Comparative Proteomics Indicates That Redox Homeostasis Is Involved in High- and Low-Temperature Stress Tolerance in a Novel Wucai (Brassica campestris L.) Genotype

 , , and
, , and
Abstract
:1. Introduction
2. Results
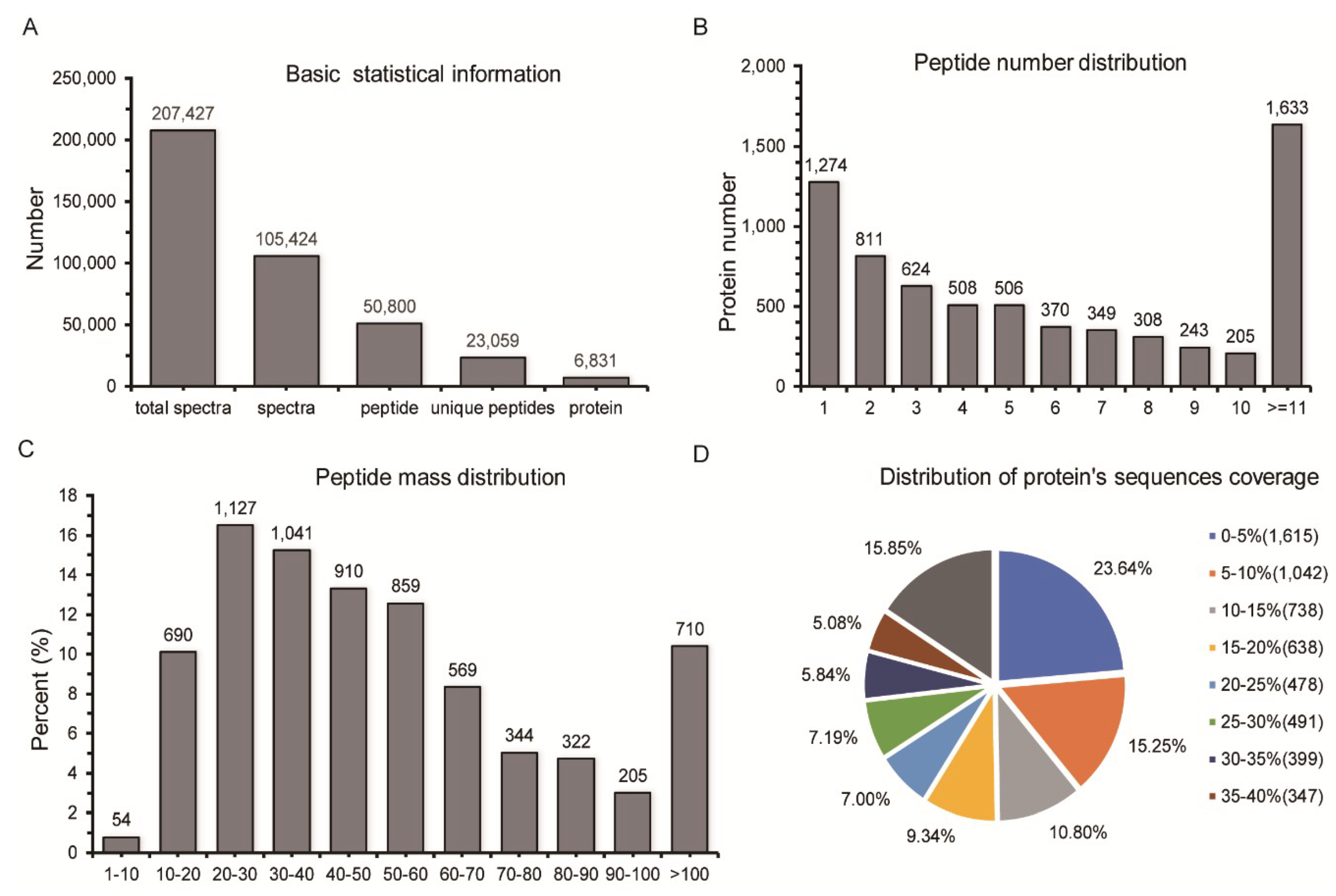
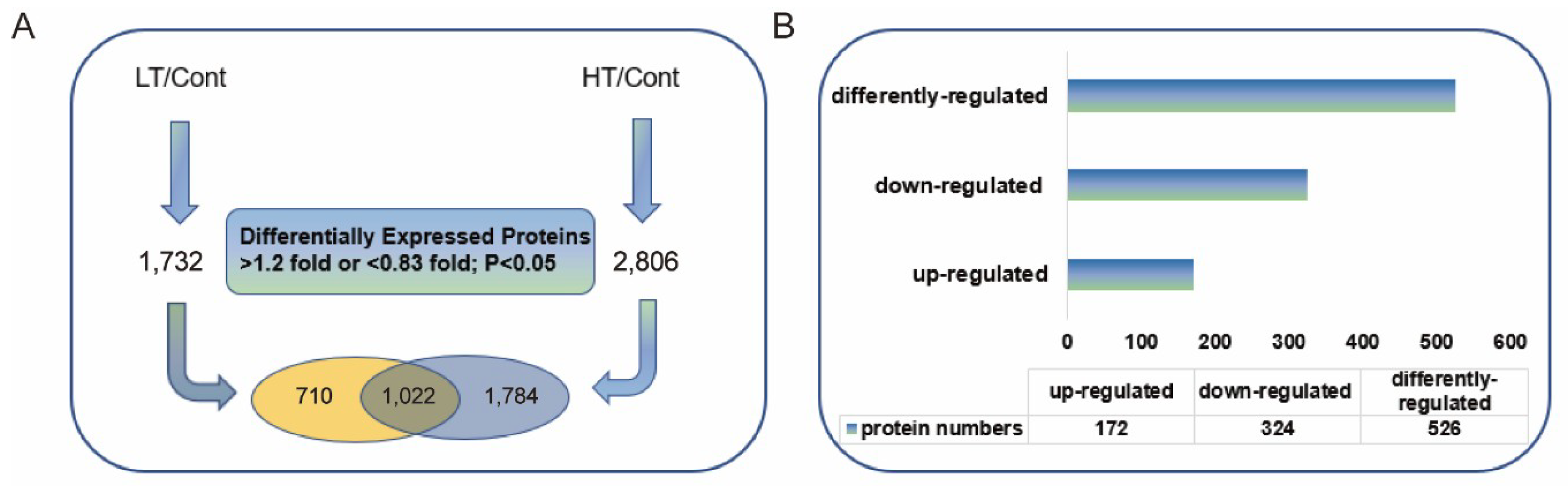
2.1. Identification of Differentially Expressed Proteins (DEPs) by Quantitative Proteomic Analysis
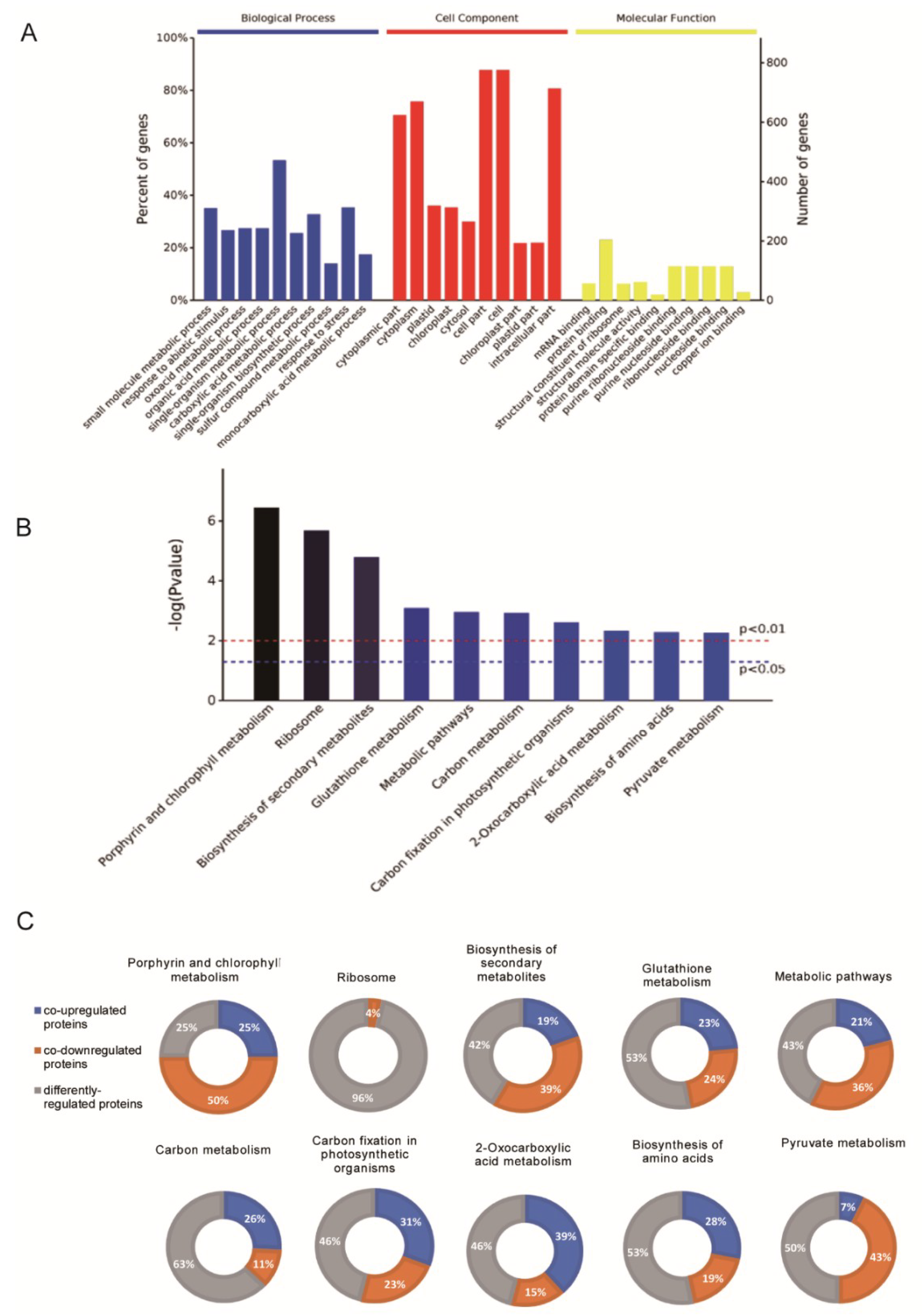
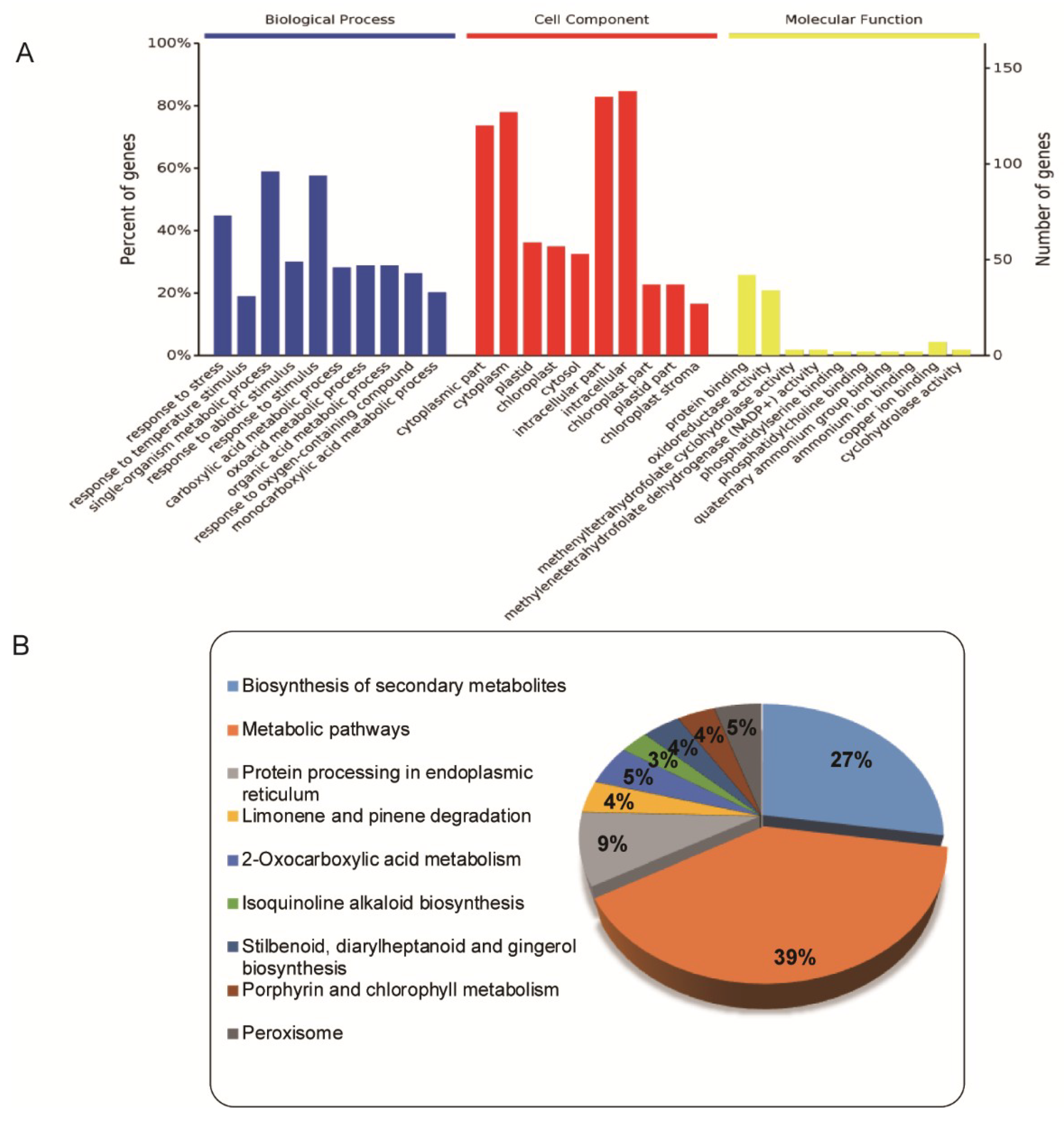
2.2. Functional Cataloging of DEPs Common to Both Low-Temperature (LT) and High-Temperature (HT) Groups
2.3. Functional Cataloging of DEPs That Were Upregulated under both LT and HT
2.3.1. Proteins Involved in Redox Homeostasis
2.3.2. Proteins Involved in Photosynthesis
2.3.3. Proteins Involved in Carbohydrate Metabolism
2.3.4. Heat-Shock Proteins and Chaperones
2.3.5. Proteins Relevant to Stimulus and Signaling Transduction
2.3.6. Metabolic-Related Proteins
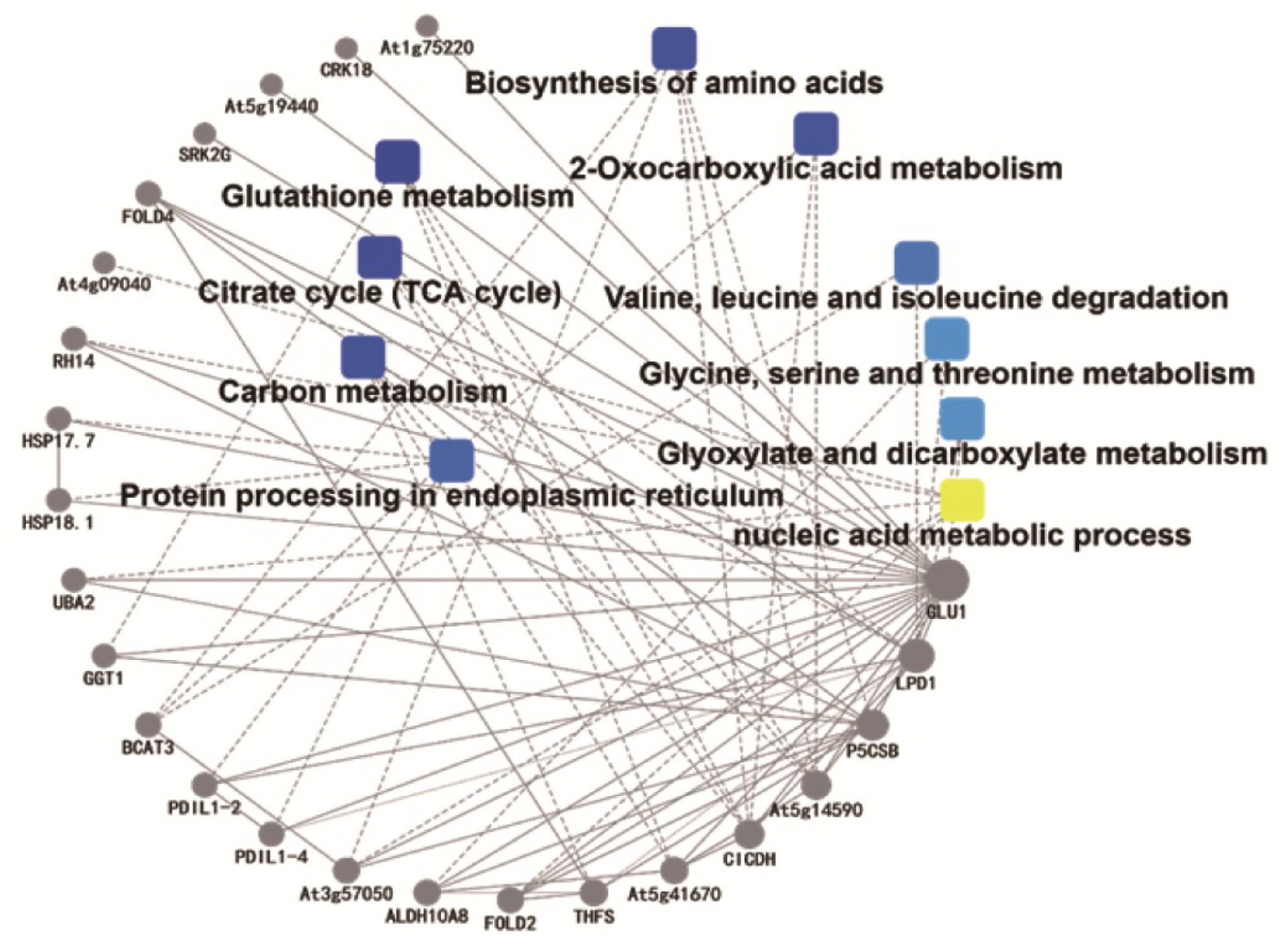
2.3.7. Protein–Protein Interaction (PPI) Analysis
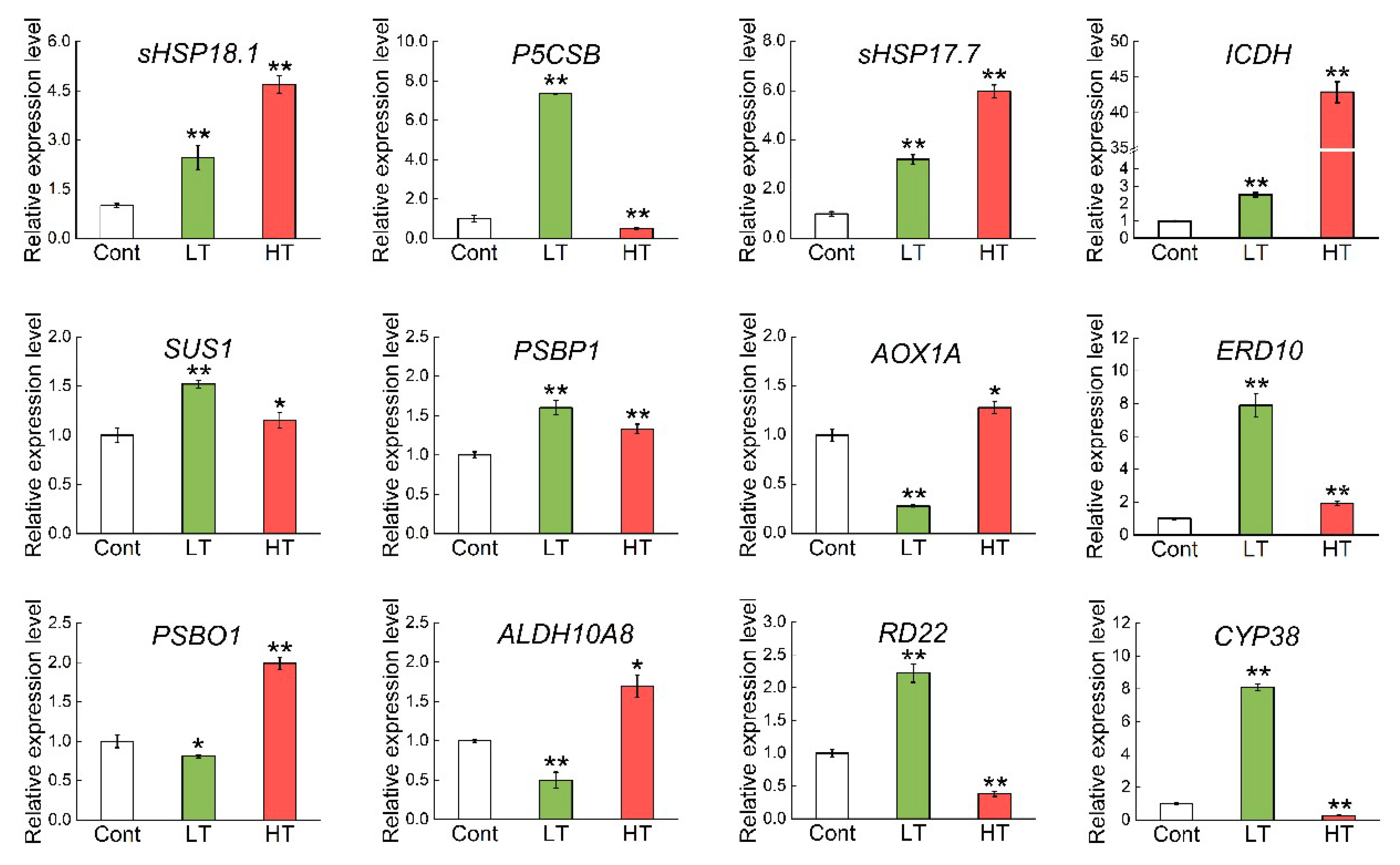
2.4. Expression Profiles of Genes Involved in DEPs That Were Upregulated under Both LT and HT
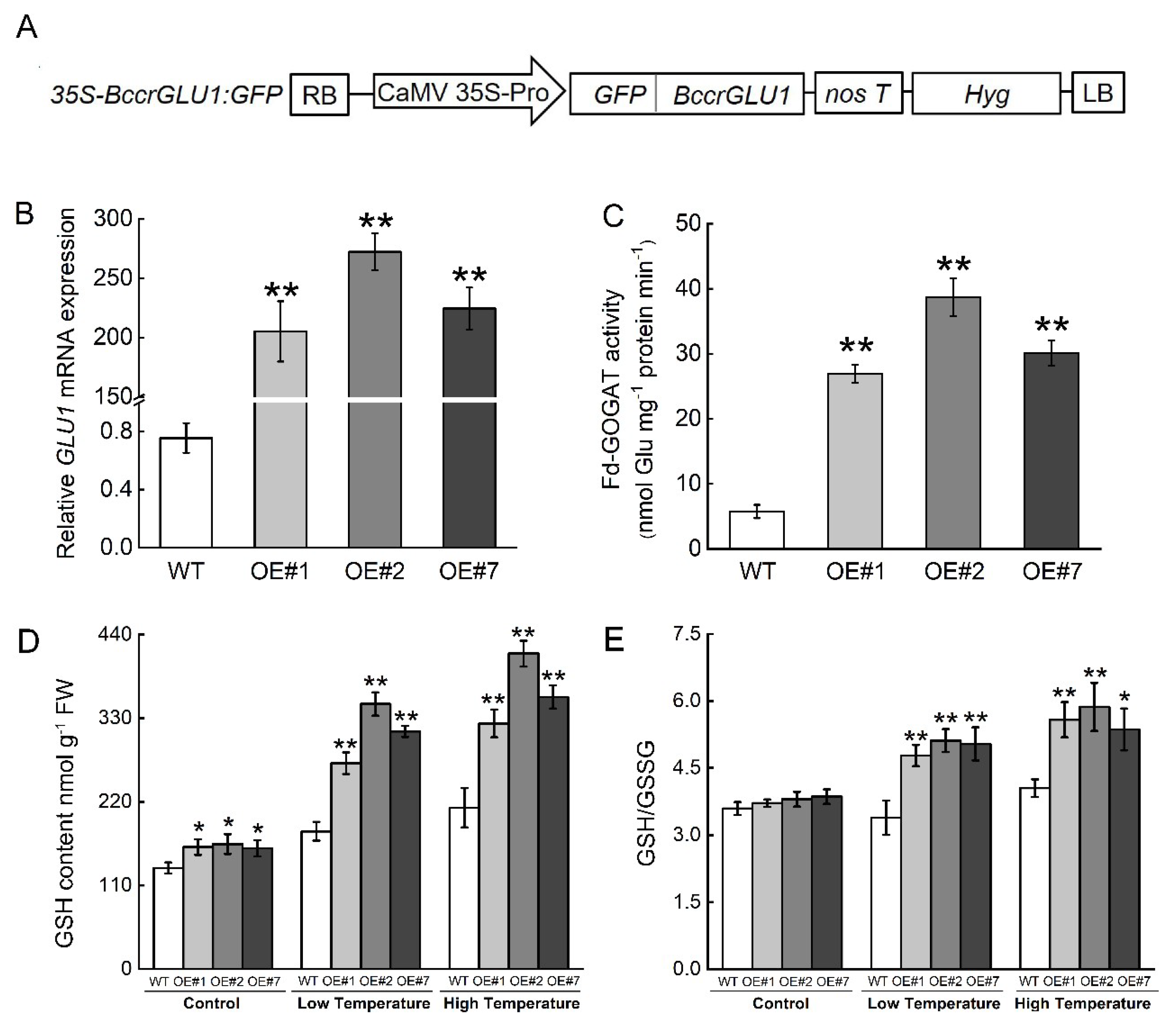
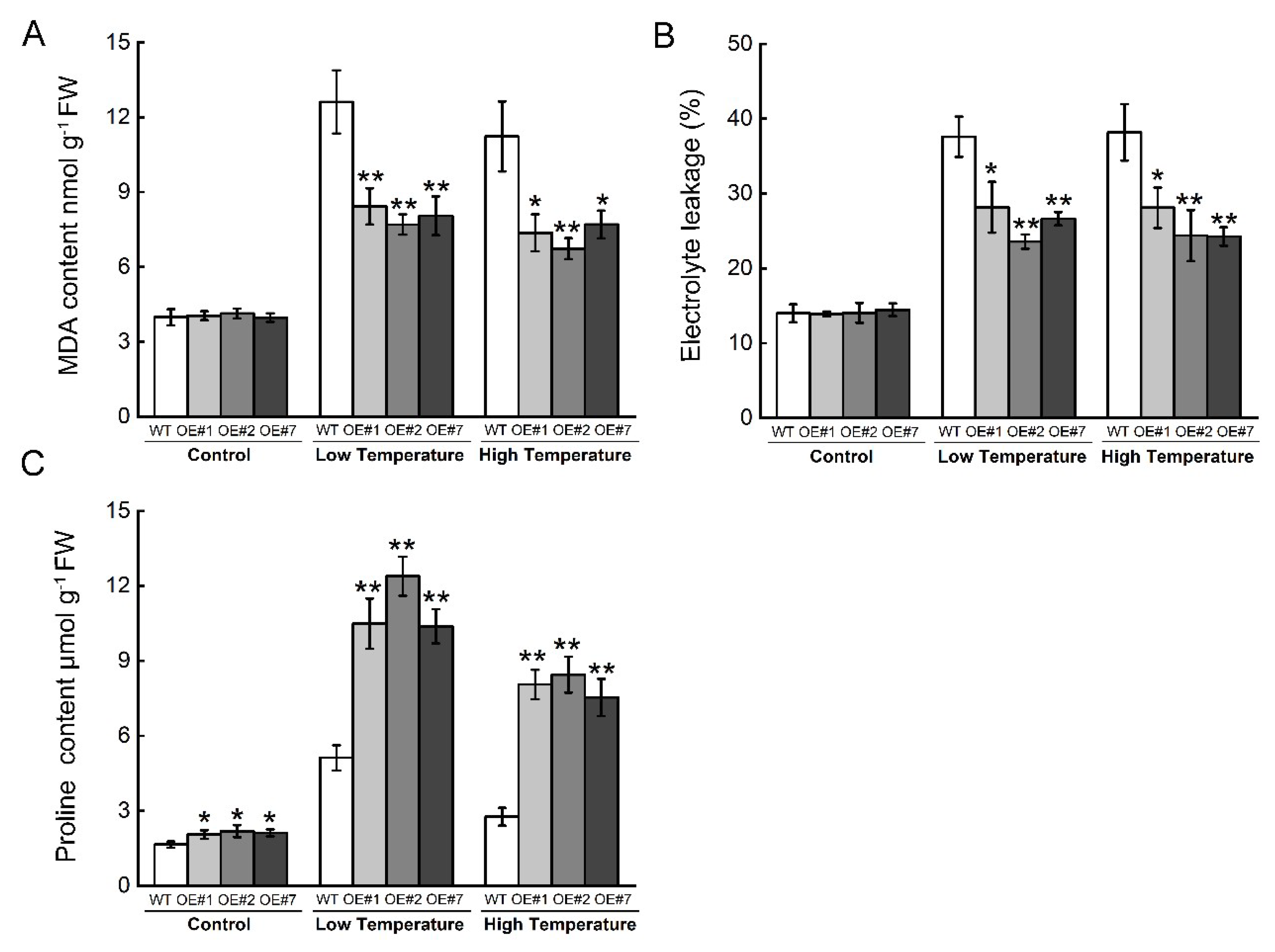
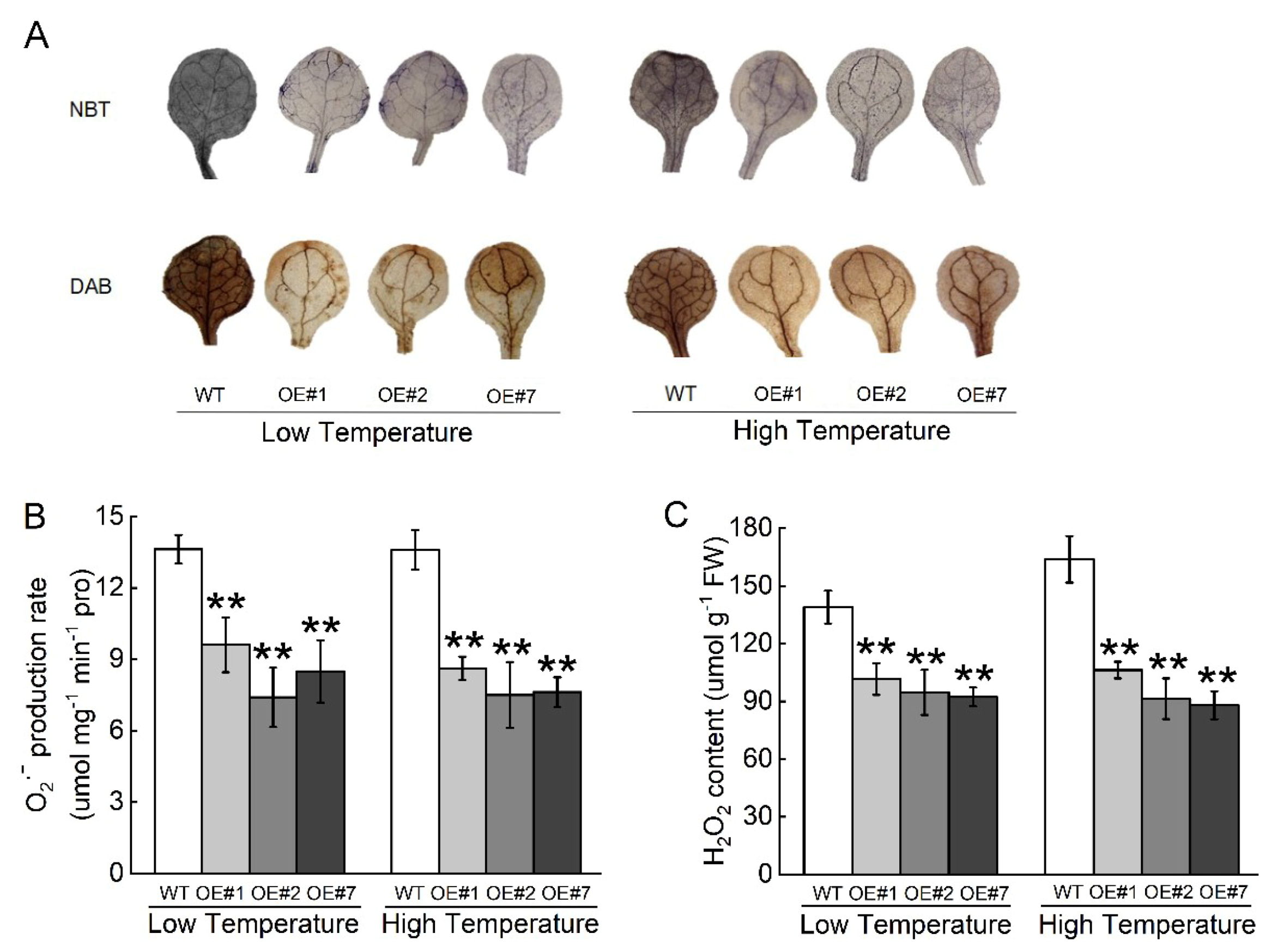
2.5. Overexpression of BccrGLU1 in Arabidopsis Leads to Temperature Stress Tolerance
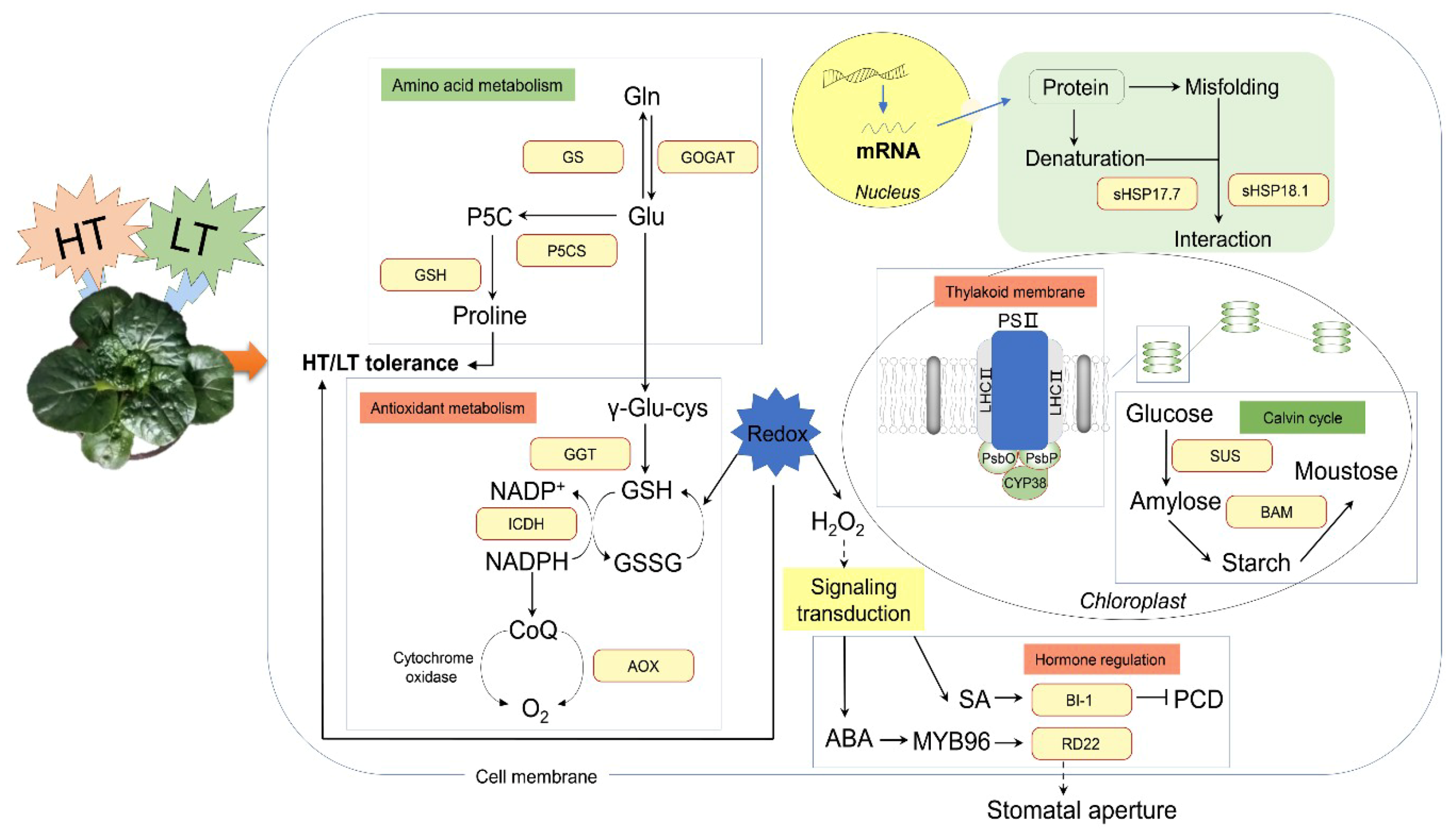
3. Discussion
4. Materials and Methods
4.1. Plant Materials and Growth Conditions
4.2. Protein Extraction
4.3. Protein Reduction, Alkylation, Digestion, and TMT Labeling
4.4. Reversed-phase liquid chromatography (RPLC) Analysis
4.5. Liquid Chromatography-Mass Spectrometry Analyses
4.6. Protein Identification and Quantification
4.7. Bioinformatics Analysis of Proteins
4.8. Transcriptional Expression Analysis by Quantitative RT-PCR
4.9. Tolerance Assay in Transgenic Arabidopsis under Temperature Stress
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| DAB | 3,3-diaminobenzidine |
| H2O2 | Hydrogen peroxide |
| HT | High temperature |
| LC-MS/MS | Liquid chromatography–mass spectrometry |
| LT | Low temperature |
| MDA | Malondialdehyde |
| NBT | Nitro blue tetrazolium |
| NHCC | Non-heading Chinese cabbage |
| O2•− | Superoxide anion |
| ROS | Reactive oxygen species |
| TMT | Tandem mass tag |
References
- Niu, Y.; Xiang, Y. An Overview of Biomembrane Functions in Plant Responses to High-Temperature Stress. Front. Plant Sci. 2018, 9, 915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tripathy, B.C.; Oelmüller, R. Reactive oxygen species generation and signaling in plants. Plant Signal. Behav. 2012, 7, 1621–1633. [Google Scholar] [CrossRef] [PubMed]
- Awasthi, R.; Bhandari, K.; Nayyar, H. Temperature stress and redox homeostasis in agricultural crops. Front. Env. Sci. 2015, 3, 11. [Google Scholar] [CrossRef]
- Feleciano, D.R.; Arnsburg, K.; Kirstein, J. Interplay between redox and protein homeostasis. Worm 2016, 5, e1170273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paciolla, C.; Paradiso, A.; de Pinto, M.C. Cellular Redox Homeostasis as Central Modulator in Plant Stress Response. In Redox State as a Central Regulator of Plant-Cell Stress Responses; Springer International Publishing: Berlin, Germany, 2016; pp. 1–23. [Google Scholar]
- Yuan, L.; Liu, S.; Zhu, S.; Chen, G.; Liu, F.; Zou, M.; Wang, C. Comparative response of two wucai (Brassica campestris L.) genotypes to heat stress on antioxidative system and cell ultrastructure in root. Acta Physiol. Plant. 2016, 38, 223. [Google Scholar] [CrossRef]
- Yuan, L.; Tang, L.; Zhu, S.; Hou, J.; Chen, G.; Liu, F.; Wang, C. Influence of heat stress on leaf morphology and nitrogen-carbohydrate metabolisms in two wucai (Brassica campestris L.) genotypes. Acta Soc. Bot. Pol. 2017, 86, 16. [Google Scholar] [CrossRef]
- Zou, M.; Yuan, L.; Zhu, S.; Liu, S.; Ge, J.; Wang, C. Response of osmotic adjustment and ascorbate-glutathione cycle to heat stress in a heat-sensitive and a heat-tolerant genotype of wucai (Brassica campestris L.). Sci. Hortic. 2016, 211, 87–94. [Google Scholar] [CrossRef]
- Shao, L.; Wang, C.; Song, J.; Zhang, H.; Zou, M. Effects of low temperature stress on physiological characteristics of savoy cultivars and assessment of their cold tolerance at seedling stage. J. Anhui Agric. Univ. 2014, 41, 265–269. [Google Scholar]
- Gao, F.; Ma, P.; Wu, Y.; Zhou, Y.; Zhang, G. Quantitative Proteomic Analysis of the Response to Cold Stress in Jojoba, a Tropical Woody Crop. Int. J. Mol. Sci. 2019, 20, 243. [Google Scholar] [CrossRef]
- Benešová, M.; Holá, D.; Fischer, L.; Jedelský, P.L.; Hnilička, F.; Wilhelmová, N.; Rothová, O.; Kočová, M.; Procházková, D.; Honnerová, J.; et al. The physiology and proteomics of drought tolerance in maize: Early stomatal closure as a cause of lower tolerance to short-term dehydration? PLoS ONE 2012, 7, e38017. [Google Scholar] [CrossRef]
- Mustafa, G.; Komatsu, S. Quantitative proteomics reveals the effect of protein glycosylation in soybean root under flooding stress. Front. Plant Sci. 2014, 5, 627. [Google Scholar] [CrossRef] [PubMed]
- Echevarría-Zomeño, S.; Fernández-Calvino, L.; Castro-Sanz, A.B.; López, J.A.; Vázquez, J.; Castellano, M.M. Dissecting the proteome dynamics of the early heat stress response leading to plant survival or death in Arabidopsis. Plant Cell Environ. 2016, 39, 1264–1278. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Guo, S.; Li, X.; Ren, X. Comparative analysis of salt-responsive phosphoproteins in maize leaves using Ti(4+)–IMAC enrichment and ESI-Q-TOF MS. Electrophoresis 2012, 34, 485–492. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.G.; Ahsan, N.; Lee, S.H.; Kang, K.Y.; Bahk, J.D.; Lee, I.J.; Lee, B.H. A proteomic approach in analyzing heat-responsive proteins in rice leaves. Proteomics 2007, 7, 3369–3383. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.K. Salt and drought stress signal transduction in plants. Annu. Rev. Plant Biol. 2002, 53, 247–273. [Google Scholar] [CrossRef]
- Parankusam, S.; Bhatnagar-Mathur, P.; Sharma, K.K. Heat responsive proteome changes reveal molecular mechanisms underlying heat tolerance in chickpea. Environ. Exp. Bot. 2017, 141, 132–144. [Google Scholar] [CrossRef]
- Zhang, Y.; Xu, L.; Zhu, X.; Gong, Y.; Xiang, F.; Sun, X.; Liu, L. Proteomic Analysis of Heat Stress Response in Leaves of Radish (Raphanus sativus L.). Plant Mol. Biol. Rep. 2013, 31, 195–203. [Google Scholar] [CrossRef]
- Gharechahi, J.; Alizadeh, H.; Naghavi, M.R.; Sharifi, G. A proteomic analysis to identify cold acclimation associated proteins in wild wheat (Triticum urartu L.). Mol. Biol. Rep. 2014, 41, 3897–3905. [Google Scholar] [CrossRef]
- Rocco, M.; Arena, S.; Renzone, G.; Scippa, G.S.; Lomaglio, T.; Verrillo, F.; Scaloni, A.; Marra, M. Proteomic analysis of temperature stress-responsive proteins in Arabidopsis thaliana rosette leaves. Mol. Biosyst. 2013, 9, 1257–1267. [Google Scholar] [CrossRef]
- Crosatti, C.; Pagani, D.; Cattivelli, L.; Stanca, A.M.; Rizza, F. Effects of growth stage and hardening conditions on the association between frost resistance and the expression of the cold-induced protein COR14b in barley. Environ. Exp. Bot. 2008, 62, 93–100. [Google Scholar] [CrossRef]
- Yuan, L.; Yuan, Y.; Liu, S.; Wang, J.; Zhu, S.; Chen, G.; Hou, J.; Wang, C. Influence of High Temperature on Photosynthesis, Antioxidative Capacity of Chloroplast, and Carbon Assimilation among Heat-tolerant and Heat-susceptible Genotypes of Non-heading Chinese Cabbage. HortScience 2017, 52, 1464–1470. [Google Scholar] [CrossRef]
- Song, X.; Liu, G.; Huang, Z.; Duan, W.; Tan, H.; Li, Y.; Hou, X. Temperature expression patterns of genes and their coexpression with LncRNAs revealed by RNA-Seq in non-heading Chinese cabbage. BMC Genom. 2016, 17, 297. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.Y.; Xu, Y.P.; Cai, X.Z. TMT-based quantitative proteomics analyses reveal novel defense mechanisms of Brassica napus against the devastating necrotrophic pathogen Sclerotinia sclerotiorum. J. Proteom. 2016, 143, 265–277. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Cao, D.; Ma, L.; Jin, X.; Yang, P.; Ye, F.; Liu, P.; Gong, Z.; Wei, C. TMT-based quantitative proteomics analysis reveals the response of tea plant (Camellia sinensis) to fluoride. J. Proteom. 2018, 176, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Takabayashi, A.; Niwata, A.; Tanaka, A. Direct interaction with ACR11 is necessary for post-transcriptional control of GLU1-encoded ferredoxin-dependent glutamate synthase in leaves. Sci. Rep. 2016, 6, 29668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, A.; Knaff, D.B. Glutamate synthase: Structural, mechanistic and regulatory properties, and role in the amino acid metabolism. Photosynth. Res. 2005, 83, 191–217. [Google Scholar] [CrossRef] [PubMed]
- Masi, A.; Trentin, A.R.; Agrawal, G.K.; Randeep, R. Gamma-glutamyl cycle in plants: A bridge connecting the environment to the plant cell? Front. Plant Sci. 2015, 6, 16–252. [Google Scholar] [CrossRef] [PubMed]
- Mhamdi, A.; Noctor, G. Analysis of the roles of the Arabidopsis peroxisomal isocitrate dehydrogenase in leaf metabolism and oxidative stress. Environ. Exp. Bot. 2015, 114, 22–29. [Google Scholar] [CrossRef]
- Lee, M.R.; Kim, C.S.; Park, T.; Choi, Y.; Lee, K. Optimization of the ninhydrin reaction and development of a multiwell plate-based high-throughput proline detection assay. Anal. Biochem. 2018, 556, 57–62. [Google Scholar] [CrossRef]
- Szabados, L.; Savoure, A. Proline: A multifunctional amino acid. Trends Plant Sci. 2010, 15, 89–97. [Google Scholar] [CrossRef]
- Missihoun, T.D.; Schmitz, J.; Klug, R.; Kirch, H.H.; Bartels, D. Betaine aldehyde dehydrogenase genes from Arabidopsis with different sub-cellular localization affect stress responses. Planta 2011, 233, 369–382. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.Y.; Liang, C.; Wang, G.P.; Luo, Y.; Wang, W. The protection of wheat plasma membrane under cold stress by glycine betaine overproduction. Biol. Plant. 2010, 54, 83–88. [Google Scholar] [CrossRef]
- Nanjo, T.; Fujita, M.; Seki, M.; Kato, T.; Tabata, S.; Shinozaki, K. Toxicity of free proline revealed in an Arabidopsis T-DNA-tagged mutant deficient in proline dehydrogenase. Plant Cell Physiol. 2003, 44, 541–548. [Google Scholar] [CrossRef] [PubMed]
- Fabro, G.; Kovacs, I.; Pavet, V.; Szabados, L.; Alvarez, M.E. Proline accumulation and AtP5CS2 gene activation are induced by plant-pathogen incompatible interactions in Arabidopsis. Mol. Plant Microbe Interact. 2004, 17, 343–350. [Google Scholar] [CrossRef] [PubMed]
- Vanlerberghe, G.C.; Ordog, S.H. Alternative Oxidase: Integrating Carbon Metabolism and Electron Transport in Plant Respiration. In Photosynthetic Nitrogen Assimilation and Associated Carbon and Respiratory Metabolism; Kluwer Academic Publishers: Dordrecht, The Netherlands, 2002; Volume 12, pp. 173–191. [Google Scholar]
- Murakami, R.; Ifuku, K.; Takabayashi, A.; Shikanai, T.; Endo, T.; Sato, F. Characterization of an Arabidopsis thaliana mutant with impaired psbO, one of two genes encoding extrinsic 33-kDa proteins in photosystem II. FEBS Lett. 2002, 523, 138–142. [Google Scholar] [CrossRef]
- Li, Q.; Chang, R.; Sun, Y.; Li, B. iTRAQ-Based Quantitative Proteomic Analysis of Spirulina platensis in Response to Low Temperature Stress. PLoS ONE 2016, 11, e0166876. [Google Scholar] [CrossRef] [PubMed]
- Allahverdiyeva, Y.; Mamedov, F.; Holmström, M.; Nurmi, M.; Lundin, B.; Styring, S.; Spetea, C.; Aro, E.M. Comparison of the electron transport properties of the psbo1 and psbo2 mutants of Arabidopsis thaliana. Biochim. Biophys. Acta Bioenerg. 2009, 1787, 1230–1237. [Google Scholar] [CrossRef]
- Nishimura, T.; Nagao, R.; Noguchi, T.; Nield, J.; Sato, F.; Ifuku, K. The N-terminal sequence of the extrinsic PsbP protein modulates the redox potential of Cyt b559 in photosystem II. Sci. Rep. 2016, 6, 21490. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, M.; Shigeto, J.; Sakamoto, A.; Morikawa, H. Selective nitration of PsbO1, PsbO2, and PsbP1 decreases PSII oxygen evolution and photochemical efficiency in intact leaves of Arabidopsis. Plant Signal. Behav. 2017, 12, e1376157. [Google Scholar] [CrossRef]
- Hesse, H.; Willmitzer, L. Expression analysis of a sucrose synthase gene from sugar beet (Beta vulgaris L.). Plant Mol. Biol. 1996, 30, 863–872. [Google Scholar] [CrossRef]
- Peng, T.; Zhu, X.; Duan, N.; Liu, J.H. PtrBAM1, a β-amylase-coding gene of Poncirus trifoliata, is a CBF regulon member with function in cold tolerance by modulating soluble sugar levels. Plant Cell Environ. 2015, 37, 2754–2767. [Google Scholar] [CrossRef]
- Xu, C.; Huang, B. Root proteomic responses to heat stress in two Agrostis grass species contrasting in heat tolerance. J. Exp. Bot. 2008, 59, 4183–4194. [Google Scholar] [CrossRef]
- Wang, W.; Vinocur, B.; Shoseyov, O.; Altman, A. Role of plant heat-shock proteins and molecular chaperones in the abiotic stress response. Trends Plant Sci. 2004, 9, 244–252. [Google Scholar] [CrossRef]
- Mamedov, T.G.; Shono, M. Molecular chaperone activity of tomato (Lycopersicon esculentum) endoplasmic reticulum-located small heat shock protein. J. Plant Res. 2008, 121, 235–243. [Google Scholar] [CrossRef]
- Sun, W.; Montagu, M.V.; Verbruggen, N. Small heat shock proteins and stress tolerance in plants. Biochim. Biophys. Acta Gene Struct. Expr. 2002, 1577, 1–9. [Google Scholar] [CrossRef]
- Lee, G.J.; Vierling, E. A small heat shock protein cooperates with heat shock protein 70 systems to reactivate a heat-denatured protein. Plant Physiol. 2000, 122, 189–198. [Google Scholar] [CrossRef]
- Gammulla, C.G.; Pascovici, D.; Atwell, B.J.; Haynes, P.A. Differential metabolic response of cultured rice (Oryza sativa) cells exposed to high- and low-temperature stress. Proteomics 2010, 10, 3001–3019. [Google Scholar] [CrossRef]
- Matsumura, H.; Nirasawa, S.; Kiba, A.; Urasaki, N.; Saitoh, H.; Ito, M.; Kawai-Yamada, M.; Uchimiya, H.; Terauchi, R. Overexpression of Bax inhibitor suppresses the fungal elicitor-induced cell death in rice (Oryza sativa L.) cells. Plant J. 2010, 33, 425–434. [Google Scholar] [CrossRef]
- Kawai-Yamada, M.; Ohori, Y.; Uchimiya, H. Dissection of Arabidopsis Bax inhibitor-1 suppressing Bax-, hydrogen peroxide-, and salicylic acid-induced cell death. Plant Cell 2004, 16, 21–32. [Google Scholar] [CrossRef]
- Wang, H.; Zhou, L.; Fu, Y.; Cheung, M.Y.; Wong, F.L.; Phang, T.H.; Sun, Z.; Lam, H.M. Expression of an apoplast-localized BURP-domain protein from soybean (GmRD22) enhances tolerance towards abiotic stress. Plant Cell Environ. 2012, 35, 1932–1947. [Google Scholar] [CrossRef]
- Matus, J.T.; Aquea, F.; Espinoza, C.; Vega, A.; Cavallini, E.; Santo, S.D.; Cañón, P.; de la Guardia, A.R.; Serrano, J.; Tornielli, G.B.; et al. Inspection of the Grapevine BURP Superfamily Highlights an Expansion of RD22 Genes with Distinctive Expression Features in Berry Development and ABA-Mediated Stress Responses. PLoS ONE 2014, 9, e110372. [Google Scholar] [CrossRef]
- Simon, C.; Langlois-Meurinne, M.; Didierlaurent, L.; Chaouch, S.; Bellvert, F.; Massoud, K.; Garmier, M.; Thareau, V.; Comte, G.; Noctor, G.; et al. The secondary metabolism glycosyltransferases UGT73B3 and UGT73B5 are components of redox status in resistance of Arabidopsis to Pseudomonas syringae pv. tomato. Plant Cell Environ. 2014, 37, 1114–1129. [Google Scholar] [CrossRef]
- Machado, C.R.; de Oliveira, R.L.; Boiteux, S.; Praekelt, U.M.; Meacock, P.A.; Menck, C.F. Thi1, a thiamine biosynthetic gene in Arabidopsis thaliana, complements bacterial defects in DNA repair. Plant Mol. Biol. 1996, 31, 585–593. [Google Scholar] [CrossRef]
- Rapala-Kozik, M.; Wolak, N.; Kujda, M.; Banas, A.K. The upregulation of thiamine (vitamin B1) biosynthesis in Arabidopsis thaliana seedlings under salt and osmotic stress conditions is mediated by abscisic acid at the early stages of this stress response. BMC Plant Biol. 2012, 12, 2. [Google Scholar] [CrossRef]
- Foyer, C.H.; Noctor, G. Stress-triggered redox signalling: What’s in pROSpect? Plant Cell Environ. 2016, 39, 951–964. [Google Scholar] [CrossRef]
- Kamrun, N.; Mirza, H.; Md, M.A.; Masayuki, F. Exogenous Spermidine Alleviates Low Temperature Injury in Mung Bean (Vigna radiata L.) Seedlings by Modulating Ascorbate-Glutathione and Glyoxalase Pathway. Int. J. Mol. Sci. 2015, 16, 30117–30132. [Google Scholar]
- Kocsy, G.; Kobrehel, K.; Szalai, G.; Duviau, M.P.; Buzás, Z.; Galiba, G. Abiotic stress-induced changes in glutathione and thioredoxin h levels in maize. Environ. Exp. Bot. 2004, 52, 101–112. [Google Scholar] [CrossRef]
- Turan, Ö.; Ekmekci, Y. Activities of photosymtem II and antioxidant enzymes in chickpea (Cicer arietinum L.) cultivars exposed to chilling temperatures. Acta Physiol. Plant. 2011, 33, 67–78. [Google Scholar] [CrossRef]
- Han, Y.; Chaouch, S.; Mhamdi, A.; Queval, G.; Zechmann, B.; Noctor, G. Functional analysis of Arabidopsis mutants points to novel roles for glutathione in coupling H2O2 to activation of salicylic acid accumulation and signaling. Antioxid. Redox Signal. 2013, 18, 2106–2121. [Google Scholar] [CrossRef]
- Han, Y.; Mhamdi, A.; Chaouch, S.; Noctor, G. Regulation of basal and oxidative stress-triggered jasmonic acid-related gene expression by glutathione. Plant Cell Environ. 2013, 36, 1135–1146. [Google Scholar] [CrossRef]
- Verbruggen, N.; Hermans, C. Proline accumulation in plants: A review. Amino Acids 2008, 35, 753–759. [Google Scholar] [CrossRef]
- Smith, P.K.; Krohn, R.I.; Hermanson, G.T.; Mallia, A.K.; Gartner, F.H.; Provenzano, M.D.; Fujimoto, E.K.; Goeke, N.M.; Olson, B.J.; Klenk, D.C. Measurement of protein using bicinchoninic acid. Anal. Biochem. 1985, 150, 76–85. [Google Scholar] [CrossRef]
- Wisniewski, J.R.; Zougman, A.; Nagaraj, N.; Mann, M. Universal sample preparation method for proteome analysis. Nat. Methods 2009, 6, 359–362. [Google Scholar] [CrossRef]
- Cen, W.; Liu, J.; Lu, S.; Jia, P.; Yu, K.; Han, Y.; Li, R.; Luo, J. Comparative proteomic analysis of QTL CTS-12 derived from wild rice (Oryza rufipogon Griff.), in the regulation of cold acclimation and de-acclimation of rice (Oryza sativa L.) in response to severe chilling stress. BMC Plant Biol. 2018, 18, 163. [Google Scholar] [CrossRef]
- Xie, S.; Nie, L.; Zheng, Y.; Wang, J.; Zhao, M.; Zhu, S.; Hou, J.; Chen, G.; Wang, C.; Yuan, L. Comparative proteomic analysis reveals that chlorophyll metabolism contributes to leaf color changes in wucai (Brassica campestris L.) responding to cold acclimation. J. Proteome Res. 2019, 18, 2478–2492. [Google Scholar] [CrossRef]
- Yang, L.T.; Qi, Y.P.; Lu, Y.B.; Guo, P.; Sang, W.; Feng, H.; Zhang, H.X.; Chen, L.S. iTRAQ protein profile analysis of Citrus sinensis roots in response to long-term boron-deficiency. J. Proteomics 2013, 93, 179–206. [Google Scholar] [CrossRef]
- Zhang, N.; Zhang, L.; Zhao, L.; Ren, Y.; Cui, D.; Chen, J.; Wang, Y.; Yu, P.; Chen, F. iTRAQ and virus-induced gene silencing revealed three proteins involved in cold response in bread wheat. Sci. Rep. 2017, 7, 7524. [Google Scholar] [CrossRef]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative CT method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef]
- Oliveira, I.C.; Lam, H.; Coschigano, K.; Melooliveira, R.; Coruzzi, G.M. Molecular-genetic dissection of ammonium assimilation in Arabidopsis thaliana. Plant Physiol. Biochem. 1997, 35, 185–198. [Google Scholar]
- Coschigano, K.T.; Melo-Oliveira, R.; Lim, J.; Coruzzi, G.M. Arabidopsis gls mutants and distinct Fd-GOGAT genes: Implications for photorespiration and primary nitrogen assimilation. Plant Cell 1998, 10, 741–752. [Google Scholar] [CrossRef]
- Clough, S.J.; Bent, A.F. Floral dip: A simplified method for Agrobacterium-mediated transformation of Arabidopsis thaliana. Plant J. 1998, 16, 735–743. [Google Scholar] [CrossRef]
- Baker, M.A.; Cerniglia, G.J.; Zaman, A. Microtiter plate assay for the measurement of glutathione and glutathione disulfide in large numbers of biological samples. Anal. Biochem. 1990, 190, 360–365. [Google Scholar] [CrossRef]
- Lin, C.C.; Kao, C.H. Disturbed ammonium assimilation is associated with growth inhibition of roots in rice seedlings caused by NaCl. Plant Growth Regul. 1996, 18, 233–238. [Google Scholar] [CrossRef]
- Heath, R.L.; Packer, L. Photoperoxidation in isolated chloroplasts: I. Kinetics and stoichiometry of fatty acid peroxidation. Arch. Biochem. Biophys. 1968, 125, 189–198. [Google Scholar] [CrossRef]
- Lutts, S.; Kinet, J.M.; Bouharmont, J. NaCl-induced Senescence in Leaves of Rice (Oryza sativa L.) Cultivars Differing in Salinity Resistance. Ann. Bot. 1996, 78, 389–398. [Google Scholar] [CrossRef]
- Bates, L.S.; Waldren, R.P.; Teare, I.D. Proline content Ref: Rapid determination of free proline for water-stress studies. Plant Soil 1973, 39, 205–207. [Google Scholar] [CrossRef]
- Patterson, B.D.; Macrae, E.A.; Ferguson, I.B. Estimation of hydrogen peroxide in plant extracts using titanium(IV). Anal. Biochem. 1984, 139, 487–492. [Google Scholar] [CrossRef]
- Elstner, E.F.; Heupel, A. Inhibition of nitrite formation from hydroxylammoniumchloride: A simple assay for superoxide dismutase. Anal. Biochem. 1976, 70, 616–620. [Google Scholar] [CrossRef]
- Juszczak, I.; Cvetkovic, J.; Zuther, E.; Hincha, D.K.; Baier, M. Natural Variation of Cold Deacclimation Correlates with Variation of Cold-Acclimation of the Plastid Antioxidant System in Arabidopsis thaliana Accessions. Front. Plant Sci. 2016, 7, 305. [Google Scholar] [CrossRef]




 , GO/KEGG term;
, GO/KEGG term;  , protein. Symbol color corresponds to degree of interaction.
, GO/KEGG term; , protein. Symbol color corresponds to degree of interaction.
, protein. Symbol color corresponds to degree of interaction.
, GO/KEGG term; , protein. Symbol color corresponds to degree of interaction.






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Accession | Name | Forward Primer (5′→3′) | Reverse Primer (5′→3′) |
|---|---|---|---|
| XM_009106490.1 | ALDH10A8 | TCGTCAATCCAGCAACCCAA | TCAGTCACCTTAGCGGCAAT |
| XM_013806620.2 | AOX1A | AGCCATCTCTTGAAACCTGC | AGCGATTCCTTTGTTACCTCC |
| XM_009151277.2 | ERD10 | ACTGTTTGACTTCTTGG | GAGGAGAGTAGGCTTATG |
| XM_022705499.1 | RD22 | CAAACACTCCCATACCA | TACACCTCCCTTTCCAA |
| XM_013841006.2 | PSBP1 | TTTCACTCTCCAAACCCGTCCA | AGCTTCACCATAGGCGGCATC |
| XM_013830831.2 | PSBO1 | CAACCTCTGCTCTCGTCGTC | CTTGCTAACACTCTCGGCCT |
| XM_009129212.2 | ICDH | AGTGAGGGAGGCTATGTGTG | CTATGCTGTTTGTGCTGGT |
| XM_009122518.1 | SUS1 | ATTTCATCATCACCAG | AGTCAATCTACGCTTC |
| XM_009118014.2 | P5CSB | CCTTTTCCACCAAGATGCAC | CCCAGGCTTCATAACTAAACGA |
| XR_002653900.1 | CYP38 | CGGGAACTTTGTGGACTTGG | GCGTTTTCTTCCCAGTCACC |
| XM_002873500.2 | sHSP17.7 | AAGACCCGTAACAACCCT | CTTTTTCCACTCACCACA |
| XM_013858088.2 | sHSP18.1 | CAGCATTCACAAACGC | CCTCCTCATAAACTTC |
| XM_009127097.2 | Actin | TGGGTTTGCTGGTGACGAT | TGCCTAGGACGACCAACAATACT |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yuan, L.; Wang, J.; Xie, S.; Zhao, M.; Nie, L.; Zheng, Y.; Zhu, S.; Hou, J.; Chen, G.; Wang, C. Comparative Proteomics Indicates That Redox Homeostasis Is Involved in High- and Low-Temperature Stress Tolerance in a Novel Wucai (Brassica campestris L.) Genotype. Int. J. Mol. Sci. 2019, 20, 3760. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20153760
Yuan L, Wang J, Xie S, Zhao M, Nie L, Zheng Y, Zhu S, Hou J, Chen G, Wang C. Comparative Proteomics Indicates That Redox Homeostasis Is Involved in High- and Low-Temperature Stress Tolerance in a Novel Wucai (Brassica campestris L.) Genotype. International Journal of Molecular Sciences. 2019; 20(15):3760. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20153760
Chicago/Turabian StyleYuan, Lingyun, Jie Wang, Shilei Xie, Mengru Zhao, Libing Nie, Yushan Zheng, Shidong Zhu, Jinfeng Hou, Guohu Chen, and Chenggang Wang. 2019. "Comparative Proteomics Indicates That Redox Homeostasis Is Involved in High- and Low-Temperature Stress Tolerance in a Novel Wucai (Brassica campestris L.) Genotype" International Journal of Molecular Sciences 20, no. 15: 3760. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20153760