Cardiac Cx43 and ECM Responses to Altered Thyroid Status Are Blunted in Spontaneously Hypertensive versus Normotensive Rats

 ,
,
Abstract
:1. Introduction
2. Results
2.1. Characteristics of Experimental Rats
2.2. Myocardial MYH7, SERCA2, and PPARγ mRNA Transcripts
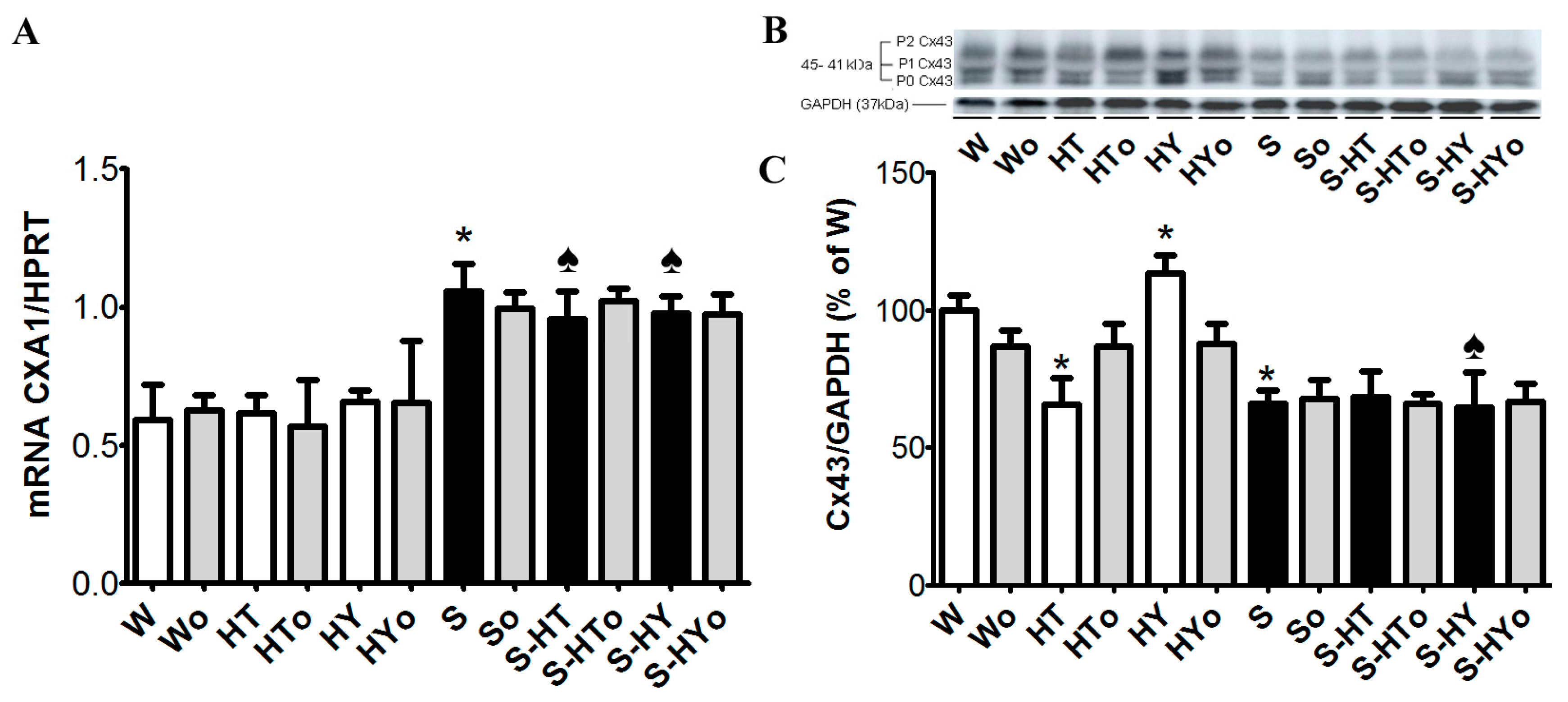
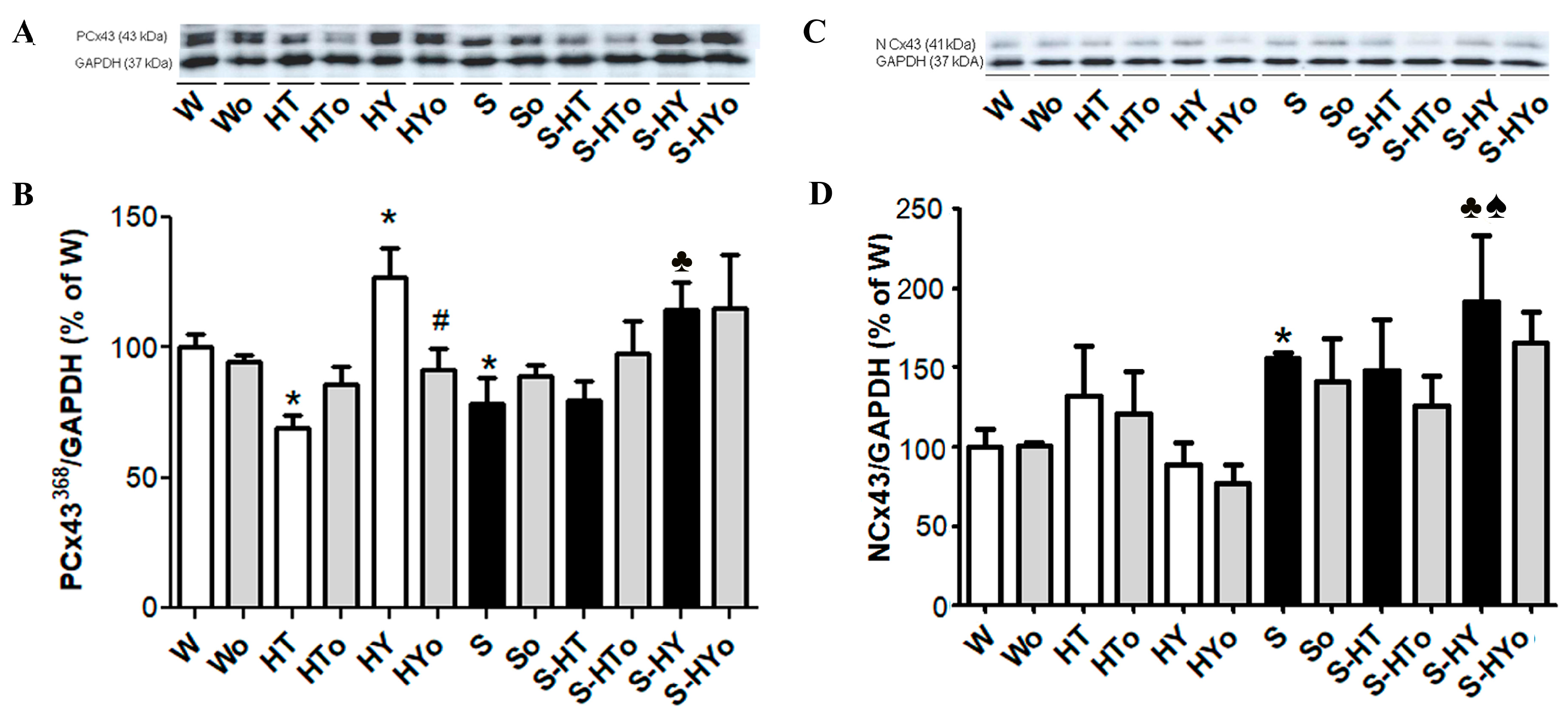
2.3. Myocardial Cx43 mRNA Transcript and Protein Levels
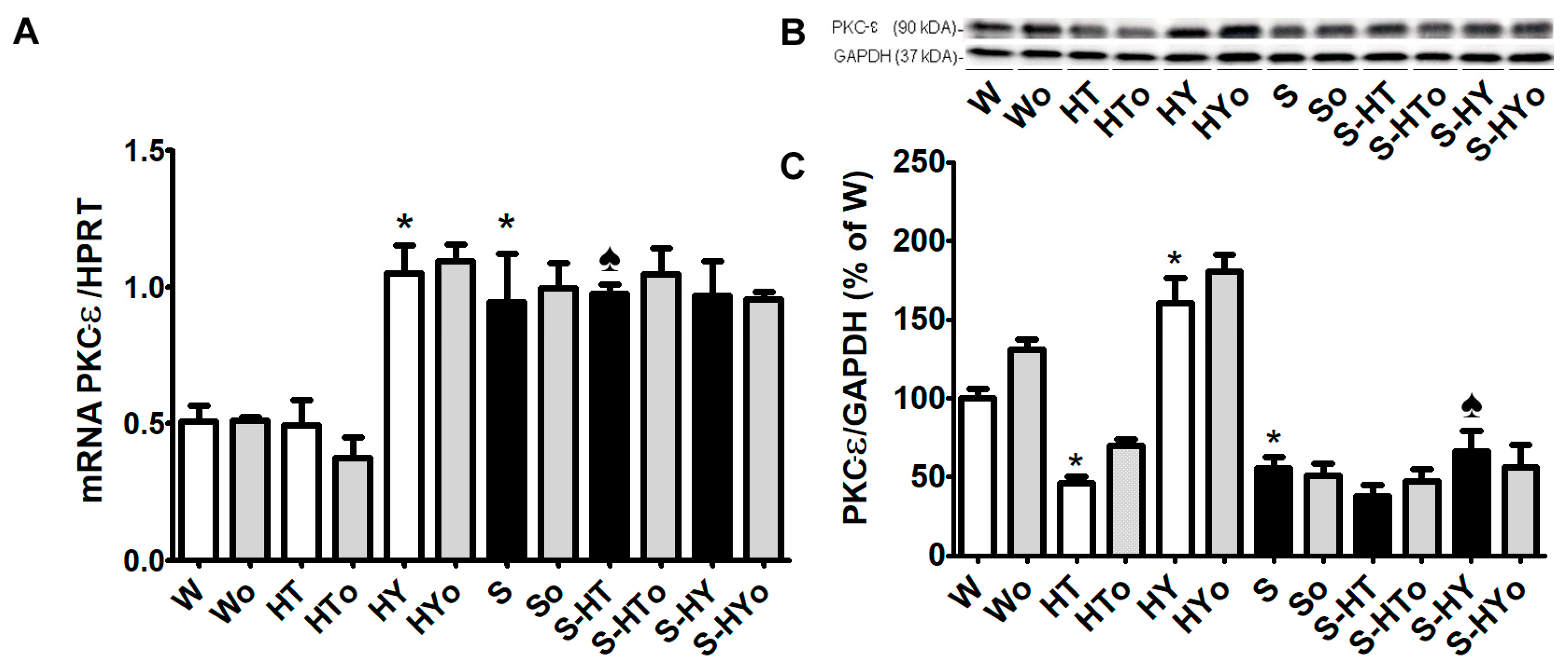
2.4. Myocardial PKC-ε mRNA Transcript and Protein Levels
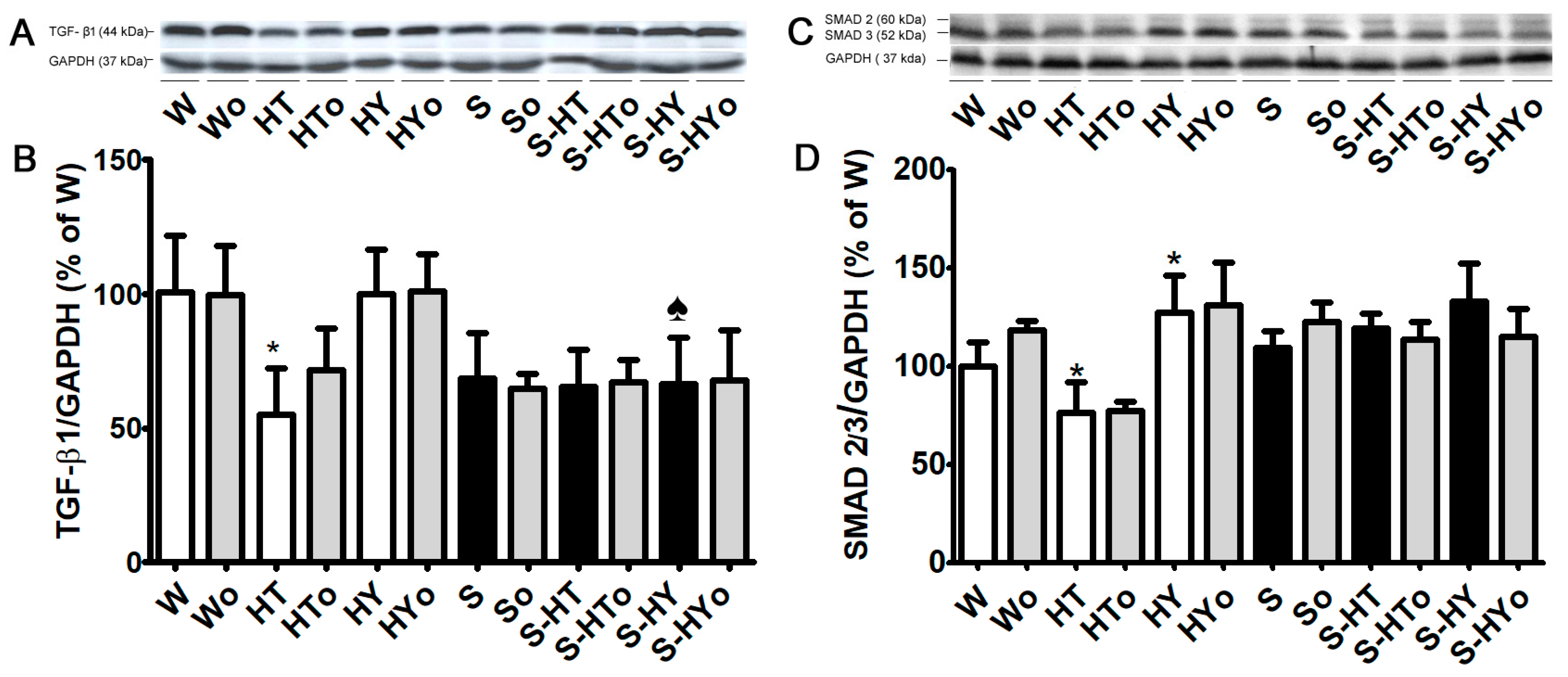
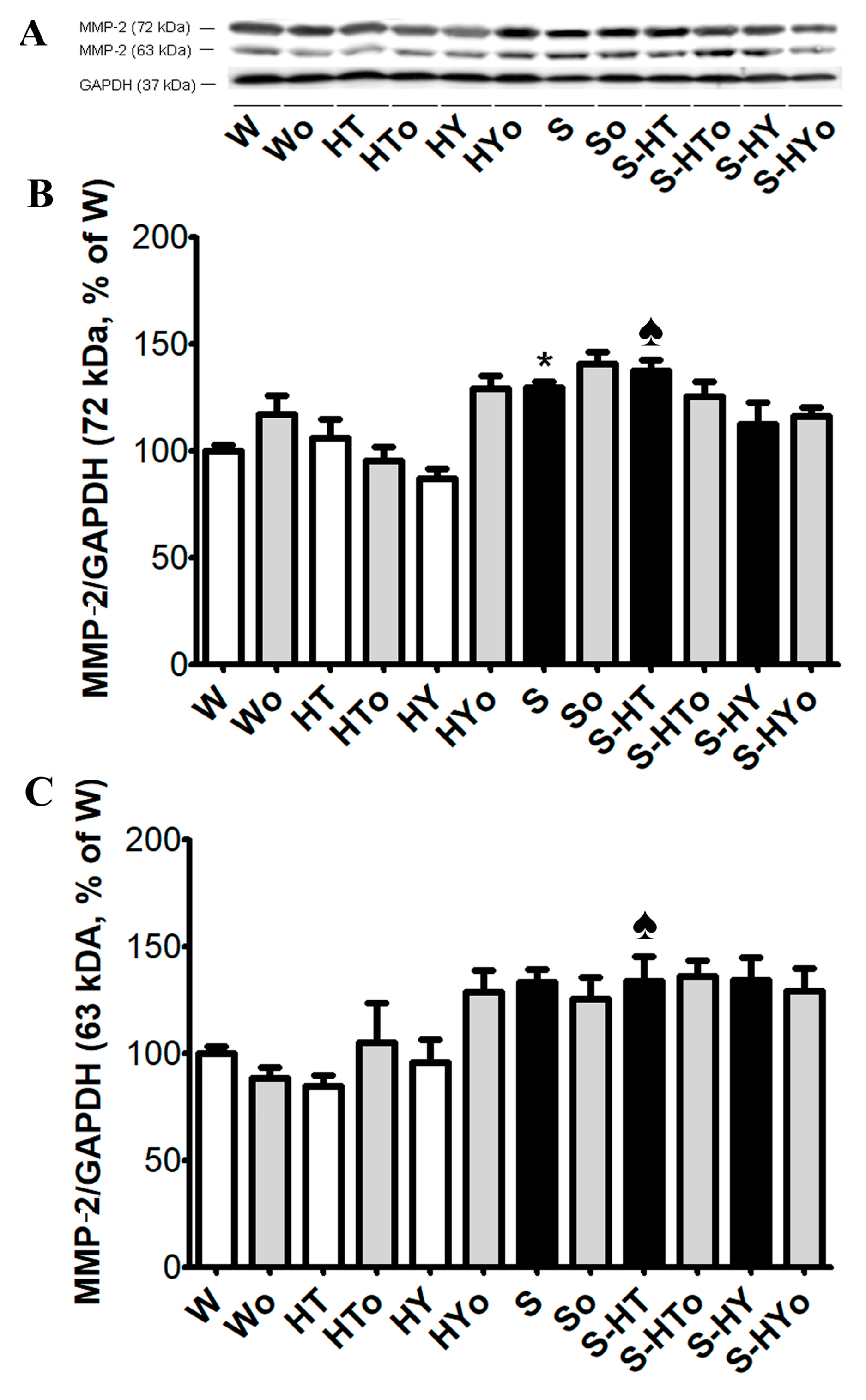
2.5. Myocardial TGF-β1, SMAD2/3, and MMP-2 Protein Levels
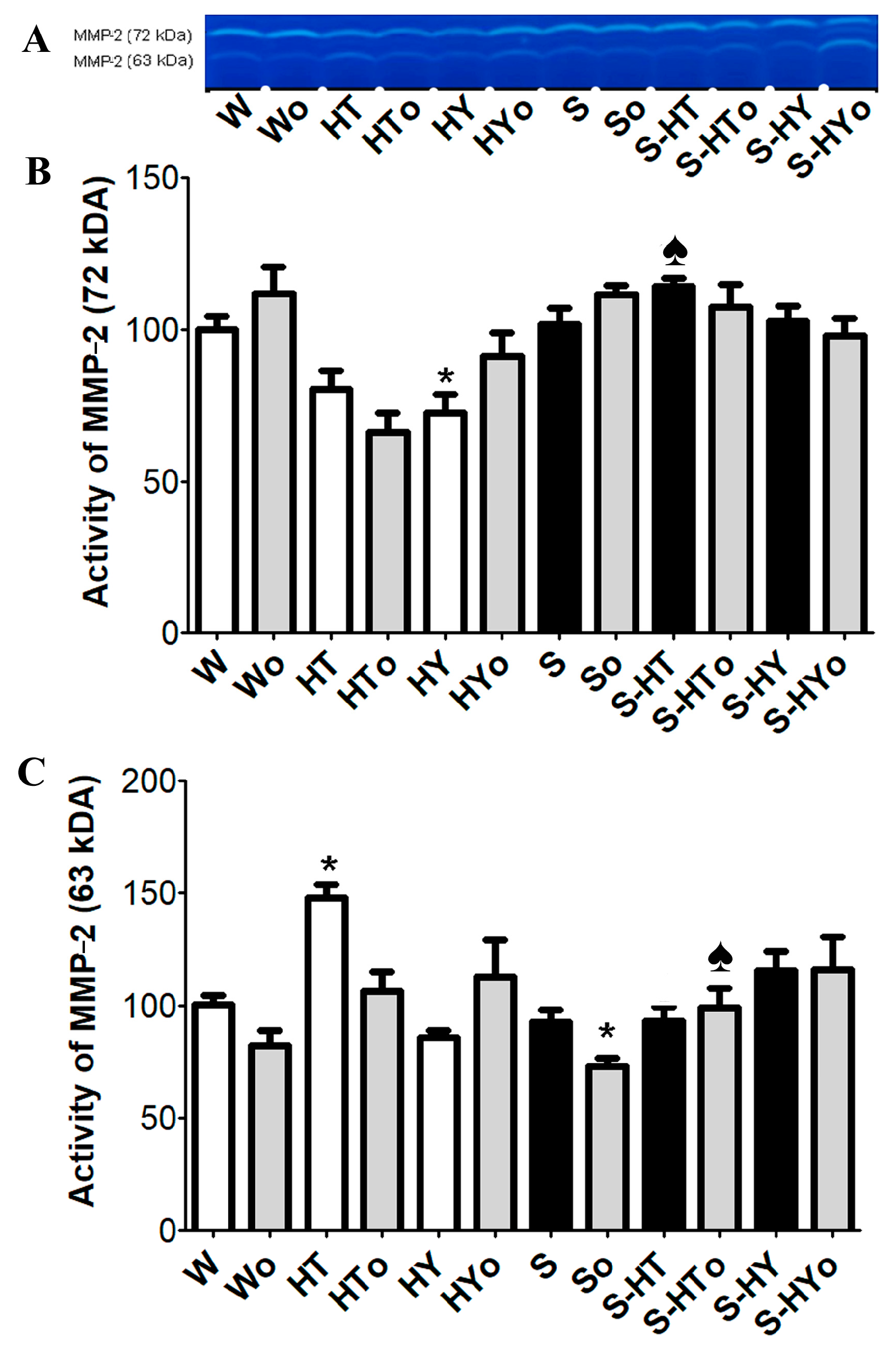
2.6. Myocardial MMP-2 Activity
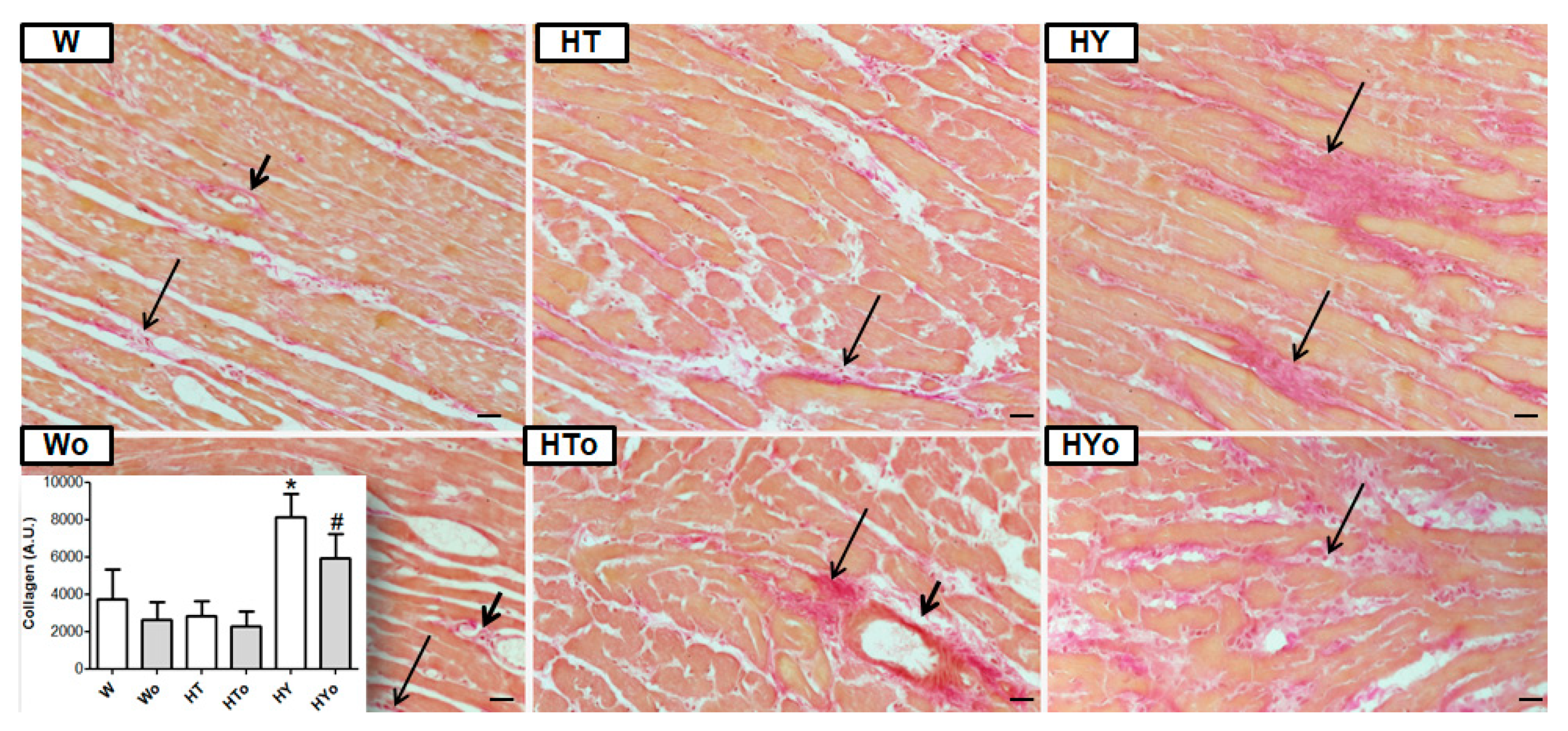
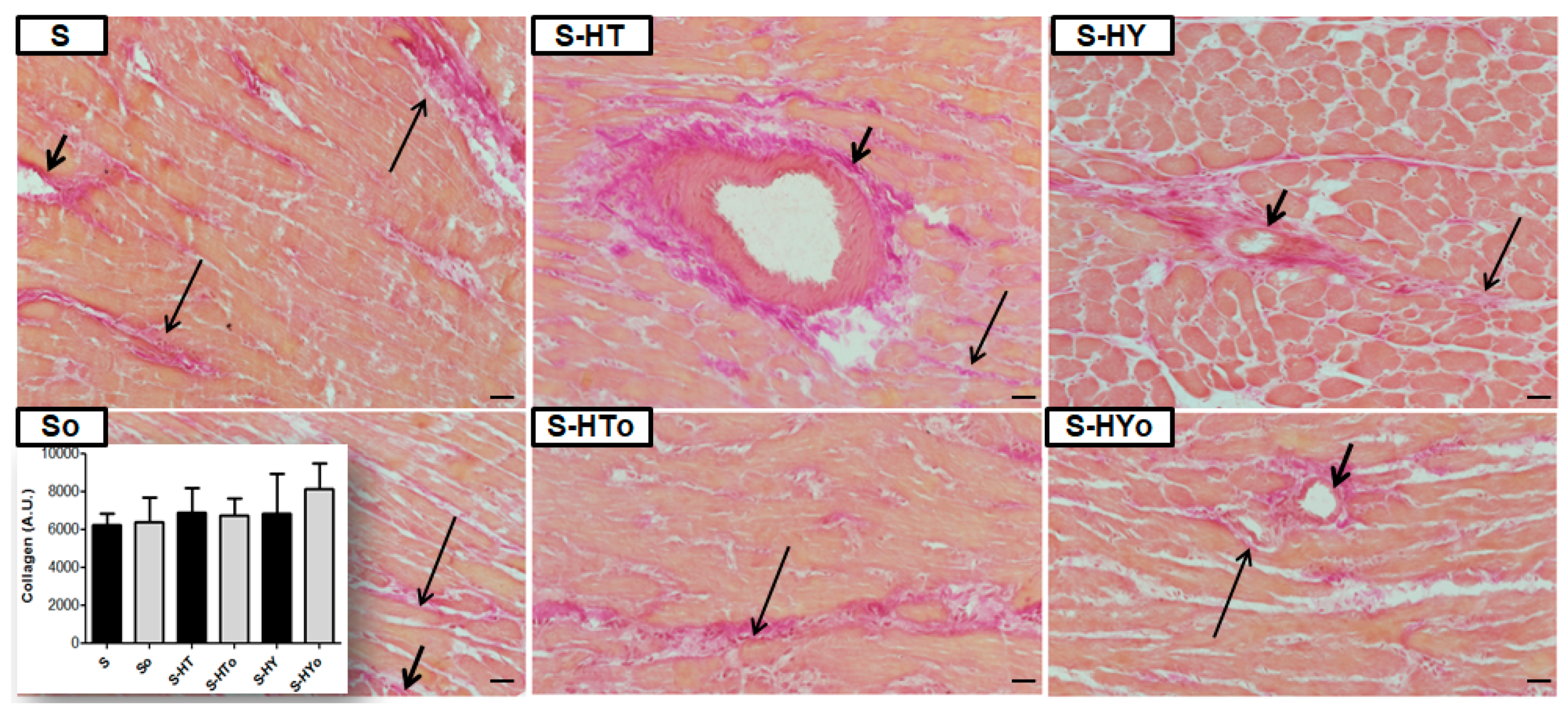
2.7. Myocardial Collagen Deposition
3. Discussion
Limitations of the Study
4. Materials and Methods
4.1. Experimental Design
4.2. Bioactive Parameters Assessed in Blood Samples
4.3. Lipid Peroxidation Assay
4.4. Determination of mRNA by Real-Time PCR
4.5. SDS-PAGE and WB analysis of proteins
4.6. Gelatin Zymography
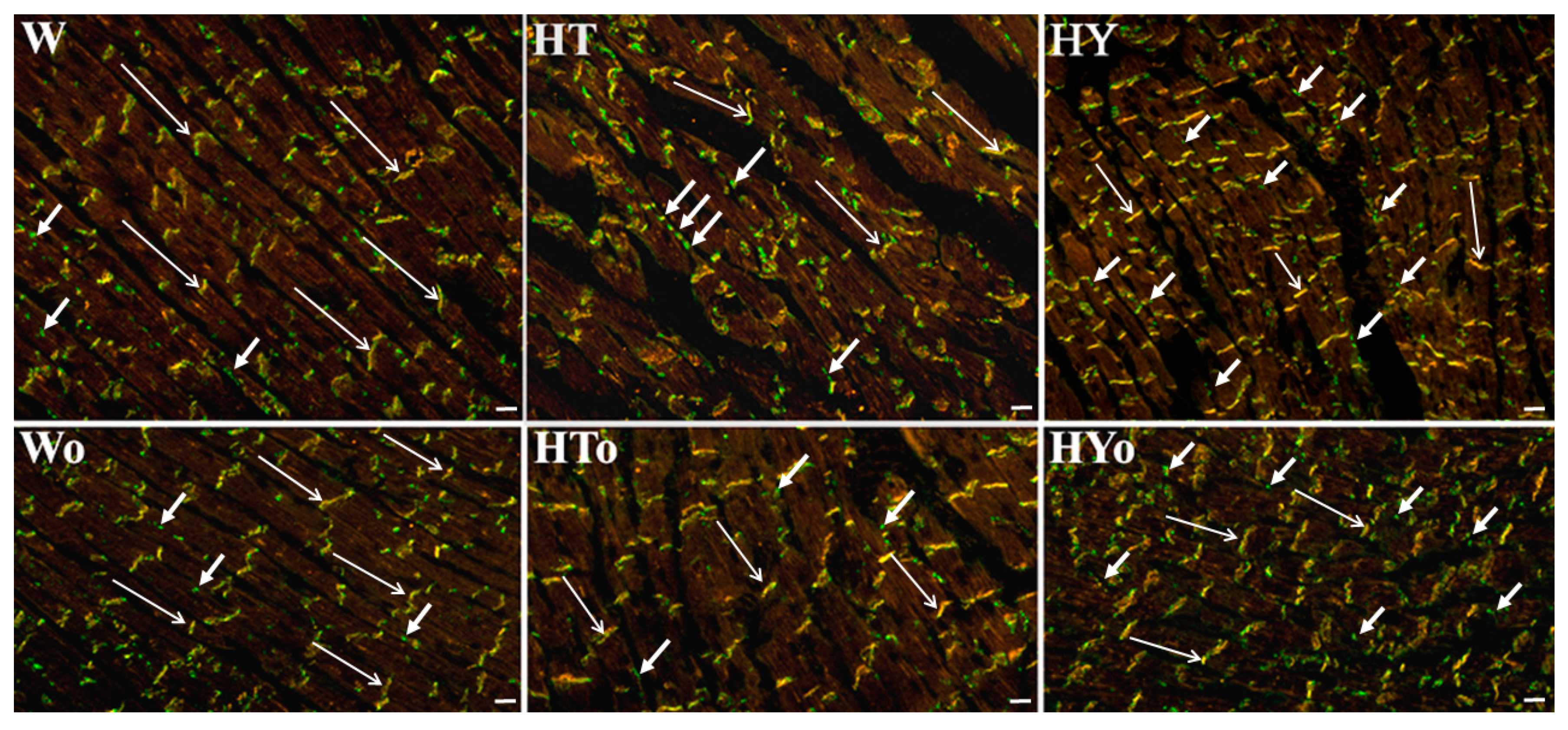
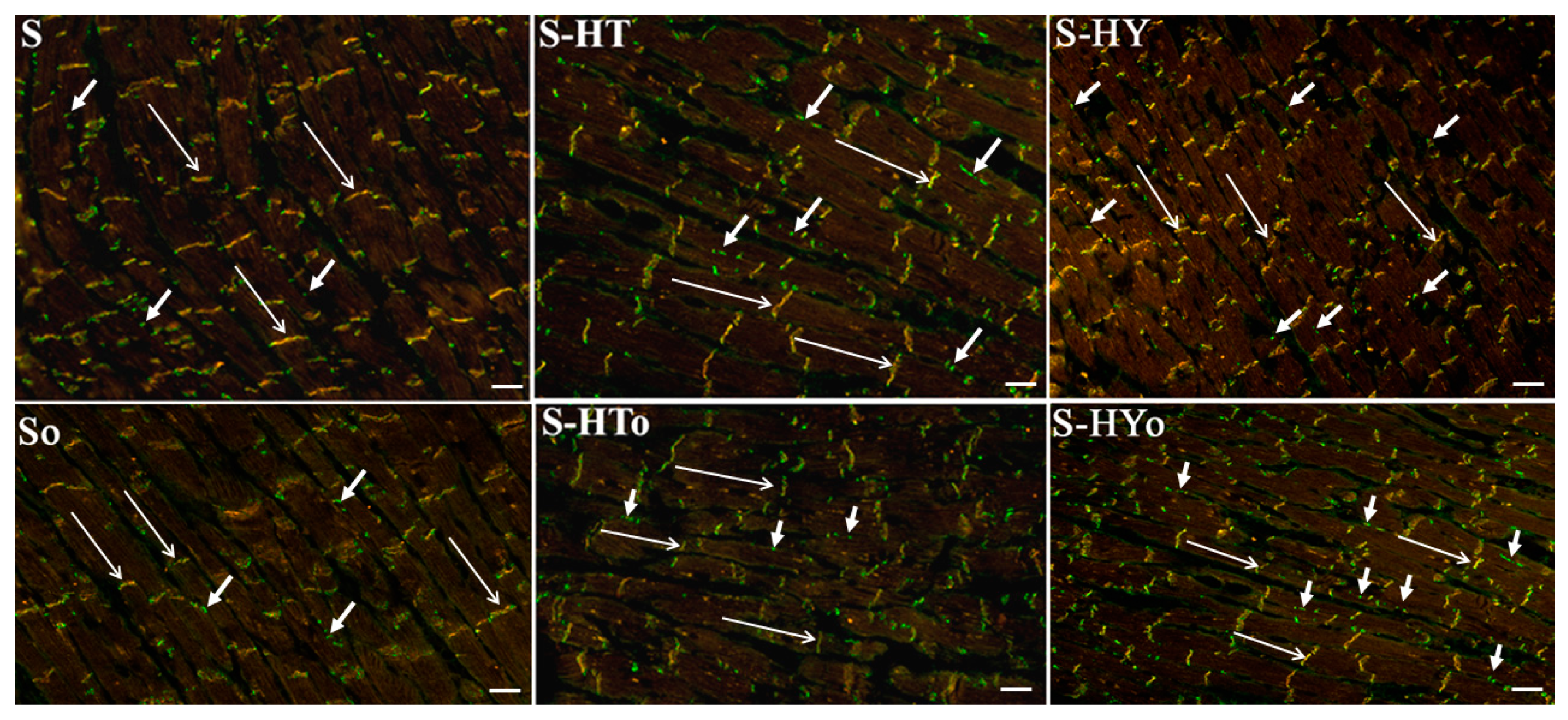
4.7. Immunolabeling of Cx43
4.8. Quantitative Image Analysis of Collagen Deposition
4.9. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Dillmann, W.H. Cellular action of thyroid hormone on the heart. Thyroid 2002, 12, 447–452. [Google Scholar] [CrossRef] [PubMed]
- Tribulova, N.; Knezl, V.; Shainberg, A.; Seki, S.; Soukup, T. Thyroid hormones and cardiac arrhythmias. Vascul. Pharmacol. 2010, 52, 102–112. [Google Scholar] [CrossRef] [PubMed]
- Soukup, T.; Smerdu, V. Effect of altered innervation and thyroid hormones on myosin heavy chain expression and fiber type transitions: A mini-review. Histochem. Cell Biol. 2014, 143, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Klein, I.; Danzi, S. Thyroid disease and the heart. Curr. Probl. Cardiol. 2016, 41, 65–92. [Google Scholar] [CrossRef] [PubMed]
- Razvi, S.; Jabbar, A.; Pingitore, A.; Danzi, S.; Biondi, B.; Klein, I.; Peeters, R.; Zaman, A.; Iervasi, G. Thyroid hormones and cardiovascular function and diseases. J. Am. Coll. Cardiol. 2018, 71, 1781–1796. [Google Scholar] [CrossRef] [PubMed]
- Dillmann, W. Cardiac hypertrophy and thyroid hormone signaling. Heart Fail. Rev. 2010, 15, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Rybin, V.; Steinberg, S.F. Thyroid hormone represses protein kinase C isoform expression and activity in rat cardiac myocytes. Circ. Res. 1996, 79, 388–398. [Google Scholar] [CrossRef]
- Zinman, T.; Shneyvays, V.; Tribulova, N.; Manoach, M.; Shainberg, A. Acute, nongenomic effect of thyroid hormones in preventing calcium overload in newborn rat cardiocytes. J. Cell. Physiol. 2006, 207, 220–231. [Google Scholar] [CrossRef]
- Biondi, B.; Palmieri, E.A.; Fazio, S.; Cosco, C.; Nocera, M.; Saccà, L.; Filetti, S.; Lombardi, G.; Perticone, F. Endogenous subclinical hyperthyroidism affects quality of life and cardiac morphology and function in young and middle-aged patients. J. Clin. Endocrinol. Metab. 2000, 85, 4701–4705. [Google Scholar]
- Pantos, C.; Mourouzis, I. Translating thyroid hormone effects into clinical practice: The relevance of thyroid hormone receptor α1 in cardiac repair. Heart Fail. Rev. 2015, 20, 273–282. [Google Scholar] [CrossRef]
- Knezl, V.; Soukup, T.; Okruhlicová, L.; Slezák, J.; Tribulová, N. Thyroid hormones modulate occurrence and termination of ventricular fibrillation by both long-term and acute actions. Physiol. Res. 2008, 57, 91–96. [Google Scholar]
- Ellervik, C.; Roselli, C.; Christophersen, I.E.; Alonso, A.; Pietzner, M.; Sitlani, C.M.; Trompet, S.; Arking, D.E.; Geelhoed, B.; Guo, X.; et al. Assessment of the relationship between genetic determinants of thyroid function and atrial fibrillation: A mendelian randomization study. JAMA Cardiol. 2019, 4, 144–152. [Google Scholar] [CrossRef] [PubMed]
- Bačová, B.; Viczenczová, C.; Žurmanová, J.; Kašparová, D.; Knezl, V.; Radošinská, J.; Beňová, T.; Pavelka, S.; Soukup, T.; Tribulová, N. Susceptibility of rats with altered thyroid status to malignant arrhythmias is primarily related to myocardial levels of connexin-43 and can be partially ameliorated by supplementation with red palm oil. Exp. Cardiol. 2013, 18, 41–46. [Google Scholar]
- Szeiffová Bačova, B.; Egan Beňová, T.; Viczenczová, C.; Soukup, T.; Rauchová, H.; Pavelka, S.; Knezl, V.; Barancík, M.; Tribulová, N. Cardiac connexin-43 and PKC signaling in rats with altered thyroid status without and with omega-3 fatty acids intake. Physiol. Res. 2016, 65, 77–90. [Google Scholar]
- Lin, H.; Mitasikova, M.; Dlugosova, K.; Okruhlicova, L.; Imanaga, I.; Ogawa, K.; Weismann, P.; Tribulova, N. Thyroid hormones suppress ε-PKC signalling, down-regulate connexin-43 and increase lethal arrhythmia susceptibility in non-diabetic and diabetic rat hearts. J. Physiol. Pharmacol. 2008, 59, 271–285. [Google Scholar] [PubMed]
- Mitašíková, M.; Lin, H.; Soukup, T.; Imanaga, I.; Tribulová, N. Diabetes and thyroid hormones affect connexin-43 and PKC-ε expression in rat heart atria. Physiol. Res. 2009, 58, 211–217. [Google Scholar] [PubMed]
- Bačová, B.S.; Vinczenzová, C.; Žurmanová, J.; Kašparová, D.; Knezl, V.; Beňová, T.E.; Pavelka, S.; Soukup, T.; Tribulová, N. Altered thyroid status affects myocardial expression of connexin-43 and susceptibility of rat heart to malignant arrhythmias that can be partially normalized by red palm oil intake. Histochem. Cell Biol. 2017, 147, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Saffitz, J.E.; Laing, J.G.; Yamada, K.A. Connexin expression and turnover implications for cardiac excitability. Circ. Res. 2000, 86, 723–728. [Google Scholar] [CrossRef]
- Danik, S.B.; Liu, F.; Zhang, J.; Suk, H.J.; Morley, G.E.; Fishman, G.I.; Gutstein, D.E. Modulation of cardiac gap junction expression and arrhythmic susceptibility. Circ. Res. 2004, 95, 1035–1041. [Google Scholar] [CrossRef]
- Yao, J.; Eghbali, M. Decreased collagen gene expression and absence of fibrosis in thyroid hormone-induced myocardial hypertrophy: Response of cardiac fibroblasts to thyroid hormone in vitro. Circ. Res. 1992, 71, 831–839. [Google Scholar] [CrossRef]
- Ziegelhöffer-Mihalovičová, B.; Briest, W.; Baba, H.A.; Raßler, B.; Zimmer, H.G. The expression of mRNA of cytokines and of extracellular matrix proteins in triiodothyronine-treated rat hearts. Mol. Cell. Biochem. 2003, 247, 61–68. [Google Scholar] [CrossRef]
- Tribulová, N.; Knezl, V.; Okruhlicová, L.; Slezák, J. Myocardial gap junctions: Targets for novel approaches in the prevention of life-threatening cardiac arrhythmias. Physiol. Res. 2008, 57, 1–13. [Google Scholar]
- Salvarani, N.; Maguy, A.; De Simone, S.A.; Miragoli, M.; Jousset, F.; Rohr, S. TGF-β1 (transforming growth factor-β1) plays a pivotal role in cardiac myofibroblast arrhythmogenicity. Circ. Arrhythmia Electrophysiol. 2017, 10, 1–14. [Google Scholar] [CrossRef]
- Lüscher, T.F. Dangerous encounters: Triggers of fatal ventricular arrhythmias and safety of interventional treatment strategies. Eur. Heart, J. 2018, 39, 3909–3912. [Google Scholar] [CrossRef]
- Severs, N.J.; Bruce, A.F.; Dupont, E.; Rothery, S. Remodelling of gap junctions and connexin expression in diseased myocardium. Cardiovasc. Res. 2008, 80, 9–19. [Google Scholar] [CrossRef] [Green Version]
- Benova, T.; Knezl, V.; Viczenczova, C.; Bacova, B.S.; Radosinska, J.; Tribulova, N. Acute anti-fibrillating and defibrillating potential of atorvastatin, melatonin, eicosapentaenoic acid and docosahexaenoic acid demonstrated in isolated heart model. J. Physiol. Pharmacol. 2015, 66. [Google Scholar]
- Radosinska, J.; Bacova, B.; Knezl, V.; Benova, T.; Zurmanova, J.; Soukup, T.; Arnostova, P.; Slezak, J.; Goncalvesova, E.; Tribulova, N. Dietary omega-3 fatty acids attenuate myocardial arrhythmogenic factors and propensity of the heart to lethal arrhythmias in a rodent model of human essential hypertension. J. Hypertens. 2013, 31, 1876–1885. [Google Scholar] [CrossRef]
- Tribulova, N.; Bacova, B.S.; Benova, T.E.; Knezl, V.; Barancik, M.; Slezak, J. Omega-3 index and anti-arrhythmic potential of omega-3 PUFAs. Nutrients 2017, 9. [Google Scholar] [CrossRef]
- Bruhn, T.O.; Jackson, I.M.D. Abnormalities of the thyroid hormone negative feedback regulation of TSH secretion in spontaneously hypertensive rats. Regul. Pept. 1992, 38, 221–230. [Google Scholar] [CrossRef]
- Wang, B.; Ouyang, J.; Xia, Z. Effects of triiodo-thyronine on angiotensin-induced cardiomyocyte hypertrophy: Reversal of increased β-myosin heavy chain gene expression. Can. J. Physiol. Pharmacol. 2006, 84, 935–941. [Google Scholar] [CrossRef]
- Nattel, S.; Maguy, A.; Le Bouter, S.; Yeh, Y.-H. Arrhythmogenic ion-channel remodeling in the heart: Heart failure, myocardial infarction, and atrial fibrillation. Physiol. Rev. 2007, 87, 425–456. [Google Scholar] [CrossRef]
- Kurebayashi, N.; Nishizawa, H.; Nakazato, Y.; Kurihara, H.; Matsushita, S.; Daida, H.; Ogawa, Y. Aberrant cell-to-cell coupling in Ca 2+ -overloaded guinea pig ventricular muscles. Am. J. Physiol. Physiol. 2008, 294, C1419–C1429. [Google Scholar] [CrossRef]
- Benova, T.; Viczenczova, C.; Radosinska, J.; Bacova, B.; Knezl, V.; Dosenko, V.; Weismann, P.; Zeman, M.; Navarova, J.; Tribulova, N. Melatonin attenuates hypertension-related proarrhythmic myocardial maladaptation of connexin-43 and propensity of the heart to lethalarrhythmias. Can. J. Physiol. Pharmacol. 2013, 91, 633–639. [Google Scholar] [CrossRef]
- Bao, X.; Reuss, L.; Altenberg, G.A. Regulation of purified and reconstituted connexin 43 hemichannels by protein kinase C-mediated phosphorylation of serine 368. J. Biol. Chem. 2004, 279, 20058–20066. [Google Scholar] [CrossRef]
- Egan Benova, T.; Szeiffova Bacova, B.; Viczenczova, C.; Diez, E.; Barancik, M.; Tribulova, N. Protection of cardiac cell-to-cell coupling attenuate myocardial remodeling and proarrhythmia induced by hypertension. Physiol. Res. 2016, 65, 29–42. [Google Scholar]
- Palatinus, J.A.; Rhett, J.M.; Gourdie, R.G. Enhanced PKCε mediated phosphorylation of connexin43 at serine 368 by a carboxyl-terminal mimetic peptide is dependent on injury. Channels 2011, 5, 236–240. [Google Scholar] [CrossRef]
- Kohutova, J.; Elsnicova, B.; Holzerova, K.; Neckar, J.; Sebesta, O.; Jezkova, J.; Vecka, M.; Vebr, P.; Hornikova, D.; Szeiffova Bacova, B.; et al. Anti-arrhythmic cardiac phenotype elicited by chronic intermittent hypoxia is associated with alterations in connexin-43 expression, phosphorylation, and distribution. Front. Endocrinol. (Lausanne). 2019, 9, 1–10. [Google Scholar] [CrossRef]
- Tribulova, N.; Szeiffova Bacova, B.; Benova, T.; Viczenczova, C. Can we protect from malignant arrhythmias by modulation of cardiac cell-to-cell coupling? J. Electrocardiol. 2015, 48, 434–440. [Google Scholar] [CrossRef]
- Solan, J.L.; Lampe, P.D. Spatio-temporal regulation of connexin43 phosphorylation and gap junction dynamics. Biochim Biophys Acta Biomembr. 2018, 1860, 83–90. [Google Scholar] [CrossRef]
- Peters, N.S.; Coromilas, J.; Severs, N.J.; Wit, A.L. Disturbed connexin43 gap junction distribution correlates with the location of reentrant circuits in the epicardial border zone of healing canine infarcts that cause ventricular tachycardia. Circulation 1997, 95, 988–996. [Google Scholar] [CrossRef]
- Duffy, H. The molecular mechanisms of gap junction remodeling. Hear. Rhythm. 2012, 9, 1331–1334. [Google Scholar] [CrossRef]
- Severs, N.J. Gap junction remodeling and cardiac arrhythmogenesis: Cause or coincidence? J. Cell. Mol. Med. 2001, 5, 355–366. [Google Scholar] [CrossRef]
- Hajje, G.; Saliba, Y.; Itani, T.; Moubarak, M.; Aftimos, G.; Farès, N. Hypothyroidism and its rapid correction alter cardiac remodeling. PLoS ONE 2014, 9, 1–13. [Google Scholar] [CrossRef]
- Chen, W.-J.; Lin, K.-H.; Lee, Y.-S. Molecular characterization of myocardial fibrosis during hypothyroidism: Evidence for negative regulation of the pro-α1(I) collagen gene expression by thyroid hormone receptor. Mol. Cell. Endocrinol. 2000, 162, 45–55. [Google Scholar] [CrossRef]
- Karim, M.A.; Ferguson, A.G.; Wakim, B.T.; Samarel, A.M. In vivo collagen turnover during development of thyroxine-induced left ventricular hypertrophy. Am. J. Physiol. Physiol. 1991, 260, C316–C326. [Google Scholar] [CrossRef]
- Ghose Roy, S.; Mishra, S.; Ghosh, G.; Bandyopadhyay, A. Thyroid hormone induces myocardial matrix degradation by activating matrix metalloproteinase-1. Matrix Biol. 2007, 26, 269–279. [Google Scholar] [CrossRef]
- Boulaksil, M.; Winckels, S.K.G.; Engelen, M.A.; Stein, M.; Van Veen, T.A.B.; Jansen, J.A.; Linnenbank, A.C.; Bierhuizen, M.F.A.; Groenewegen, W.A.; Van Oosterhout, M.F.M.; et al. Heterogeneous connexin43 distribution in heart failure is associated with dispersed conduction and enhanced susceptibility to ventricular arrhythmias. Eur. J. Heart Fail. 2010, 12, 913–921. [Google Scholar] [CrossRef]
- Kurtz TW, M.R.J. Biological variability in Wistar-Kyoto rats. Implications for research with the spontaneously hypertensive rat. Hypertension 1987, 10, 127–131. [Google Scholar] [CrossRef]
- Zhang-james, Y.; Middleton, F.A.; Faraone, S. V Genetic architecture of Wistar-Kyoto rat and spontaneously hypertensive rat substrains from different sources. Physiol Genomics 2013, 45, 528–538. [Google Scholar] [CrossRef]
- Rauchová, H.; Vokurková, M.; Pavelka, S.; Behuliak, M.; Tribulová, N.; Soukup, T. N-3 polyunsaturated fatty acids supplementation does not affect changes of lipid metabolism induced in rats by altered thyroid status. Horm. Metab. Res. 2013, 45, 507–512. [Google Scholar] [CrossRef]
- Holzerová, K.; Hlaváčková, M.; Žurmanová, J.; Borchert, G.; Neckář, J.; Kolář, F.; Novák, F.; Nováková, O. Involvement of PKCε in cardioprotection induced by adaptation to chronic continuous hypoxia. Physiol. Res. 2015, 64, 191–201. [Google Scholar]
- Barancik, M.; Bohacova, V.; Gibalova, L.; Sedlak, J.; Sulova, Z.; Breier, A. Potentiation of anticancer drugs: Effects of pentoxifylline on neoplastic cells. Int. J. Mol. Sci. 2012, 13, 369–382. [Google Scholar] [CrossRef]
- Sykora, M.; Kamocsaiova, L.; Benova, T.E.; Frimmel, K.; Ujhazy, E.; Mach, M.; Barancik, M.; Tribulova, N.; Bacova, B.S. Alterations in myocardial connexin-43 and matrix metalloproteinase-2 signaling in response to pregnancy and oxygen deprivation of Wistar rats: a pilot study. Can J Physiol Pharmacol. 2019. [Epub ahead of print]. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variables | Intake | WKRs | WKR-HTs | WKR-HYs | SHRs | SHR-HTs | SHR-HYs |
|---|---|---|---|---|---|---|---|
| T3 (nmol/L) | - | 0.88 ± 0.29 | 1.67 ± 0.55 * | 0.26 ± 0.11 * | 0.86 ± 0.21 | 1.61 ± 0.48 ♣ | 0.21 ± 0.04 ♣ |
| Omacor | 1.03 ± 0.26 | 1.25 ± 0.24 | 0.20 ± 0.05 | 0.97 ± 0.23 | 1.30 ± 0.28 | 0.16 ± 0.04 | |
| T4 (nmol/L) | - | 60.52 ± 7.91 | 101.71 ± 38.51 * | 8.76 ± 4.55 * | 39.17 ± 11.29 | 78.39 ± 20.91 ♣ | 11.71 ± 2.11 |
| Omacor | 54.27 ± 15.81 | 67.26 ± 10.95 # | 5.95 ± 3.32 | 55.02 ± 10.11 | 52.63 ± 10.77 | 6.98 ± 2.65 | |
| BG (mmol/L) | - | 4.97 ± 0.59 | 5.53 ± 0.40 | 5.97 ± 0.61 | 6.85 ± 0.58 | 5.57 ± 0.06 | 6.50 ± 0.72 |
| Omacor | 5.43±1.00 | 4.80 ± 0.61 | 5.48 ± 0.59 | 7.02 ± 0.63 | 5.85 ± 0.69 | 5.38 ± 0.58 | |
| TGW (mg) | - | 38 ± 3 | 33 ± 8 | 94 ± 24 * | 22 ± 5 | 18 ± 4 | 54 ± 10♣♠ |
| Omacor | 51 ± 8 | 38 ± 10 | 106 ± 15 | 31 ± 8 | 17 ± 6 | 45 ± 4 | |
| BW (g) | - | 444 ± 36 | 405 ± 27 | 431 ± 21 | 410 ± 25 | 368 ± 22 | 387 ± 39 |
| Omacor | 362 ± 38 # | 339 ± 29 # | 416 ± 24 | 433 ± 28 | 355 ± 10 | 360 ± 12 | |
| HW (g) | - | 1.18 ± 0.08 | 1.56 ± 0.05 * | 0.88 ± 0.12 * | 1.72 ± 0.18 * | 2.10 ± 0.44 ♣♠ | 1.24 ± 0.15 ♣♠ |
| Omacor | 0.92 ± 0.1 | 1.49 ± 0.15 | 0.95 ± 0.06 | 1.65 ± 0.13 | 1.88 ± 0.11 | 1.09 ± 0.15 | |
| LVW (g) | - | 0.84 ± 0.05 | 1.15 ± 0.12 * | 0.68 ± 0.05 * | 1.32 ± 0.09 * | 1.62 ± 0.29 ♣♠ | 0.98 ± 0.11 ♣♠ |
| Omacor | 0.68 ± 0.10 | 1.02 ± 0.11 | 0.69 ± 0.06 | 1.30 ± 0.17 | 1.51 ± 0.12 | 0.91 ± 0.09 | |
| TBARSs (nmol/mg) | - | 1.42 ± 0.41 | 2.74 ± 0.45 * | 1.43 ± 0.35 | 1.49 ± 0.15 | 1.35 ± 0.28 | 1.31 ± 0.21 |
| Omacor | 1.94 ± 0.25 | 1.94 ± 0.34 # | 1.45 ± 0.23 | 1.83 ± 0.62 | 1.59 ± 0.23 | 1.62 ± 0.17 |
| Variables | Intake | WKRs | WKR-HTs | WKR-HYs | SHRs | SHR-HTs | SHR-HYs |
|---|---|---|---|---|---|---|---|
| TAG (mmol/L) | - | 1.18 ± 0.34 | 0.84 ± 0.14 | 0.62 ± 0.22 * | 0.80 ± 0.19 | 0.88 ± 0.43 | 0.52 ± 0.05 |
| Omacor | 0.77 ± 0.27 | 0.53 ± 0.16 | 0.44 ± 0.05 | 0.81 ± 0.23 | 0.55 ± 0.13 | 1.51 ± 0.04 | |
| HDLchol (mmol/L) | - | 1.11 ± 0.13 | 1.13 ± 0.19 | 1.66 ± 0.14 * | 0.99 ± 0.06 | 1.07 ± 0.09 | 1.83 ± 0.41 ♣ |
| Omacor | 1.33 ± 0.28 | 0.83 ± 0.08 | 1.84 ± 0.26 | 0.95 ± 0.13 | 0.87 ± 0.15 | 2.18 ± 0.12 | |
| LDLchol (mmol/L) | - | 0.35 ± 0.08 | 0.33 ± 0.11 | 0.80 ± 0.16 * | 0.42 ± 0.09 | 0.52 ± 0.11 | 1.06 ± 0.15 ♣♠ |
| Omacor | 0.54 ± 0.33 | 0.20 ± 0.05 | 0.79 ± 0.12 | 0.44 ± 0.10 | 0.28 ± 0.10 | 1.01 ± 0.23 | |
| Tchol/HLDchol | - | 1.53 ± 0.11 | 1.46 ± 0.16 | 1.56 ± 0.09 | 1.38 ± 0.57 | 1.65 ± 0.16 | 1.65 ± 0.12 |
| Omacor | 1.67 ± 0.14 | 1.38 ± 0.07 | 1.48 ± 0.05 | 1.64 ± 0.10 | 1.44 ± 0.06 | 1.57 ± 0.09 |
| Variables | Intake | WKRs | WKR-HTs | WKR-HYs | SHRs | SHR-HTs | SHR-HYs |
|---|---|---|---|---|---|---|---|
| mRNA MYH7 | - | 37.78 ± 4.39 | 3.55 ± 0.95 * | 132.60 ± 41.59 * | 237.40 ± 67.27 * | 48.161 ± 25.781 ♣ | 368.82 ± 97.55 ♣♠ |
| Omacor | 22.94 ± 3.95 | 4.64 ± 2.15 | 81.24 ± 6.78 | 246.10 ± 91.50 | 68.462 ± 22.53 | 441.72 ± 39.92 | |
| mRNA SERCA2 | - | 43.77 ± 7.60 | 61.52 ± 16.26 | 20.08 ± 3.95 * | 36.07 ± 2.36 | 45.07 ± 5.09 | 13.42 ± 5.09 |
| Omacor | 40.95 ± 9.99 | 46.71 ± 12.72 | 19.22 ± 5.04 | 31.20 ± 13.47 | 32.47 ± 5.94 | 15.35 ± 4.44 | |
| mRNA PPARγ | - | 0.03 ± 0.01 | 0.06 ± 0.02 * | 0.02 ± 0.01 | 0.03 ± 0.01 | 0.05 ± 0.002 | 0.02 ± 0.002 |
| Omacor | 0.05 ± 0.01 | 0.05 ± 0.02 | 0.03 ± 0.01 | 0.02 ± 0.01 | 0.04 ± 0.004 | 0.02 ± 0.003 |
| Transcript | Forward (5′→3′) | Reverse (5′→3′) |
|---|---|---|
| Connexin-43 | AGCCTGAACTCTCATTTTTCCTT | CCATGTCTGGGCACCTCT |
| Protein kinase C epsilon | AAACACCCTTATCTAACCCAACTCT | CATATTCCATGACGAAGAAGAGC |
| β-myosin heavy chain | GCACCGTGGACTACAATATCCT | 5CTTAGGAGCTTGAGGGAGGAC |
| Sarcoplasmic reticulum Ca2+ATPase | ACCTGGAAGATTCTGCGAAC | AATCCTGGGAGGGTCCAG |
| Peroxisome proliferator-activated receptor-γ | CCCAATGGTTGCTGATTACA | GGACGCAGGCTCTACTTTGA |
| Hypoxanthine guanine phosphoribosyl transferase | GACCGGTTCTGTCATGTCG | ACCTGGTTCATCATCACTAATCAC |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sykora, M.; Szeiffova Bacova, B.; Egan Benova, T.; Barancik, M.; Zurmanova, J.; Rauchova, H.; Weismann, P.; Pavelka, S.; Kurahara, L.H.; Slezak, J.; et al. Cardiac Cx43 and ECM Responses to Altered Thyroid Status Are Blunted in Spontaneously Hypertensive versus Normotensive Rats. Int. J. Mol. Sci. 2019, 20, 3758. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20153758
Sykora M, Szeiffova Bacova B, Egan Benova T, Barancik M, Zurmanova J, Rauchova H, Weismann P, Pavelka S, Kurahara LH, Slezak J, et al. Cardiac Cx43 and ECM Responses to Altered Thyroid Status Are Blunted in Spontaneously Hypertensive versus Normotensive Rats. International Journal of Molecular Sciences. 2019; 20(15):3758. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20153758
Chicago/Turabian StyleSykora, Matus, Barbara Szeiffova Bacova, Tamara Egan Benova, Miroslav Barancik, Jitka Zurmanova, Hana Rauchova, Peter Weismann, Stanislav Pavelka, Lin Hai Kurahara, Jan Slezak, and et al. 2019. "Cardiac Cx43 and ECM Responses to Altered Thyroid Status Are Blunted in Spontaneously Hypertensive versus Normotensive Rats" International Journal of Molecular Sciences 20, no. 15: 3758. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20153758