Anti-Proliferative and Pro-Apoptotic Effects of Licochalcone A through ROS-Mediated Cell Cycle Arrest and Apoptosis in Human Bladder Cancer Cells

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
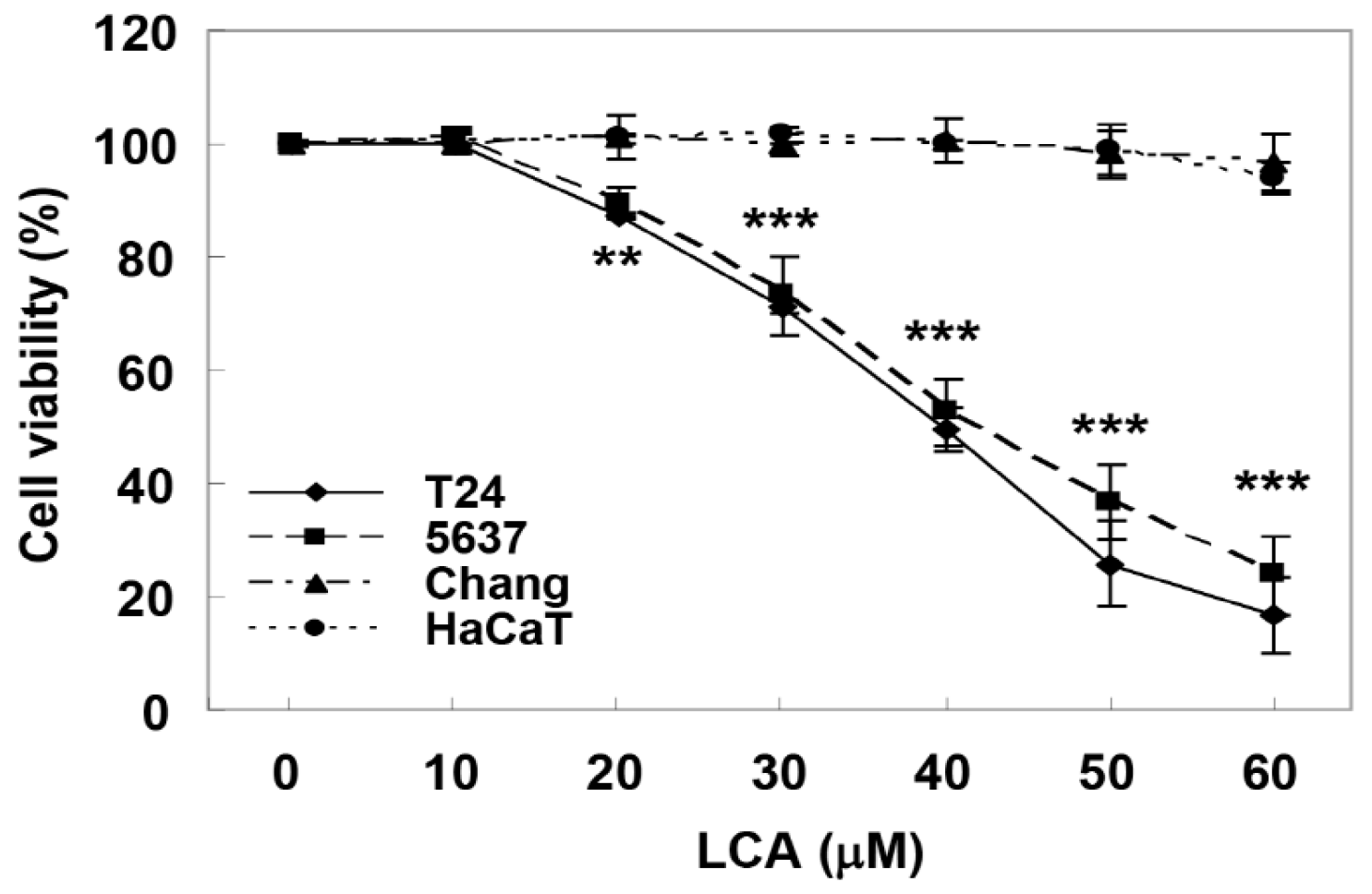
2.1. LCA Causes Cell Growth Inhibition in Human Bladder Cancer Cells
2.2. LCA Induces G2/M Phase Arrest and Apoptosis in Bladder Cancer T24 Cells
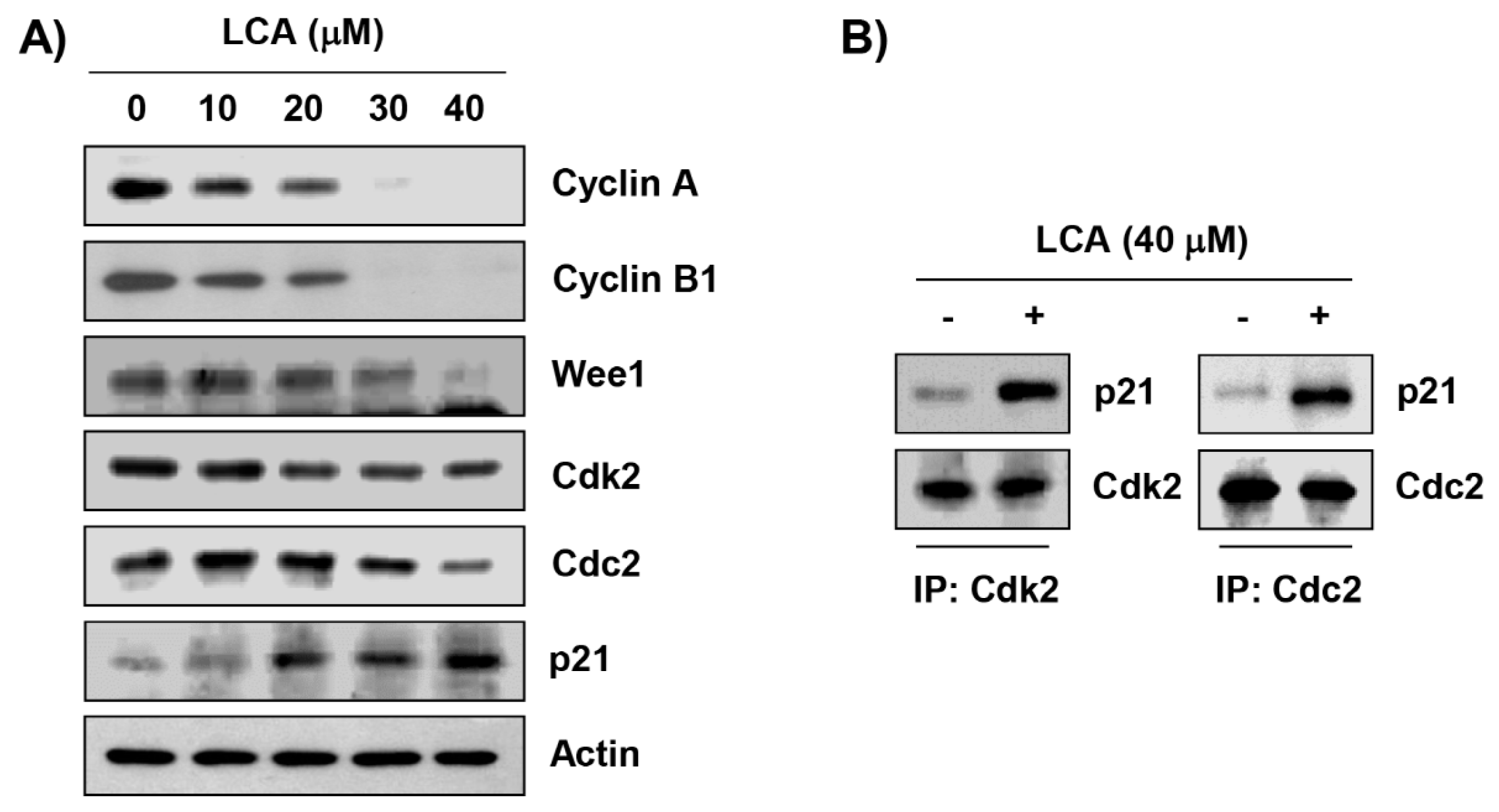
2.3. LCA Regulates the Expression of G2/M Phase-Associated Proteins in T24 Cells
2.4. LCA Activates Caspases in T24 Cells
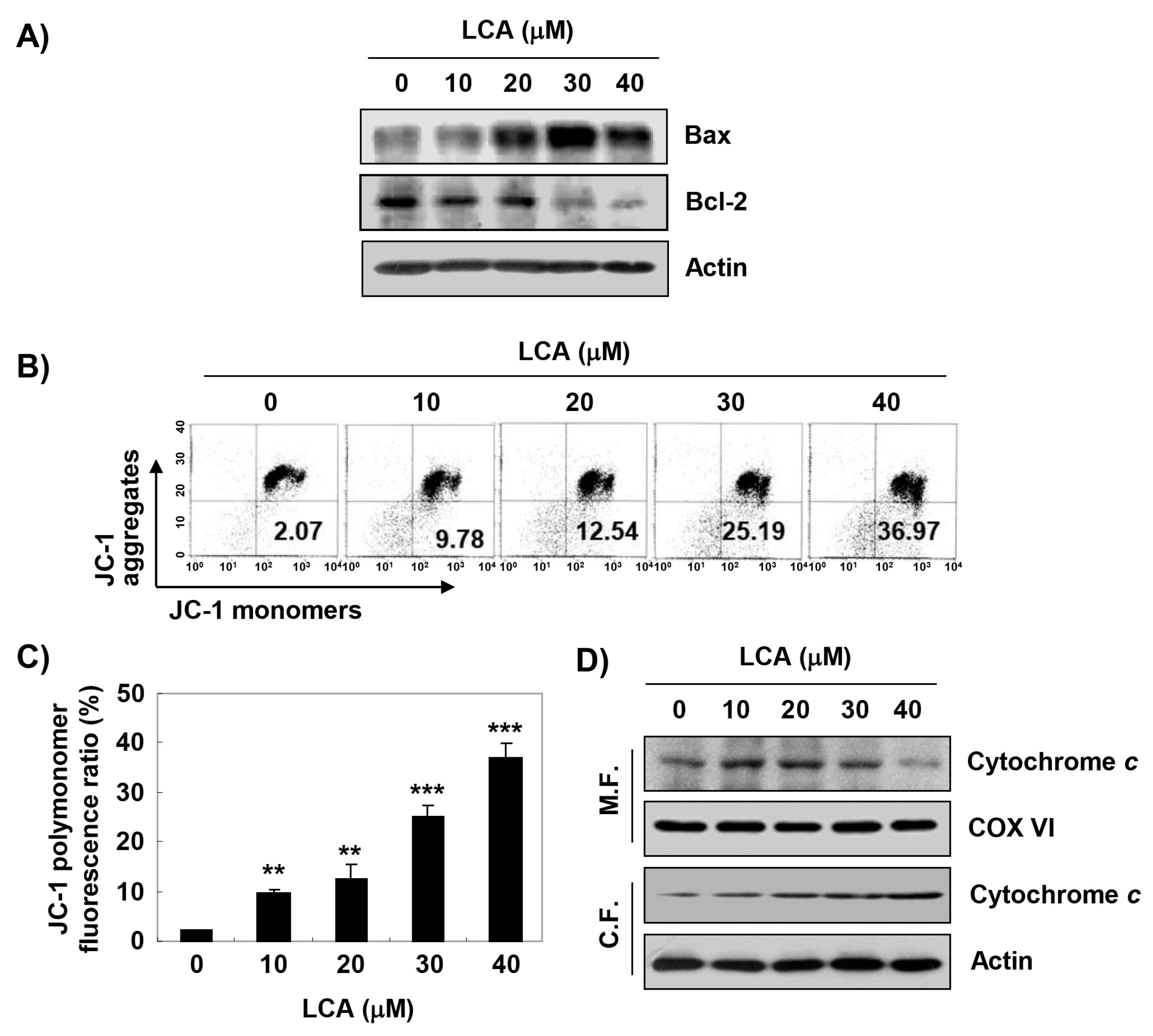
2.5. LCA Increases Mitochondrial Dysfunction in T24 Cells
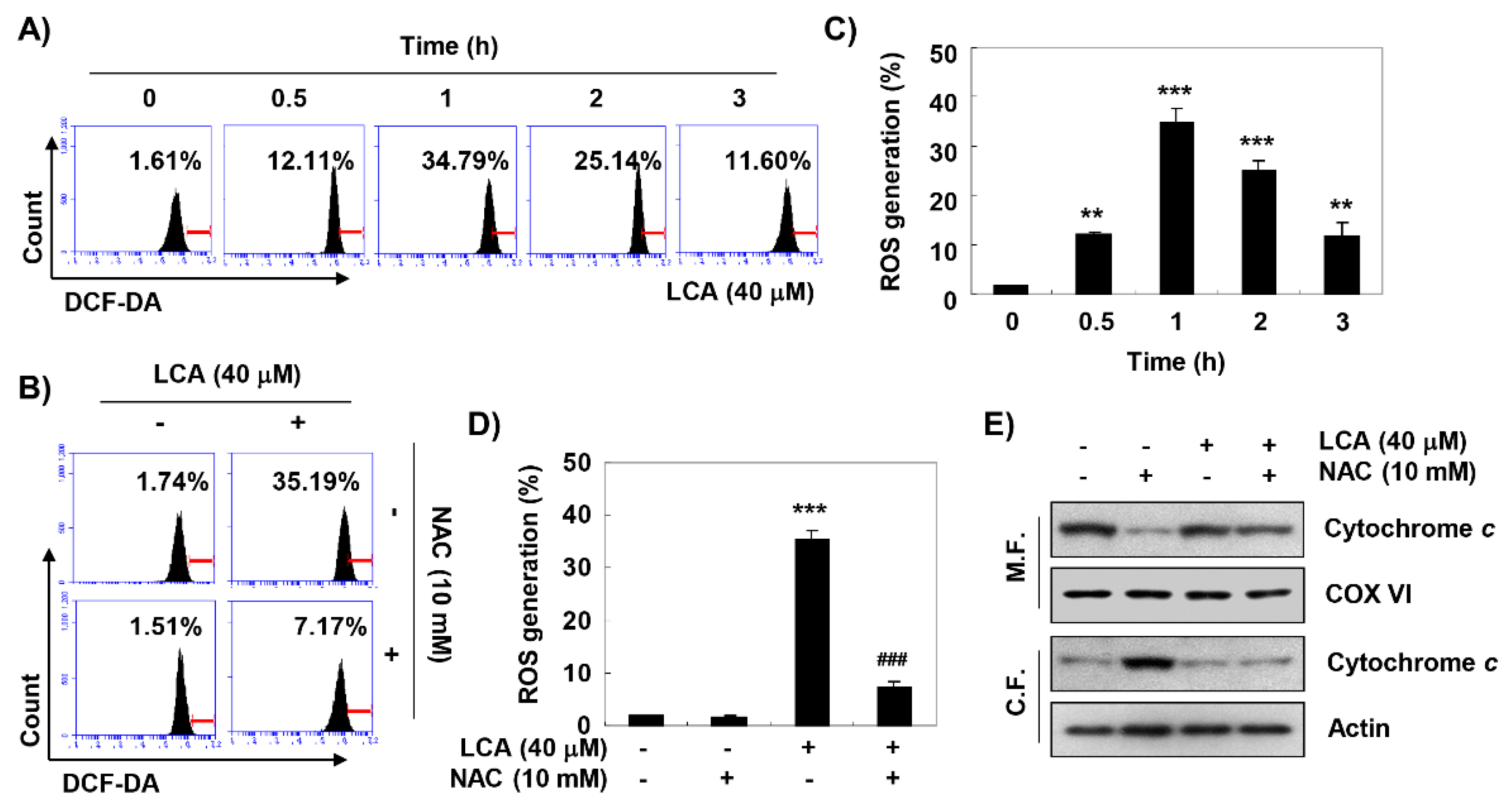
2.6. LCA Induces ROS-Dependent Mitochondrial Dysfunction in T24 Cells
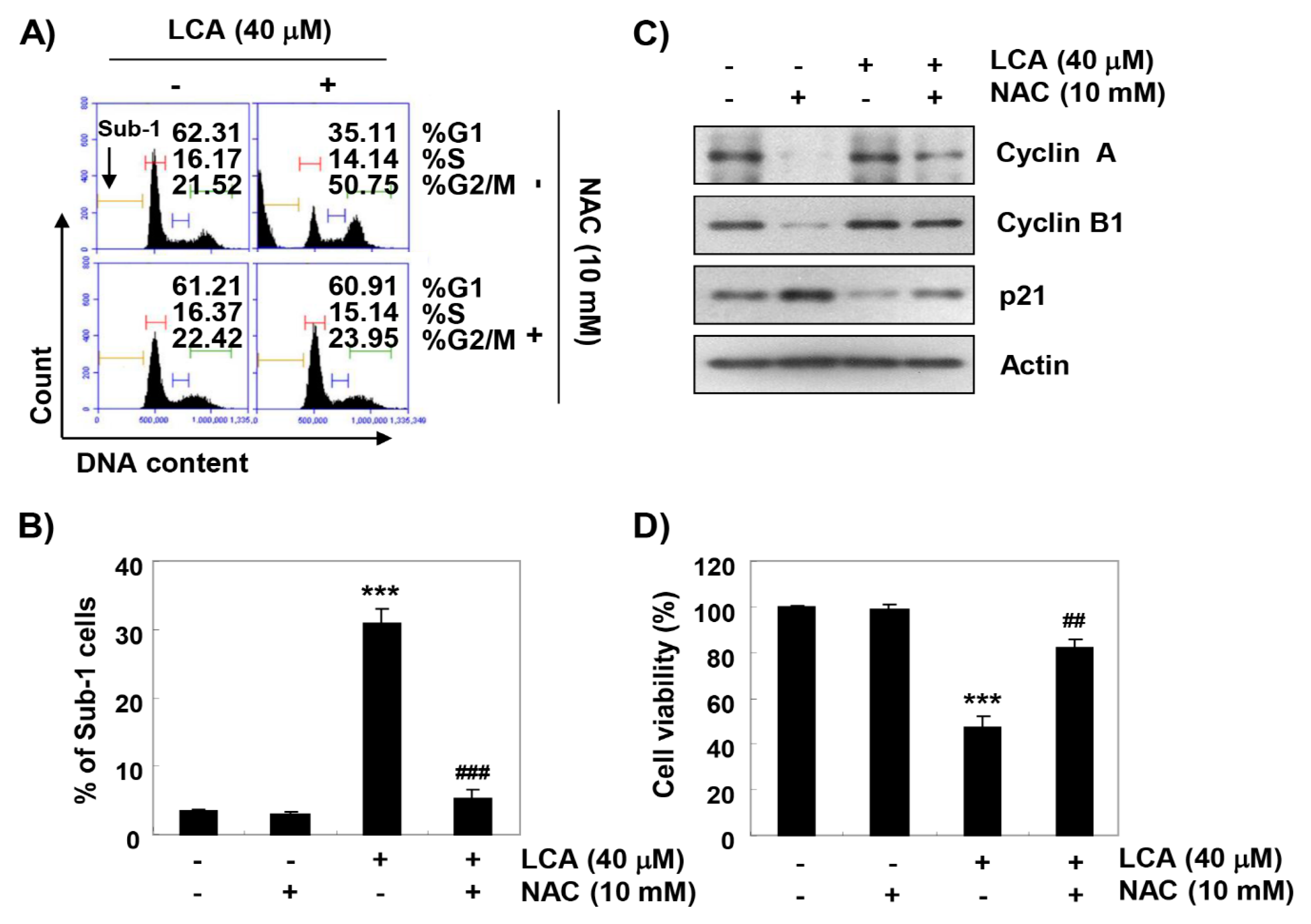
2.7. ROS Acts as an Upstream Regulator of LCA-Induced Growth Arrest and Apoptosis in T24 Cells
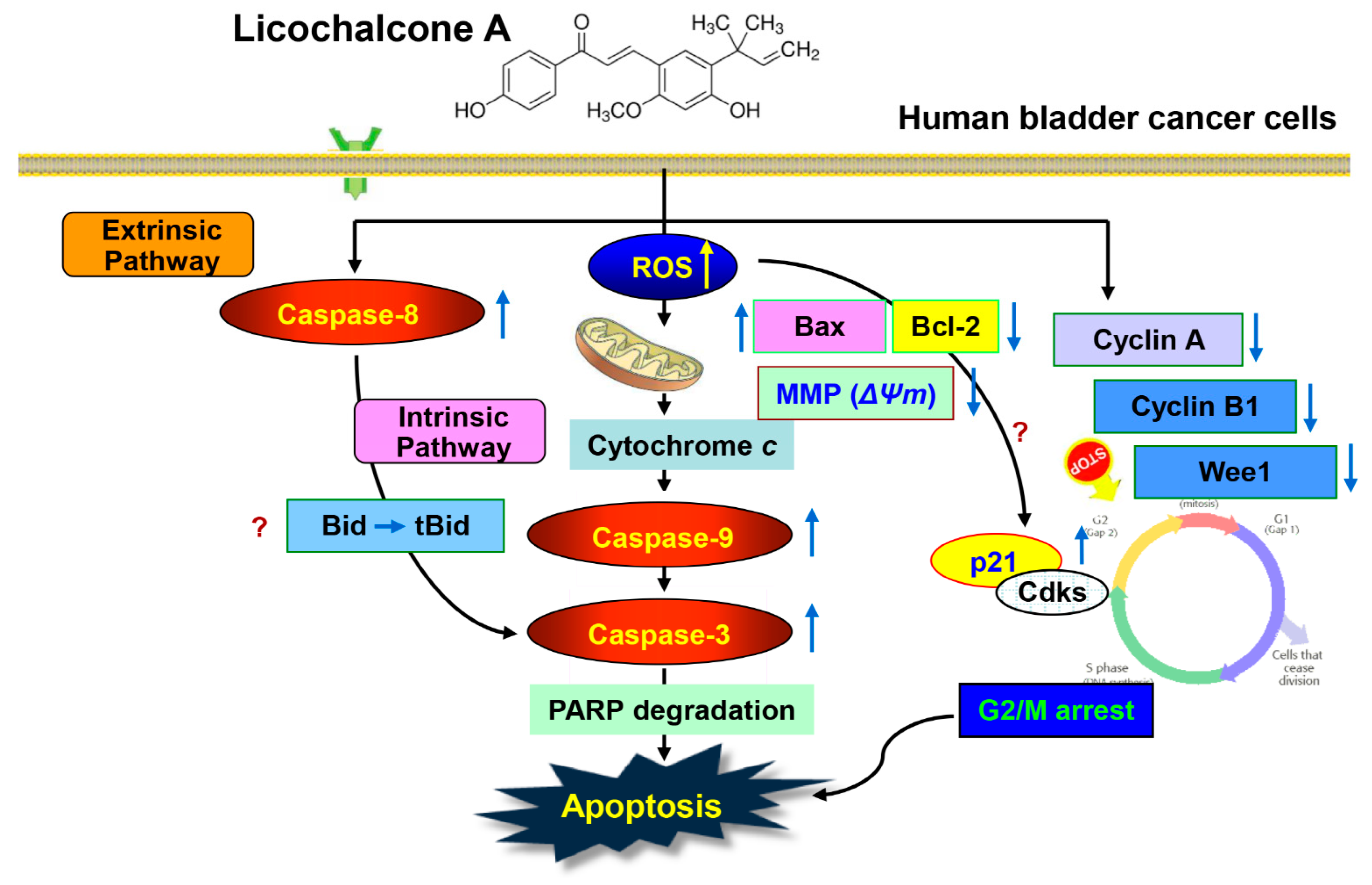
3. Discussion
4. Materials and Methods
4.1. Cell Culture and LCA Treatment
4.2. Cell Viability
4.3. Determination of Cell Cycle Distribution by Flow Cytometric Analysis
4.4. Detection of Apoptotic Morphological Changes
4.5. Determination of Apoptotic Cell Death by Flow Cytometric Analysis
4.6. Protein Extraction, Co-Iimmunoprecipitation, and Western Blot Analysis
4.7. Caspase Activity Assay
4.8. Measurement of MMP (ΔΨm) and ROS Production
4.9. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| Cdc | Cell division cycle |
| Cdk | Cyclin-dependent kinase |
| COX VI | Cytochrome oxidase subunit VI |
| DAPI | 4′,6-diamidino-2-phenylindole |
| DCF-DA | 5,6-carboxy-2′,7′-dichlorodihydrofluorescein diacetate |
| DMSO | Dimethyl sulfoxide |
| DR | Ddeath receptor |
| ECL | Enhanced chemiluminescence |
| FBS | Fetal bovine serum |
| FITC | Fluorescein isothiocyanate |
| IP | Immunoprecipitation |
| JC-1 | 5,5‘,6,6’-tetrachloro-1,1’,3,3’-tetraethyl-imidacarbocyanine iodide |
| LCA | Licochalcone A |
| MMP | Mitochondrial membrane potential |
| MTT | 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyltetra-zolium bromide |
| NAC | N-acetyl-L-cysteine |
| p-NA | P-nitroaniline |
| PARP | Poly(ADP-ribose) polymerase |
| PBS | Phosphate-buffered saline |
| PI | Propidium iodide |
| PVDF | Polyvinylidene difluoride |
| ROS | Reactive oxygen species |
| SD | Standard deviation |
| SDS | Sodium dodecyl sulfate |
References
- Kumar, A.; Jaitak, V. Natural products as multidrug resistance modulators in cancer. Eur. J. Med. Chem. 2019, 176, 268–291. [Google Scholar] [CrossRef] [PubMed]
- de Oliveira Júnior, R.G.; Christiane Adrielly, A.F.; da Silva Almeida, J.R.G.; Grougnet, R.; Thiéry, V.; Picot, L. Sensitization of tumor cells to chemotherapy by natural products: A systematic review of preclinical data and molecular mechanisms. Fitoterapia 2018, 129, 383–400. [Google Scholar] [CrossRef] [PubMed]
- Tewari, D.; Rawat, P.; Singh, P.K. Adverse drug reactions of anticancer drugs derived from natural sources. Food Chem. Toxicol. 2019, 123, 522–535. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Ledesma, B.; Hsieh, C.C. Chemopreventive role of food-derived proteins and peptides: A review. Crit. Rev. Food Sci. Nutr. 2017, 57, 2358–2376. [Google Scholar] [CrossRef] [PubMed]
- Hassan, M.; Watari, H.; AbuAlmaaty, A.; Ohba, Y.; Sakuragi, N. Apoptosis and molecular targeting therapy in cancer. Biomed. Res. Int. 2014, 2014, 150845. [Google Scholar] [CrossRef] [PubMed]
- Medema, R.H.; Macůrek, L. Checkpoint control and cancer. Oncogene 2012, 31, 2601–2613. [Google Scholar] [CrossRef] [PubMed]
- Bortolotto, L.F.; Barbosa, F.R.; Silva, G.; Bitencourt, T.A.; Beleboni, R.O.; Baek, S.J.; Marins, M.; Fachin, A.L. Cytotoxicity of trans-chalcone and licochalcone A against breast cancer cells is due to apoptosis induction and cell cycle arrest. Biomed. Pharmacother. 2017, 85, 425–433. [Google Scholar] [CrossRef]
- Schnekenburger, M.; Dicato, M.; Diederich, M. Plant-derived epigenetic modulators for cancer treatment and prevention. Biotechnol. Adv. 2014, 32, 1123–1132. [Google Scholar] [CrossRef]
- Khan, T.; Gurav, P. PhytoNanotechnology: Enhancing delivery of plant based anti-cancer drugs. Front. Pharmacol. 2018, 8, 1002. [Google Scholar] [CrossRef]
- Hansen, E.; Andersen, J.H. Screening for marine natural products with potential as chemotherapeutics for acute myeloid leukemia. Curr. Pharm. Biotechnol. 2016, 17, 71–77. [Google Scholar] [CrossRef]
- Niemeijer, N.D.; Alblas, G.; van Hulsteijn, L.T.; Dekkers, O.M.; Corssmit, E.P. Chemotherapy with cyclophosphamide, vincristine and dacarbazine for malignant paraganglioma and pheochromocytoma: Systematic review and meta-analysis. Clin. Endocrinol. 2014, 81, 642–651. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Hsieh, T.C.; Guo, J.; Kunicki, J.; Lee, M.Y.; Darzynkiewicz, Z.; Wu, J.M. Licochalcone-A, a novel flavonoid isolated from licorice root (Glycyrrhiza glabra), causes G2 and late-G1 arrests in androgen-independent PC-3 prostate cancer cells. Biochem. Biophys. Res. Commun. 2004, 322, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Messier, C.; Grenier, D. Effect of licorice compounds licochalcone A, glabridin and glycyrrhizic acid on growth and virulence properties of Candida albicans. Mycoses 2011, 54, e801–e806. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, H.L.; Hansen, H.S.; Staerk, D.; Christensen, S.B.; Hägerstrand, H.; Jaroszewski, J.W. The antiparasitic compound licochalcone a is a potent echinocytogenic agent that modifies the erythrocyte membrane in the concentration range where antiplasmodial activity is observed. Antimicrob. Agents Chemother. 2004, 48, 4067–4071. [Google Scholar] [CrossRef] [PubMed]
- Kwon, H.S.; Park, J.H.; Kim, D.H.; Kim, Y.H.; Park, J.H.; Shin, H.K.; Kim, J.K. Licochalcone A isolated from licorice suppresses lipopolysaccharide-stimulated inflammatory reactions in RAW264.7 cells and endotoxin shock in mice. J. Mol. Med. 2008, 86, 1287–1295. [Google Scholar] [CrossRef] [PubMed]
- Feldman, M.; Grenier, D. Cranberry proanthocyanidins act in synergy with licochalcone A to reduce Porphyromonas gingivalis growth and virulence properties, and to suppress cytokine secretion by macrophages. J. Appl. Microbiol. 2012, 113, 438–447. [Google Scholar] [CrossRef] [PubMed]
- Liang, M.; Li, X.; Ouyang, X.; Xie, H.; Chen, D. Antioxidant mechanisms of echinatin and licochalcone A. Molecules 2018, 24, E3. [Google Scholar] [CrossRef] [PubMed]
- Su, X.; Li, T.; Liu, Z.; Huang, Q.; Liao, K.; Ren, R.; Lu, L.; Qi, X.; Wang, M.; Chen, J.; et al. Licochalcone A activates Keap1-Nrf2 signaling to suppress arthritis via phosphorylation of p62 at serine 349. Free Radic. Biol. Med. 2018, 115, 471–483. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.J.; Liang, W.M.; Chen, C.J.; Tsang, H.; Chiou, J.S.; Liu, X.; Cheng, C.F.; Lin, T.H.; Liao, C.C.; Huang, S.M.; et al. Network analysis and mechanisms of action of Chinese herb-related natural compounds in lung cancer cells. Phytomedicine 2019, 58, 152893. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhang, Y.S.; Thakur, K.; Hussain, S.S.; Zhang, J.G.; Xiao, G.R.; Wei, Z.J. Licochalcone A from licorice root, an inhibitor of human hepatoma cell growth via induction of cell apoptosis and cell cycle arrest. Food Chem. Toxicol. 2018, 120, 407–417. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.J.; Wu, G.J.; Chen, R.J.; Chang, C.C.; Lien, L.M.; Chiu, C.C.; Tseng, M.F.; Huang, L.T.; Lin, K.H. Licochalcone A attenuates glioma cell growth in vitro and in vivo through cell cycle arrest. Food Funct. 2018, 9, 4500–4507. [Google Scholar] [CrossRef] [PubMed]
- Qiu, C.; Zhang, T.; Zhang, W.; Zhou, L.; Yu, B.; Wang, W.; Yang, Z.; Liu, Z.; Zou, P.; Liang, G. Licochalcone A inhibits the proliferation of human lung cancer cell lines A549 and H460 by inducing G2/M cell cycle arrest and ER stress. Int. J. Mol. Sci. 2017, 18, E1761. [Google Scholar] [CrossRef] [PubMed]
- Chuang, C.Y.; Tang, C.M.; Ho, H.Y.; Hsin, C.H.; Weng, C.J.; Yang, S.F.; Chen, P.N.; Lin, C.W. Licochalcone A induces apoptotic cell death via JNK/p38 activation in human nasopharyngeal carcinoma cells. Environ. Toxicol. 2019, 34, 853–860. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.C.; Su, H.H.; Fang, L.W.; Wu, S.J.; Liou, C.J. Licochalcone A inhibits cellular motility by suppressing E-cadherin and MAPK signaling in breast cancer. Cells 2019, 8, E218. [Google Scholar] [CrossRef]
- Kang, T.H.; Seo, J.H.; Oh, H.; Yoon, G.; Chae, J.I.; Shim, J.H. Licochalcone A suppresses specificity protein 1 as a novel target in human breast cancer cells. J. Cell. Biochem. 2017, 118, 4652–4663. [Google Scholar] [CrossRef]
- Yang, X.; Jiang, J.; Yang, X.; Han, J.; Zheng, Q. Licochalcone A induces T24 bladder cancer cell apoptosis by increasing intracellular calcium levels. Mol. Med. Rep. 2016, 14, 911–919. [Google Scholar] [CrossRef] [Green Version]
- Tsai, J.P.; Lee, C.H.; Ying, T.H.; Lin, C.L.; Lin, C.L.; Hsueh, J.T.; Hsieh, Y.H. Licochalcone A induces autophagy through PI3K/Akt/mTOR inactivation and autophagy suppression enhances licochalcone A-induced apoptosis of human cervical cancer cells. Oncotarget 2015, 6, 28851–28866. [Google Scholar] [CrossRef]
- Kim, K.H.; Yoon, G.; Cho, J.J.; Cho, J.H.; Cho, Y.S.; Chae, J.I.; Shim, J.H. Licochalcone A induces apoptosis in malignant pleural mesothelioma through downregulation of Sp1 and subsequent activation of mitochondria-related apoptotic pathway. Int. J. Oncol. 2015, 46, 1385–1392. [Google Scholar] [CrossRef]
- Kim, J.S.; Park, M.R.; Lee, S.Y.; Kim, D.K.; Moon, S.M.; Kim, C.S.; Cho, S.S.; Yoon, G.; Im, H.J.; You, J.S.; et al. Licochalcone A induces apoptosis in KB human oral cancer cells via a caspase-dependent FasL signaling pathway. Oncol. Rep. 2014, 31, 755–762. [Google Scholar] [CrossRef]
- Cho, J.J.; Chae, J.I.; Yoon, G.; Kim, K.H.; Cho, J.H.; Cho, S.S.; Cho, Y.S.; Shim, J.H. Licochalcone A, a natural chalconoid isolated from Glycyrrhiza inflata root, induces apoptosis via Sp1 and Sp1 regulatory proteins in oral squamous cell carcinoma. Int. J. Oncol. 2014, 45, 667–674. [Google Scholar] [CrossRef]
- Choi, A.Y.; Choi, J.H.; Hwang, K.Y.; Jeong, Y.J.; Choe, W.; Yoon, K.S.; Ha, J.; Kim, S.S.; Youn, J.H.; Yeo, E.J.; et al. Licochalcone A induces apoptosis through endoplasmic reticulum stress via a phospholipase Cγ1-, Ca2+-, and reactive oxygen species-dependent pathway in HepG2 human hepatocellular carcinoma cells. Apoptosis 2014, 19, 682–697. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.; Li, D.; Zhao, H.; Jiang, J.; Wang, P.; Ma, X.; Sun, X.; Zheng, Q. Licochalcone A-induced human bladder cancer T24 cells apoptosis triggered by mitochondria dysfunction and endoplasmic reticulum stress. Biomed. Res. Int. 2013, 2013, 474272. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.S.; Kwak, S.W.; Kim, Y.J.; Lee, S.A.; Park, E.S.; Myung, S.C.; Kim, W.; Lee, M.S.; Lee, J.J. Guanylate cyclase activator YC-1 potentiates apoptotic effect of licochalcone A on human epithelial ovarian carcinoma cells via activation of death receptor and mitochondrial pathways. Eur. J. Pharmacol. 2012, 683, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Hao, W.; Yuan, X.; Yu, L.; Gao, C.; Sun, X.; Wang, D.; Zheng, Q. Licochalcone A-induced human gastric cancer BGC-823 cells apoptosis by regulating ROS-mediated MAPKs and PI3K/AKT signaling pathways. Sci. Rep. 2015, 5, 10336. [Google Scholar] [CrossRef] [PubMed]
- Park, M.R.; Kim, S.G.; Cho, I.A.; Oh, D.; Kang, K.R.; Lee, S.Y.; Moon, S.M.; Cho, S.S.; Yoon, G.; Kim, C.S.; et al. Licochalcone-A induces intrinsic and extrinsic apoptosis via ERK1/2 and p38 phosphorylation-mediated TRAIL expression in head and neck squamous carcinoma FaDu cells. Food Chem. Toxicol. 2015, 77, 34–43. [Google Scholar] [CrossRef] [PubMed]
- Niu, Q.; Zhao, W.; Wang, J.; Li, C.; Yan, T.; Lv, W.; Wang, G.; Duan, W.; Zhang, T.; Wang, K.; et al. LicA induces autophagy through ULK1/Atg13 and ROS pathway in human hepatocellular carcinoma cells. Int. J. Mol. Med. 2018, 41, 2601–2608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, J.; Yuan, X.; Zhao, H.; Yan, X.; Sun, X.; Zheng, Q. Licochalcone A inhibiting proliferation of bladder cancer T24 cells by inducing reactive oxygen species production. Biomed. Mater. Eng. 2014, 24, 1019–1025. [Google Scholar] [PubMed]
- Bai, J.; Li, Y.; Zhang, G. Cell cycle regulation and anticancer drug discovery. Cancer Biol. Med. 2017, 14, 348–362. [Google Scholar]
- de Graaf, A.O.; de Witte, T.; Jansen, J.H. Inhibitor of apoptosis proteins: New therapeutic targets in hematological cancer? Leukemia 2004, 18, 1751–1759. [Google Scholar] [CrossRef]
- Sánchez-Martínez, C.; Gelbert, L.M.; Lallena, M.J.; de Dios, A. Cyclin dependent kinase (CDK) inhibitors as anticancer drugs. Bioorg. Med. Chem. Lett. 2015, 25, 3420–3435. [Google Scholar] [CrossRef]
- Matheson, C.J.; Backos, D.S.; Reigan, P. Targeting WEE1 kinase in cancer. Trends Pharmacol. Sci. 2016, 37, 872–881. [Google Scholar] [CrossRef] [PubMed]
- Bulavin, D.V.; Demidenko, Z.N.; Phillips, C.; Moody, S.A.; Fornace, A.J., Jr. Phosphorylation of Xenopus Cdc25C at Ser285 interferes with ability to activate a DNA damage replication checkpoint in pre-midblastula embryos. Cell Cycle 2003, 2, 263–266. [Google Scholar] [CrossRef] [PubMed]
- Karimian, A.; Ahmadi, Y.; Yousefi, B. Multiple functions of p21 in cell cycle, apoptosis and transcriptional regulation after DNA damage. DNA Repair 2016, 42, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Reinhardt, H.C.; Schumacher, B. The p53 network: Cellular and systemic DNA damage responses in aging and cancer. Trends Genet. 2012, 28, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Hydbring, P.; Malumbres, M.; Sicinski, P. Non-canonical functions of cell cycle cyclins and cyclin-dependent kinases. Nat. Rev. Mol. Cell Biol. 2016, 17, 280–292. [Google Scholar] [CrossRef] [PubMed]
- Cooper, M.J.; Haluschak, J.J.; Johnson, D.; Schwartz, S.; Morrison, L.J.; Lippa, M.; Hatzivassiliou, G.; Tan, J. p53 mutations in bladder carcinoma cell lines. Oncol. Res. 1994, 6, 569–579. [Google Scholar] [PubMed]
- Edlich, F. BCL-2 proteins and apoptosis: Recent insights and unknowns. Biochem. Biophys. Res. Commun. 2018, 500, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, C.M.; Singh, A.T.K. Apoptosis: A target for anticancer therapy. Int. J. Mol. Sci. 2018, 19, 448. [Google Scholar] [CrossRef]
- Kantari, C.; Walczak, H. Caspase-8 and bid: Caught in the act between death receptors and mitochondria. Biochim. Biophys. Acta. 2011, 1813, 558–563. [Google Scholar] [CrossRef]
- Birkinshaw, R.W.; Czabotar, P.E. The BCL-2 family of proteins and mitochondrial outer membrane permeabilisation. Semin. Cell Dev. Biol. 2017, 72, 152–162. [Google Scholar] [CrossRef]
- Badrinath, N.; Yoo, S.Y. Mitochondria in cancer: In the aspects of tumorigenesis and targeted therapy. Carcinogenesis 2018, 39, 1419–1430. [Google Scholar] [CrossRef] [PubMed]
- Bolhassani, A. Cancer chemoprevention by natural carotenoids as an efficient strategy. Anticancer Agents Med. Chem. 2015, 15, 1026–1231. [Google Scholar] [CrossRef] [PubMed]
- NavaneethaKrishnan, S.; Rosales, J.L.; Lee, K.Y. ROS-mediated cancer cell killing through dietary phytochemicals. Oxid. Med. Cell. Longev. 2019, 2019, 9051542. [Google Scholar] [CrossRef] [PubMed]
- Vallejo, M.J.; Salazar, L.; Grijalva, M. Oxidative stress modulation and ROS-mediated toxicity in cancer: A review on in vitro models for plant-derived compounds. Oxid. Med. Cell. Longev. 2017, 2017, 4586068. [Google Scholar] [CrossRef] [PubMed]
- Ranjan, A.; Fofaria, N.M.; Kim, S.H.; Srivastava, S.K. Modulation of signal transduction pathways by natural compounds in cancer. Chin. J. Nat. Med. 2015, 13, 730–742. [Google Scholar] [CrossRef]
- Moloney, J.N.; Cotter, T.G. ROS signalling in the biology of cancer. Semin. Cell Dev. Biol. 2018, 80, 50–64. [Google Scholar] [CrossRef] [PubMed]
- Galadari, S.; Rahman, A.; Pallichankandy, S.; Thayyullathil, F. Reactive oxygen species and cancer paradox: To promote or to suppress? Free Radic. Biol. Med. 2017, 104, 144–164. [Google Scholar] [CrossRef] [PubMed]
- Park, C.; Jeong, N.Y.; Kim, G.Y.; Han, M.H.; Chung, I.M.; Kim, W.J.; Yoo, Y.H.; Choi, Y.H. Momilactone B induces apoptosis and G1 arrest of the cell cycle in human monocytic leukemia U937 cells through downregulation of pRB phosphorylation and induction of the cyclin-dependent kinase inhibitor p21Waf1/Cip1. Oncol. Rep. 2014, 31, 1653–1660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.Y.; Kim, J.H.; Lee, J.C.; Won, M.H.; Yang, S.R.; Kim, H.C.; Wie, M.B. Zinc oxide nanoparticles exhibit both cyclooxygenase- and lipoxygenase-mediated apoptosis in human bone marrow-derived mesenchymal stem cells. Toxicol. Res. 2019, 35, 83–91. [Google Scholar] [CrossRef]








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hong, S.H.; Cha, H.-J.; Hwang-Bo, H.; Kim, M.Y.; Kim, S.Y.; Ji, S.Y.; Cheong, J.; Park, C.; Lee, H.; Kim, G.-Y.; et al. Anti-Proliferative and Pro-Apoptotic Effects of Licochalcone A through ROS-Mediated Cell Cycle Arrest and Apoptosis in Human Bladder Cancer Cells. Int. J. Mol. Sci. 2019, 20, 3820. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20153820
Hong SH, Cha H-J, Hwang-Bo H, Kim MY, Kim SY, Ji SY, Cheong J, Park C, Lee H, Kim G-Y, et al. Anti-Proliferative and Pro-Apoptotic Effects of Licochalcone A through ROS-Mediated Cell Cycle Arrest and Apoptosis in Human Bladder Cancer Cells. International Journal of Molecular Sciences. 2019; 20(15):3820. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20153820
Chicago/Turabian StyleHong, Su Hyun, Hee-Jae Cha, Hyun Hwang-Bo, Min Yeong Kim, So Young Kim, Seon Yeong Ji, JaeHun Cheong, Cheol Park, Hyesook Lee, Gi-Young Kim, and et al. 2019. "Anti-Proliferative and Pro-Apoptotic Effects of Licochalcone A through ROS-Mediated Cell Cycle Arrest and Apoptosis in Human Bladder Cancer Cells" International Journal of Molecular Sciences 20, no. 15: 3820. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20153820