Olmesartan Attenuates Kidney Fibrosis in a Murine Model of Alport Syndrome by Suppressing Tubular Expression of TGFβ

 , , and
, , and
Abstract
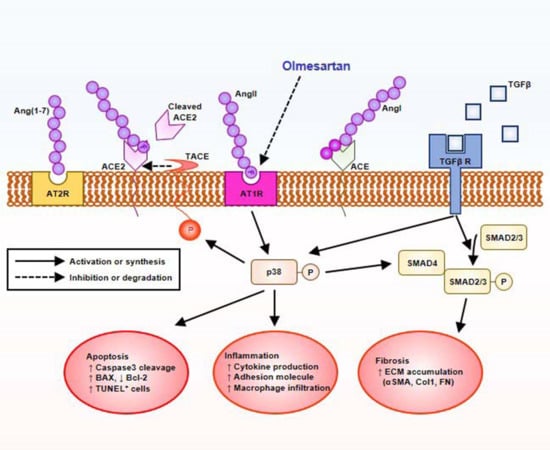
:
1. Introduction
2. Results
2.1. Olmesartan Ameliorates Kidney Fibrosis in Col4a3–/– Mice
2.2. Preferential Upregulation of TGFβ in Tubulointerstitium in Col4a3–/– Mice is Attenuated by Olmesartan
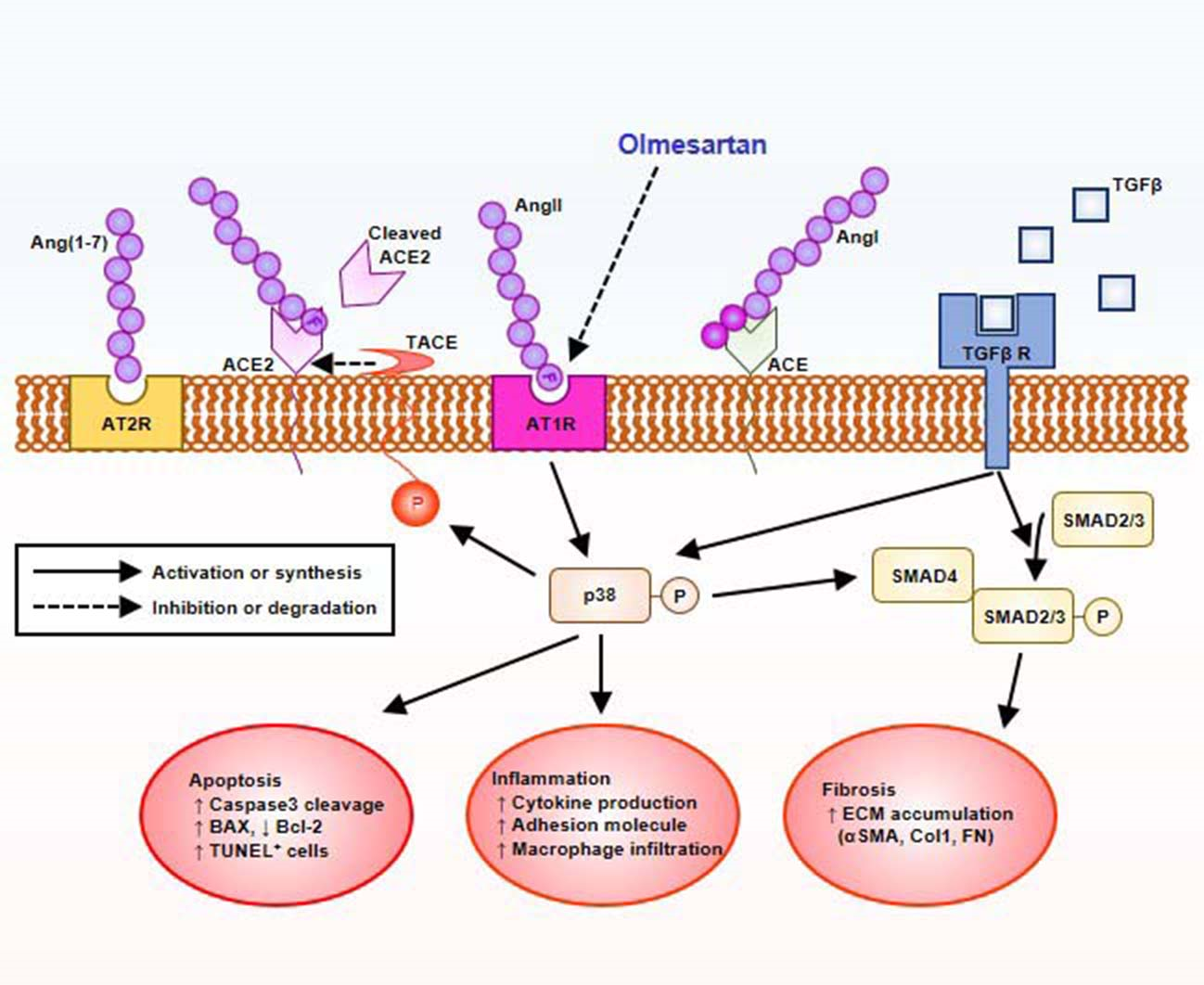
2.3. Downregulation of ACE2 in Col4a3–/– Mice Is Counteracted by Olmesartan
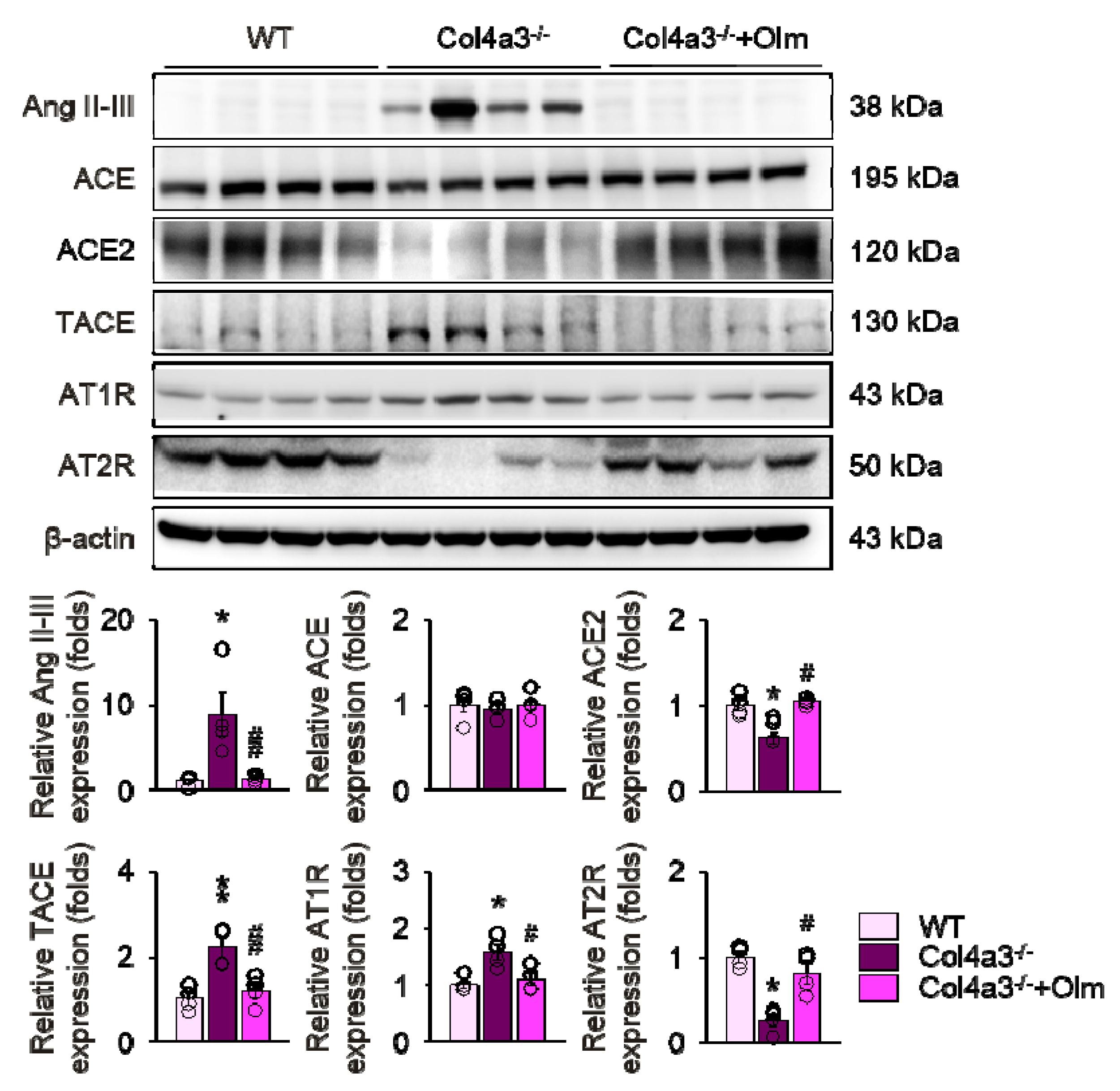
2.4. Olmesartan Prevents Apoptosis and Inflammation in the Kidneys of Col4a3–/– Mice
2.5. Olmesartan Targets RAS-TGFβ Feedback Loop to Regulate HK-2 Cell Fibrosis
3. Discussion
4. Materials and Methods
4.1. Experimental Animals and Protocols
4.2. Serum and urine NGAL measurement by ELISA
4.3. Histology and Immunohistochemistry
4.4. Detection of Apoptosis with Tunel Staining
4.5. Cell Culture
4.6. Semi-quantitative Immunoblotting
4.7. Real-Time qPCR
4.8. Measurement of BP
4.9. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Hudson, B.G.; Tryggvason, K.; Sundaramoorthy, M.; Neilson, E.G. Alport’s Syndrome, Goodpasture’s Syndrome, and Type IV Collagen. New Engl. J. Med. 2003, 348, 2543–2556. [Google Scholar] [CrossRef] [PubMed]
- Boutaud, A.; Borza, D.B.; Bondar, O.; Gunwar, S.; Netzer, K.O.; Singh, N.; Ninomiya, Y.; Sado, Y.; Noelken, M.E.; Hudson, B.G. Type IV collagen of the glomerular basement membrane. Evidence that the chain specificity of network assembly is encoded by the noncollagenous NC1 domains. J. Biol. Chem. 2000, 275, 30716–30724. [Google Scholar] [CrossRef] [PubMed]
- Abrahamson, D.R.; Prettyman, A.C.; Robert, B.; St John, P.L. Laminin-1 reexpression in Alport mouse glomerular basement membranes. Kidney Int. 2003, 63, 826–834. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Shield, C.F.; Todd, P.; Hudson, B.G.; Neilson, E.G. Isoform switching of type IV collagen is developmentally arrested in X-linked Alport syndrome leading to increased susceptibility of renal basement membranes to endoproteolysis. J. Clin. Invest. 1997, 99, 2470–2478. [Google Scholar] [CrossRef] [PubMed]
- Cosgrove, D.; Meehan, D.T.; Grunkemeyer, J.A.; Kornak, J.M.; Sayers, R.; Hunter, W.J.; Samuelson, G.C. Collagen COL4A3 knockout: A mouse model for autosomal Alport syndrome. Genes Dev. 1996, 10, 2981–2992. [Google Scholar] [CrossRef] [PubMed]
- Gross, O.; Beirowski, B.; Koepke, M.L.; Kuck, J.; Reiner, M.; Addicks, K.; Smyth, N.; Schulze-Lohoff, E.; Weber, M. Preemptive ramipril therapy delays renal failure and reduces renal fibrosis in COL4A3-knockout mice with Alport syndrome. Kidney Int. 2003, 63, 438–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gross, O.; Schulze-Lohoff, E.; Koepke, M.L.; Beirowski, B.; Addicks, K.; Bloch, W.; Smyth, N.; Weber, M. Antifibrotic, nephroprotective potential of ACE inhibitor vs AT1 antagonist in a murine model of renal fibrosis. Nephrol. Dial. Transpl. 2004, 19, 1716–1723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savige, J.; Gregory, M.; Gross, O.; Kashtan, C.; Ding, J.; Flinter, F. Expert guidelines for the management of Alport syndrome and thin basement membrane nephropathy. J. Am. Soc. Nephrol. 2013, 24, 364–375. [Google Scholar] [CrossRef] [PubMed]
- Webb, N.J.; Shahinfar, S.; Wells, T.G.; Massaad, R.; Gleim, G.W.; McCrary Sisk, C.; Lam, C. Losartan and enalapril are comparable in reducing proteinuria in children with Alport syndrome. Pediatr. Nephrol. 2013, 28, 737–743. [Google Scholar] [CrossRef]
- Donoghue, M.; Hsieh, F.; Baronas, E.; Godbout, K.; Gosselin, M.; Stagliano, N.; Donovan, M.; Woolf, B.; Robison, K.; Jeyaseelan, R.; et al. A Novel Angiotensin-Converting Enzyme–Related Carboxypeptidase (ACE2) Converts Angiotensin I to Angiotensin 1-9. Circ. Res. 2000, 87, e1–e9. [Google Scholar] [CrossRef] [PubMed]
- Vickers, C.; Hales, P.; Kaushik, V.; Dick, L.; Gavin, J.; Tang, J.; Godbout, K.; Parsons, T.; Baronas, E.; Hsieh, F.; et al. Hydrolysis of biological peptides by human angiotensin-converting enzyme-related carboxypeptidase. J. Biol. Chem. 2002, 277, 14838–14843. [Google Scholar] [CrossRef] [PubMed]
- Ferrario, C.M.; Chappell, M.C.; Tallant, E.A.; Brosnihan, K.B.; Diz, D.I. Counterregulatory Actions of Angiotensin-(1-7). Hypertension 1997, 30, 535–541. [Google Scholar] [CrossRef] [PubMed]
- Ferrario, C.M.; Averill, D.B.; Brosnihan, K.B.; Chappell, M.C.; Iskandar, S.S.; Dean, R.H.; Diz, D.I. Vasopeptidase inhibition and Ang-(1-7) in the spontaneously hypertensive rat. Kidney Int. 2002, 62, 1349–1357. [Google Scholar] [CrossRef] [PubMed]
- Oudit, G.Y.; Herzenberg, A.M.; Kassiri, Z.; Wong, D.; Reich, H.; Khokha, R.; Crackower, M.A.; Backx, P.H.; Penninger, J.M.; Scholey, J.W. Loss of angiotensin-converting enzyme-2 leads to the late development of angiotensin II-dependent glomerulosclerosis. Am. J. Pathol. 2006, 168, 1808–1820. [Google Scholar] [CrossRef] [PubMed]
- Wong, D.W.; Oudit, G.Y.; Reich, H.; Kassiri, Z.; Zhou, J.; Liu, Q.C.; Backx, P.H.; Penninger, J.M.; Herzenberg, A.M.; Scholey, J.W. Loss of angiotensin-converting enzyme-2 (Ace2) accelerates diabetic kidney injury. Am. J. Pathol. 2007, 171, 438–451. [Google Scholar] [CrossRef] [PubMed]
- Bae, E.H.; Fang, F.; Williams, V.R.; Konvalinka, A.; Zhou, X.; Patel, V.B.; Song, X.; John, R.; Oudit, G.Y.; Pei, Y.; et al. Murine recombinant angiotensin-converting enzyme 2 attenuates kidney injury in experimental Alport syndrome. Kidney Int. 2017, 91, 1347–1361. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.S.; Kim, I.J.; Bae, E.H.; Ma, S.K.; Lee, J.; Kim, S.W. Angiotensin-(1-7) Attenuates Kidney Injury Due to Obstructive Nephropathy in Rats. PLoS ONE 2015, 10, e0142664. [Google Scholar] [CrossRef]
- Nishio, M.; Sakata, Y.; Mano, T.; Yoshida, J.; Ohtani, T.; Takeda, Y.; Miwa, T.; Masuyama, T.; Yamamoto, K.; Hori, M. Therapeutic effects of angiotensin II type 1 receptor blocker at an advanced stage of hypertensive diastolic heart failure. J. Hypertens. 2007, 25, 455–461. [Google Scholar] [CrossRef]
- Yoshida, K.; Kohzuki, M. Clinical and Experimental Aspects of Olmesartan Medoxomil, a New Angiotensin II Receptor Antagonist. Cardiovasc. Drug Rev. 2004, 22, 285–308. [Google Scholar] [CrossRef]
- Agata, J.; Ura, N.; Yoshida, H.; Shinshi, Y.; Sasaki, H.; Hyakkoku, M.; Taniguchi, S.; Shimamoto, K. Olmesartan Is an Angiotensin II Receptor Blocker with an Inhibitory Effect on Angiotensin-Converting Enzyme. Hypertens. Res. 2006, 29, 865–874. [Google Scholar] [CrossRef] [Green Version]
- Ishiyama, Y.; Gallagher, P.E.; Averill, D.B.; Tallant, E.A.; Brosnihan, K.B.; Ferrario, C.M. Upregulation of angiotensin-converting enzyme 2 after myocardial infarction by blockade of angiotensin II receptors. Hypertension 2004, 43, 970–976. [Google Scholar] [CrossRef] [PubMed]
- Sukumaran, V.; Veeraveedu, P.T.; Lakshmanan, A.P.; Gurusamy, N.; Yamaguchi, K.I.; Ma, M.; Suzuki, K.; Kodama, M.; Watanabe, K. Olmesartan medoxomil treatment potently improves cardiac myosin-induced dilated cardiomyopathy via the modulation of ACE-2 and ANG 1–7 mas receptor. Free Radic. Res. 2012, 46, 850–860. [Google Scholar] [CrossRef] [PubMed]
- Tanno, T.; Tomita, H.; Narita, I.; Kinjo, T.; Nishizaki, K.; Ichikawa, H.; Kimura, Y.; Tanaka, M.; Osanai, T.; Okumura, K. Olmesartan Inhibits Cardiac Hypertrophy in Mice Overexpressing Renin Independently of Blood Pressure: Its Beneficial Effects on ACE2/Ang(1-7)/Mas Axis and NADPH Oxidase Expression. J. Cardiovasc Pharm. 2016, 67, 503–509. [Google Scholar] [CrossRef] [PubMed]
- Sayers, R.; Kalluri, R.; Rodgers, K.D.; Shield, C.F.; Meehan, D.T.; Cosgrove, D. Role for transforming growth factor-beta1 in alport renal disease progression. Kidney Int. 1999, 56, 1662–1673. [Google Scholar] [CrossRef] [PubMed]
- Patel, V.B.; Clarke, N.; Wang, Z.; Fan, D.; Parajuli, N.; Basu, R.; Putko, B.; Kassiri, Z.; Turner, A.J.; Oudit, G.Y. Angiotensin II induced proteolytic cleavage of myocardial ACE2 is mediated by TACE/ADAM-17: A positive feedback mechanism in the RAS. J. Mol. Cell. Cardiol. 2014, 66, 167–176. [Google Scholar] [CrossRef]
- Ross, M.J.; Nangaku, M. ACE2 as therapy for glomerular disease: The devil is in the detail. Kidney Int. 2017, 91, 1269–1271. [Google Scholar] [CrossRef]
- Wysocki, J.; Ye, M.; Khattab, A.M.; Fogo, A.; Martin, A.; David, N.V.; Kanwar, Y.; Osborn, M.; Batlle, D. Angiotensin-converting enzyme 2 amplification limited to the circulation does not protect mice from development of diabetic nephropathy. Kidney Int. 2017, 91, 1336–1346. [Google Scholar] [CrossRef]
- Kralova, J.; Dvorak, M.; Koc, M.; Kral, V. p38 MAPK plays an essential role in apoptosis induced by photoactivation of a novel ethylene glycol porphyrin derivative. Oncogene 2008, 27, 3010–3020. [Google Scholar] [CrossRef]
- Wada, T.; Penninger, J.M. Mitogen-activated protein kinases in apoptosis regulation. Oncogene 2004, 23, 2838–2849. [Google Scholar] [CrossRef] [Green Version]
- Daemen, M.A.; van ‘t Veer, C.; Denecker, G.; Heemskerk, V.H.; Wolfs, T.G.; Clauss, M.; Vandenabeele, P.; Buurman, W.A. Inhibition of apoptosis induced by ischemia-reperfusion prevents inflammation. J. Clin. Invest. 1999, 104, 541–549. [Google Scholar] [CrossRef] [Green Version]
- Cauwels, A.; Janssen, B.; Waeytens, A.; Cuvelier, C.; Brouckaert, P. Caspase inhibition causes hyperacute tumor necrosis factor–induced shock via oxidative stress and phospholipase A2. Nat. Immunol. 2003, 4, 387–393. [Google Scholar] [CrossRef] [PubMed]
- Bellon, T.; Martinez, V.; Lucendo, B.; del Peso, G.; Castro, M.J.; Aroeira, L.S.; Rodriguez-Sanz, A.; Ossorio, M.; Sanchez-Villanueva, R.; Selgas, R.; et al. Alternative activation of macrophages in human peritoneum: Implications for peritoneal fibrosis. Nephrol. Dial. Transplant. 2011, 26, 2995–3005. [Google Scholar] [CrossRef] [PubMed]
- Fogo, A.B.; Lusco, M.A.; Najafian, B.; Alpers, C.E. AJKD Atlas of Renal Pathology: Alport Syndrome. Am. J. Kidney Dis. 2016, 68, e15–e16. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.S.; Song, J.H.; Kim, I.J.; Joo, S.Y.; Eom, G.H.; Kim, I.; Cha, H.; Cho, J.M.; Ma, S.K.; Kim, S.W.; et al. Histone deacetylase inhibitor, CG200745 attenuates renal fibrosis in obstructive kidney disease. Sci. Rep. 2018, 8, 11546. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| WT | Col4a3–/– | Col4a3-/-+Olm | |
|---|---|---|---|
| Body weight (g) | 19.7 ± 2.6 | 20.0 ± 0.4 | 20.7 ± 1.0 |
| Kidney weight (g) | 0.1 ± 0.0 | 0.1 ± 0.0 | 0.1 ± 0.0 |
| Kidney weight / body weight (g/kg) | 7.6 ± 1.6 | 9.3 ± 0.3 | 8.3 ± 0.4 |
| Urine creatinine (mg/dL) | 49.0 ± 10.5 | 21.2 ± 3.5 * | 9.1 ± 3.3 ## |
| Urine albumin (μg/mL) | 7.2 ± 2.0 | 234.5 ± 32.5 * | 72.0 ± 45.1 # |
| Urine albumin-to-creatinine ratio | 15.1 ± 4.4 | 1119.0 ± 188.5 ** | 426.3 ± 105.1 **,## |
| Serum NGAL (ng/mL) | 77.2 ± 7.5 | 335.5 ± 126.1 * | 43.8 ± 6.4 # |
| Urine NGAL (ng/mL) | 61.1 ± 3.8 | 848.8 ± 12.7 ** | 27.8 ± 2.8 ## |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Suh, S.H.; Choi, H.S.; Kim, C.S.; Kim, I.J.; Ma, S.K.; Scholey, J.W.; Kim, S.W.; Bae, E.H. Olmesartan Attenuates Kidney Fibrosis in a Murine Model of Alport Syndrome by Suppressing Tubular Expression of TGFβ. Int. J. Mol. Sci. 2019, 20, 3843. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20153843
Suh SH, Choi HS, Kim CS, Kim IJ, Ma SK, Scholey JW, Kim SW, Bae EH. Olmesartan Attenuates Kidney Fibrosis in a Murine Model of Alport Syndrome by Suppressing Tubular Expression of TGFβ. International Journal of Molecular Sciences. 2019; 20(15):3843. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20153843
Chicago/Turabian StyleSuh, Sang Heon, Hong Sang Choi, Chang Seong Kim, In Jin Kim, Seong Kwon Ma, James W. Scholey, Soo Wan Kim, and Eun Hui Bae. 2019. "Olmesartan Attenuates Kidney Fibrosis in a Murine Model of Alport Syndrome by Suppressing Tubular Expression of TGFβ" International Journal of Molecular Sciences 20, no. 15: 3843. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20153843