Sirtuin 1 Regulates Mitochondrial Biogenesis and Provides an Endogenous Neuroprotective Mechanism Against Seizure-Induced Neuronal Cell Death in the Hippocampus Following Status Epilepticus

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
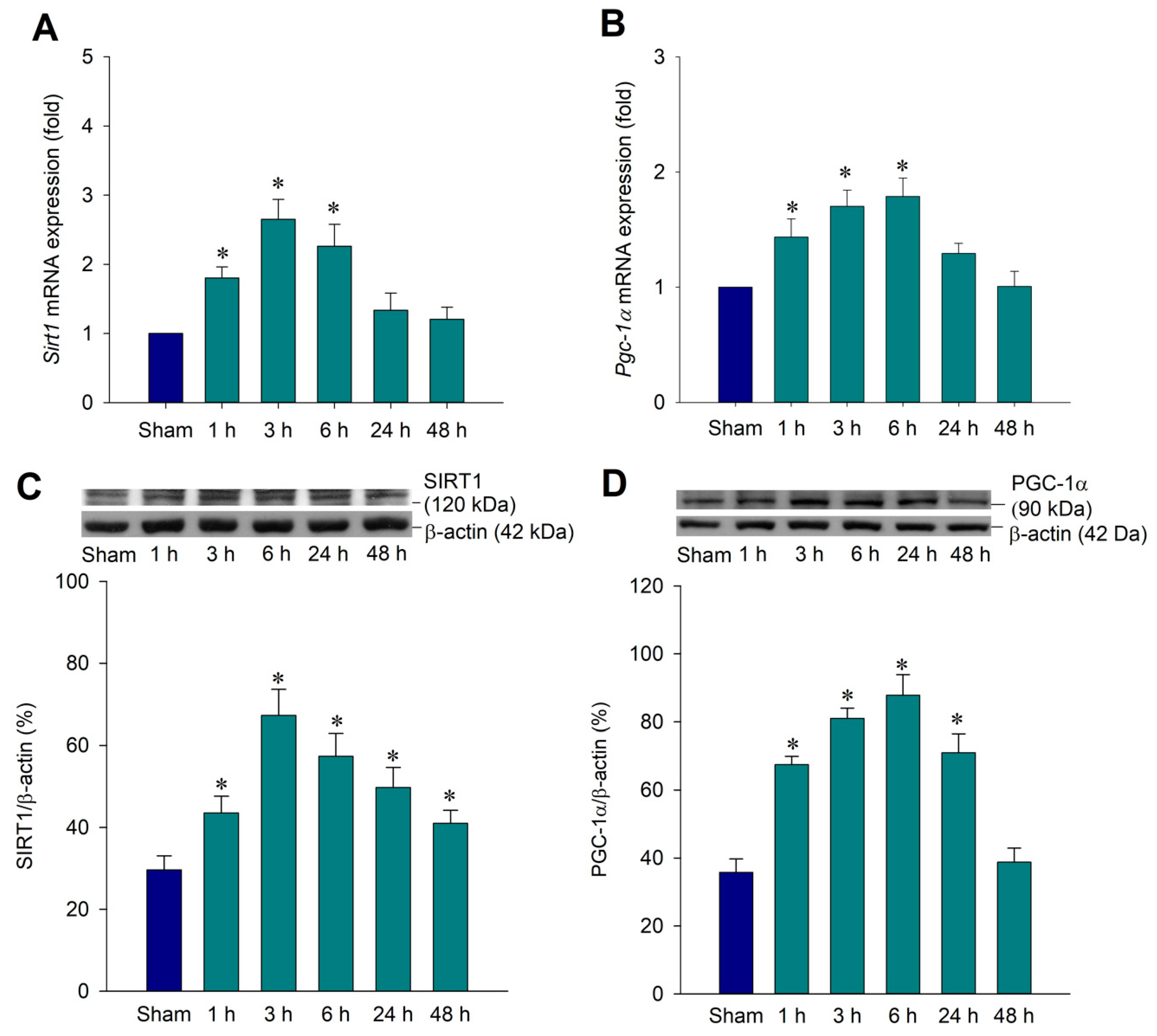
2.1. Temporal Changes of SIRT1 and PGC-1α Expression in the Hippocampal CA3 Region Following Status Epilepticus
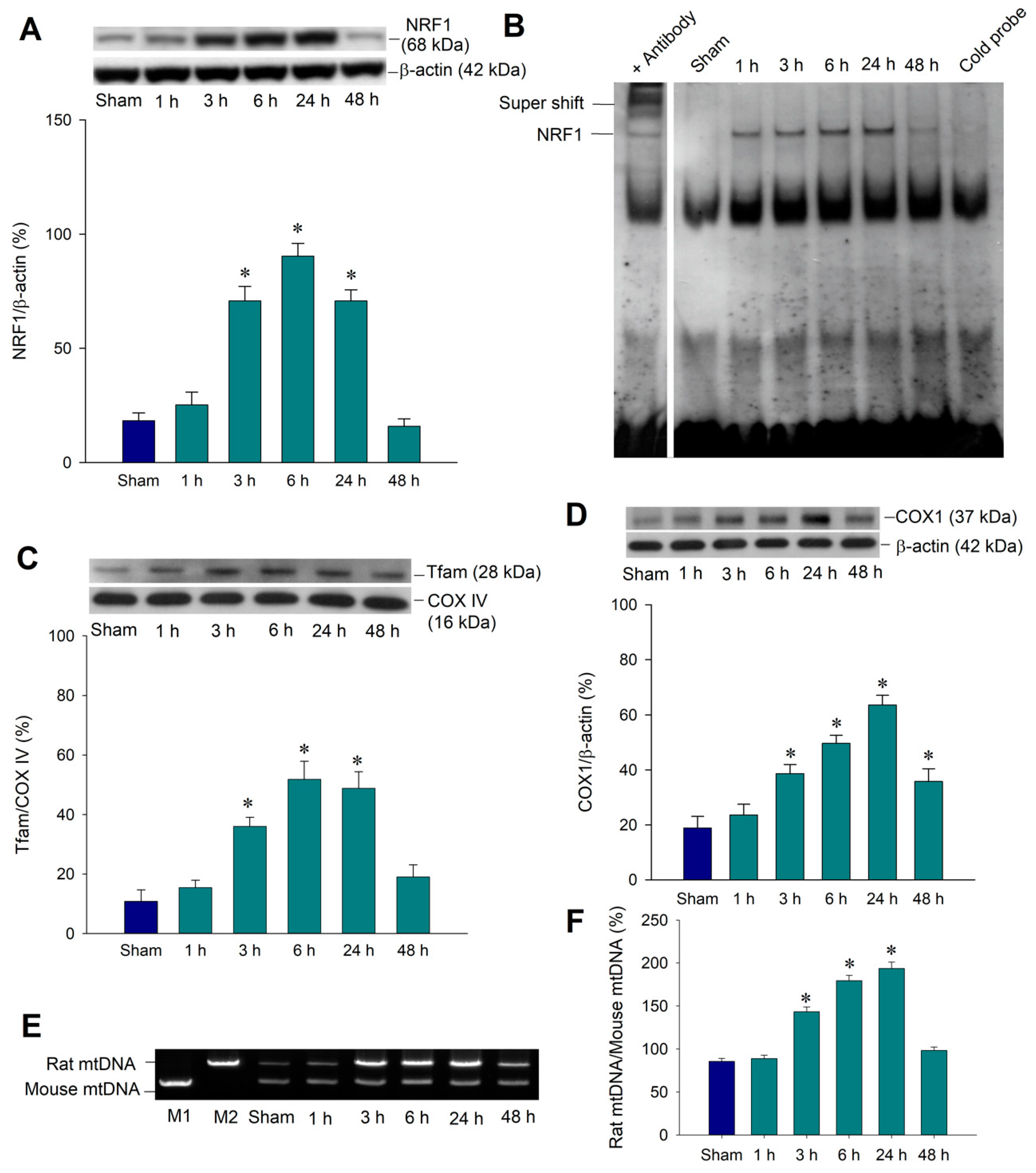
2.2. Temporal Changes of Mitochondrial Biogenesis Machinery Expression in the Hippocampal CA3 Region Following Status Epilepticus
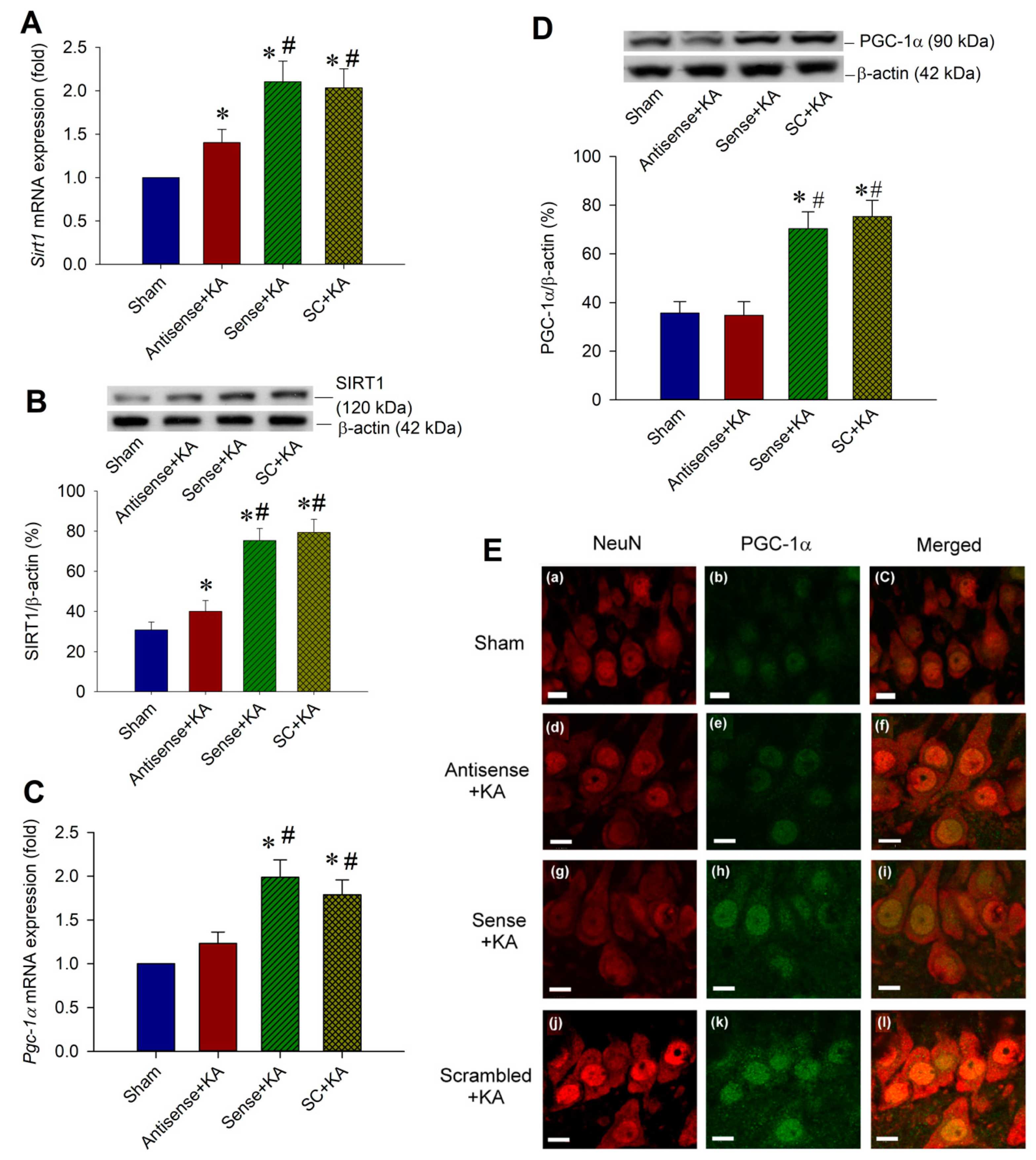
2.3. Effect of Sirt1 Knock-Down by Antisense Strategy on PGC-1α Expressions in the Hippocampal CA3 Region Following Status Epilepticus
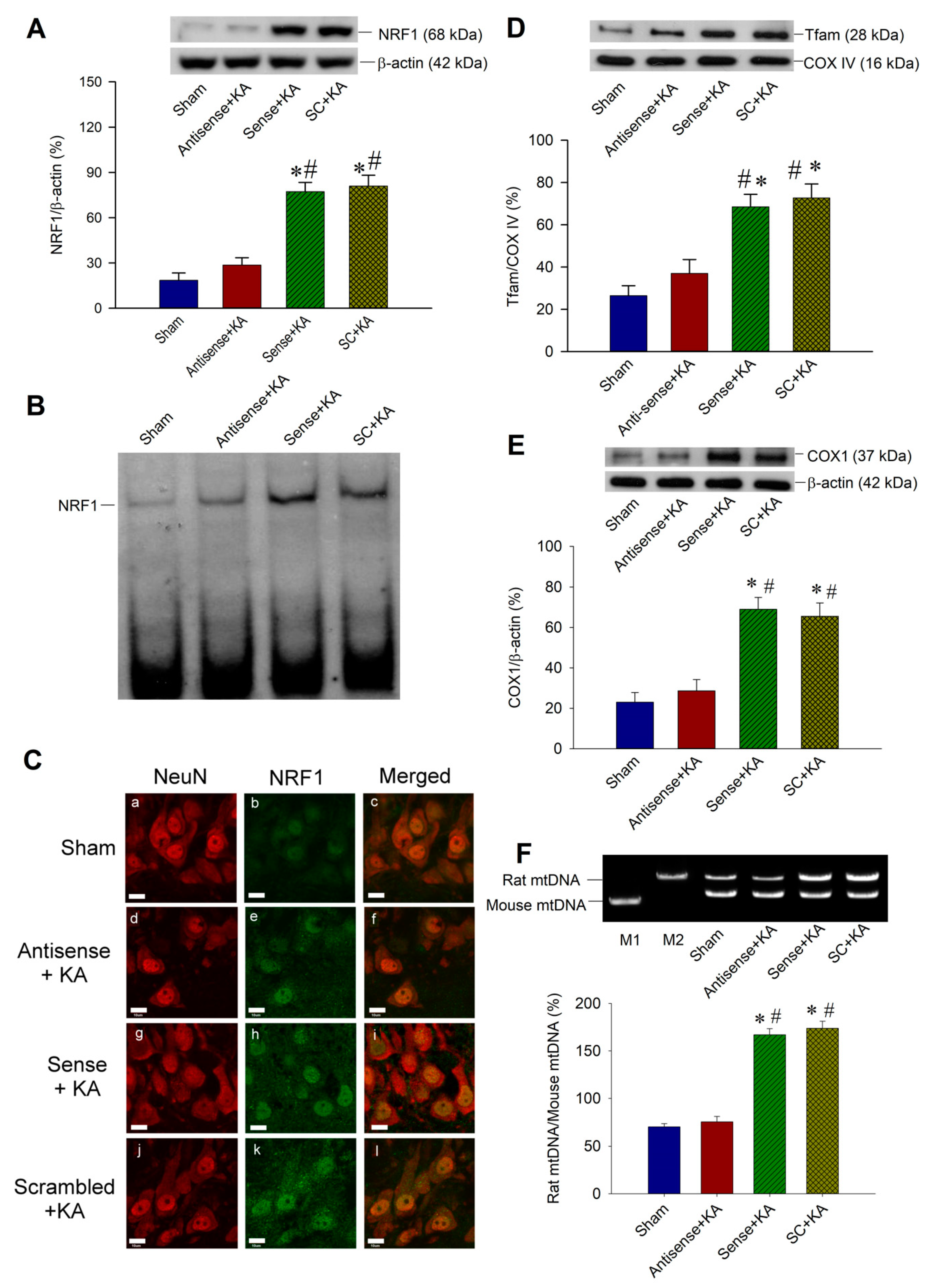
2.4. Effect of Sirt1 Knock-Down by Antisense Strategy on Mitochondrial Biogenesis Machinery Expression in the Hippocampal CA3 Region Following Status Epilepticus
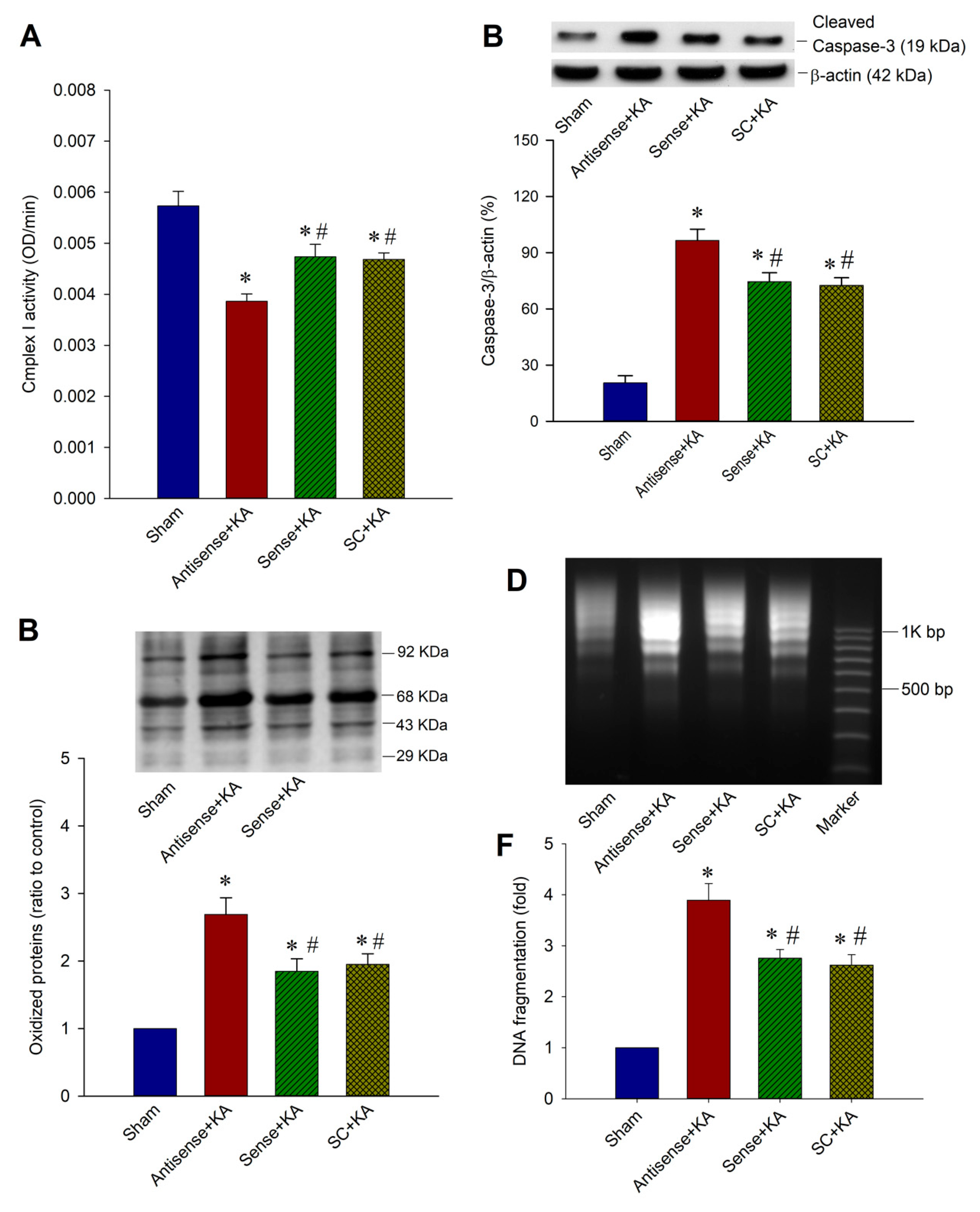
2.5. Effect of SIRT1/PGC-1α Signaling Pathway on Mitochondrial Respiratory Chain Function, Protein Oxidation, and Neuronal Cell Survival in the Hippocampal CA3 Region Following Status Epilepticus
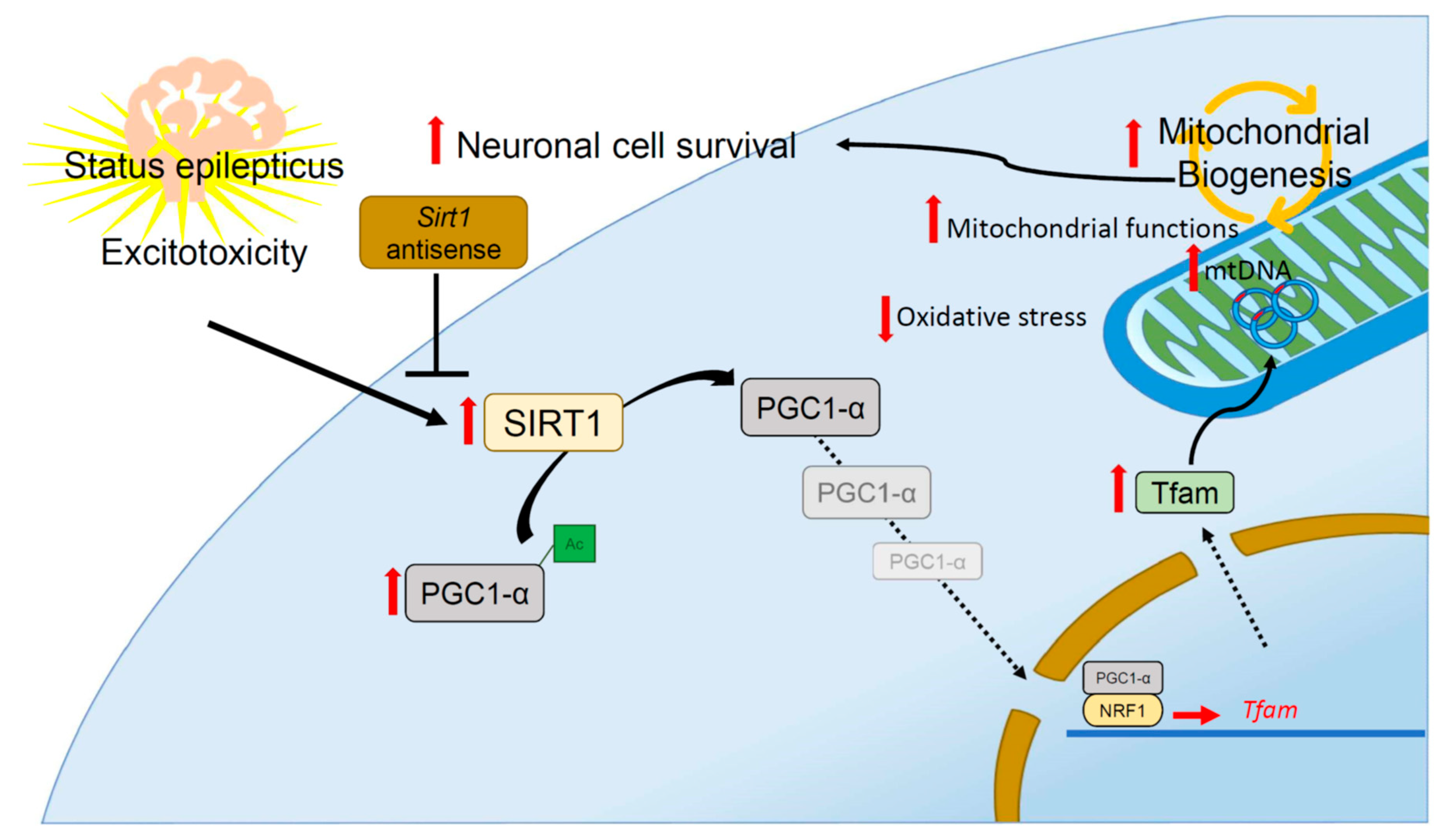
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Experimental Status Epilepticus
4.3. Gene Knockdown by Microinjection of Oligonucleotides into the Hippocampus
4.4. Collection of the Hippocampal Tissue
4.5. RNA Isolation and Reverse Transcription Real-Time Polymerase Chain Reaction
- Sirt1:
- forward: 5’-TCGTGGAGACATTTTTAATCAGG-3’;reverse: 5’-CAGTGTCCGAGTCTGAATCCT-3’.
- Pgc-1α:
- forward: 5’-GTTTCATTACCTACCGTTACAC-3’;reverse: 5’-ATCGTCTGAGTTTGAATCTAGG-3’.
- GAPDH:
- forward: 5′-CAACTCCCATTCTTCCACCT-3′;reverse: 5′-GCCATATTCATTGTCATACCAG-3′.
4.6. Western Blot Analysis
4.7. Double Immunofluorescence Staining and Laser Confocal Microscopy
4.8. Electrophoretic Mobility Shift Assay (EMSA)
4.9. Long Polymerase Chain Reaction (PCR) For Quantitation of Mitochondrial DNA
4.10. Assays for the Activity of Complex I of Mitochondrial Respiratory Chain Enzyme
4.11. Measurement of Protein Oxidation (Oxidative Stress)
4.12. Qualitative and Quantitative Analysis of DNA Fragmentation
4.13. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| SIRT1 | Sirtuin 1 |
| PPARγ | Peroxisome proliferator-activated receptors γ |
| PGC-1α | Peroxisome proliferator-activated receptor γ-1α |
| ODN | Oligodeoxynucleotide |
| EMSA | Electrophoretic mobility shift assay |
| COX1 | Cytochrome c oxidase 1 |
| NRF | Nuclear respiratory factor |
| Tfam | Mitochondrial transcription factor A |
| mtDNA | Mitochondrial DNA |
| UCP2 | Mitochondrial uncoupling protein 2 |
| Drp1 | Dynamin-related protein 1 |
References
- Bell, G.S.; Neligan, A.; Sander, J.W. An unknown quantity--the worldwide prevalence of epilepsy. Epilepsia 2014, 55, 958–962. [Google Scholar] [CrossRef] [PubMed]
- Trinka, E.; Brigo, F.; Shorvon, S. Recent advances in status epilepticus. Curr. Opin. Neurol. 2016, 29, 189–198. [Google Scholar] [CrossRef] [PubMed]
- Haut, S.R.; Veliskova, J.; Moshe, S.L. Susceptibility of immature and adult brains to seizure effects. Lancet. Neurol. 2004, 3, 608–617. [Google Scholar] [CrossRef]
- Henshall, D.C.; Simon, R.P. Epilepsy and apoptosis pathways. J. Cereb. Blood Flow Metab. 2005, 25, 1557–1572. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.D.; Chang, A.Y.; Chuang, Y.C. The potential role of mitochondrial dysfunction in seizure-associated cell death in the hippocampus and epileptogenesis. J. Bioenerg. Biomembr. 2010, 42, 461–465. [Google Scholar] [CrossRef] [PubMed]
- Power, K.N.; Gramstad, A.; Gilhus, N.E.; Hufthammer, K.O.; Engelsen, B.A. Cognitive function after status epilepticus versus after multiple generalized tonic-clonic seizures. Epilepsy Res. 2018, 140, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Falco-Walter, J.J.; Bleck, T. Treatment of Established Status Epilepticus. J. Clin. Med. 2016, 5, 49. [Google Scholar] [CrossRef] [PubMed]
- Osorio, C.M.; Lin, K.; Guarnieri, R.; de Oliveira Thais, M.E.R.; Dresch Vascouto, H.; Remor, A.P.; Lopes, M.W.; Linhares, M.N.; Ben, J.; de Paula Martins, R.; et al. Mitochondrial respiratory chain complex enzyme activities of limbic structures and psychiatric diagnosis in temporal lobe epilepsy patients: Preliminary results. CNS Neurosci. Ther. 2017, 23, 700–702. [Google Scholar] [CrossRef] [PubMed]
- Folbergrova, J.; Jesina, P.; Haugvicova, R.; Lisy, V.; Houstek, J. Sustained deficiency of mitochondrial complex I activity during long periods of survival after seizures induced in immature rats by homocysteic acid. Neurochem. Int. 2010, 56, 394–403. [Google Scholar] [CrossRef]
- Chuang, Y.C.; Chang, A.Y.; Lin, J.W.; Hsu, S.P.; Chan, S.H. Mitochondrial dysfunction and ultrastructural damage in the hippocampus during kainic acid-induced status epilepticus in the rat. Epilepsia 2004, 45, 1202–1209. [Google Scholar] [CrossRef]
- Chuang, Y.C.; Chen, S.D.; Liou, C.W.; Lin, T.K.; Chang, W.N.; Chan, S.H.; Chang, A.Y. Contribution of nitric oxide, superoxide anion, and peroxynitrite to activation of mitochondrial apoptotic signaling in hippocampal CA3 subfield following experimental temporal lobe status epilepticus. Epilepsia 2009, 50, 731–746. [Google Scholar] [CrossRef] [PubMed]
- Kudin, A.P.; Zsurka, G.; Elger, C.E.; Kunz, W.S. Mitochondrial involvement in temporal lobe epilepsy. Exp. Neurol. 2009, 218, 326–332. [Google Scholar] [CrossRef] [PubMed]
- Kunz, W.S.; Kudin, A.P.; Vielhaber, S.; Blumcke, I.; Zuschratter, W.; Schramm, J.; Beck, H.; Elger, C.E. Mitochondrial complex I deficiency in the epileptic focus of patients with temporal lobe epilepsy. Ann. Neurol. 2000, 48, 766–773. [Google Scholar] [CrossRef]
- Huang, C.W.; Lin, K.M.; Hung, T.Y.; Chuang, Y.C.; Wu, S.N. Multiple Actions of Rotenone, an Inhibitor of Mitochondrial Respiratory Chain, on Ionic Currents and Miniature End-Plate Potential in Mouse Hippocampal (mHippoE-14) Neurons. Cell Physiol. Biochem. 2018, 47, 330–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.D.; Zhen, Y.Y.; Lin, J.W.; Lin, T.K.; Huang, C.W.; Liou, C.W.; Chan, S.H.; Chuang, Y.C. Dynamin-Related Protein 1 Promotes Mitochondrial Fission and Contributes to The Hippocampal Neuronal Cell Death Following Experimental Status Epilepticus. CNS Neurosci. Ther. 2016, 22, 988–999. [Google Scholar] [CrossRef] [PubMed]
- Chuang, Y.C.; Chen, S.D.; Lin, T.K.; Liou, C.W.; Chang, W.N.; Chan, S.H.; Chang, A.Y. Upregulation of nitric oxide synthase II contributes to apoptotic cell death in the hippocampal CA3 subfield via a cytochrome c/caspase-3 signaling cascade following induction of experimental temporal lobe status epilepticus in the rat. Neuropharmacology 2007, 52, 1263–1273. [Google Scholar] [CrossRef]
- Balog, J.; Mehta, S.L.; Vemuganti, R. Mitochondrial fission and fusion in secondary brain damage after CNS insults. J. Cereb. Blood Flow Metab. 2016, 36, 2022–2033. [Google Scholar] [CrossRef]
- Kim, H.; Lee, J.Y.; Park, K.J.; Kim, W.H.; Roh, G.S. A mitochondrial division inhibitor, Mdivi-1, inhibits mitochondrial fragmentation and attenuates kainic acid-induced hippocampal cell death. BMC Neurosci. 2016, 17, 33. [Google Scholar] [CrossRef]
- Houtkooper, R.H.; Pirinen, E.; Auwerx, J. Sirtuins as regulators of metabolism and healthspan. Nat. Rev. Mol. Cell Biol. 2012, 13, 225–238. [Google Scholar] [CrossRef] [Green Version]
- Al Massadi, O.; Quinones, M.; Lear, P.; Dieguez, C.; Nogueiras, R. The brain: A new organ for the metabolic actions of SIRT1. Horm. Metab. Res. 2013, 45, 960–966. [Google Scholar] [CrossRef]
- Nemoto, S.; Fergusson, M.M.; Finkel, T. SIRT1 functionally interacts with the metabolic regulator and transcriptional coactivator PGC-1{alpha}. J. Biol. Chem. 2005, 280, 16456–16460. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.J.; Zhao, X.H.; Chen, W.; Bo, N.; Wang, X.J.; Chi, Z.F.; Wu, W. Sirtuin 1 activation enhances the PGC-1alpha/mitochondrial antioxidant system pathway in status epilepticus. Mol. Med. Rep. 2015, 11, 521–526. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Lin, Y.; Xie, N.; Xue, Y.; Tao, H.; Rui, C.; Xu, J.; Cao, L.; Liu, X.; Jiang, H.; et al. Impaired mitochondrial biogenesis in hippocampi of rats with chronic seizures. Neuroscience 2011, 194, 234–240. [Google Scholar] [CrossRef] [PubMed]
- Scarpulla, R.C. Transcriptional paradigms in mammalian mitochondrial biogenesis and function. Physiol. Rev. 2008, 88, 611–638. [Google Scholar] [CrossRef]
- Yin, K.J.; Chen, S.D.; Lee, J.M.; Xu, J.; Hsu, C.Y. ATM gene regulates oxygen-glucose deprivation-induced nuclear factor-kappaB DNA-binding activity and downstream apoptotic cascade in mouse cerebrovascular endothelial cells. Stroke 2002, 33, 2471–2477. [Google Scholar] [CrossRef]
- Chen, H.; Hu, C.J.; He, Y.Y.; Yang, D.I.; Xu, J.; Hsu, C.Y. Reduction and restoration of mitochondrial dna content after focal cerebral ischemia/reperfusion. Stroke 2001, 32, 2382–2387. [Google Scholar] [CrossRef]
- Chuang, Y.C.; Lin, T.K.; Huang, H.Y.; Chang, W.N.; Liou, C.W.; Chen, S.D.; Chang, A.Y.; Chan, S.H. Peroxisome proliferator-activated receptors gamma/mitochondrial uncoupling protein 2 signaling protects against seizure-induced neuronal cell death in the hippocampus following experimental status epilepticus. J. Neuroinflammation 2012, 9, 184. [Google Scholar] [CrossRef] [PubMed]
- Haigis, M.C.; Sinclair, D.A. Mammalian sirtuins: Biological insights and disease relevance. Annu. Rev. Pathol. 2010, 5, 253–295. [Google Scholar] [CrossRef]
- Chen, B.; Zang, W.; Wang, J.; Huang, Y.; He, Y.; Yan, L.; Liu, J.; Zheng, W. The chemical biology of sirtuins. Chem. Soc. Rev. 2015, 44, 5246–5264. [Google Scholar] [CrossRef]
- Cormio, A.; Guerra, F.; Cormio, G.; Pesce, V.; Fracasso, F.; Loizzi, V.; Cantatore, P.; Selvaggi, L.; Gadaleta, M.N. The PGC-1alpha-dependent pathway of mitochondrial biogenesis is upregulated in type I endometrial cancer. Biochem. Biophys. Res. Commun. 2009, 390, 1182–1185. [Google Scholar] [CrossRef]
- Ramadori, G.; Lee, C.E.; Bookout, A.L.; Lee, S.; Williams, K.W.; Anderson, J.; Elmquist, J.K.; Coppari, R. Brain SIRT1: Anatomical distribution and regulation by energy availability. J. Neurosci. 2008, 28, 9989–9996. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Si, P.; Qin, H.; Yin, L.; Yan, L.J.; Zhang, C. The Neuroprotective Effects of SIRT1 on NMDA-Induced Excitotoxicity. Oxid. Med. Cell Longev. 2017, 2017, 2823454. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Xu, S.; Qian, Y.; Xiao, Q. Resveratrol regulates microglia M1/M2 polarization via PGC-1alpha in conditions of neuroinflammatory injury. Brain Behav. Immun. 2017, 64, 162–172. [Google Scholar] [CrossRef] [PubMed]
- Sun, A.Y.; Wang, Q.; Simonyi, A.; Sun, G.Y. Resveratrol as a therapeutic agent for neurodegenerative diseases. Mol. Neurobiol. 2010, 41, 375–383. [Google Scholar] [CrossRef]
- Oppenheimer, H.; Gabay, O.; Meir, H.; Haze, A.; Kandel, L.; Liebergall, M.; Gagarina, V.; Lee, E.J.; Dvir-Ginzberg, M. 75-kd sirtuin 1 blocks tumor necrosis factor alpha-mediated apoptosis in human osteoarthritic chondrocytes. Arthritis. Rheum. 2012, 64, 718–728. [Google Scholar] [CrossRef]
- Dvir-Ginzberg, M.; Gagarina, V.; Lee, E.J.; Booth, R.; Gabay, O.; Hall, D.J. Tumor necrosis factor alpha-mediated cleavage and inactivation of SirT1 in human osteoarthritic chondrocytes. Arthritis. Rheum. 2011, 63, 2363–2373. [Google Scholar] [CrossRef]
- Takayama, K.; Ishida, K.; Matsushita, T.; Fujita, N.; Hayashi, S.; Sasaki, K.; Tei, K.; Kubo, S.; Matsumoto, T.; Fujioka, H.; et al. SIRT1 regulation of apoptosis of human chondrocytes. Arthritis. Rheum. 2009, 60, 2731–2740. [Google Scholar] [CrossRef]
- Zhang, F.; Wang, S.; Gan, L.; Vosler, P.S.; Gao, Y.; Zigmond, M.J.; Chen, J. Protective effects and mechanisms of sirtuins in the nervous system. Prog. Neurobiol. 2011, 95, 373–395. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Jackson, C.W.; Khoury, N.; Escobar, I.; Perez-Pinzon, M.A. Brain SIRT1 Mediates Metabolic Homeostasis and Neuroprotection. Front Endocrinol. (Lausanne) 2018, 9, 702. [Google Scholar] [CrossRef]
- Rizzi, L.; Roriz-Cruz, M. Sirtuin 1 and Alzheimer’s disease: An up-to-date review. Neuropeptides 2018, 71, 54–60. [Google Scholar] [CrossRef]
- Wan, D.; Zhou, Y.; Wang, K.; Hou, Y.; Hou, R.; Ye, X. Resveratrol provides neuroprotection by inhibiting phosphodiesterases and regulating the cAMP/AMPK/SIRT1 pathway after stroke in rats. Brain Res. Bull 2016, 121, 255–262. [Google Scholar] [CrossRef]
- Zhang, J.F.; Zhang, Y.L.; Wu, Y.C. The Role of Sirt1 in Ischemic Stroke: Pathogenesis and Therapeutic Strategies. Front Neurosci. 2018, 12, 833. [Google Scholar] [CrossRef] [PubMed]
- Folbergrova, J.; Jesina, P.; Kubova, H.; Otahal, J. Effect of Resveratrol on Oxidative Stress and Mitochondrial Dysfunction in Immature Brain during Epileptogenesis. Mol. Neurobiol. 2018, 55, 7512–7522. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Xu, Q.; Zhang, L.; Kong, D.; Ma, R.; Wang, L. Protective effect of resveratrol against kainate-induced temporal lobe epilepsy in rats. Neurochem. Res. 2009, 34, 1393–1400. [Google Scholar] [CrossRef] [PubMed]
- Castro, O.W.; Upadhya, D.; Kodali, M.; Shetty, A.K. Resveratrol for Easing Status Epilepticus Induced Brain Injury, Inflammation, Epileptogenesis, and Cognitive and Memory Dysfunction-Are We There Yet? Front Neurol. 2017, 8, 603. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; You, Z.; Li, M.; Pang, L.; Cheng, J.; Wang, L. Protective Effect of Resveratrol on the Brain in a Rat Model of Epilepsy. Neurosci. Bull 2017, 33, 273–280. [Google Scholar] [CrossRef]
- Folbergrova, J.; Kunz, W.S. Mitochondrial dysfunction in epilepsy. Mitochondrion 2012, 12, 35–40. [Google Scholar] [CrossRef]
- Scharfman, H.E. The neurobiology of epilepsy. Curr. Neurol. Neurosci. Rep. 2007, 7, 348–354. [Google Scholar] [CrossRef]
- Belenguer, P.; Duarte, J.M.N.; Schuck, P.F.; Ferreira, G.C. Mitochondria and the Brain: Bioenergetics and Beyond. Neurotox. Res. 2019. [Google Scholar] [CrossRef]
- Yang, J.L.; Mukda, S.; Chen, S.D. Diverse roles of mitochondria in ischemic stroke. Redox. Biol. 2018, 16, 263–275. [Google Scholar] [CrossRef]
- Simon, R.; Henshall, D.; Stoehr, S.; Meller, R. Endogenous mechanisms of neuroprotection. Epilepsia 2007, 48 (Suppl. 8), 72–73. [Google Scholar] [CrossRef]
- Berkeley, J.L.; Decker, M.J.; Levey, A.I. The role of muscarinic acetylcholine receptor-mediated activation of extracellular signal-regulated kinase 1/2 in pilocarpine-induced seizures. J. Neurochem. 2002, 82, 192–201. [Google Scholar] [CrossRef] [PubMed]
- Nicoletti, J.N.; Lenzer, J.; Salerni, E.A.; Shah, S.K.; Elkady, A.; Khalid, S.; Quinteros, D.; Rotella, F.; Betancourth, D.; Croll, S.D. Vascular endothelial growth factor attenuates status epilepticus-induced behavioral impairments in rats. Epilepsy Behav. 2010, 19, 272–277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jimenez-Mateos, E.M.; Henshall, D.C. Seizure preconditioning and epileptic tolerance: Models and mechanisms. Int. J. Physiol. Pathophysiol Pharmacol. 2009, 1, 180–191. [Google Scholar] [PubMed]
- Sanchez, P.E.; Fares, R.P.; Risso, J.J.; Bonnet, C.; Bouvard, S.; Le-Cavorsin, M.; Georges, B.; Moulin, C.; Belmeguenai, A.; Bodennec, J.; et al. Optimal neuroprotection by erythropoietin requires elevated expression of its receptor in neurons. Proc. Natl. Acad. Sci. USA 2009, 106, 9848–9853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, H.C.; Guarente, L. SIRT1 mediates central circadian control in the SCN by a mechanism that decays with aging. Cell 2013, 153, 1448–1460. [Google Scholar] [CrossRef] [PubMed]
- Ng, F.; Wijaya, L.; Tang, B.L. SIRT1 in the brain-connections with aging-associated disorders and lifespan. Front Cell Neurosci. 2015, 9, 64. [Google Scholar] [CrossRef]
- Singh, P.; Hanson, P.S.; Morris, C.M. SIRT1 ameliorates oxidative stress induced neural cell death and is down-regulated in Parkinson’s disease. BMC Neurosci. 2017, 18, 46. [Google Scholar] [CrossRef]
- Zhou, Y.; Wang, S.; Li, Y.; Yu, S.; Zhao, Y. SIRT1/PGC-1alpha Signaling Promotes Mitochondrial Functional Recovery and Reduces Apoptosis after Intracerebral Hemorrhage in Rats. Front Mol. Neurosci. 2017, 10, 443. [Google Scholar] [CrossRef]
- Makela, J.; Tselykh, T.V.; Kukkonen, J.P.; Eriksson, O.; Korhonen, L.T.; Lindholm, D. Peroxisome proliferator-activated receptor-gamma (PPARgamma) agonist is neuroprotective and stimulates PGC-1alpha expression and CREB phosphorylation in human dopaminergic neurons. Neuropharmacology 2016, 102, 266–275. [Google Scholar] [CrossRef]
- Liu, P.; Zou, D.; Yi, L.; Chen, M.; Gao, Y.; Zhou, R.; Zhang, Q.; Zhou, Y.; Zhu, J.; Chen, K.; et al. Quercetin ameliorates hypobaric hypoxia-induced memory impairment through mitochondrial and neuron function adaptation via the PGC-1alpha pathway. Restor. Neurol. Neurosci. 2015, 33, 143–157. [Google Scholar] [CrossRef] [PubMed]
- Chuang, Y.C.; Chen, S.D.; Hsu, C.Y.; Chen, S.F.; Chen, N.C.; Jou, S.B. Resveratrol Promotes Mitochondrial Biogenesis and Protects against Seizure-Induced Neuronal Cell Damage in the Hippocampus Following Status Epilepticus by Activation of the PGC-1alpha Signaling Pathway. Int. J. Mol. Sci. 2019, 20, 998. [Google Scholar] [CrossRef] [PubMed]
- Hall, A.M.; Brennan, G.P.; Nguyen, T.M.; Singh-Taylor, A.; Mun, H.S.; Sargious, M.J.; Baram, T.Z. The Role of Sirt1 in Epileptogenesis. eNeuro 2017, 4. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Cruzat, V.F.; Newsholme, P.; Cheng, J.; Chen, Y.; Lu, Y. Regulation of SIRT1 in aging: Roles in mitochondrial function and biogenesis. Mech Ageing Dev 2016, 155, 10–21. [Google Scholar] [CrossRef] [PubMed]
- Tang, B.L. Sirt1 and the Mitochondria. Mol. Cells 2016, 39, 87–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Y.; Wang, H.; Fang, J.; Dai, W.; Zhou, J.; Wang, X.; Zhou, M. SS-31 Provides Neuroprotection by Reversing Mitochondrial Dysfunction after Traumatic Brain Injury. Oxid. Med. Cell Longev. 2018, 2018, 4783602. [Google Scholar] [CrossRef] [PubMed]
- Ye, Q.; Ye, L.; Xu, X.; Huang, B.; Zhang, X.; Zhu, Y.; Chen, X. Epigallocatechin-3-gallate suppresses 1-methyl-4-phenyl-pyridine-induced oxidative stress in PC12 cells via the SIRT1/PGC-1alpha signaling pathway. BMC Complement Altern. Med. 2012, 12, 82. [Google Scholar] [CrossRef] [PubMed]
- Scarpulla, R.C.; Vega, R.B.; Kelly, D.P. Transcriptional integration of mitochondrial biogenesis. Trends Endocrinol Metab. 2012, 23, 459–466. [Google Scholar] [CrossRef] [Green Version]
- Jornayvaz, F.R.; Shulman, G.I. Regulation of mitochondrial biogenesis. Essays Biochem. 2010, 47, 69–84. [Google Scholar] [CrossRef] [Green Version]
- Gadaleta, M.N.; Cormio, A.; Pesce, V.; Lezza, A.M.; Cantatore, P. Aging and mitochondria. Biochimie 1998, 80, 863–870. [Google Scholar] [CrossRef]
- Wu, Z.; Puigserver, P.; Andersson, U.; Zhang, C.; Adelmant, G.; Mootha, V.; Troy, A.; Cinti, S.; Lowell, B.; Scarpulla, R.C.; et al. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell 1999, 98, 115–124. [Google Scholar] [CrossRef]
- Chen, S.D.; Yang, D.I.; Lin, T.K.; Shaw, F.Z.; Liou, C.W.; Chuang, Y.C. Roles of oxidative stress, apoptosis, PGC-1alpha and mitochondrial biogenesis in cerebral ischemia. Int. J. Mol. Sci. 2011, 12, 7199–7215. [Google Scholar] [CrossRef] [PubMed]
- Villena, J.A. New insights into PGC-1 coactivators: Redefining their role in the regulation of mitochondrial function and beyond. FEBS J. 2015, 282, 647–672. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.C.; Chen, S.D.; Lin, T.K.; Chang, W.N.; Liou, C.W.; Chang, A.Y.; Chan, S.H.; Chuang, Y.C. Heat shock protein 70 protects against seizure-induced neuronal cell death in the hippocampus following experimental status epilepticus via inhibition of nuclear factor-kappaB activation-induced nitric oxide synthase II expression. Neurobiol. Dis. 2014, 62, 241–249. [Google Scholar] [CrossRef] [PubMed]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chuang, Y.-C.; Chen, S.-D.; Jou, S.-B.; Lin, T.-K.; Chen, S.-F.; Chen, N.-C.; Hsu, C.-Y. Sirtuin 1 Regulates Mitochondrial Biogenesis and Provides an Endogenous Neuroprotective Mechanism Against Seizure-Induced Neuronal Cell Death in the Hippocampus Following Status Epilepticus. Int. J. Mol. Sci. 2019, 20, 3588. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20143588
Chuang Y-C, Chen S-D, Jou S-B, Lin T-K, Chen S-F, Chen N-C, Hsu C-Y. Sirtuin 1 Regulates Mitochondrial Biogenesis and Provides an Endogenous Neuroprotective Mechanism Against Seizure-Induced Neuronal Cell Death in the Hippocampus Following Status Epilepticus. International Journal of Molecular Sciences. 2019; 20(14):3588. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20143588
Chicago/Turabian StyleChuang, Yao-Chung, Shang-Der Chen, Shuo-Bin Jou, Tsu-Kung Lin, Shu-Fang Chen, Nai-Ching Chen, and Chung-Yao Hsu. 2019. "Sirtuin 1 Regulates Mitochondrial Biogenesis and Provides an Endogenous Neuroprotective Mechanism Against Seizure-Induced Neuronal Cell Death in the Hippocampus Following Status Epilepticus" International Journal of Molecular Sciences 20, no. 14: 3588. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20143588