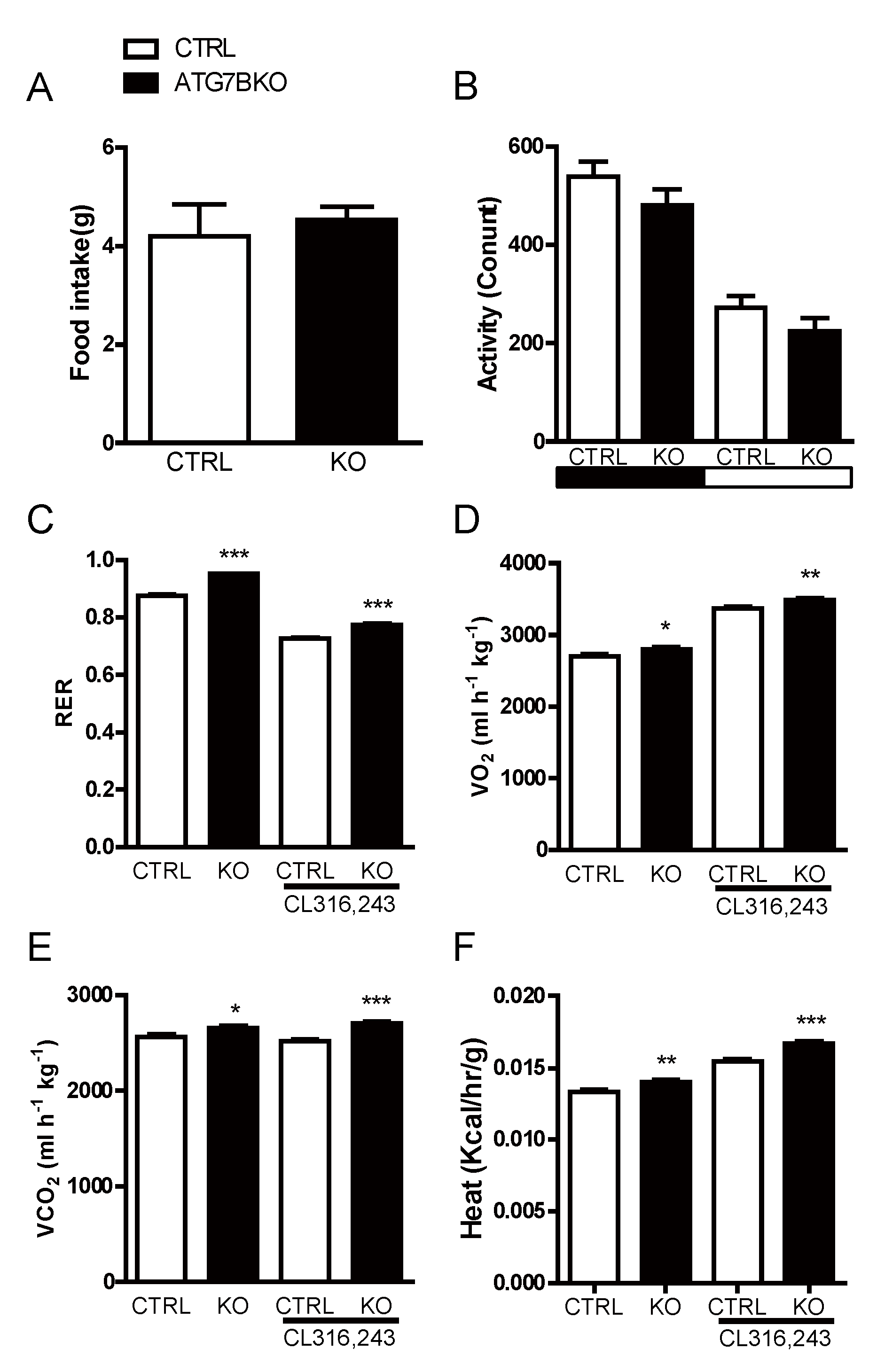
2.3. Body Weight Reduction in ATG7B KO Mice Is Due to Enhanced Energy Expenditure
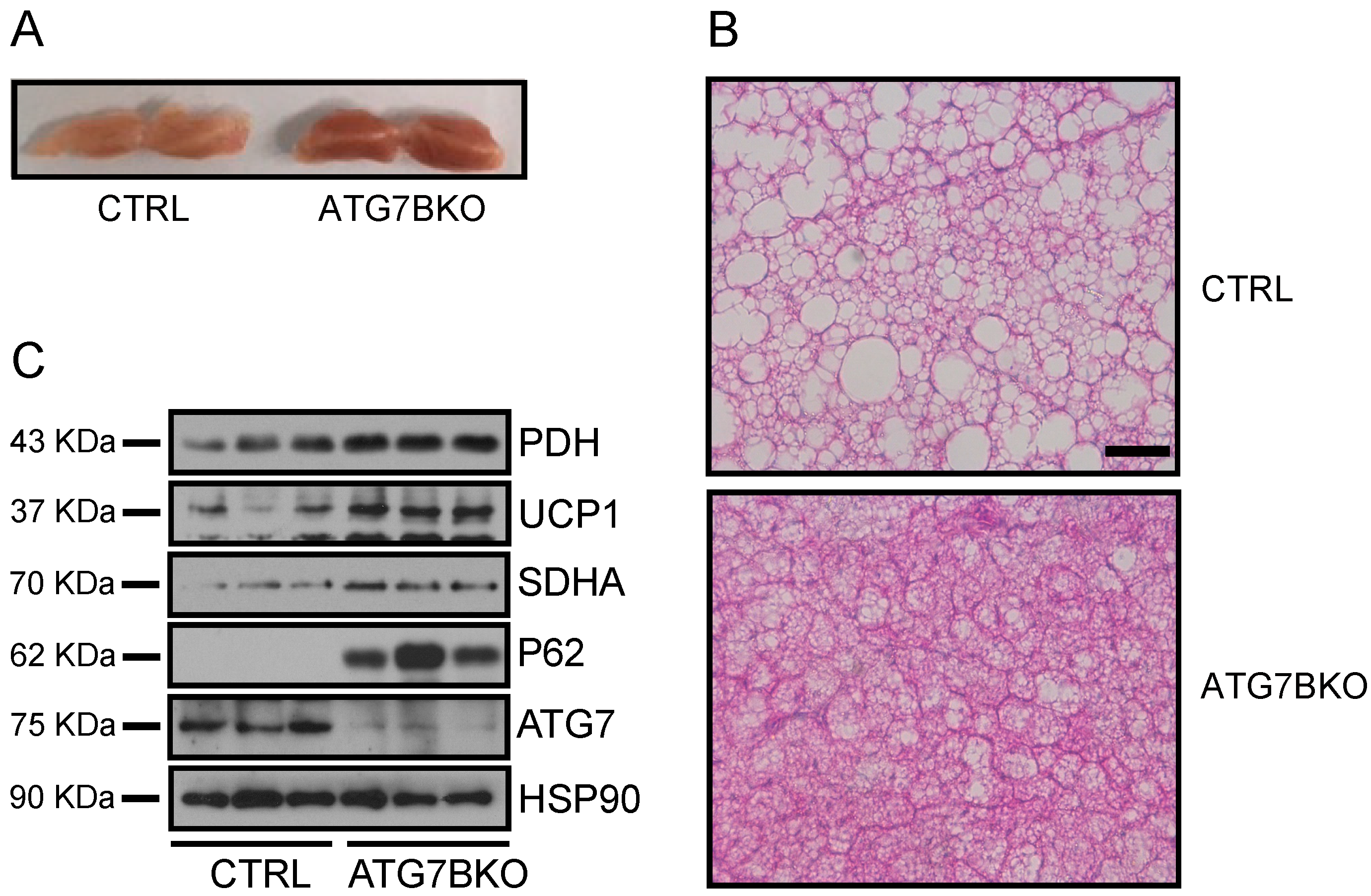
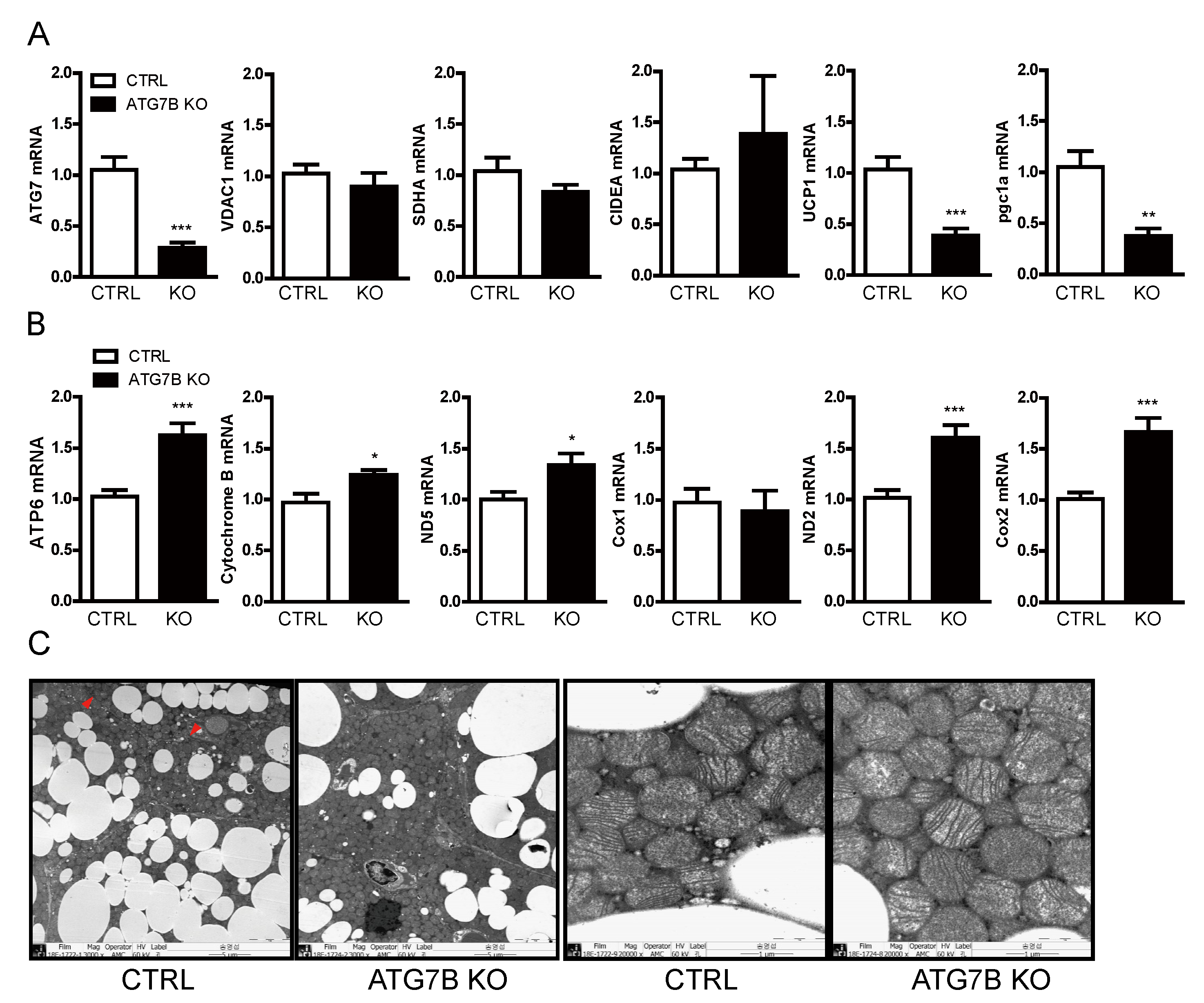
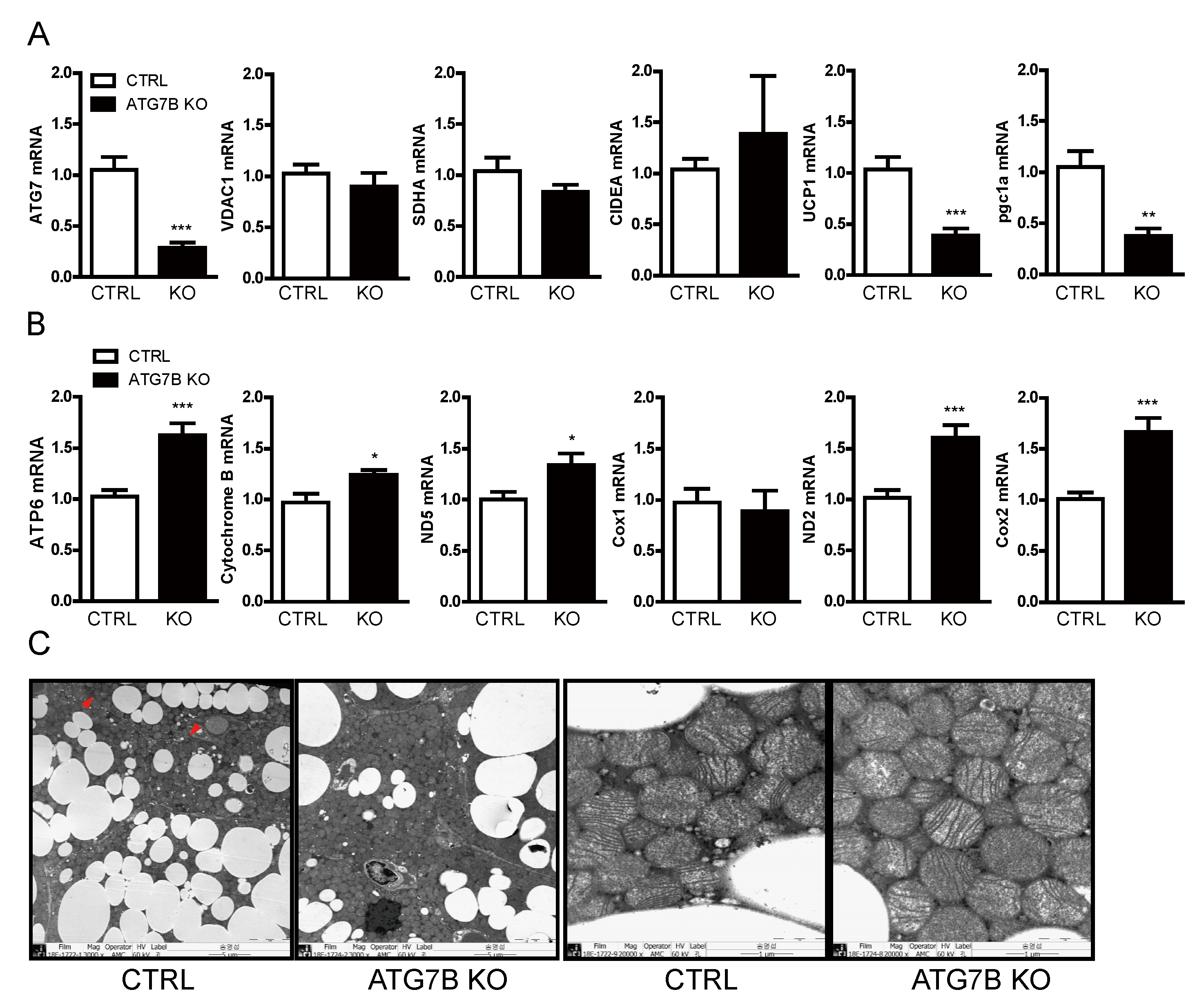
2.4. Increased Mitochondrial Content in BAT of ATG7B KO Mice
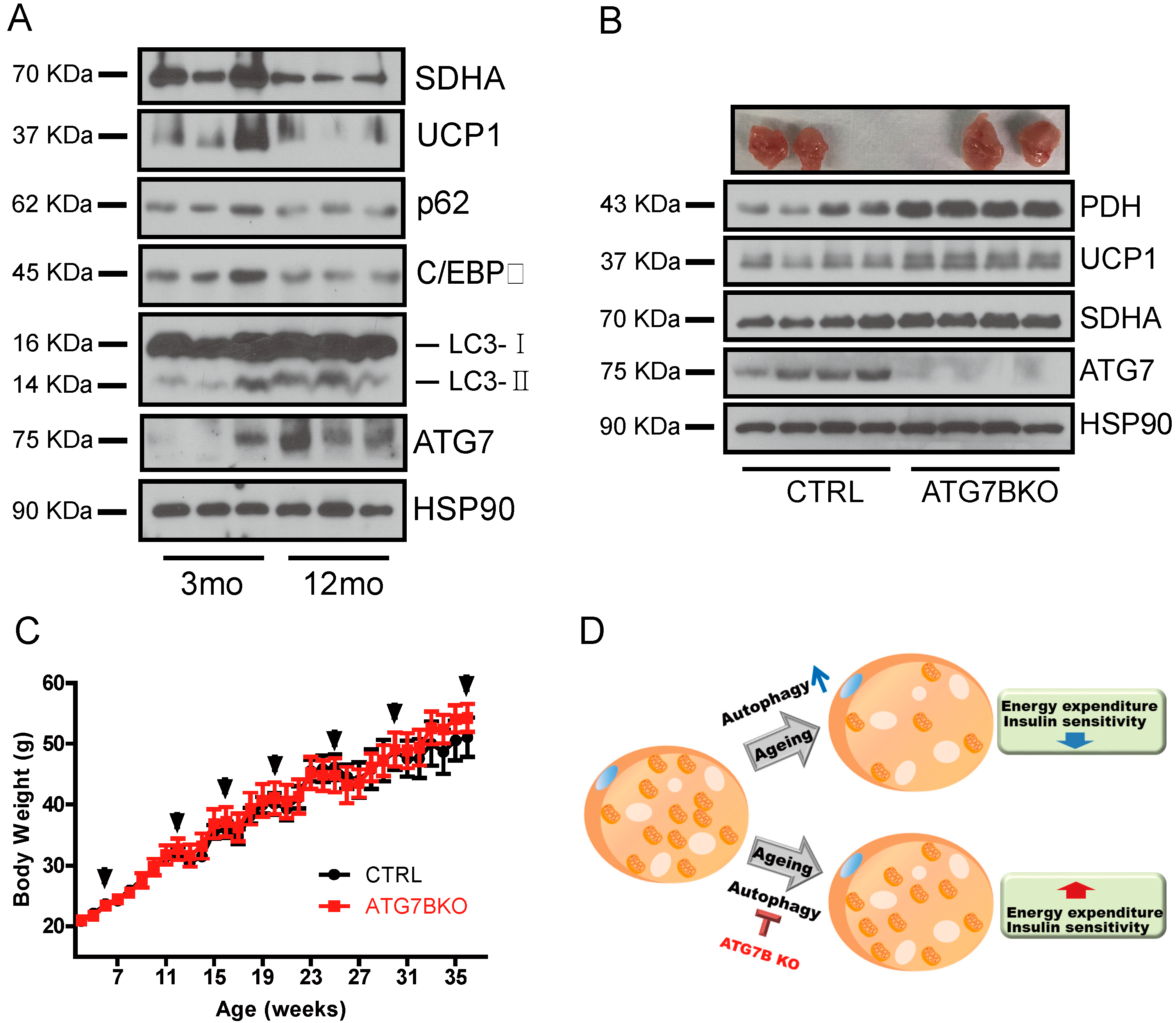
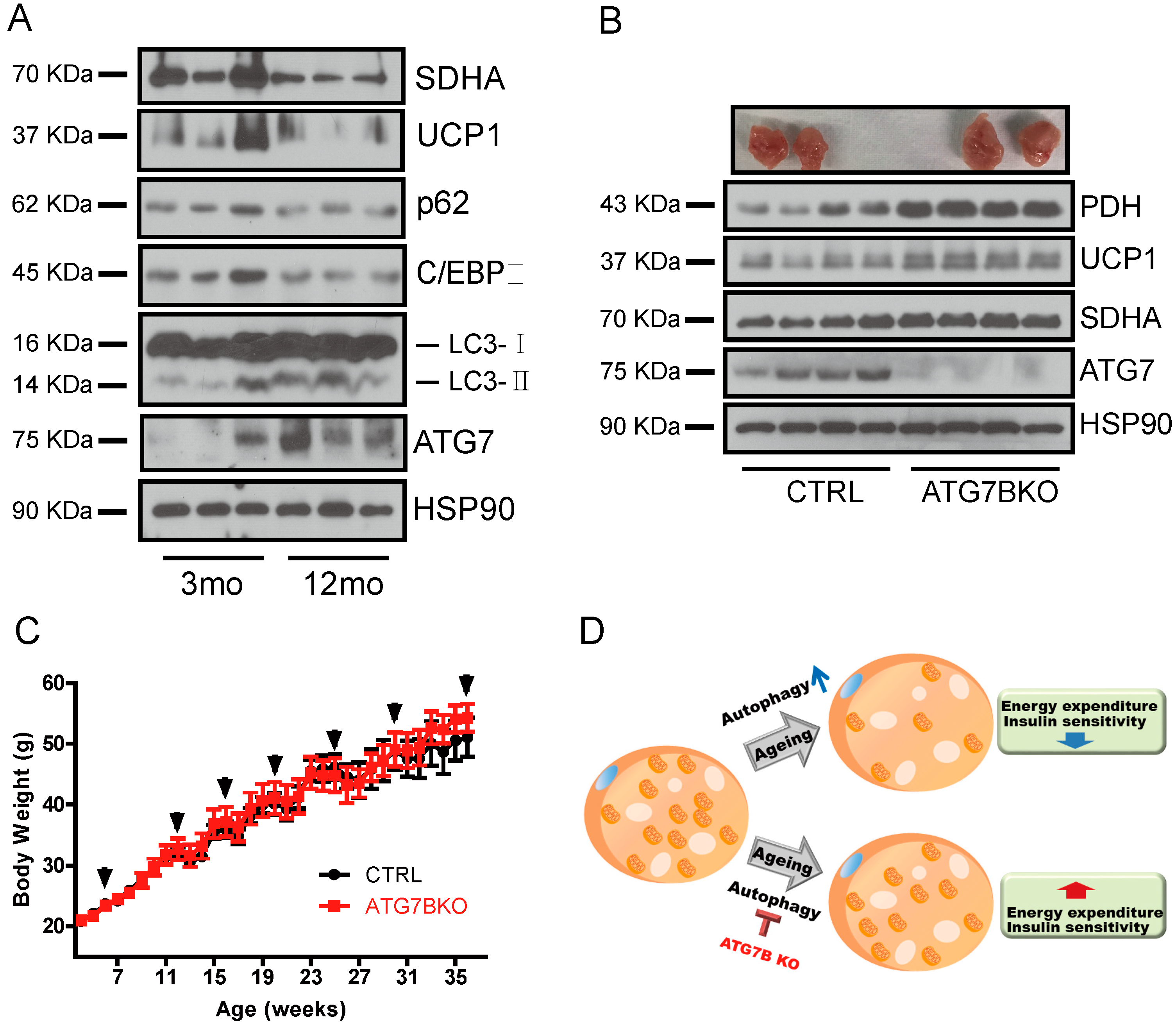
2.5. Age-Associated Increase in Brown Adipocyte Autophagy
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Indirect Mouse Calorimetry Study
4.3. Insulin Tolerance Test
4.4. Histology
4.5. mRNA Analysis
4.6. Antibodies and Immunoblots
4.7. Statistics
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| BAT | brown adipose tissue |
| eWAT | epididymal white adipose tissue |
| iWAT | inguinal white adipose tissue |
| UCP1 | uncoupling protein 1 |
| Atg7 | autophagy related 7 |
| RER | respiratory exchange ratio |
| KO | knock out |
References
- Cannon, B.; Nedergaard, J. Brown adipose tissue: Function and physiological significance. Physiol. Rev. 2004, 84, 277–359. [Google Scholar] [CrossRef]
- Smith, R.E.; Roberts, J.C.; Hittelman, K.J. Nonphosphorylating respiration of mitochondria from brown adipose tissue of rats. Science 1966, 154, 653–654. [Google Scholar] [CrossRef] [PubMed]
- Aquila, H.; Link, T.A.; Klingenberg, M. The uncoupling protein from brown fat mitochondria is related to the mitochondrial ADP/ATP carrier. Analysis of sequence homologies and of folding of the protein in the membrane. EMBO J. 1985, 4, 2369–2376. [Google Scholar] [CrossRef] [PubMed]
- Echtay, K.S.; Bienengraeber, M.; Mayinger, P.; Heimpel, S.; Winkler, E.; Druhmann, D.; Frischmuth, K.; Kamp, F.; Huang, S.G. Uncoupling proteins: Martin Klingenberg’s contributions for 40 years. Arch. Biochem. Biophys. 2018, 657, 41–55. [Google Scholar] [CrossRef]
- Chouchani, E.T.; Kazak, L.; Spiegelman, B.M. New Advances in Adaptive Thermogenesis: UCP1 and Beyond. Cell Metab. 2019, 29, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.W.; Nadeau, B.E.; Foster, D.O. A nonocclusive cannula for sampling venous blood from the interscapular brown fat of conscious rats. Can. J. Physiol. Pharmacol. 1986, 64, 1434–1439. [Google Scholar] [CrossRef] [PubMed]
- Cinti, S. Between brown and white: Novel aspects of adipocyte differentiation. Ann. Med. 2011, 43, 104–115. [Google Scholar] [CrossRef]
- Cao, Y. Angiogenesis and vascular functions in modulation of obesity, adipose metabolism, and insulin sensitivity. Cell Metab. 2013, 18, 478–489. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Klionsky, D.J. Development by self-digestion: Molecular mechanisms and biological functions of autophagy. Dev. Cell 2004, 6, 463–477. [Google Scholar] [CrossRef]
- Singh, R.; Kaushik, S.; Wang, Y.; Xiang, Y.; Novak, I.; Komatsu, M.; Tanaka, K.; Cuervo, A.M.; Czaja, M.J. Autophagy regulates lipid metabolism. Nature 2009, 458, 1131–1135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of cells and tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, S.; Rodriguez-Navarro, J.A.; Arias, E.; Kiffin, R.; Sahu, S.; Schwartz, G.J.; Cuervo, A.M.; Singh, R. Autophagy in hypothalamic AgRP neurons regulates food intake and energy balance. Cell Metab. 2011, 14, 173–183. [Google Scholar] [CrossRef] [PubMed]
- Quan, W.; Kim, H.K.; Moon, E.Y.; Kim, S.S.; Choi, C.S.; Komatsu, M.; Jeong, Y.T.; Lee, M.K.; Kim, K.W.; Kim, M.S.; et al. Role of hypothalamic proopiomelanocortin neuron autophagy in the control of appetite and leptin response. Endocrinology 2012, 153, 1817–1826. [Google Scholar] [CrossRef] [PubMed]
- Coupe, B.; Ishii, Y.; Dietrich, M.O.; Komatsu, M.; Horvath, T.L.; Bouret, S.G. Loss of autophagy in pro-opiomelanocortin neurons perturbs axon growth and causes metabolic dysregulation. Cell Metab. 2012, 15, 247–255. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Li, P.; Fu, S.; Calay, E.S.; Hotamisligil, G.S. Defective hepatic autophagy in obesity promotes ER stress and causes insulin resistance. Cell Metab. 2010, 11, 467–478. [Google Scholar] [CrossRef] [PubMed]
- Ebato, C.; Uchida, T.; Arakawa, M.; Komatsu, M.; Ueno, T.; Komiya, K.; Azuma, K.; Hirose, T.; Tanaka, K.; Kominami, E.; et al. Autophagy is important in islet homeostasis and compensatory increase of beta cell mass in response to high-fat diet. Cell Metab. 2008, 8, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Fujitani, Y.; Kawamori, R.; Watada, H. The role of autophagy in pancreatic beta-cell and diabetes. Autophagy 2009, 5, 280–282. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.H.; Jeong, Y.T.; Oh, H.; Kim, S.H.; Cho, J.M.; Kim, Y.N.; Kim, S.S.; Kim do, H.; Hur, K.Y.; Kim, H.K.; et al. Autophagy deficiency leads to protection from obesity and insulin resistance by inducing Fgf21 as a mitokine. Nat. Med. 2013, 19, 83–92. [Google Scholar] [CrossRef]
- Martinez-Lopez, N.; Athonvarangkul, D.; Sahu, S.; Coletto, L.; Zong, H.; Bastie, C.C.; Pessin, J.E.; Schwartz, G.J.; Singh, R. Autophagy in Myf5+ progenitors regulates energy and glucose homeostasis through control of brown fat and skeletal muscle development. EMBO Rep. 2013, 14, 795–803. [Google Scholar] [CrossRef]
- Komatsu, M.; Waguri, S.; Chiba, T.; Murata, S.; Iwata, J.; Tanida, I.; Ueno, T.; Koike, M.; Uchiyama, Y.; Kominami, E.; et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 2006, 441, 880–884. [Google Scholar] [CrossRef]
- Rosenwald, M.; Perdikari, A.; Rulicke, T.; Wolfrum, C. Bi-directional interconversion of brite and white adipocytes. Nat. Cell Biol. 2013, 15, 659–667. [Google Scholar] [CrossRef] [PubMed]
- Rosenwald, M.; Wolfrum, C. The origin and definition of brite versus white and classical brown adipocytes. Adipocyte 2014, 3, 4–9. [Google Scholar] [CrossRef] [PubMed]
- Joplin, R.; Wallace, L.L.; Johnson, G.D.; Lindsay, J.G.; Yeaman, S.J.; Palmer, J.M.; Strain, A.J.; Neuberger, J.M. Subcellular localization of pyruvate dehydrogenase dihydrolipoamide acetyltransferase in human intrahepatic biliary epithelial cells. J. Pathol. 1995, 176, 381–390. [Google Scholar] [CrossRef] [PubMed]
- Jezek, P.; Jaburek, M.; Porter, R.K. Uncoupling mechanism and redox regulation of mitochondrial uncoupling protein 1 (UCP1). Biochim. Biophys. Acta Bioenerg. 2019, 1860, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Puigserver, P.; Andersson, U.; Zhang, C.; Adelmant, G.; Mootha, V.; Troy, A.; Cinti, S.; Lowell, B.; Scarpulla, R.C.; et al. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell 1999, 98, 115–124. [Google Scholar] [CrossRef]
- Cypess, A.M.; Lehman, S.; Williams, G.; Tal, I.; Rodman, D.; Goldfine, A.B.; Kuo, F.C.; Palmer, E.L.; Tseng, Y.H.; Doria, A.; et al. Identification and importance of brown adipose tissue in adult humans. N. Engl. J. Med. 2009, 360, 1509–1517. [Google Scholar] [CrossRef] [PubMed]
- Ouellet, V.; Routhier-Labadie, A.; Bellemare, W.; Lakhal-Chaieb, L.; Turcotte, E.; Carpentier, A.C.; Richard, D. Outdoor temperature, age, sex, body mass index, and diabetic status determine the prevalence, mass, and glucose-uptake activity of 18F-FDG-detected BAT in humans. J. Clin. Endocrinol. Metab. 2011, 96, 192–199. [Google Scholar] [CrossRef] [PubMed]
- Kopecky, J.; Clarke, G.; Enerback, S.; Spiegelman, B.; Kozak, L.P. Expression of the mitochondrial uncoupling protein gene from the aP2 gene promoter prevents genetic obesity. J. Clin. Investig. 1995, 96, 2914–2923. [Google Scholar] [CrossRef] [PubMed]
- Stefl, B.; Janovska, A.; Hodny, Z.; Rossmeisl, M.; Horakova, M.; Syrovy, I.; Bemova, J.; Bendlova, B.; Kopecky, J. Brown fat is essential for cold-induced thermogenesis but not for obesity resistance in aP2-Ucp mice. Am. J. Physiol. 1998, 274, E527–E533. [Google Scholar] [CrossRef]
- Mantzoros, C.S.; Frederich, R.C.; Qu, D.; Lowell, B.B.; Maratos-Flier, E.; Flier, J.S. Severe leptin resistance in brown fat-deficient uncoupling protein promoter-driven diphtheria toxin A mice despite suppression of hypothalamic neuropeptide Y and circulating corticosterone concentrations. Diabetes 1998, 47, 230–238. [Google Scholar] [CrossRef]
- Hamann, A.; Flier, J.S.; Lowell, B.B. Obesity after genetic ablation of brown adipose tissue. Z. Ernahrungswiss 1998, 37, 1–7. [Google Scholar] [PubMed]
- Nedergaard, J.; Bengtsson, T.; Cannon, B. Unexpected evidence for active brown adipose tissue in adult humans. Am. J. Physiol. Endocrinol. Metab. 2007, 293, E444–E452. [Google Scholar] [CrossRef] [PubMed]
- van Marken Lichtenbelt, W.D.; Vanhommerig, J.W.; Smulders, N.M.; Drossaerts, J.M.; Kemerink, G.J.; Bouvy, N.D.; Schrauwen, P.; Teule, G.J. Cold-activated brown adipose tissue in healthy men. N. Engl. J. Med. 2009, 360, 1500–1508. [Google Scholar] [CrossRef] [PubMed]
- Saito, M.; Okamatsu-Ogura, Y.; Matsushita, M.; Watanabe, K.; Yoneshiro, T.; Nio-Kobayashi, J.; Iwanaga, T.; Miyagawa, M.; Kameya, T.; Nakada, K.; et al. High incidence of metabolically active brown adipose tissue in healthy adult humans: Effects of cold exposure and adiposity. Diabetes 2009, 58, 1526–1531. [Google Scholar] [CrossRef]
- Virtanen, K.A.; Lidell, M.E.; Orava, J.; Heglind, M.; Westergren, R.; Niemi, T.; Taittonen, M.; Laine, J.; Savisto, N.J.; Enerback, S.; et al. Functional brown adipose tissue in healthy adults. N. Engl. J. Med. 2009, 360, 1518–1525. [Google Scholar] [CrossRef] [PubMed]
- Vijgen, G.H.; Bouvy, N.D.; Teule, G.J.; Brans, B.; Schrauwen, P.; van Marken Lichtenbelt, W.D. Brown adipose tissue in morbidly obese subjects. PLoS ONE 2011, 6, e17247. [Google Scholar] [CrossRef]
- Richard, D.; Picard, F. Brown fat biology and thermogenesis. Front. Biosci. (Landmark Ed) 2011, 16, 1233–1260. [Google Scholar] [CrossRef]
- Puigserver, P.; Wu, Z.; Park, C.W.; Graves, R.; Wright, M.; Spiegelman, B.M. A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell 1998, 92, 829–839. [Google Scholar] [CrossRef]
- Kelly, D.P.; Scarpulla, R.C. Transcriptional regulatory circuits controlling mitochondrial biogenesis and function. Genes Dev. 2004, 18, 357–368. [Google Scholar] [CrossRef] [Green Version]
- Kleiner, S.; Mepani, R.J.; Laznik, D.; Ye, L.; Jurczak, M.J.; Jornayvaz, F.R.; Estall, J.L.; Chatterjee Bhowmick, D.; Shulman, G.I.; Spiegelman, B.M. Development of insulin resistance in mice lacking PGC-1alpha in adipose tissues. Proc. Natl. Acad. Sci. USA 2012, 109, 9635–9640. [Google Scholar] [CrossRef]
- Vernochet, C.; Damilano, F.; Mourier, A.; Bezy, O.; Mori, M.A.; Smyth, G.; Rosenzweig, A.; Larsson, N.G.; Kahn, C.R. Adipose tissue mitochondrial dysfunction triggers a lipodystrophic syndrome with insulin resistance, hepatosteatosis, and cardiovascular complications. FASEB J. 2014, 28, 4408–4419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vives-Bauza, C.; Zhou, C.; Huang, Y.; Cui, M.; de Vries, R.L.; Kim, J.; May, J.; Tocilescu, M.A.; Liu, W.; Ko, H.S.; et al. PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc. Natl. Acad. Sci. USA 2010, 107, 378–383. [Google Scholar] [CrossRef] [PubMed]
- Vives-Bauza, C.; Przedborski, S. Mitophagy: The latest problem for Parkinson’s disease. Trends Mol. Med. 2011, 17, 158–165. [Google Scholar] [CrossRef] [PubMed]
- Ashrafi, G.; Schwarz, T.L. The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ. 2013, 20, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Masiero, E.; Agatea, L.; Mammucari, C.; Blaauw, B.; Loro, E.; Komatsu, M.; Metzger, D.; Reggiani, C.; Schiaffino, S.; Sandri, M. Autophagy is required to maintain muscle mass. Cell Metab. 2009, 10, 507–515. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.H.; Cho, M.H.; Kim, J.Y.; Kwon, M.S.; Peak, J.J.; Kang, S.W.; Yoon, S.Y.; Song, Y. Impaired macrophage autophagy induces systemic insulin resistance in obesity. Oncotarget 2016, 7, 35577–35591. [Google Scholar] [CrossRef]
- Kwon, M.; Eom, J.; Kim, D.; Kim, J.; Heredia, J.; Kang, S.W.; Song, Y. Skeletal Muscle Tissue Trib3 Links Obesity with Insulin Resistance by Autophagic Degradation of AKT2. Cell Physiol. Biochem. 2018, 48, 1543–1555. [Google Scholar] [CrossRef]
- Altshuler-Keylin, S.; Kajimura, S. Mitochondrial homeostasis in adipose tissue remodeling. Sci. Signal. 2017, 10, eaai9248. [Google Scholar] [CrossRef] [Green Version]
- Short, K.R.; Bigelow, M.L.; Kahl, J.; Singh, R.; Coenen-Schimke, J.; Raghavakaimal, S.; Nair, K.S. Decline in skeletal muscle mitochondrial function with aging in humans. Proc. Natl. Acad. Sci. USA 2005, 102, 5618–5623. [Google Scholar] [CrossRef] [Green Version]
- Tate, E.L.; Herbener, G.H. A morphometric study of the density of mitochondrial cristae in heart and liver of aging mice. J. Gerontol. 1976, 31, 129–134. [Google Scholar] [CrossRef]
- Yoneshiro, T.; Aita, S.; Matsushita, M.; Okamatsu-Ogura, Y.; Kameya, T.; Kawai, Y.; Miyagawa, M.; Tsujisaki, M.; Saito, M. Age-related decrease in cold-activated brown adipose tissue and accumulation of body fat in healthy humans. Obesity (Silver Spring) 2011, 19, 1755–1760. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, H.; Sato, Y.; Mori, N. Difference in induction of uncoupling protein genes in adipose tissues between young and old rats during cold exposure. FEBS Lett. 1999, 458, 157–161. [Google Scholar] [CrossRef]
- McDonald, R.B.; Horwitz, B.A. Brown adipose tissue thermogenesis during aging and senescence. J. Bioenerg. Biomembr. 1999, 31, 507–516. [Google Scholar] [CrossRef] [PubMed]
- Ost, A.; Svensson, K.; Ruishalme, I.; Brannmark, C.; Franck, N.; Krook, H.; Sandstrom, P.; Kjolhede, P.; Stralfors, P. Attenuated mTOR signaling and enhanced autophagy in adipocytes from obese patients with type 2 diabetes. Mol. Med. 2010, 16, 235–246. [Google Scholar] [CrossRef] [PubMed]
- Kovsan, J.; Bluher, M.; Tarnovscki, T.; Kloting, N.; Kirshtein, B.; Madar, L.; Shai, I.; Golan, R.; Harman-Boehm, I.; Schon, M.R.; et al. Altered autophagy in human adipose tissues in obesity. J. Clin. Endocrinol. Metab. 2011, 96, E268–E277. [Google Scholar] [CrossRef] [PubMed]
- Jansen, H.J.; van Essen, P.; Koenen, T.; Joosten, L.A.; Netea, M.G.; Tack, C.J.; Stienstra, R. Autophagy activity is up-regulated in adipose tissue of obese individuals and modulates proinflammatory cytokine expression. Endocrinology 2012, 153, 5866–5874. [Google Scholar] [CrossRef]
- Kosacka, J.; Kern, M.; Kloting, N.; Paeschke, S.; Rudich, A.; Haim, Y.; Gericke, M.; Serke, H.; Stumvoll, M.; Bechmann, I.; et al. Autophagy in adipose tissue of patients with obesity and type 2 diabetes. Mol. Cell. Endocrinol. 2015, 409, 21–32. [Google Scholar] [CrossRef]
- Leu, S.Y.; Tsai, Y.C.; Chen, W.C.; Hsu, C.H.; Lee, Y.M.; Cheng, P.Y. Raspberry ketone induces brown-like adipocyte formation through suppression of autophagy in adipocytes and adipose tissue. J. Nutr. Biochem. 2018, 56, 116–125. [Google Scholar] [CrossRef]
- Zhang, Y.; Goldman, S.; Baerga, R.; Zhao, Y.; Komatsu, M.; Jin, S. Adipose-specific deletion of autophagy-related gene 7 (atg7) in mice reveals a role in adipogenesis. Proc. Natl. Acad. Sci. USA 2009, 106, 19860–19865. [Google Scholar] [CrossRef]
- Shimizu, I.; Aprahamian, T.; Kikuchi, R.; Shimizu, A.; Papanicolaou, K.N.; MacLauchlan, S.; Maruyama, S.; Walsh, K. Vascular rarefaction mediates whitening of brown fat in obesity. J. Clin. Investig. 2014, 124, 2099–2112. [Google Scholar] [CrossRef] [Green Version]
- Altshuler-Keylin, S.; Shinoda, K.; Hasegawa, Y.; Ikeda, K.; Hong, H.; Kang, Q.; Yang, Y.; Perera, R.M.; Debnath, J.; Kajimura, S. Beige Adipocyte Maintenance Is Regulated by Autophagy-Induced Mitochondrial Clearance. Cell Metab. 2016, 24, 402–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wikstrom, J.D.; Mahdaviani, K.; Liesa, M.; Sereda, S.B.; Si, Y.; Las, G.; Twig, G.; Petrovic, N.; Zingaretti, C.; Graham, A.; et al. Hormone-induced mitochondrial fission is utilized by brown adipocytes as an amplification pathway for energy expenditure. EMBO J. 2014, 33, 418–436. [Google Scholar] [CrossRef] [PubMed]
- Cairo, M.; Villarroya, J.; Cereijo, R.; Campderros, L.; Giralt, M.; Villarroya, F. Thermogenic activation represses autophagy in brown adipose tissue. Int. J. Obes. (Lond.) 2016, 40, 1591–1599. [Google Scholar] [CrossRef] [PubMed]
- Shu, J.; Matarese, A.; Santulli, G. Diabetes, body fat, skeletal muscle, and hypertension: The ominous chiasmus? J. Clin. Hypertens. (Greenwich) 2019, 21, 239–242. [Google Scholar] [CrossRef] [PubMed]
- Ruan, C.C.; Kong, L.R.; Chen, X.H.; Ma, Y.; Pan, X.X.; Zhang, Z.B.; Gao, P.J. A2A Receptor Activation Attenuates Hypertensive Cardiac Remodeling via Promoting Brown Adipose Tissue-Derived FGF21. Cell Metab. 2018, 28, 476–489. [Google Scholar] [CrossRef] [PubMed]
- He, J.L.; Zhao, M.; Xia, J.J.; Guan, J.; Liu, Y.; Wang, L.Q.; Song, D.X.; Qu, M.Y.; Zuo, M.; Wen, X.; et al. FGF21 ameliorates the neurocontrol of blood pressure in the high fructose-drinking rats. Sci. Rep. 2016, 6, 29582. [Google Scholar] [CrossRef] [PubMed]
- Facemire, C.S.; Nixon, A.B.; Griffiths, R.; Hurwitz, H.; Coffman, T.M. Vascular endothelial growth factor receptor 2 controls blood pressure by regulating nitric oxide synthase expression. Hypertension 2009, 54, 652–658. [Google Scholar] [CrossRef]
- Must, A.; Spadano, J.; Coakley, E.H.; Field, A.E.; Colditz, G.; Dietz, W.H. The disease burden associated with overweight and obesity. JAMA 1999, 282, 1523–1529. [Google Scholar] [CrossRef]
- Hruby, A.; Hu, F.B. The Epidemiology of Obesity: A Big Picture. Pharmacoeconomics 2015, 33, 673–689. [Google Scholar] [CrossRef]
- Garrison, R.J.; Kannel, W.B.; Stokes, J., 3rd; Castelli, W.P. Incidence and precursors of hypertension in young adults: The Framingham Offspring Study. Prev. Med. 1987, 16, 235–251. [Google Scholar] [CrossRef]
- Kong, L.R.; Zhou, Y.P.; Chen, D.R.; Ruan, C.C.; Gao, P.J. Decrease of Perivascular Adipose Tissue Browning Is Associated With Vascular Dysfunction in Spontaneous Hypertensive Rats During Aging. Front. Physiol. 2018, 9, 400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bang, S.; Won, K.H.; Moon, H.R.; Yoo, H.; Hong, A.; Song, Y.; Chang, S.E. Novel regulation of melanogenesis by adiponectin via the AMPK/CRTC pathway. Pigment Cell Melanoma Res. 2017, 30, 553–557. [Google Scholar] [CrossRef] [PubMed]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, D.; Kim, J.-H.; Kang, Y.-H.; Kim, J.S.; Yun, S.-C.; Kang, S.-W.; Song, Y. Suppression of Brown Adipocyte Autophagy Improves Energy Metabolism by Regulating Mitochondrial Turnover. Int. J. Mol. Sci. 2019, 20, 3520. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20143520
Kim D, Kim J-H, Kang Y-H, Kim JS, Yun S-C, Kang S-W, Song Y. Suppression of Brown Adipocyte Autophagy Improves Energy Metabolism by Regulating Mitochondrial Turnover. International Journal of Molecular Sciences. 2019; 20(14):3520. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20143520
Chicago/Turabian StyleKim, Donghwan, Ji-Hye Kim, Young-Ho Kang, Je Seong Kim, Sung-Cheol Yun, Sang-Wook Kang, and Youngsup Song. 2019. "Suppression of Brown Adipocyte Autophagy Improves Energy Metabolism by Regulating Mitochondrial Turnover" International Journal of Molecular Sciences 20, no. 14: 3520. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20143520