Comparison and Phylogenetic Analysis of Chloroplast Genomes of Three Medicinal and Edible Amomum Species

 , ,
, ,
Abstract
:1. Introduction
2. Results and Discussion
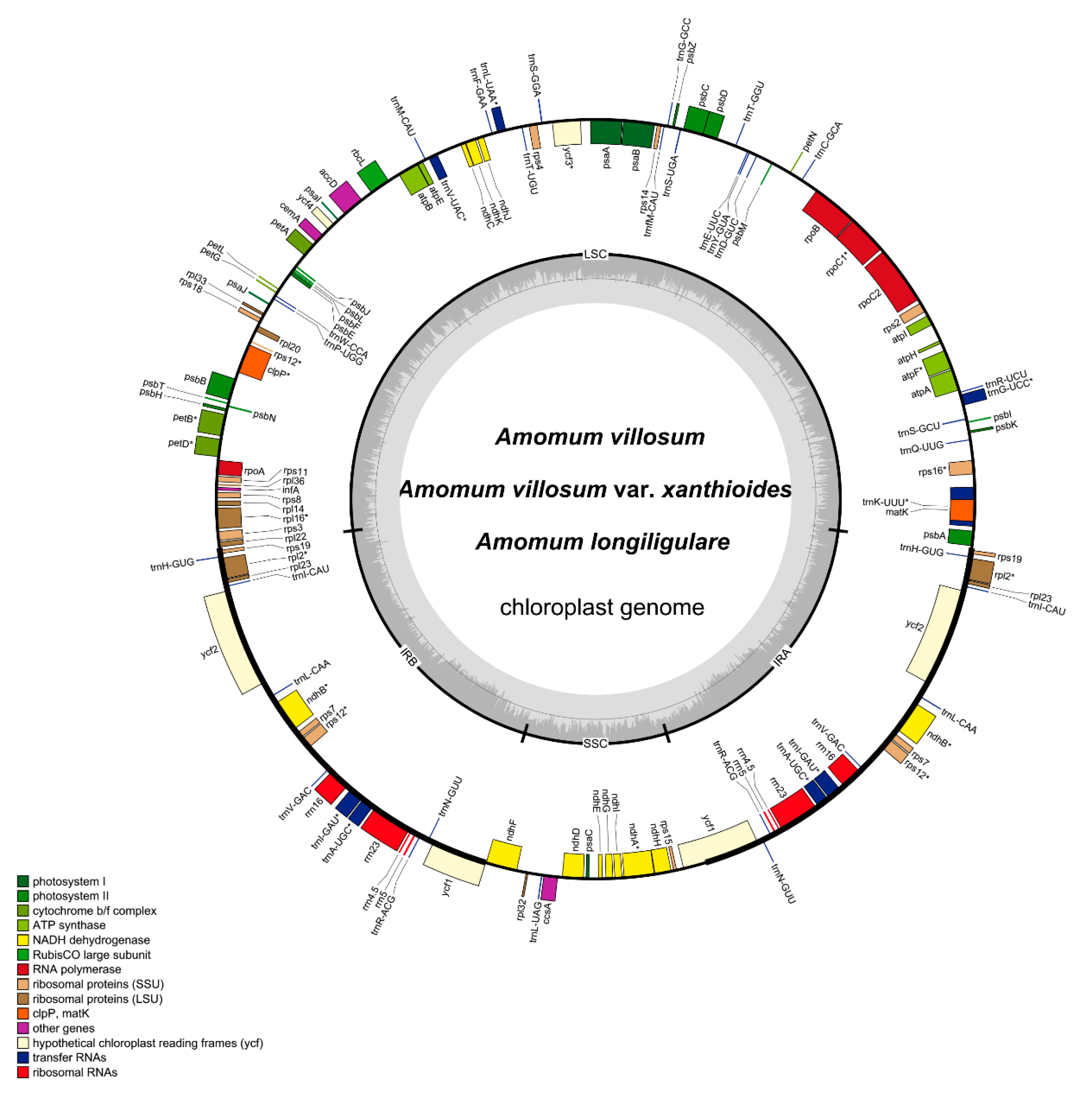
2.1. Genome Length and Features
2.2. Codon Usage
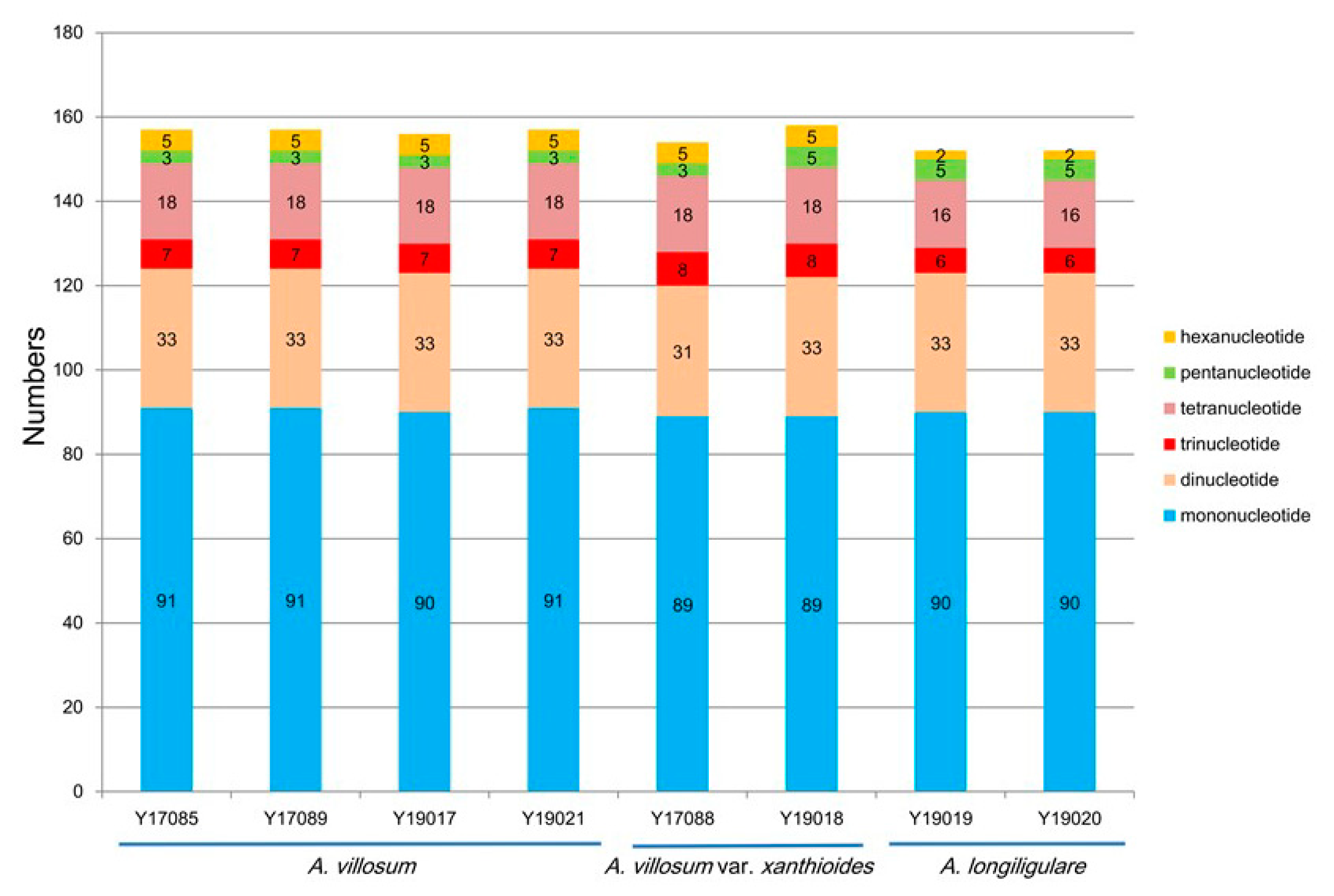
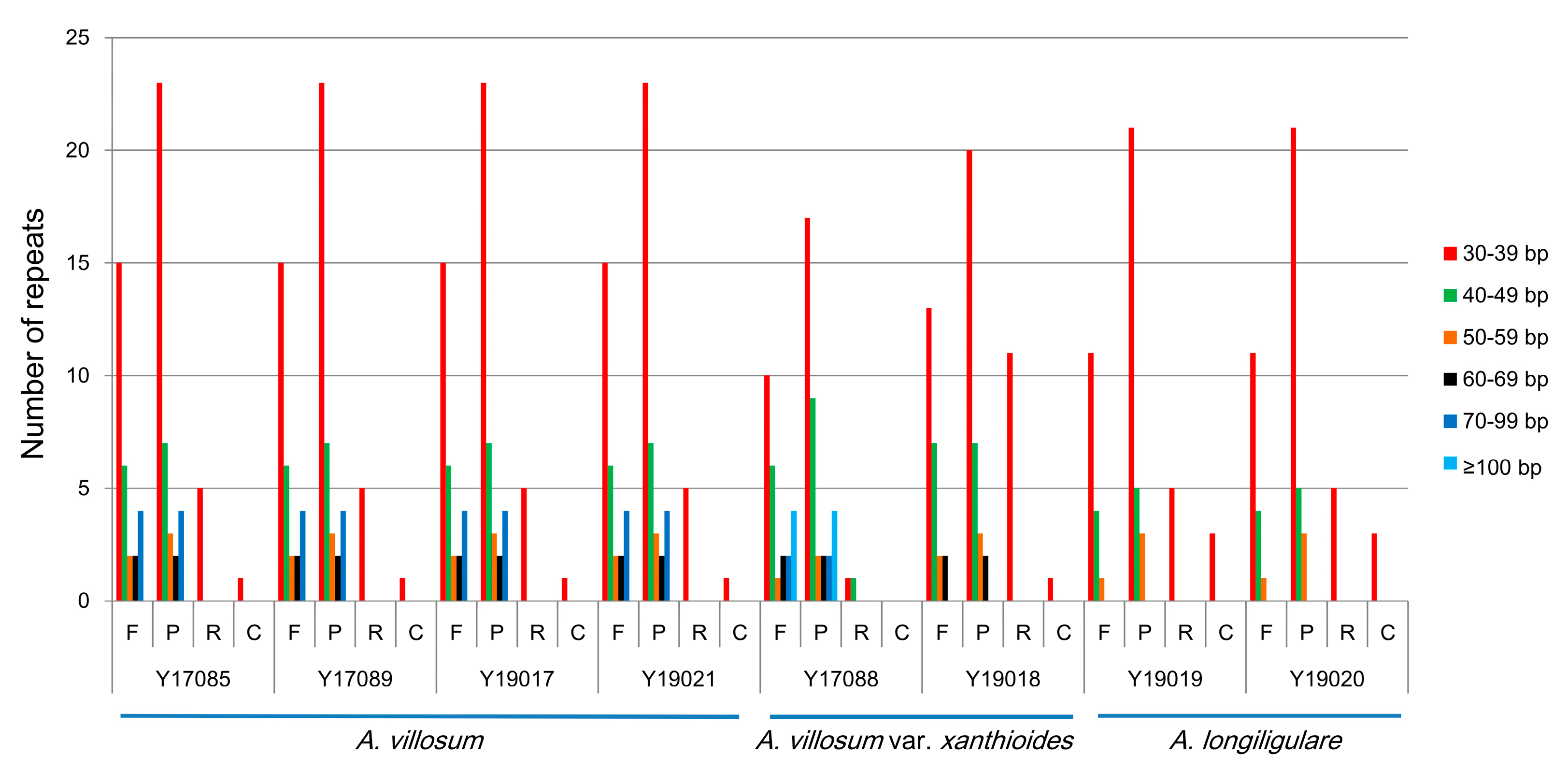
2.3. SSRs Analyses and Repeat Structures
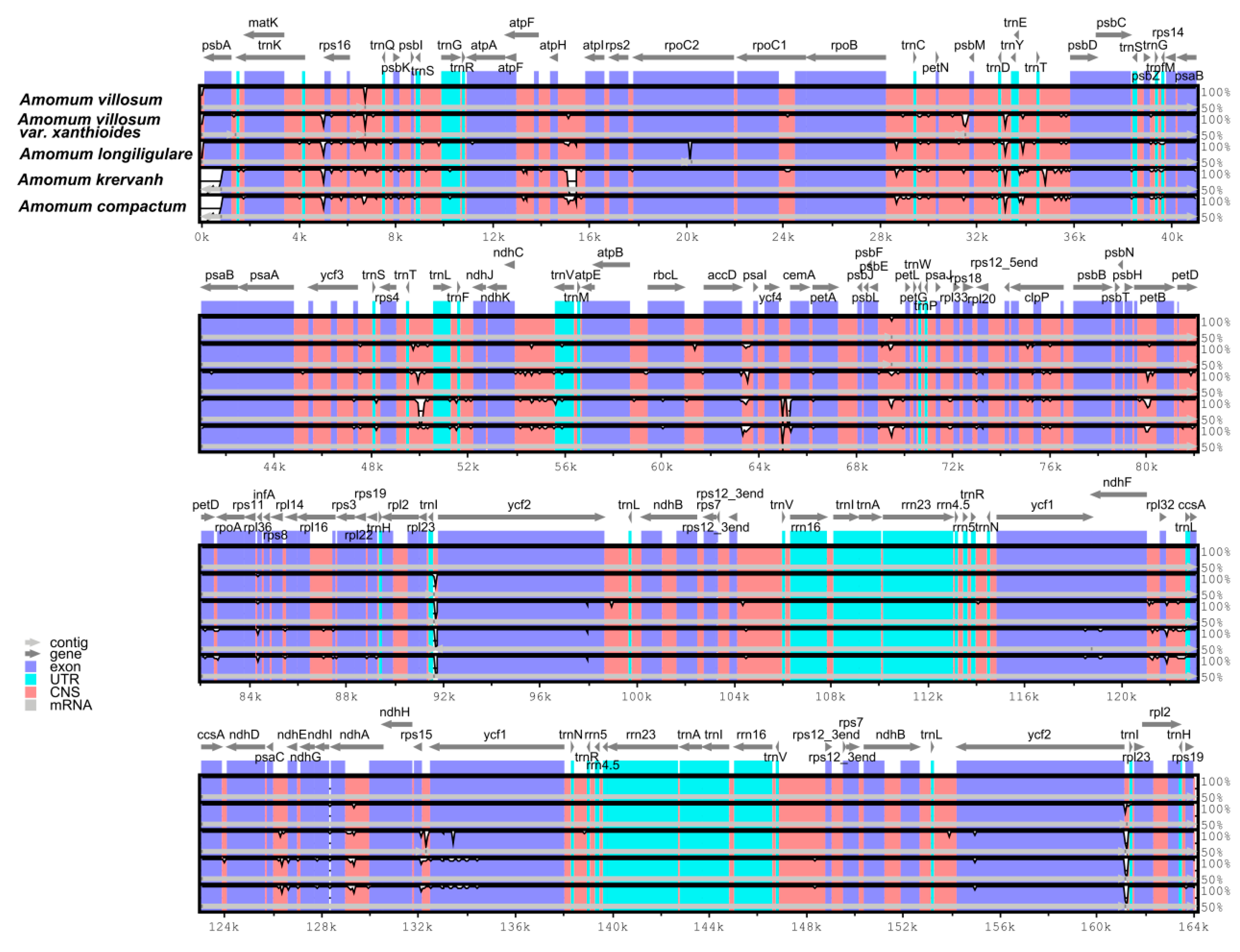
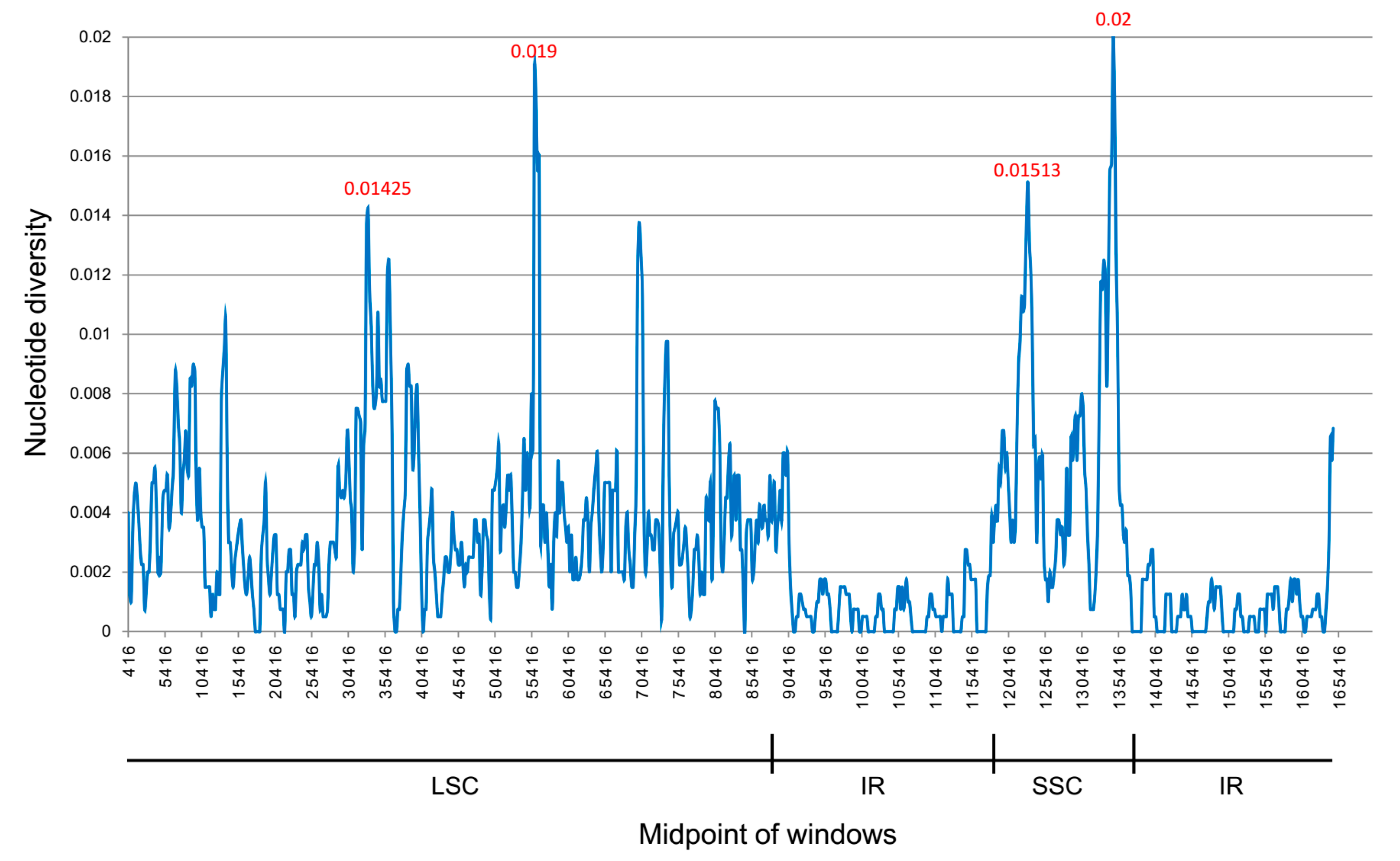
2.4. Interspecific Comparison
2.5. Species Authentication Analyses Based on cp Highly Divergent Regions
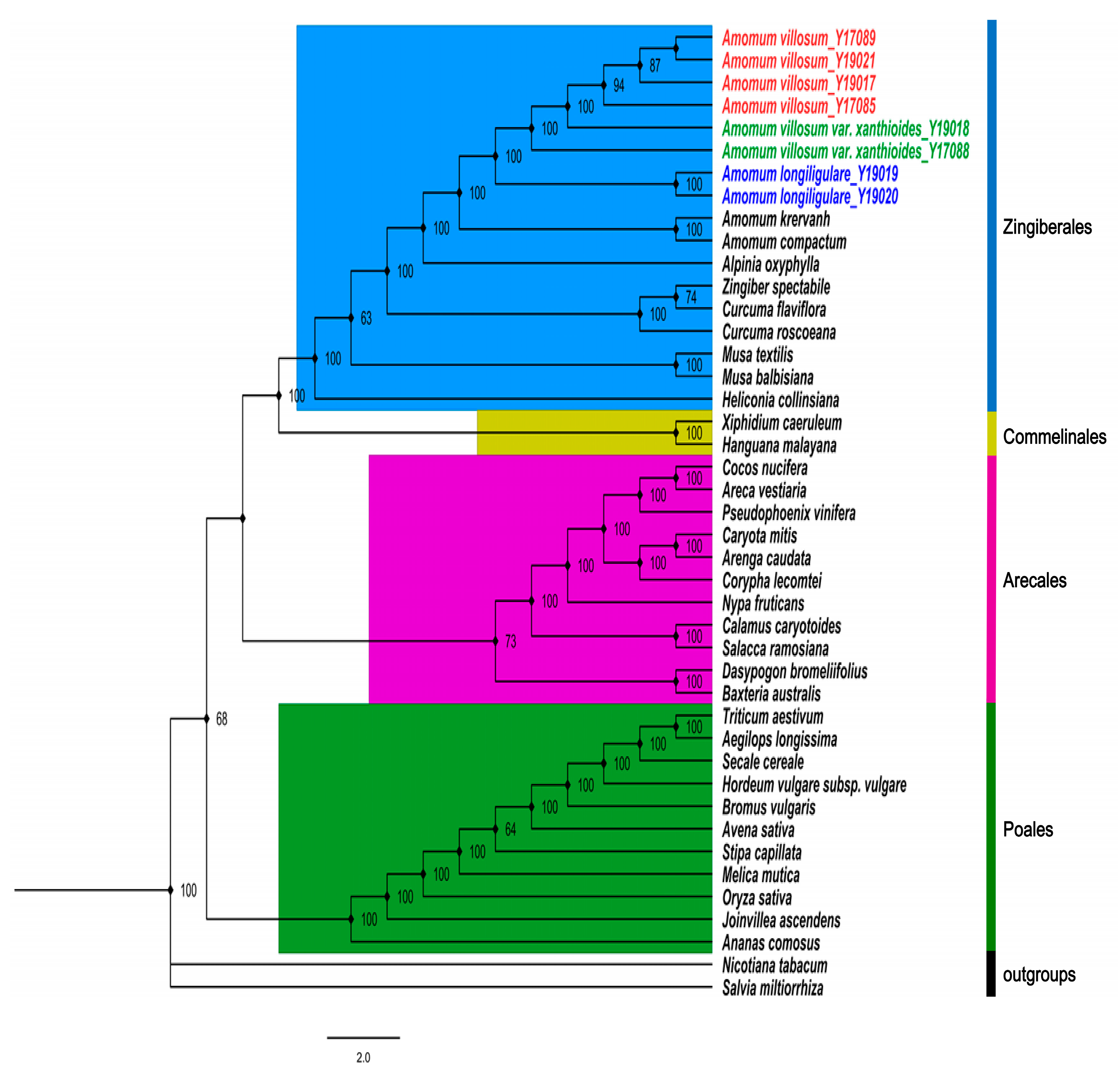
2.6. Phylogenetic Analysis
3. Materials and Methods
3.1. Plant Material, DNA Extraction, and Sequencing
3.2. Cp Genome Assembly
3.3. Cp genome Annotation and Structure Analysis
3.4. Cp genome Comparisons and Species Authentication
3.5. Phylogenetic Analyses
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| LSC | Large single copy |
| SSC | Small single copy |
| IR | Inverted repeat |
| ML | Maximum likelihood |
| SSR | Simple sequence repeats |
| ATP | Adenosine triphosphate |
| NADH | Nicotinamide adenine dinucleotide |
References
- Duan, L.S.; Zhang, L.X.; Peng, J.M.; Jie, M.A. Original Report on Investigation of Ammomum Villosum Germplasm Resources in Xishuangbanna. Lishizhen Med. Mater. Med. Res. 2009, 20, 627–628. [Google Scholar]
- Commission, C.P. Pharmacopoeia of the Peoples Republic of China; China Medical Science Press, 2015. Available online: https://www.tsoshop.co.uk/Medical/Pharmacopoeia/Chinese-Pharmacopoeia/ (accessed on 18 August 2019).
- Chen, Z.; Ni, W.; Yang, C.; Zhang, T.; Lu, S.; Zhao, R.; Mao, X.; Yu, J. Therapeutic Effect of Amomum villosum on Inflammatory Bowel Disease in Rats. Front. Pharmacol. 2018, 9, 639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, C.L.; Zhao, H.Y.; Jiang, J.G. Evaluation of multi-activities of 14 edible species from Zingiberaceae. Int. J. Food Sci. Nutr. 2013, 64, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Li, X.; Xiao, F.; Zhang, Z.; Xu, Z.; Wang, H. Studies on the analgesic and anti-inflammatory effect of bornyl acetate in volatile oil from Amomum villosum. J. Chin. Med. Mater. 2004, 27, 438. [Google Scholar]
- Tang, L.; He, G.; Su, J.; Xu, H. The Strategy to Promote the Development of Industry of Genuine Medicinal Material of Amomum villosum. Chin. Agric. Sci. Bull. 2012, 28, 94–99. [Google Scholar]
- Quan, Q.; Lin, J.C.; Liang, J.M.; Hu, Z.L.; Huang, J.X. Compare on the Content of Bronyl Acetate and Total Volatile Oil in Fructus Amomi from Different Producing Area. Guid. J. Tradit. Chin. Med. Pharm. 2017, 23, 70–73. [Google Scholar]
- Chen, J.; Ding, P.; Xu, X.; Xu, H. A resource investigation and commodity identification of Fructus Amomi. J. Chin. Med. Mater. 2001, 24, 18. [Google Scholar]
- Han, J.P.; Li, M.N.; Shi, L.C. Identification of Amomi fructus and its adulterants based on ITS2 sequences. Glob. Tradit. Chin. Med. 2011, 4, 99–102. [Google Scholar]
- Zhou, L.; Wang, P.; Huang, F.; Cao, L.; Liang, R. ITS sequence analysis of Amomum villosum. Chin. Tradit. Herb. Drugs 2002, 33, 72–75. [Google Scholar]
- Wang, P.; Huang, F.; Zhou, L.; Cao, L.; Liang, S.; Xu, H.; Liu, J. Analysis of Amomun villosum species and some adulterants of zingiberaceae by RAPD. J. Chin. Med. Mater. 2000, 23, 71–74. [Google Scholar]
- Huang, Q.; Duan, Z.; Yang, J.; Ma, X.; Zhan, R.; Xu, H.; Chen, W. SNP typing for germplasm identification of Amomum villosum Lour. Based on DNA barcoding markers. PLoS ONE 2014, 9, e114940. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhou, J.; Cui, Y.; Wang, Y.; Duan, B.; Yao, H. Identification of Ligularia Herbs Using the Complete Chloroplast Genome as a Super-Barcode. Front. Pharmacol. 2018, 9, 695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, I.; Yang, S.; Kim, W.; Noh, P.; Lee, H.; Moon, B. Authentication of Herbal Medicines Dipsacus asper and Phlomoides umbrosa Using DNA Barcodes, Chloroplast Genome, and Sequence Characterized Amplified Region (SCAR) Marker. Molecules 2018, 23, 1748. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Qi, Z.C.; Zhao, Y.P.; Fu, C.X.; Jenny Xiang, Q.Y. Complete cpDNA genome sequence of Smilax china and phylogenetic placement of Liliales--influences of gene partitions and taxon sampling. Mol. Phylogenetics Evol. 2012, 64, 545–562. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Li, Y.; Yang, H.; Zhou, B. Chloroplast Genome of the Folk Medicine and Vegetable Plant Talinum paniculatum (Jacq.) Gaertn.: Gene Organization, Comparative and Phylogenetic Analysis. Molecules 2018, 23, 857. [Google Scholar]
- Green, B.R. Chloroplast genomes of photosynthetic eukaryotes. Plant J. 2011, 66, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.-H.; Yin, W.-B.; Hu, Z.-M. Advances in chloroplast engineering. J. Genet. Genom. 2009, 36, 387–398. [Google Scholar] [CrossRef]
- Zhou, J.; Chen, X.; Cui, Y.; Sun, W.; Li, Y.; Wang, Y.; Song, J.; Yao, H. Molecular Structure and Phylogenetic Analyses of Complete Chloroplast Genomes of Two Aristolochia Medicinal Species. Int. J. Mol. Sci. 2017, 18, 1839. [Google Scholar] [CrossRef] [PubMed]
- Jansen, R.K.; Cai, Z.; Raubeson, L.A.; Daniell, H.; Depamphilis, C.W.; Leebensmack, J.; Müller, K.F.; Guisingerbellian, M.; Haberle, R.C.; Hansen, A.K. Analysis of 81 genes from 64 plastid genomes resolves relationships in angiosperms and identifies genome-scale evolutionary patterns. Proc. Natl. Acad. Sci. USA 2007, 104, 19369–19374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, L.; Qian, J.; Li, X.; Sun, Z.; Xu, X.; Chen, S. Complete Chloroplast Genome of Medicinal Plant Lonicera japonica: Genome Rearrangement, Intron Gain and Loss, and Implications for Phylogenetic Studies. Molecules 2017, 22, 249. [Google Scholar] [CrossRef]
- Daniell, H.; Lin, C.S.; Ming, Y.; Chang, W.J. Chloroplast genomes: Diversity, evolution, and applications in genetic engineering. Genome Biol. 2016, 17, 134. [Google Scholar] [CrossRef] [PubMed]
- Peter, H.R.; Zhang, L.B.; Ihsan, A.; Nicholas, J.; Zhu, G.H. Flora of China; Science Press: Beijing, China; Missouri Botanical Garden Press: St. Louis, MO, USA, 2000; Volume 24, pp. 347–356. [Google Scholar]
- Xiang, B.; Li, X.; Qian, J.; Wang, L.; Ma, L.; Tian, X.; Wang, Y. The Complete Chloroplast Genome Sequence of the Medicinal Plant Swertia mussotii Using the PacBio RS II Platform. Molecules 2016, 21, 1029. [Google Scholar] [CrossRef] [PubMed]
- Jiang, D.; Zhao, Z.; Zhang, T.; Zhong, W.; Liu, C.; Yuan, Q.; Huang, L. The Chloroplast Genome Sequence of Scutellaria baicalensis Provides Insight into Intraspecific and Interspecific Chloroplast Genome Diversity in Scutellaria. Genes 2017, 8, 227. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Dang, Y.; Li, Q.; Lu, J.; Li, X.; Wang, Y. Complete Chloroplast Genome Sequence of Poisonous and Medicinal Plant Datura stramonium: Organizations and Implications for Genetic Engineering. PLoS ONE 2014, 9, e110656. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Li, Q.; Hu, Z.; Li, X.; Chen, S. The Complete Amomum kravanh Chloroplast Genome Sequence and Phylogenetic Analysis of the Commelinids. Molecules 2017, 22, 1875. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.L.; Li, Q.; Xu, J.; Li, X.W. Complete chloroplast genome of the medicinal plant Amomum compactum: Gene organization, comparative analysis, and phylogenetic relationships within Zingiberales. Chin. Med. 2018, 13, 10. [Google Scholar] [CrossRef]
- Guo, H.; Liu, J.; Luo, L.; Wei, X.; Zhang, J.; Qi, Y.; Zhang, B.; Liu, H.; Xiao, P. Complete chloroplast genome sequences of Schisandra chinensis: Genome structure, comparative analysis, and phylogenetic relationship of basal angiosperms. Sci. China Life Sci. 2017, 60, 1286–1290. [Google Scholar] [CrossRef]
- Xu, J.; Feng, D.; Song, G.; Wei, X.; Chen, L.; Wu, X.; Li, X.; Zhu, Z. The first intron of rice EPSP synthase enhances expression of foreign gene. Sci. China 2003, 46, 561–569. [Google Scholar] [CrossRef]
- Kelchner, S.A. Group II introns as phylogenetic tools: Structure, function, and evolutionary constraints. Am. J. Bot. 2002, 89, 1651–1669. [Google Scholar] [CrossRef]
- Mattick, J.S. Introns: Evolution and function. Curr. Opin. Genet. Dev. 1994, 4, 823–831. [Google Scholar] [CrossRef]
- Chorev, M.; Carmel, L. The Function of Introns. Front. Genet. 2012, 3, 55. [Google Scholar] [CrossRef] [Green Version]
- Butt, A.M.; Nasrullah, I.; Qamar, R.; Tong, Y. Evolution of codon usage in Zika virus genomes is host and vector specific. Emerg. Microbes Infect. 2016, 5, e107. [Google Scholar] [CrossRef]
- Powell, W.; Morgante, M.; Mcdevitt, R.; Vendramin, G.G.; Rafalski, J.A. Polymorphic simple sequence repeat regions in chloroplast genomes: Applications to the population genetics of pines. Proc. Natl. Acad. Sci. USA 1995, 92, 7759–7763. [Google Scholar] [CrossRef]
- Ai-Hong, Y.; Jin-Ju, Z.; Xiao-Hong, Y.; Hong-Wen, H. Chloroplast microsatellite markers in Liriodendron tulipifera (Magnoliaceae) and cross-species amplification in L. chinense. Am. J. Bot. 2011, 98, 123–126. [Google Scholar]
- Jiao, Y. Development of simple sequence repeat (SSR) markers from a genome survey of Chinese bayberry (Myrica rubra). Bmc Genom. 2012, 13, 201. [Google Scholar] [CrossRef]
- Echt, C.S.; Deverno, L.L.; Anzidei, M.; Vendramin, G.G. Chloroplast microsatellites reveal population genetic diversity in red pine, Pinus resinosa Ait. Mol. Ecol. 2010, 7, 307–316. [Google Scholar] [CrossRef]
- Navascués, M.; Vaxevanidou, Z.; Climent, J.; Gil, L.; Emerson, B.C. Chloroplast microsatellites reveal colonization and metapopulation dynamics in the Canary Island pine. Mol. Ecol. 2010, 15, 2691–2698. [Google Scholar] [CrossRef]
- Baneh, H.D.; Mohammadi, S.A.; Labra, M.; Nazemieh, A.; Mardi, M. Chloroplast microsatellites markers to assess genetic diversity in wild and cultivated grapevines of Iran. Pak. J. Biol. Sci. 2007, 10, 1855–1859. [Google Scholar]
- Park, H.; Kim, C.; Lee, Y.M.; Kim, J.H.J.A.i.P.S. Development of Chloroplast Microsatellite Markers for the Endangered Maianthemum bicolor (Asparagaceae s.l.). Appl. Plant. Sci. 2016, 4, apps.1600032. [Google Scholar] [CrossRef]
- Yan, Y.D.; Li, X.Y.; Worth, J.R.P.; Lin, X.Y.; Ruhsam, M.; Chen, L.; Wu, X.T.; Wang, M.Q.; Thomas, P.I.; Wen, Y.F. Development of chloroplast microsatellite markers for Glyptostrobus pensilis (Cupressaceae). Appl. Plant. Sci. 2019, 7, e11277. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Nie, J.; Xiao, L.; Hu, Z.; Wang, B. Comparative Chloroplast Genome Analysis of Rhubarb Botanical Origins and the Development of Specific Identification Markers. Molecules 2018, 23, 2811. [Google Scholar] [CrossRef]
- Niu, Z.; Xue, Q.; Zhu, S.; Sun, J.; Liu, W.; Ding, X. The Complete Plastome Sequences of Four Orchid Species: Insights into the Evolution of the Orchidaceae and the Utility of Plastomic Mutational Hotspots. Front. Plant Sci. 2017, 8, 715. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.B.; Tang, M.; Li, H.T.; Zhang, Z.R.; Li, D.Z. Complete chloroplast genome of the genus Cymbidium: Lights into the species identification, phylogenetic implications and population genetic analyses. Bmc Evol. Biol. 2013, 13, 84. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Li, Y.; Zhou, J.-g.; Chen, X.-l.; Cui, Y.-x.; Xu, Z.-c.; Li, Y.-h.; Song, J.-y.; Duan, B.-z.; Yao, H. Gene losses and partial deletion of small single-copy regions of the chloroplast genomes of two hemiparasitic Taxillus species. Sci. Rep. 2017, 7, 12834. [Google Scholar] [CrossRef]
- Gogniashvili, M.; Naskidashvili, P.; Bedoshvili, D.; Kotorashvili, A.; Kotaria, N.; Beridze, T. Complete chloroplast DNA sequences of Zanduri wheat (Triticum spp.). Genet. Resour. Crop. Evol. 2015, 62, 1269–1277. [Google Scholar] [CrossRef]
- Boetzer, M.; Henkel, C.V.; Jansen, H.J.; Butler, D.; Pirovano, W. Scaffolding pre-assembled contigs using SSPACE. Bioinformatics 2011, 27, 578–579. [Google Scholar] [CrossRef]
- Acemel, R.D.; Tena, J.J.; Irastorza-Azcarate, I.; Marletaz, F.; Gomez-Marin, C.; De, l.C.-M.E.; Bertrand, S.; Diaz, S.G.; Aldea, D.; Aury, J.M. A single three-dimensional chromatin compartment in amphioxus indicates a stepwise evolution of vertebrate Hox bimodal regulation. Nat. Genet. 2016, 48, 336–341. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Shi, L.; Zhu, Y.; Chen, H.; Zhang, J.; Lin, X.; Guan, X.J.B.G. CpGAVAS, an integrated web server for the annotation, visualization, analysis, and GenBank submission of completely sequenced chloroplast genome sequences. Bmc Genom. 2012, 13, 715. [Google Scholar] [CrossRef]
- Wyman, S.K.; Jansen, R.K.; Boore, J.L. Automatic annotation of organellar genomes with DOGMA. Bioinformatics 2004, 20, 3252–3255. [Google Scholar] [CrossRef] [Green Version]
- Schattner, P.; Brooks, A.N.; Lowe, T.M. The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res. 2005, 33, 686–689. [Google Scholar] [CrossRef]
- Lohse, M.; Drechsel, O.; Bock, R. OrganellarGenomeDRAW (OGDRAW): A tool for the easy generation of high-quality custom graphical maps of plastid and mitochondrial genomes. Curr. Genet. 2007, 52, 267–274. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol. Biol Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef]
- Kurtz, S.; Choudhuri, J.V.; Ohlebusch, E.; Schleiermacher, C.; Stoye, J.; Giegerich, R. REPuter: The manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001, 29, 4633–4642. [Google Scholar] [CrossRef]
- Yang, X.M.; Sun, J.T.; Xue, X.F.; Zhu, W.C.; Hong, X.Y. Development and characterization of 18 novel EST-SSRs from the western flower thrips, Frankliniella occidentalis (Pergande). Int. J. Mol. Sci. 2012, 13, 2863–2876. [Google Scholar] [CrossRef]
- Frazer, K.A.; Pachter, L.; Poliakov, A.; Rubin, E.M.; Dubchak, I. VISTA: Computational tools for comparative genomics. Nucleic Acids Res. 2004, 32, W273. [Google Scholar] [CrossRef]
- Nakamura, T.; Yamada, K.D.; Tomii, K.; Katoh, K. Parallelization of MAFFT for large-scale multiple sequence alignments. Bioinformatics 2018, 34, 2490–2492. [Google Scholar] [CrossRef] [Green Version]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef]
- Katoh, K.; Rozewicki, J.; Yamada, K.D. MAFFT online service: Multiple sequence alignment, interactive sequence choice and visualization. Brief. Bioinform. 2017. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type | A. Villosum | A. Villosum var. Xanthioides | A. Longiligulare |
|---|---|---|---|
| Accession Number | MH161416 MH161418 MN067431 MN067432 | MH161417 MN067433 | MN067434 MN067435 |
| Total Length (bp) | 164,068–164,069 | 163,981–163,985 | 163,608 |
| LSC Length (bp) | 88,797–88,798 | 88,720–88,857 | 88,680 |
| IR Length (bp) | 29,959 | 29,886–29,948 | 29,820 |
| SSC Length (bp) | 135,352–135,353 | 15,352–15,369 | 15,288 |
| CDS Length (bp) | 83,190 | 83,178–83,196 | 83,160 |
| Total GC content | 36.0% | 36.0% | 36.1% |
| GC content of LSC | 33.7% | 33.7% | 33.7% |
| GC content of IRa | 41.1% | 41.1% | 41.1% |
| GC content of IRb | 41.1% | 41.1% | 41.1% |
| GC content of SSC | 30.0% | 30.0% | 30.1% |
| AT content at 1st position | 55.4% | 55.4% | 55.4% |
| AT content at 2nd position | 62.6% | 62.6% | 62.6% |
| AT content at 3rd position | 71.2% | 71.2% | 71.2% |
| Group of Genes | Gene Names | Number of Genes |
|---|---|---|
| Photosystem I | psaA, psaB, psaC, psaI, psaJ | 5 |
| Photosystem II | psbA, psbB, psbC, psbD, psbE, psbF, psbH, psbI, psbJ, psbK, psbL, psbM, psbN, psbT, psbZ | 15 |
| Cytochrome b/f complex | petA, petB *, petD *, petG, petL, petN | 6 |
| ATP synthase | atpA, atpB, atpE, atpF *, atpH, atpI | 6 |
| NADH dehydrogenase | ndhA *, ndhB(×2) *, ndhC, ndhD, ndhE, ndhF, ndhG, ndhH, ndhI, ndhJ, ndhK | 12 |
| RubisCO large subunit | rbcL | 1 |
| RNA polymerase | rpoA, rpoB, rpoC1 *, rpoC2 | 4 |
| Ribosomal proteins (SSU) | rps2, rps3, rps4, rps7(×2), rps8, rps11, rps12(×2) **, rps14, rps15, rps16 *, rps18, rps19(×2) | 15 |
| Ribosomal proteins (LSU) | rpl2(×2) *, rpl14, rpl16 *, rpl20, rpl22, rpl23(×2), rpl32, rpl33, rpl36 | 11 |
| Proteins of unknown function | ycf1(×2), ycf2(×2), ycf3 **, ycf4 | 6 |
| Transfer RNAs | 38 tRNAs (8 in the IRs(×2), 6 contain one intron) | 38 |
| Ribosomal RNAs | rrn4.5(×2), rrn5(×2), rrn16(×2), rrn23(×2) | 8 |
| Other genes | accD, clpP **, matK, ccsA, cemA, infA | 6 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cui, Y.; Chen, X.; Nie, L.; Sun, W.; Hu, H.; Lin, Y.; Li, H.; Zheng, X.; Song, J.; Yao, H. Comparison and Phylogenetic Analysis of Chloroplast Genomes of Three Medicinal and Edible Amomum Species. Int. J. Mol. Sci. 2019, 20, 4040. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20164040
Cui Y, Chen X, Nie L, Sun W, Hu H, Lin Y, Li H, Zheng X, Song J, Yao H. Comparison and Phylogenetic Analysis of Chloroplast Genomes of Three Medicinal and Edible Amomum Species. International Journal of Molecular Sciences. 2019; 20(16):4040. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20164040
Chicago/Turabian StyleCui, Yingxian, Xinlian Chen, Liping Nie, Wei Sun, Haoyu Hu, Yulin Lin, Haitao Li, Xilong Zheng, Jingyuan Song, and Hui Yao. 2019. "Comparison and Phylogenetic Analysis of Chloroplast Genomes of Three Medicinal and Edible Amomum Species" International Journal of Molecular Sciences 20, no. 16: 4040. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20164040