The Atherogenic Role of Circulating Modified Lipids in Atherosclerosis

 , and
, and
Abstract
:1. Introduction
Development of Atherosclerotic Lesions: Background
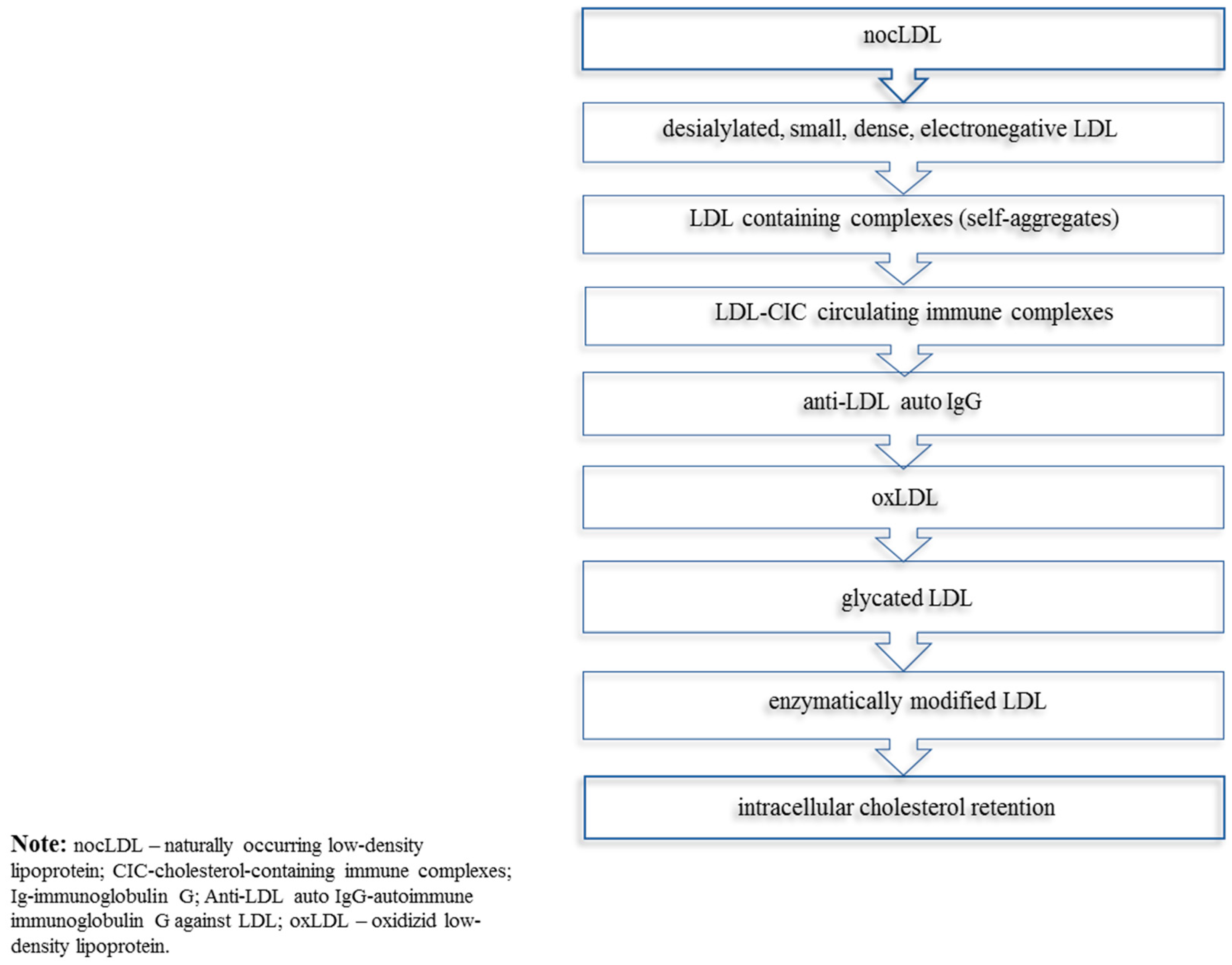
2. The Concept of Multiple Modifications of LDL in Atherogenesis
Desialylation of LDL Augments Its Atherogenicity
3. The Role of Sialidases in Atherosclerosis
4. Diagnostic and Therapeutic Approaches
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- World Health Organization. World Heart Day 2017; WHO: Geneva, Switzerland, 2017. [Google Scholar]
- Bloom, D.E.; Cafiero, E.T.; Jané-Llopis, E.; Abrahams-Gessel, S.; Bloom, L.R.; Fathima, S.; Feigl, A.B.; Gaziano, T.; Mowafi, M.; Pandya, A.; et al. The Global Economic Burden of Noncommunicable Disease; World Economic Forum: Geneva, Switzerland, 2011. [Google Scholar]
- Wang, S.; Petzold, M.; Cao, J.; Zhang, Y.; Wang, W. Direct medical costs of hospitalizations for cardiovascular diseases in Shanghai, China: Trends and projections. Medicine 2015, 94, e837. [Google Scholar] [CrossRef] [PubMed]
- Board, J.B.S. Joint British Societies’ consensus recommendations for the prevention of cardiovascular disease (JBS3). Heart 2014, 100, ii1–ii67. [Google Scholar]
- Kaplan, H.; Thompson, R.C.; Trumble, B.C.; Wann, L.S.; Allam, A.H.; Beheim, B.; Frohlich, B.; Sutherland, M.L.; Sutherland, J.D.; Stieglitz, J.; et al. Coronary atherosclerosis in indigenous South American Tsimane: A cross-sectional cohort study. Lancet 2017, 389, 1730–1739. [Google Scholar] [CrossRef]
- Orekhov, A.N.; Ivanova, E.A. Introduction of the special issue “Atherosclerosis and Related Diseases”. Vessel Plus 2017, 1, 163–165. [Google Scholar] [CrossRef]
- Rohali, K.; Likitha, G.; Alekhya, A.; Polamraju, V.S.; Srinivas, D.P. A Review Based Study on Risk factors for Coronary Heart Disease. J. Med. Sci. Clin. Res. 2015, 3, 5051–5069. [Google Scholar]
- Wadhera, R.K.; Steen, D.L.; Khan, I.; Giugliano, R.P.; Foody, J.M. A review of low-density lipoprotein cholesterol, treatment strategies, and its impact on cardiovascular disease morbidity and mortality. J. Clin. Lipidol. 2016, 10, 472–489. [Google Scholar] [CrossRef] [PubMed]
- Orakzai, S.H.; Orakzai, R.H.; Nasir, K.; Santos, R.D.; Edmundowicz, D.; Budoff, M.J.; Blumenthal, R.S. Subclinical coronary atherosclerosis: Racial profiling is necessary! Am. Heart J. 2006, 152, 819–827. [Google Scholar] [CrossRef]
- McMahan, C.A.; Gidding, S.S.; Fayad, Z.A.; Zieske, A.W.; Malcom, G.T.; Tracy, R.E.; Strong, J.P.; McGill, H.C., Jr.; Pathobiological Determinants of Atherosclerosis in Youth Research Group. Risk Scores Predict Atherosclerotic Lesions in Young People. Arch. Intern. Med. 2005, 165, 883–890. [Google Scholar] [CrossRef] [Green Version]
- Tzou, W.S.; Douglas, P.S.; Srinivasan, S.R.; Bond, M.G.; Tang, R.; Chen, W.; Berenson, G.S.; Stein, J.H. Increased Subclinical Atherosclerosis in Young Adults with Metabolic Syndrome. J. Am. Coll. Cardiol. 2005, 46, 457. [Google Scholar] [CrossRef]
- Insull, W. The Pathology of Atherosclerosis: Plaque Development and Plaque Responses to Medical Treatment. Am. J. Med. 2009, 122, S3–S14. [Google Scholar] [CrossRef]
- Spring, B.; Moller, A.C.; Colangelo, L.A.; Siddique, J.; Roehrig, M.; Daviglus, M.L.; Polak, J.F.; Reis, J.P.; Sidney, S.; Liu, K. Healthy Lifestyle Change and Subclinical Atherosclerosis in Young Adults. Circulation 2014, 130, 10–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eikendal, A.L.; Groenewegen, K.A.; Bots, M.L.; Peters, S.A.; Uiterwaal, C.S.; den Ruijter, H.M. Relation Between Adolescent Cardiovascular Risk Factors and Carotid Intima-Media Echogenicity in Healthy Young Adults: The Atherosclerosis Risk in Young Adults (ARYA) Study. J. Am. Heart Assoc. 2016, 5, e002941. [Google Scholar] [CrossRef] [PubMed]
- Orekhov, A.N.; Ivanova, E.A. Intracellular Cholesterol Lowering as Novel Target for Anti-Atherosclerotic Therapy. Cholest. Low. Ther. Drugs 2016. [Google Scholar] [CrossRef]
- Rubin, J.B.; Borden, W.B. Coronary Heart Disease in Young Adults. Curr. Atheroscler. Rep. 2012, 14, 140–149. [Google Scholar] [CrossRef] [PubMed]
- Chatzizisis, Y.S.; Coskun, A.U.; Jonas, M.; Edelman, E.R.; Feldman, C.L.; Stone, P.H. Role of Endothelial Shear Stress in the Natural History of Coronary Atherosclerosis and Vascular Remodeling. J. Am. Coll. Cardiol. 2007, 49, 2379. [Google Scholar] [CrossRef] [PubMed]
- Orekhov, A.N.; Ivanova, E.A. Cellular models of atherosclerosis and their implication for testing natural substances with anti-atherosclerotic potential. Phytomedicine 2016, 23, 1190–1197. [Google Scholar] [CrossRef] [PubMed]
- Sukhorukov, V.N.; Karagodin, V.P.; Orekhov, A.N. Atherogenic modification of low-density lipoproteins. Biomed. Khim. 2016, 62, 391–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orekhov, A.N. Anti-atherosclerotic Drugs from Natural Products. Nat. Prod. Chem. Res. 2013. [Google Scholar] [CrossRef]
- Orekhov, A.N.; Tertov, V.V.; Kudryashov, S.A.; Smirnov, V.N. Triggerlike Stimulation of Cholesterol Accumulation and DNA and Extracellular Matrix Synthesis Induced by Atherogenic Serum or Low-Density Lipoprotein in Cultured Cells. Circ. Res. 1990, 66, 311–320. [Google Scholar] [CrossRef]
- Martin, S.S.; Blumenthal, R.S.; Miller, M. LDL Cholesterol: The Lower the Better. Med. Clin. 2012, 96, 13–26. [Google Scholar] [CrossRef]
- Sala, F.; Catapano, A.L.; Norata, G.D. High density lipoproteins and atherosclerosis: Emerging aspects. J. Geriatr. Cardiol. 2012, 9, 401–407. [Google Scholar] [PubMed]
- Harangi, M.; Szentpéteri, A.; Nádró, B.; Lőrincz, H.; Seres, I.; Páll, D.; Paragh, G. HDL subfraction distribution and HDL function in untreated dyslipidemic patients. Vessel Plus 2017, 1, 166–173. [Google Scholar]
- Venugopal, S.K.; Jialal, I. Biochemistry, Low Density Lipoprotein. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2019. [Google Scholar]
- Zakiev, E.R.; Sukhorukov, V.N.; Melnichenko, A.A.; Sobenin, I.A.; Ivanova, E.A.; Orekhov, A.N. Lipid composition of circulating multiple-modified low density lipoprotein. Lipids Health Dis. 2016, 15, 134. [Google Scholar] [CrossRef] [PubMed]
- Orekhov, A.N.; Tertov, V.V.; Mukhin, D.N.; Mikhailenko, I.A. Modification of low density lipoprotein by desialylation causes lipid accumulation in cultured cells: Discovery of desialylated lipoprotein with altered cellular metabolism in the blood of atherosclerotic patients. Biochem. Biophys. Res. Commun. 1989, 162, 206–211. [Google Scholar] [CrossRef]
- Tertov, V.V.; Kaplun, V.V.; Sobenin, I.A.; Orekhov, A.N. Low-density lipoprotein modification occurring in human plasma: Possible mechanism of in vivo lipoprotein desialylation as a primary step of atherogenic modification. Atherosclerosis 1998, 138, 183–195. [Google Scholar] [CrossRef]
- Jaakkola, O.; Solakivi, T.; Tertov, V.V.; Orekhov, A.N.; Miettinen, T.A.; Nikkari, T. Characteristics of low-density lipoprotein subfractions from patients with coronary artery disease. Coron. Artery Dis. 1993, 4, 379–385. [Google Scholar] [CrossRef] [PubMed]
- Tertov, V.V.; Kaplun, V.V.; Sobenin, I.A.; Boytsova, E.Y.; Bovin, N.V.; Orekhov, A.N. Human plasma trans-sialidase causes atherogenic modification of low density lipoprotein. Atherosclerosis 2001, 159, 103–115. [Google Scholar] [CrossRef]
- Tertov, V.V.; Sobenin, I.A.; Orekhov, A.N. Modified (desialylated) low-density lipoprotein measured in serum by lectin-sorbent assay. Clin. Chem. 1995, 41, 1018–1021. [Google Scholar]
- Tertov, V.V.; Sobenin, I.A.; Tonevitsky, A.G.; Orekhov, A.N.; Smirnov, V.N. Isolation of atherogenic modified (desialylated) low density lipoprotein from blood of atherosclerotic patients: Separation from native lipoprotein by affinity chromatography. Biochem. Biophys. Res. Commun. 1990, 167, 1122–1127. [Google Scholar] [CrossRef]
- Tertov, V.V.; Sobenin, I.A.; Orekhov, A.N. Characterization of desialylated low-density lipoproteins which cause intracellular lipid accumulation. Int. J. Tissue React. 1992, 14, 155–162. [Google Scholar]
- Ruelland, A.; Gallou, G.; Legras, B.; Paillard, F.; Cloarec, L. LDL sialic acid content in patients with coronary artery disease. Clin. Chim. Acta 1993, 221, 127–133. [Google Scholar] [CrossRef]
- Orekhov, A.N.; Elizova, N.V.; Melnichenko, A.A.; Karagodin, V.P.; Myasoedova, V.A.; Zhelankin, A.V.; Trubinov, S.S.; Orekhova, V.A.; Sinyov, V.V.; Barinova, V.A.; et al. GW26-e0208 Naturally occurring multiple-modified low density lipoprotein (LDL). J. Am. Coll. Cardiol. 2019, 66, C71. [Google Scholar] [CrossRef]
- Tertov, V.V.; Sobenin, I.A.; Gabbasov, Z.A.; Popov, E.G.; Jaakkola, O.; Solakivi, T.; Nikkari, T.; Smirnov, V.N.; Orekhov, A.N. Multiple-modified desialylated low density lipoproteins that cause intracellular lipid accumulation. Isolation, fractionation and characterization. Lab. Investig. 1992, 67, 665–675. [Google Scholar] [PubMed]
- Ivanova, E.A.; Bobryshev, Y.V.; Orekhov, A.N. LDL electronegativity index: A potential novel index for predicting cardiovascular disease. Vasc. Health Risk Manag. 2015, 11, 525–532. [Google Scholar] [PubMed]
- Tertov, V.V.; Sobenin, I.A.; Orekhov, A.N. Similarity Between Naturally Occurring Modified Desialylated, Electronegative and Aortic Low Density Lipoprotein. Free Radic. Res. 1996, 25, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Pentikainen, M.O.; Öörni, K.; Ala-Korpela, M.; Kovanen, P.T. Modified LDL—Trigger of atherosclerosis and inflammation in the arterial intima. J. Intern. Med. 2000, 247, 359–370. [Google Scholar] [CrossRef] [PubMed]
- Orekhov, A.N.; Tertov, V.V.; Mukhin, D.N. Desialylated low density lipoprotein—Naturally occurring modified lipoprotein with atherogenic potency. Atheroscler 1991, 86, 153–161. [Google Scholar] [CrossRef]
- Tertov, V.V.; Sobenin, I.A.; Kaplun, V.V.; Orekhov, A.N. Antioxidant content in low density lipoprotein and lipoprotein oxidation in vivo and in vitro. Free Radic. Res. 1998, 29, 165–173. [Google Scholar] [CrossRef]
- Younis, N.; Charlton-Menys, V.; Sharma, R.; Soran, H.; Durrington, P.N. Glycation of LDL in non-diabetic people: Small dense LDL is preferentially glycated both in vivo and in vitro. Atheroscler 2009, 202, 162–168. [Google Scholar] [CrossRef]
- Soran, H.; Durrington, P.N. Susceptibility of LDL and its subfractions to glycation. Curr. Opin. Lipidol. 2011, 22, 254. [Google Scholar] [CrossRef]
- Matsui, H.; Okumura, K.; Toki, Y.; Hayakawa, T. Low-density lipoprotein particle size as an independent predictor of glycated low-density lipoprotein level. Diabetes Care 1999, 22, 1220. [Google Scholar] [CrossRef] [PubMed]
- Avogaro, P.; Bon, G.B.; Cazzolato, G. Presence of a modified low density lipoprotein in humans. Arteriosclerosis 1988, 8, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Krauss, R.M. All Low-Density Lipoprotein Particles Are Not Created Equal. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 959–961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sobenin, I.A.; Tertov, V.V.; Orekhov, A.N. Atherogenic Modified LDL in Diabetes. Diabetes 1996, 45, S35–S39. [Google Scholar] [CrossRef] [PubMed]
- Vekic, J.; Zeljkovic, A.; Jelic-Ivanovic, Z.; Spasojevic-Kalimanovska, V.; Bogavac-Stanojevic, N.; Memon, L.; Spasic, S. Small, dense LDL cholesterol and apolipoprotein B: Relationship with serum lipids and LDL size. Atheroscler 2009, 207, 496–501. [Google Scholar] [CrossRef] [PubMed]
- Griffin, B.A. Lipoprotein atherogenicity: An overview of current mechanisms. Proc. Nutr. Soc. 1999, 58, 163–169. [Google Scholar] [CrossRef]
- Younis, N.; Sharma, R.; Soran, H.; Charlton-Menys, V.; Elseweidy, M.; Durrington, P.N. Glycation as an atherogenic modification of LDL. Curr. Opin. Lipidol. 2008, 19, 378. [Google Scholar] [CrossRef] [PubMed]
- Tsutomu, H.; Yasuki, I.; Shinji, K.; Miwako, T.; Ayako, I.; Haruhisa, S.; Junichi, Y.; Gen, Y. Clinical Significance of Small Dense Low-Density Lipoprotein Cholesterol Levels Determined by the Simple Precipitation Method. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 558–563. [Google Scholar] [Green Version]
- Fukushima, Y.; Hirayama, S.; Ueno, T.; Dohi, T.; Miyazaki, T.; Ohmura, H.; Mokuno, H.; Miyauchi, K.; Miida, T.; Daida, H. Small dense LDL cholesterol is a robust therapeutic marker of statin treatment in patients with acute coronary syndrome and metabolic syndrome. Clin. Chim. Acta 2011, 412, 1423–1427. [Google Scholar] [CrossRef]
- Toledo, F.G.S.; Sniderman, A.D.; Kelley, D.E. Influence of Hepatic Steatosis (Fatty Liver) on Severity and Composition of Dyslipidemia in Type 2 Diabetes. Diabetes Care 2006, 29, 1845–1850. [Google Scholar] [CrossRef] [Green Version]
- Cali, A.M.G.; Zern, T.L.; Taksali, S.E.; de Oliveira, A.M.; Dufour, S.; Otvos, J.D.; Caprio, S. Intrahepatic Fat Accumulation and Alterations in Lipoprotein Composition in Obese Adolescents: A perfect proatherogenic state. Diabetes Care 2007, 30, 3093–3098. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, M.; Pernice, V.; Frasheri, A.; Lorenzo, G.D.; Rini, G.B.; Spinas, G.A.; Berneis, K. Small, dense low-density lipoproteins (LDL) are predictors of cardio- and cerebro-vascular events in subjects with the metabolic syndrome. Clin. Endocrinol. 2009, 70, 870–875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dousset, N.; Dousset, J.C.; Taus, M.; Ferretti, G.; Curatola, G.; Soléra, M.L.; Valdiguié, P. Effect of desialylation on low density lipoproteins: Comparative study before and after oxidative stress. Biochem. Mol. Biol. Int. 1994, 32, 555–563. [Google Scholar] [PubMed]
- Tertov, V.V.; Sobenin, I.A.; Gabbasov, Z.A.; Popov, E.G.; Orekhov, A.N. Lipoprotein aggregation as an essential condition of intracellular lipid accumulation caused by modified low density lipoproteins. Biochem. Biophys. Res. Commun. 1989, 163, 489–494. [Google Scholar] [CrossRef]
- Sobenin, I.A.; Suprun, I.V.; Karagodin, V.P.; Feoktistov, A.S.; Melnichenko, A.A.; Orekhov, A.N. The Interaction of Plasma Sialylated and Desialylated Lipoproteins with Collagen from the Intima and Media of Uninvolved and Atherosclerotic Human Aorta. Available online: https://www.hindawi.com/journals/jl/2011/254267/ (accessed on 10 July 2019).
- Orekhov, A.N.; Tertov, V.V.; Mukhin, D.N.; Koteliansky, V.E.; Glukhova, M.A.; Khashimov, K.A.; Smirnov, V.N. Association of low-density lipoprotein with particulate connective tissue matrix components enhances cholesterol accumulation in cultured subendothelial cells of human aorta. Biochim. Biophys. Acta Mol. Cell Res. 1987, 928, 251–258. [Google Scholar] [CrossRef]
- Orekhov, A.N.; Tertov, V.V.; Mukhin, D.N.; Koteliansky, V.E.; Glukhova, M.A.; Frid, M.G.; Sukhova, G.K.; Khashimov, K.A.; Smirnov, V.N. Insolubilization of low density lipoprotein induces cholesterol accumulation in cultured subendothelial cells of human aorta. Atherosclerosis 1989, 79, 59–70. [Google Scholar] [CrossRef]
- Younis, N.N.; Soran, H.; Pemberton, P.; Charlton-Menys, V.; Elseweidy, M.M.; Durrington, P.N. Small dense LDL is more susceptible to glycation than more buoyant LDL in Type 2 diabetes. Clin. Sci. 2013, 124, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Rivas-Urbina, A.; Benitez, S.; Perez, A.; Sanchez-Quesada, J.L. Modified low-density lipoproteins as biomarkers in diabetes and metabolic syndrome. Front. Biosci. 2018, 23, 1220–1240. [Google Scholar] [Green Version]
- Tabas, I.; Williams, K.J.; Born, J. Subendothelial Lipoprotein Retention as the Initiating Process in Atherosclerosis. Circulation 2007, 116, 1832–1844. [Google Scholar] [CrossRef]
- Sobenin, I.A.; Galitsyna, E.V.; Grechko, A.V.; Orekhov, A.N. Small dense and desialylated low density lipoprotein in diabetic patients. Vessel Plus 2017, 1, 29–37. [Google Scholar] [CrossRef] [Green Version]
- Orekhov, A.N.; Melnichenko, A.A.; Sobenin, I.A. Approach to Reduction of Blood Atherogenicity. Available online: https://www.hindawi.com/journals/omcl/2014/738679/abs/ (accessed on 10 July 2019).
- Hirayama, S.; Miida, T. Small dense LDL: An emerging risk factor for cardiovascular disease. Clin. Chim. Acta 2012, 414, 215–224. [Google Scholar] [CrossRef] [PubMed]
- Anber, V.; Griffin, B.A.; McConnell, M.; Packard, C.J.; Shepherd, J. Influence of plasma lipid and LDL-subfraction profile on the interaction between low density lipoprotein with human arterial wall proteoglycans. Atherosclerosis 1996, 124, 261–271. [Google Scholar] [CrossRef]
- Tertov, V.V.; Orekhov, A.N. Metabolism of Native and Naturally Occurring Multiple Modified Low Density Lipoprotein in Smooth Muscle Cells of Human Aortic Intima. Exp. Mol. Pathol. 1997, 64, 127–145. [Google Scholar] [CrossRef] [PubMed]
- Harada, L.M.; Carvalho, M.D.T.; Passarelli, M.; Quintão, E.C.R. Lipoprotein desialylation simultaneously enhances the cell cholesterol uptake and impairs the reverse cholesterol transport system: In vitro evidences utilizing neuraminidase-treated lipoproteins and mouse peritoneal macrophages. Atherosclerosis 1998, 139, 65–75. [Google Scholar] [CrossRef]
- Orekhov, A.N.; Tertov, V.V.; Kabakov, A.E.; Pokrovsky, S.N.; Smirnov, V.N. Autoantibodies against modified low density lipoprotein. Nonlipid factor of blood plasma that stimulates foam cell formation. Arterioscler. Thromb. J. Vasc. Biol. 1991, 11, 316–326. [Google Scholar] [CrossRef]
- Gounopoulos, P.; Merki, E.; Hansen, L.F.; Choi, S.H.; Tsimikas, S. Antibodies to oxidized low density lipoprotein: Epidemiological studies and potential clinical applications in cardiovascular disease. Minerva Cardioangiol. 2007, 55, 821–837. [Google Scholar] [PubMed]
- Kacharava, A.G.; Tertov, V.V.; Orekhov, A.N. Autoantibodies against low-density lipoprotein and atherogenic potential of blood. Ann. Med. 1993, 25, 551–555. [Google Scholar] [PubMed]
- Tertov, V.V.; Orekhov, A.N.; Sayadyan, K.S.; Serebrennikov, S.G.; Kacharava, A.G.; Lyakishev, A.A.; Smirnov, V.N. Correlation between cholesterol content in circulating immune complexes and atherogenic properties of CHD patients’ serum manifested in cell culture. Atherosclerosis 1990, 81, 183–189. [Google Scholar] [CrossRef]
- Orekhov, A.N.; Kalenich, O.S.; Tertov, V.V.; Novikov, I.D. Lipoprotein immune complexes as markers of atherosclerosis. Int. J. Tissue React. 1991, 13, 233–236. [Google Scholar]
- Sobenin, I.A.; Karagodin, V.P.; Melnichenko, A.A.; Bobryshev, Y.V.; Orekhov, A.N. Diagnostic and Prognostic Value of Low-Density Lipoprotein-Containing Circulating Immune Complexes in Atherosclerosis. J. Clin. Immunol. 2013, 33, 489–495. [Google Scholar] [CrossRef]
- Wang, J.; Niu, D.; Meng, Y.; Han, A.; Li, K.; Zhang, C. Plasma oxidized lipoprotein(a) and its immune complexes are present in newborns and children. Clin. Chim. Acta 2009, 407, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Nikonova, E.Y.; Tertov, V.V.; Sato, C.; Kitajima, K.; Bovin, N.V. Specificity of human trans-sialidase as probed with gangliosides. Bioorg. Med. Chem. Lett. 2004, 14, 5161–5164. [Google Scholar] [CrossRef] [PubMed]
- Momiyama, Y.; Ohmori, R.; Taniguchi, H.; Nakamura, H.; Ohsuzu, F. Association of Mycoplasma pneumoniae infection with coronary artery disease and its interaction with chlamydial infection. Atherosclerosis 2004, 176, 139–144. [Google Scholar] [CrossRef] [PubMed]
- Golovanova, N.K.; Gracheva, E.V.; Il’inskaya, O.P.; Tararak, E.M.; Prokazova, N.V. Sialidase Activity in Normal and Atherosclerotic Human Aortic Intima. Biochemistry 2002, 67, 1230–1234. [Google Scholar] [PubMed]
- Garavelo, S.M.; Higuchi, M.D.L.; Pereira, J.J.; Reis, M.M.; Kawakami, J.T.; Ikegami, R.N.; Palomino, S.A.; Wadt, N.S.; Agouni, A. Comparison of the Protective Effects of Individual Components of Particulated trans-Sialidase (PTCTS), PTC and TS, against High Cholesterol Diet-Induced Atherosclerosis in Rabbits. BioMed Res. Int. 2017, 2017, 7212985. [Google Scholar] [CrossRef]
- Karagodin, V.P.; Sukhorukov, V.N.; Myasoedova, V.A.; Grechko, A.V.; Orekhov, A.N. Diagnostics and Therapy of Human Diseases—Focus on Sialidases. Curr. Pharm. Des. 2018, 24, 2870–2875. [Google Scholar] [CrossRef]
- Sukhorukov, V.N.; Karagodin, V.P.; Zakiev, E.R.; Grechko, A.V.; Orekhov, A.N. Sialidases: Therapeutic and antiatherogenic potential. Curr. Pharm. Des. 2017, 23, 4696–4701. [Google Scholar] [CrossRef]
- Yang, A.; Gyulay, G.; Mitchell, M.; White, E.; Trigatti, B.L.; Igdoura, S.A. Hypomorphic sialidase expression decreases serum cholesterol by downregulation of VLDL production in mice. J. Lipid Res. 2012, 53, 2573–2585. [Google Scholar] [CrossRef] [Green Version]
- Moon, S.-K.; Cho, S.-H.; Kim, K.-W.; Jeon, J.H.; Ko, J.-H.; Kim, B.Y.; Kim, C.-H. Overexpression of membrane sialic acid-specific sialidase Neu3 inhibits matrix metalloproteinase-9 expression in vascular smooth muscle cells. Biochem. Biophys. Res. Commun. 2007, 356, 542–547. [Google Scholar] [CrossRef]
- Orekhov, A.N.; Bobryshev, Y.V.; Sobenin, I.A.; Melnichenko, A.A.; Chistiakov, D.A. Modified Low Density Lipoprotein and Lipoprotein-Containing Circulating Immune Complexes as Diagnostic and Prognostic Biomarkers of Atherosclerosis and Type 1 Diabetes Macrovascular Disease. Int. J. Mol. Sci. 2014, 15, 12807–12841. [Google Scholar] [CrossRef] [Green Version]
- Fraley, A.E.; Schwartz, G.G.; Olsson, A.G.; Kinlay, S.; Szarek, M.; Rifai, N.; Libby, P.; Ganz, P.; Witztum, J.L.; Tsimikas, S.; et al. Relationship of oxidized phospholipids and biomarkers of oxidized low-density lipoprotein with cardiovascular risk factors, inflammatory biomarkers, and effect of statin therapy in patients with acute coronary syndromes: Results from the MIRACL (Myocardial Ischemia Reduction with Aggressive Cholesterol Lowering) trial. J. Am. Coll. Cardiol. 2009, 53, 2186–2196. [Google Scholar] [PubMed]
- Fraley, A.E.; Tsimikas, S. Clinical applications of circulating oxidized low-density lipoprotein biomarkers in cardiovascular disease. Curr. Opin. Lipidol. 2006, 17, 502–509. [Google Scholar] [CrossRef]
- Wang, J.; Qiang, H.; Zhang, C.; Liu, X.; Chen, D.; Wang, S. Detection of IgG-bound lipoprotein(a) immune complexes in patients with coronary heart disease. Clin. Chim. Acta 2003, 327, 115–122. [Google Scholar] [CrossRef]
- Orekhov, A.N.; Sobenin, I.A.; Korneev, N.V.; Kirichenko, T.V.; Myasoedova, V.A.; Melnichenko, A.A.; Balcells, M.; Edelman, E.R.; Bobryshev, Y.V. Anti-atherosclerotic therapy based on botanicals. Recent Pat. Cardiovasc. Drug Discov. 2013, 8, 56–66. [Google Scholar] [CrossRef] [PubMed]
- Doo, Y.C.; Han, S.J.; Lee, J.H.; Cho, G.Y.; Hong, K.S.; Han, K.R.; Lee, N.H.; Oh, D.J.; Ryu, K.H.; Rhim, C.Y.; et al. Associations among oxidized low-density lipoprotein antibody, C-reactive protein, interleukin-6, and circulating cell adhesion molecules in patients with unstable angina pectoris. Am. J. Cardiol. 2004, 93, 554–558. [Google Scholar] [CrossRef] [PubMed]
- Sobenin, I.A.; Salonen, J.T.; Zhelankin, A.V.; Melnichenko, A.A.; Kaikkonen, J.; Bobryshev, Y.V.; Orekhov, A.N. Low Density Lipoprotein-Containing Circulating Immune Complexes: Role in Atherosclerosis and Diagnostic Value. Available online: https://www.hindawi.com/journals/bmri/2014/205697/abs/ (accessed on 12 July 2019).
- Orekhov, A.N.; Sobenin, I.A.; Revin, V.V.; Bobryshev, Y.V. Development of Antiatherosclerotic Drugs on the basis of Natural Products Using Cell Model Approach. Oxidative Med. Cell. Longev. 2015, 2015, 463797. [Google Scholar] [CrossRef] [PubMed]
- Myasoedova, V.A.; Ivashinnikova, G.A.; Sobenin, I.A.; Ivanova, E.A.; Orekhov, A.N. Blood Serum Atherogenicity: Cellular Test for the Development of Anti-Atherosclerotic Therapy. Curr. Pharm. Des. 2017, 23, 1195–1206. [Google Scholar] [CrossRef]
- Bambauer, R.; Olbricht, C.J.; Schoeppe, E. Low-Density Lipoprotein Apheresis for Prevention and Regression of Atherosclerosis: Clinical Results. Ther. Apher. 1997, 1, 242–248. [Google Scholar] [CrossRef]


{kind=link}
| Signaling Pathway | Lipid Modification | ||
|---|---|---|---|
| Naturally Occurring | Desialylation | Oxidation | |
| EP2---> BDNF | ↑ | - | - |
| hypoxia pathways | ↑ | ↑ | - |
| insulin--->AKT-1 pathway | ↑ | - | - |
| Prostanoid receptor signaling | ↑ | ↑ | - |
| TBK1:TRIF:IKK-i--->p50:RelA | ↑ | ↓ | - |
| TNF-α--->p50:RelA-p65 | ↑ | - | - |
| Akt-1---Mdm2--->AR | ↓ | ↓ | - |
| Caspase network | ↓ | - | - |
| cyclosome--->Nek2A | ↓ | ↓ | - |
| cyclosome---/SnoN | ↓ | ↓ | - |
| dsRNA--->c-Jun | ↓ | - | - |
| E1---PIRH2---/p53 | ↓ | ↓ | - |
| Emi1---Cdc20---/cyclosome | ↓ | ↓ | - |
| Emi1---Fzr1---/cyclosome | ↓ | ↓ | - |
| ER-α pathway | ↓ | ↓ | ↑ |
| HMGCR regulation | ↓ | ↓ | - |
| Htt degradation | ↓ | ↓ | - |
| LT-betaR---NIK, RelB--->CCL19 | ↓ | ↓ | ↑ |
| MAD2---Cdc20--->cyclosome | ↓ | ↓ | - |
| MEK--->ABP-280 | ↓ | ↓ | - |
| Diagnostic Technique/Biomarker | References |
|---|---|
| Measuring total level of LDL-CIC | [75] |
| Detection of trans-sialidase activity | [82] |
| Measuring the proportion of mmLDL in serum | [85] |
| Measuring the levels of circulating oxLDL by a specific monoclonal antibody | [86,87] |
| Measuring the cholesterol content and apoB-100 in LDL-containing immune complexes | [88] |
| Simultaneous measurement of total and desialylated apoB-100 in serum and calculation of desialylated apoB-100 fraction size | [89] |
| Detection of the level of circulating anti-LDL antibodies (IgG and IgM) | [90,91] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Summerhill, V.I.; Grechko, A.V.; Yet, S.-F.; Sobenin, I.A.; Orekhov, A.N. The Atherogenic Role of Circulating Modified Lipids in Atherosclerosis. Int. J. Mol. Sci. 2019, 20, 3561. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20143561
Summerhill VI, Grechko AV, Yet S-F, Sobenin IA, Orekhov AN. The Atherogenic Role of Circulating Modified Lipids in Atherosclerosis. International Journal of Molecular Sciences. 2019; 20(14):3561. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20143561
Chicago/Turabian StyleSummerhill, Volha I., Andrey V. Grechko, Shaw-Fang Yet, Igor A. Sobenin, and Alexander N. Orekhov. 2019. "The Atherogenic Role of Circulating Modified Lipids in Atherosclerosis" International Journal of Molecular Sciences 20, no. 14: 3561. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20143561