Deciphering the Molecular Recognition Mechanism of Multidrug Resistance Staphylococcus aureus NorA Efflux Pump Using a Supervised Molecular Dynamics Approach

 , , , , ,
, , , , ,  , and
, and
Abstract
:
1. Introduction
2. Results and Discussion
2.1. Prediction and Assessment of the NorA 3D Structure
2.2. MdfA Template and NorA Comparison
2.3. Biological Assay of CLM on NorA
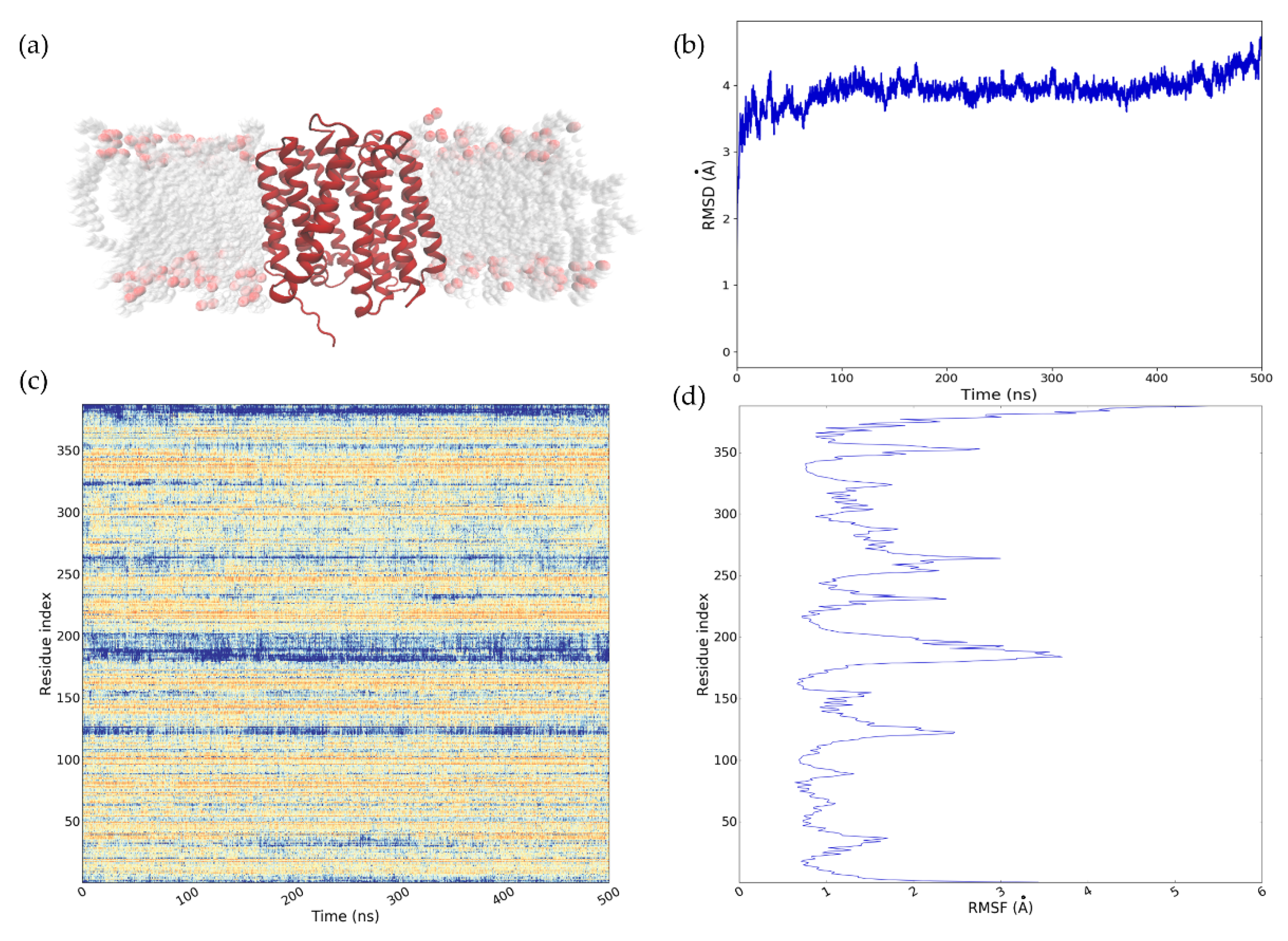
2.4. Refinement of the NorA Predicted Model Using MD
2.5. Binding Site Definition and Preliminary Docking
2.6. Substrate Binding Simulations Using SuMD
2.6.1. General Overview of SuMD Analysis
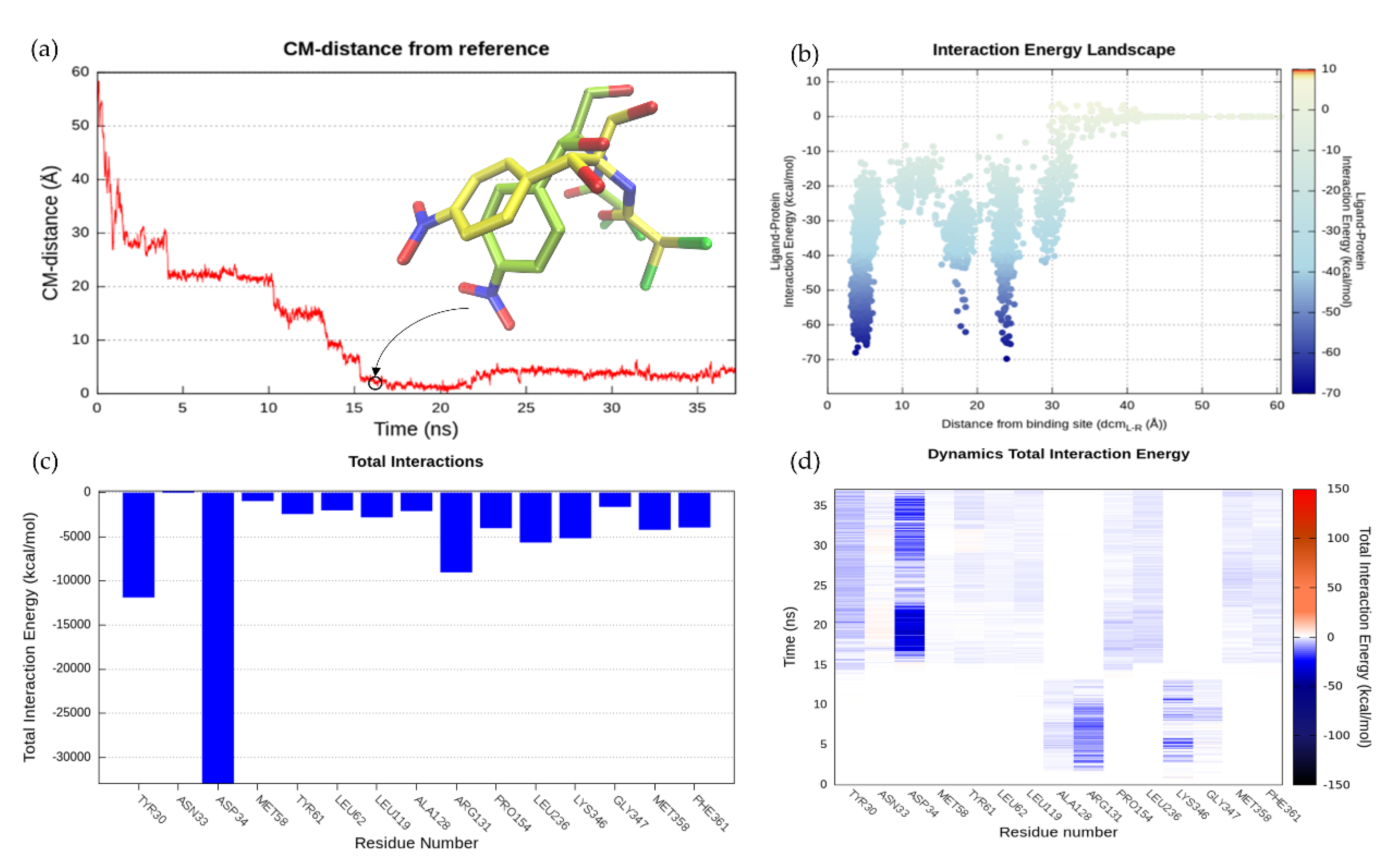
2.6.2. SuMD Validation: MdfA-CLM Recognition Pathway
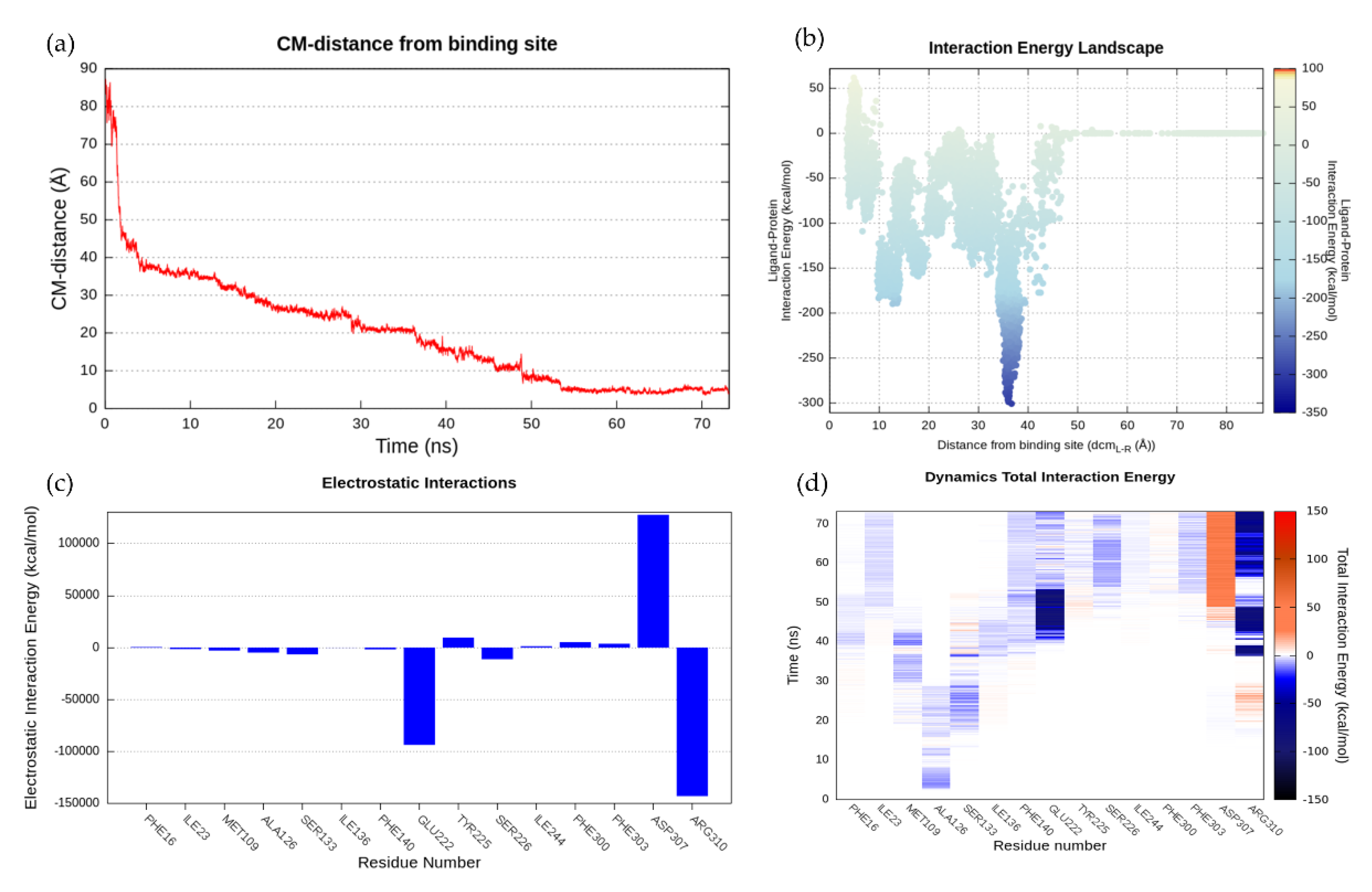
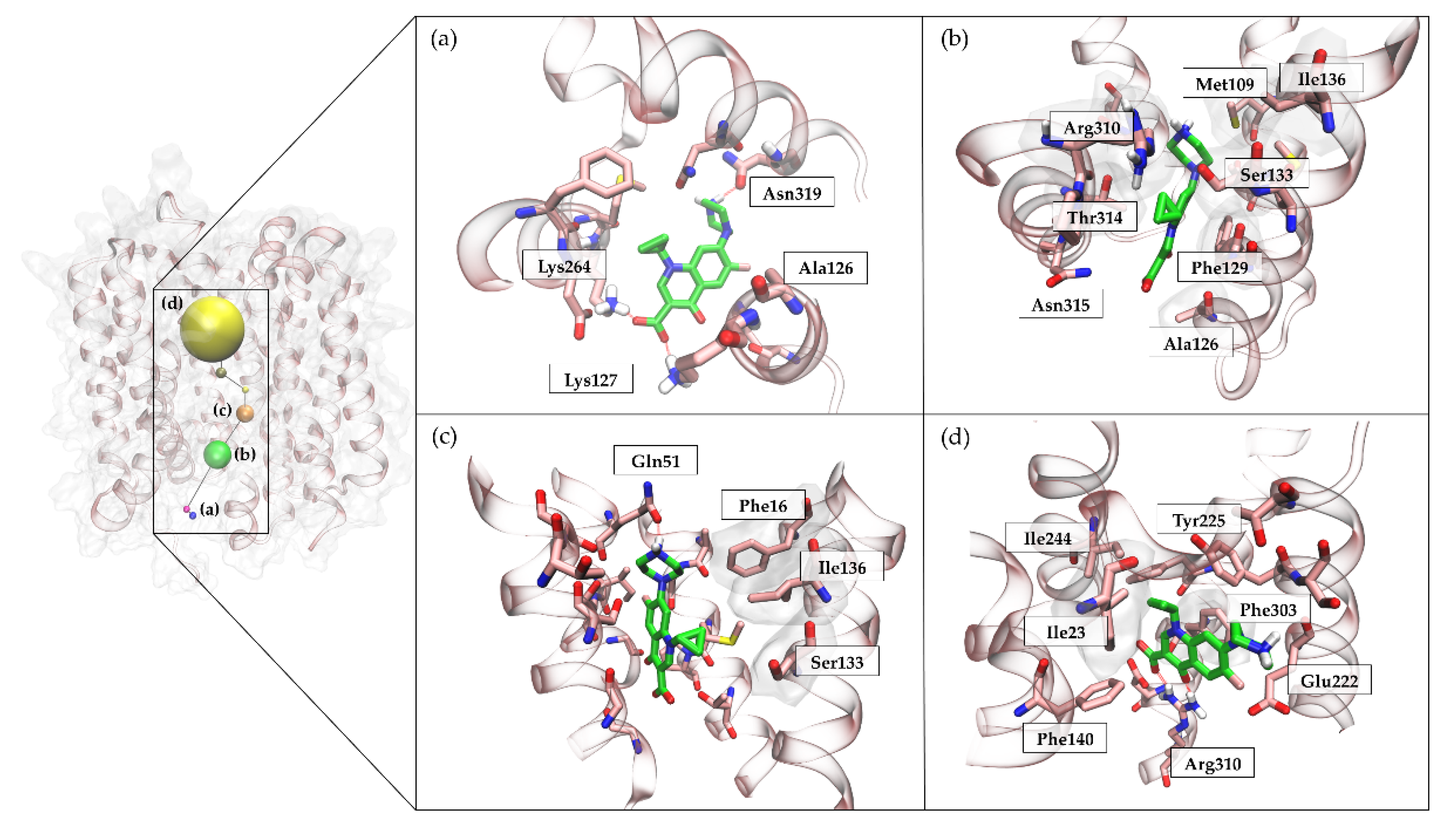
2.6.3. NorA-CPX Recognition Pathway
3. Materials and Methods
3.1. General
3.2. Protein Modeling: Preparation of the NorA Target
3.3. MdfA Crystal Structure Preparation
3.4. Ligand Preparation
3.5. Molecular Docking Experiments
3.6. Solvated System Setup and Equilibration
3.7. Molecular Dynamics (MD) Simulations
3.8. Supervised Molecular Dynamics (SuMD)
3.9. Analysis of pepSuMD Trajectories
3.10. Microbiological Assays
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| MD | Molecular Dynamics |
| TM | Transmembrane |
| PDB | Protein Data Bank |
| Cin | Inward Conformation |
| Cout | Outward Conformation |
| SuMD | Supervised Molecular Dynamics |
| MFS | Major Facilitator Superfamily |
| CPX | Ciprofloxacin |
| CLM | Chloramphenicol |
| MIC | minimum inhibitory concentration |
| EtBr | ethidium bromide |
| RMSD | Root Mean Square Deviation Standard |
| RMSF | Root Mean Square Fluctuation Standard |
| POPC | 1-palmitoyl-2-oleyl-glycerol-3-phospho-choline |
| OPM | Orientation of Proteins in Membrane |
| NPT | Isothermal-isobaric ensemble |
References
- World Health Organization. Antimicrobial Resistance Global Report on Surveillance; World Health Organization: Geneva, Switzerland, 2014; Volume 61, pp. 383–394. [Google Scholar]
- Blair, J.M.A.; Webber, M.A.; Baylay, A.J.; Ogbolu, D.O.; Piddock, L.J. Molecular mechanisms of antibiotic resistance. Nat. Rev. Microbiol. 2015, 13, 42–51. [Google Scholar] [CrossRef] [PubMed]
- Bagnoli, F.; Rappuoli, R.; Grandi, G. Staphylococcus aureus: Microbiology, Pathology, Immunology, Therapy and Prophylaxis; Springer: Cham, Switzerland, 2017; ISBN 978-3-319-72061-6. [Google Scholar]
- Costa, S.S.; Viveiros, M.; Amaral, L.; Couto, I. Multidrug Efflux Pumps in Staphylococcus aureus: An Update. Open Microbiol. J. 2013, 7, 59–71. [Google Scholar] [CrossRef] [PubMed]
- Ubukata, K.; Itoh-Yamashita, N.; Konno, M. Cloning and expression of the norA gene for fluoroquinolone resistance in Staphylococcus aureus. Antimicrob. Agents Chemother. 1989, 33, 1535–1539. [Google Scholar] [CrossRef] [PubMed]
- Pao, S.S.; Paulsen, I.A.N.T.; Saier, M.H. Amr000001. Microbiol. Mol. Biol. Rev. 2009, 62, 1–25. [Google Scholar]
- Yoshida, H.; Bogaki, M.; Nakamura, S.; Ubukata, K.; Konno, M. Nucleotide sequence and characterization of the Staphylococcus aureus norA gene, which confers resistance to quinolones. J. Bacteriol. 1990, 172, 6942–6949. [Google Scholar] [CrossRef] [PubMed]
- Neyfakh, A.A.; Borsch, C.M.; Kaatz, G.W. Fluoroquinolone resistance protein NorA of Staphylococcus aureus is a multidrug efflux transporter. Antimicrob. Agents Chemother. 1993, 37, 128–129. [Google Scholar] [CrossRef] [PubMed]
- Felicetti, T.; Cannalire, R.; Pietrella, D.; Latacz, G.; Lubelska, A.; Manfroni, G.; Barreca, M.L.; Massari, S.; Tabarrini, O.; Kieć-Kononowicz, K.; et al. 2-Phenylquinoline S. aureus NorA Efflux Pump Inhibitors: Evaluation of the Importance of Methoxy Group Introduction. J. Med. Chem. 2018, 61, 7827–7848. [Google Scholar] [CrossRef]
- Sabatini, S.; Piccioni, M.; Felicetti, T.; De Marco, S.; Manfroni, G.; Pagiotti, R.; Nocchetti, M.; Cecchetti, V.; Pietrella, D. Investigation on the effect of known potent: S. aureus NorA efflux pump inhibitors on the staphylococcal biofilm formation. RSC Adv. 2017, 7, 37007–37014. [Google Scholar] [CrossRef]
- Holler, J.G.; Slotved, H.-C.; Mølgaard, P.; Olsen, C.E.; Christensen, S.B. Chalcone inhibitors of the NorA efflux pump in Staphylococcus aureus whole cells and enriched everted membrane vesicles. Bioorg. Med. Chem. 2012, 20, 4514–4521. [Google Scholar] [CrossRef]
- Fontaine, F.; Hequet, A.; Voisin-Chiret, A.S.; Bouillon, A.; Lesnard, A.; Cresteil, T.; Jolivalt, C.; Rault, S. First identification of boronic species as novel potential inhibitors of the Staphylococcus aureus NorA efflux pump. J. Med. Chem. 2014, 57, 2536–2548. [Google Scholar] [CrossRef]
- Schmitz, F.J.; Fluit, A.C.; Lückefahr, M.; Engler, B.; Hofmann, B.; Verhoef, J.; Heinz, H.P.; Hadding, U.; Jones, M.E. The effect of reserpine, an inhibitor of multidrug efflux pumps, on the in-vitro activities of ciprofloxacin, sparfloxacin and moxifloxacin against clinical isolates of Staphylococcus aureus. J. Antimicrob. Chemother. 1998, 42, 807–810. [Google Scholar] [CrossRef]
- Sabatini, S.; Gosetto, F.; Manfroni, G.; Tabarrini, O.; Kaatz, G.W.; Patel, D.; Cecchetti, V. Evolution from a Natural Flavones Nucleus to Obtain 2-(4-Propoxyphenyl)quinoline Derivatives As Potent Inhibitors of the S. aureus NorA Efflux Pump. J. Med. Chem. 2011, 54, 5722–5736. [Google Scholar] [CrossRef]
- Sabatini, S.; Kaatz, G.W.; Rossolini, G.M.; Brandini, D.; Fravolini, A. From phenothiazine to 3-phenyl-1,4-benzothiazine derivatives as inhibitors of the Staphylococcus aureus NorA multidrug efflux pump. J. Med. Chem. 2008, 51, 4321–4330. [Google Scholar] [CrossRef]
- Brincat, J.P.; Carosati, E.; Sabatini, S.; Manfroni, G.; Fravolini, A.; Raygada, J.L.; Patel, D.; Kaatz, G.W.; Cruciani, G. Discovery of novel inhibitors of the NorA multidrug transporter of Staphylococcus aureus. J. Med. Chem. 2011, 54, 354–365. [Google Scholar] [CrossRef]
- Yin, Y.; He, X.; Szewczyk, P.; Nguyen, T.; Chang, G. Structure of the multidrug transporter EmrD from Escherichia coli. Science 2006, 312, 741–744. [Google Scholar] [CrossRef]
- Kang, X.; Feng, W.; Jiang, D.; Huang, B.; Liu, X.; Zhang, X.C.; Liu, J.; Zhao, Y.; Wang, X.; Fan, J.; et al. Structure of the YajR transporter suggests a transport mechanism based on the conserved motif A. Proc. Natl. Acad. Sci. USA 2013, 110, 14664–14669. [Google Scholar] [Green Version]
- Zhao, Y.Y.; Heng, J.; Zhao, Y.Y.; Liu, M.; Liu, Y.; Fan, J.; Wang, X.; Zhang, X.C. Substrate-bound structure of the E. coli multidrug resistance transporter MdfA. Cell Res. 2015, 25, 1060–1073. [Google Scholar]
- Nagarathinam, K.; Nakada-Nakura, Y.; Parthier, C.; Terada, T.; Juge, N.; Jaenecke, F.; Liu, K.; Hotta, Y.; Miyaji, T.; Omote, H.; et al. Outward open conformation of a Major Facilitator Superfamily multidrug/H+ antiporter provides insights into switching mechanism. Nat. Commun. 2018, 9, 4005. [Google Scholar] [CrossRef]
- Berman, H.M. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Sabbadin, D.; Moro, S. Supervised molecular dynamics (SuMD) as a helpful tool to depict GPCR-ligand recognition pathway in a nanosecond time scale. J. Chem. Inf. Model. 2014, 54, 372–376. [Google Scholar] [CrossRef]
- Zhang, Y. I-TASSER server for protein 3D structure prediction. BMC Bioinform. 2008, 9, 40. [Google Scholar] [CrossRef]
- Roy, A.; Zhang, Y.; Yang, J.; Yan, R.; Poisson, J.; Xu, D. The I-TASSER Suite: Protein structure and function prediction. Nat. Methods 2014, 12, 7–8. [Google Scholar]
- Bienert, S.; Heer, F.T.; de Beer, T.A.P.; Lepore, R.; Rempfer, C.; Gumienny, R.; Bordoli, L.; Schwede, T.; Bertoni, M.; Tauriello, G.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar]
- Peng, J.; Xu, J. Raptorx: Exploiting structure information for protein alignment by statistical inference. Proteins Struct. Funct. Bioinform. 2011, 79, 161–171. [Google Scholar] [CrossRef] [Green Version]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J.E. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef] [Green Version]
- Ishitani, R.; Doki, S.; Kato, H.E.; Nureki, O. Structural Basis for Dynamic Mechanism of Proton-Coupled Symport by the Peptide Transporter POT. Seibutsu Butsuri 2014, 54, 85–90. [Google Scholar] [CrossRef] [Green Version]
- Chemical Computing Group (CCG)|Computer-Aided Molecular Design. Available online: https://www.chemcomp.com/ (accessed on 24 March 2019).
- Kaatz, G.W.; Seo, S.M. Mechanisms of fluoroquinolone resistance in genetically related strains of Staphylococcus aureus. Antimicrob. Agents Chemother. 1997, 41, 2733–2737. [Google Scholar] [CrossRef]
- Singh, S.; Kalia, N.P.; Joshi, P.; Kumar, A.; Sharma, P.R.; Kumar, A.; Bharate, S.B.; Khan, I.A. Boeravinone B, A novel dual inhibitor of nora bacterial efflux pump of Staphylococcus aureus and Human P-Glycoprotein, reduces the biofilm formation and intracellular invasion of bacteria. Front. Microbiol. 2017, 8. [Google Scholar] [CrossRef]
- Ester, M.; Kriegel, H.P.; Sander, J.; Xu, X. A Density-Based Algorithm for Discovering Clusters in Large Spatial Databases with Noise. In Kdd; AAAI Press: Portland, OR, USA, 1996. [Google Scholar]
- Edgar, R.; Bibi, E. MdfA, an Escherichia coli Multidrug Resistance Protein with an Extraordinarily Broad Spectrum of Drug Recognition. J. Bacteriol. 1997, 179, 2274–2280. [Google Scholar] [CrossRef]
- Halgren, T.A. Identifying and characterizing binding sites and assessing druggability. J. Chem. Inf. Model. 2009, 49, 377–389. [Google Scholar] [CrossRef]
- Kalia, N.P.; Mahajan, P.; Mehra, R.; Nargotra, A.; Sharma, J.P.; Koul, S.; Khan, I.A. Capsaicin, a novel inhibitor of the NorA efflux pump, reduces the intracellular invasion of Staphylococcus aureus. J. Antimicrob. Chemother. 2012, 67, 2401–2408. [Google Scholar] [CrossRef]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Korb, O.; Stützle, T.; Exner, T.E. PLANTS: Application of Ant Colony Optimization to Structure-Based Drug Design; Springer: Berlin/Heidelberg, Germany, 2006; pp. 247–258. [Google Scholar]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 2002, 267, 727–748. [Google Scholar] [CrossRef]
- Harvey, M.J.; Giupponi, G.; De Fabritiis, G. ACEMD: Accelerating biomolecular dynamics in the microsecond time scale. J. Chem. Theory Comput. 2009, 5, 1632–1639. [Google Scholar] [CrossRef]
- Bateman, A.; Martin, M.J.; O’Donovan, C.; Magrane, M.; Apweiler, R.; Alpi, E.; Antunes, R.; Arganiska, J.; Bely, B.; Bingley, M.; et al. UniProt: A hub for protein information. Nucleic Acids Res. 2015, 43, D204–D212. [Google Scholar]
- Wang, H.; Lu, H.; Källberg, M.; Xu, J.; Peng, J.; Wang, S.; Wang, Z. Template-based protein structure modeling using the RaptorX web server. Nat. Protoc. 2012, 7, 1511–1522. [Google Scholar] [Green Version]
- Studer, G.; Biasini, M.; Schwede, T. Assessing the local structural quality of transmembrane protein models using statistical potentials (QMEANBrane). Bioinformatics 2014, 30, 505–511. [Google Scholar] [CrossRef]
- Lomize, M.A.; Lomize, A.L.; Pogozheva, I.D.; Mosberg, H.I. OPM: Orientations of proteins in membranes database. Bioinformatics 2006, 22, 623–625. [Google Scholar] [CrossRef]
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I.; et al. CHARMM General Force Field (CGenFF): A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 2010, 31, 671–690. [Google Scholar]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Grubmüller, H.; Groll, V. Max Planck Institute for Biophysical Chemistry. Solvate. Available online: https://www.mpibpc.mpg.de/grubmueller/solvate (accessed on 11 April 2019).
- Allen, W.J.; Lemkul, J.A.; Bevan, D.R. GridMAT-MD: A grid-based membrane analysis tool for use with molecular dynamics. J. Comput. Chem. 2009, 30, 1952–1958. [Google Scholar] [CrossRef]
- Van der Walt, S.; Colbert, S.C.; Varoquaux, G. The NumPy Array: A Structure for Efficient Numerical Computation. Comput. Sci. Eng. 2011, 13, 22–30. [Google Scholar] [CrossRef] [Green Version]
- McGibbon, R.T.; Beauchamp, K.A.; Harrigan, M.P.; Klein, C.; Swails, J.M.; Hernández, C.X.; Schwantes, C.R.; Wang, L.-P.; Lane, T.J.; Pande, V.S. MDTraj: A Modern Open Library for the Analysis of Molecular Dynamics Trajectories. Biophys. J. 2015, 109, 1528–1532. [Google Scholar] [CrossRef] [Green Version]
- Salmaso, V.; Sturlese, M.; Cuzzolin, A.; Moro, S. Exploring Protein-Peptide Recognition Pathways Using a Supervised Molecular Dynamics Approach. Struct. 2017, 25, 655–662. [Google Scholar] [CrossRef]
- Bakan, A.; Meireles, L.M.; Bahar, I. ProDy: Protein dynamics inferred from theory and experiments. Bioinformatics 2011, 27, 1575–1577. [Google Scholar] [CrossRef]
- Clinical Laboratory Standards Institute. Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria that Grow Aerobically, 7th ed.; Approved Standard M7-a7; Clinical and Laboratory Standards Institute: Wayne PA, USA, 2006. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SA-1199 | SA-1199B | |
|---|---|---|
| CPX | 0.25 a | 8 a |
| CLM | 4 | 4 |
| EtBr | 1 a | 32 a |
| System | Replica | Outcome | Time (ns) | Best dcm (L-R) Å |
|---|---|---|---|---|
| Subset A | ||||
| CLM-MdfA | 1 | productive | 32 | 3.1 |
| CLM-MdfA | 2 | productive | 36 | 2.9 |
| CLM-MdfA | 3 | non productive | 13 | 23.4 |
| CLM-MdfA | 4 | productive | 37 | 3.1 |
| CLM-NorA | 1 | productive | 16 | 34.4 |
| CLM-NorA | 2 | productive | 58 | 0.4 |
| CLM-NorA | 3 | productive | 47 | 1.7 |
| CLM-NorA | 4 | productive | 14 | 30.7 |
| Subset B | ||||
| CPX-MdfA | 1 | productive | 56 | 3.6 |
| CPX-MdfA | 2 | non productive | 23 | 16.3 |
| CPX-MdfA | 3 | productive | 44 | 28 |
| CPX-MdfA | 4 | productive | 47 | 2.9 |
| CPX-NorA | 1 | non productive | 16 | 25.5 |
| CPX-NorA | 2 | non productive | 19 | 26.3 |
| CPX-NorA | 3 | non productive | 26 | 26.3 |
| CPX-NorA | 4 | productive | 73 | 3.4 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Palazzotti, D.; Bissaro, M.; Bolcato, G.; Astolfi, A.; Felicetti, T.; Sabatini, S.; Sturlese, M.; Cecchetti, V.; Barreca, M.L.; Moro, S. Deciphering the Molecular Recognition Mechanism of Multidrug Resistance Staphylococcus aureus NorA Efflux Pump Using a Supervised Molecular Dynamics Approach. Int. J. Mol. Sci. 2019, 20, 4041. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20164041
Palazzotti D, Bissaro M, Bolcato G, Astolfi A, Felicetti T, Sabatini S, Sturlese M, Cecchetti V, Barreca ML, Moro S. Deciphering the Molecular Recognition Mechanism of Multidrug Resistance Staphylococcus aureus NorA Efflux Pump Using a Supervised Molecular Dynamics Approach. International Journal of Molecular Sciences. 2019; 20(16):4041. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20164041
Chicago/Turabian StylePalazzotti, Deborah, Maicol Bissaro, Giovanni Bolcato, Andrea Astolfi, Tommaso Felicetti, Stefano Sabatini, Mattia Sturlese, Violetta Cecchetti, Maria Letizia Barreca, and Stefano Moro. 2019. "Deciphering the Molecular Recognition Mechanism of Multidrug Resistance Staphylococcus aureus NorA Efflux Pump Using a Supervised Molecular Dynamics Approach" International Journal of Molecular Sciences 20, no. 16: 4041. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20164041