A Novel Method for Controlled Gene Expression via Combined Bleomycin and Plasmid DNA Electrotransfer

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
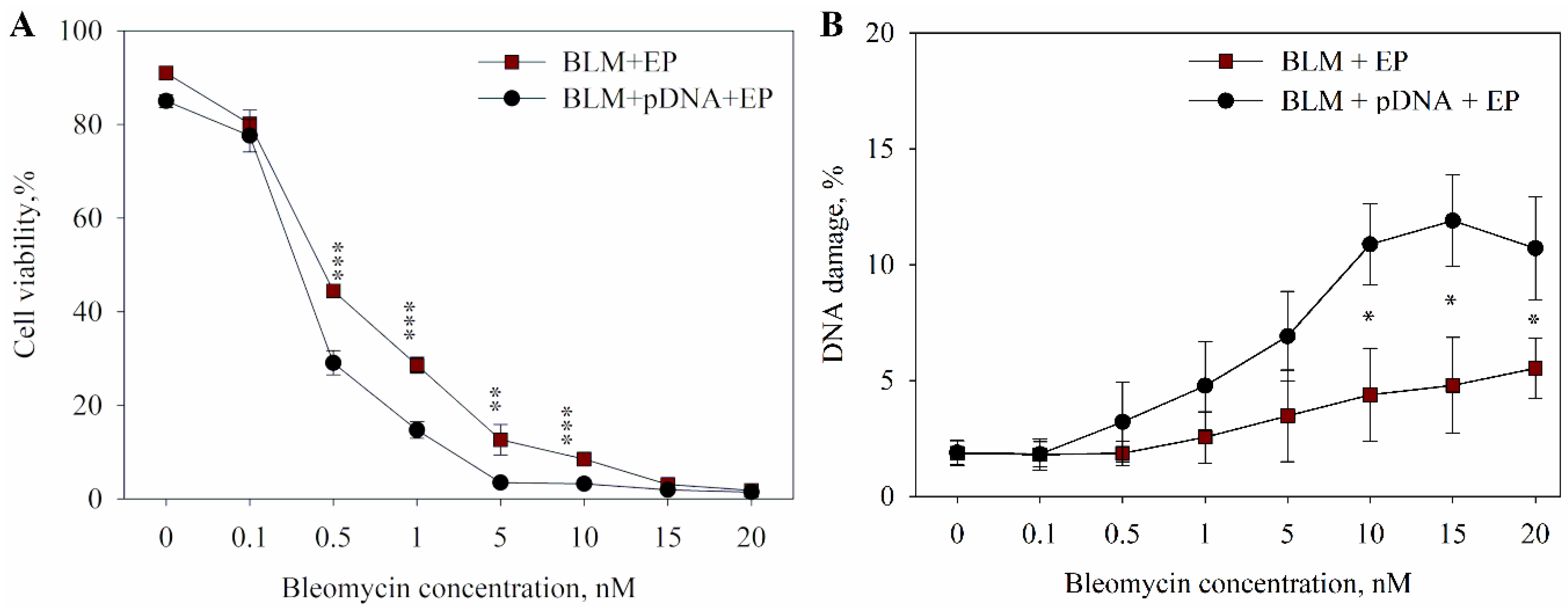
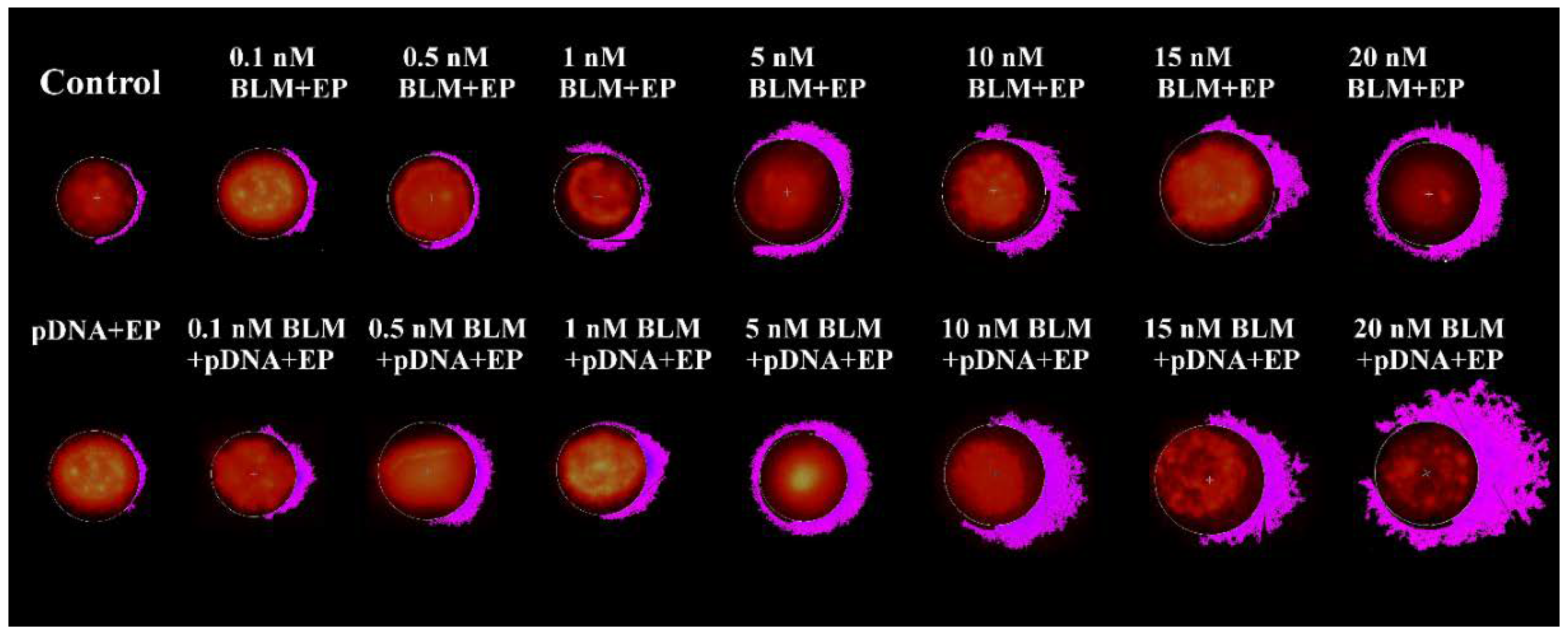
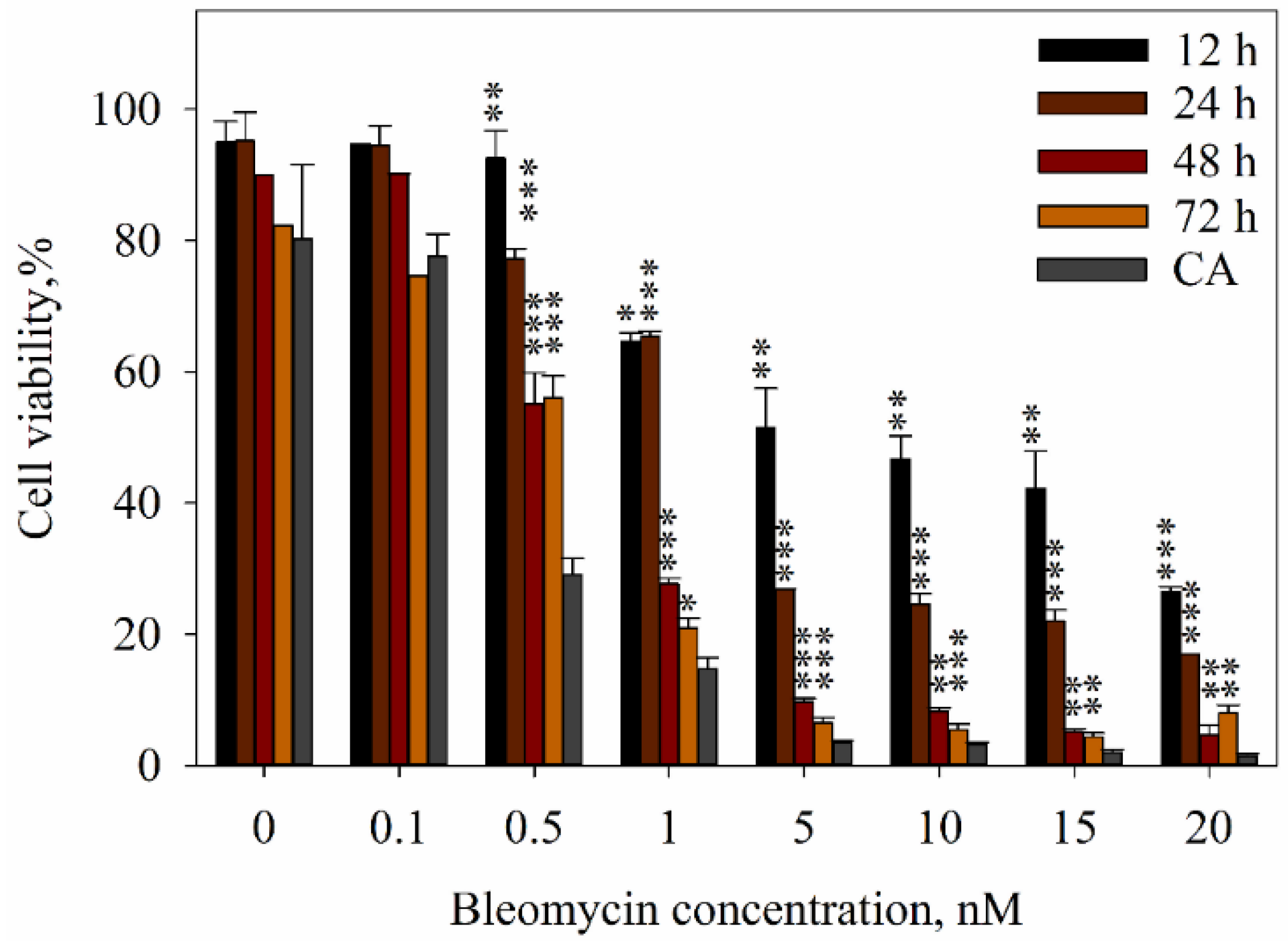
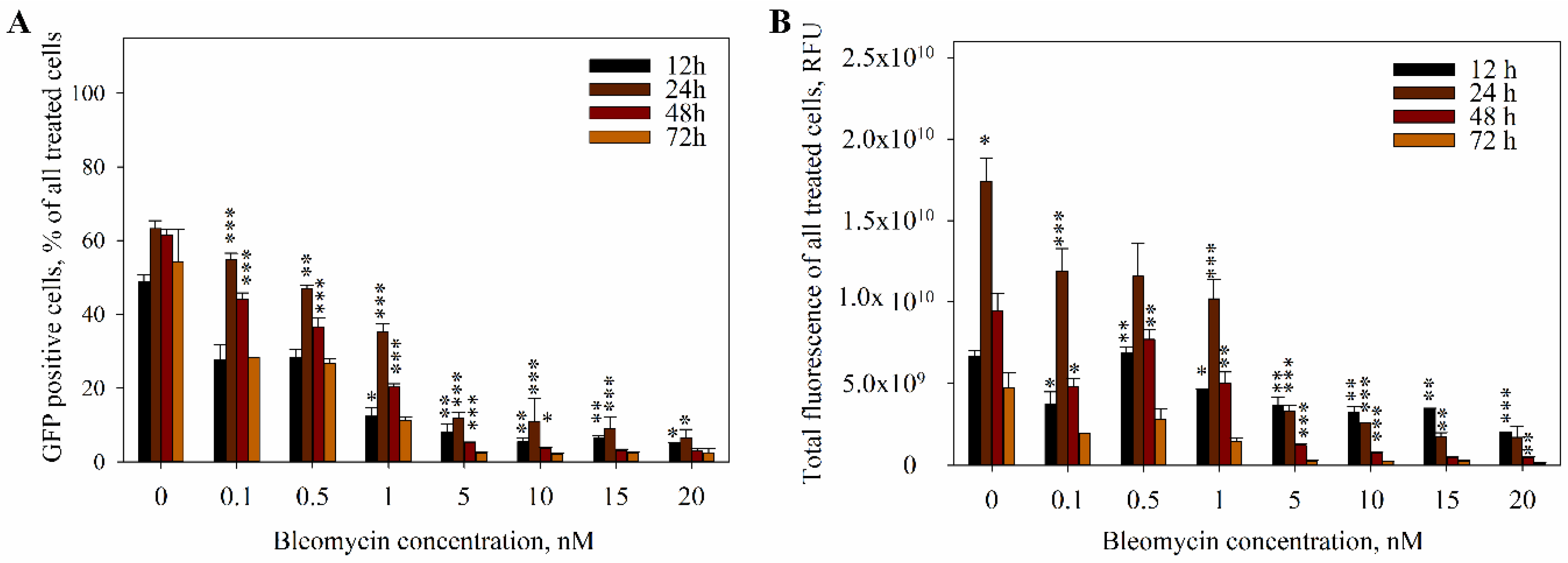
2. Results
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Plasmid DNA Preparation
4.3. Cell Electroporation
4.4. The Measurement of DNA Electrotransfer Efficiency
4.5. DNA Damage Evaluation
4.6. Evaluation of Cell Viability
4.7. Statistics
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Weaver, J.C. Electroporation theory. Concepts and mechanisms. Methods Mol. Biol. 1995, 55, 3–28. [Google Scholar] [PubMed]
- Chopra, S.; Satkauskas, S. Electrotransfer of cytokine genes for cancer treatment. In Proceedings of the CBU International Conference Proceedings, Prague, Czech Republic, 25 September 2018; Volume 6, pp. 1036–1041. [Google Scholar] [CrossRef]
- Calvet, C.Y.; Mir, L.M. The promising alliance of anti-cancer electrochemotherapy with immunotherapy. Cancer Metastasis Rev. 2016, 35, 165–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heller, L.; Pottinger, C.; Jaroszeski, M.J.; Gilbert, R.; Heller, R. In vivo electroporation of plasmids encoding GM-CSF or interleukin-2 into existing B16 melanomas combined with electrochemotherapy induces long-term antitumour immunity. Melanoma Res. 2000, 10, 577–583. [Google Scholar] [CrossRef] [PubMed]
- Kanduser, M.; Sentjurc, M.; Miklavcic, D. Cell membrane fluidity related to electroporation and resealing. Eur. Biophys. J. 2006, 35, 196–204. [Google Scholar] [CrossRef] [PubMed]
- Rols, M. Electropermeabilization, a physical method for the delivery of therapeutic molecules into cells. Biochim. Biophys. Acta Biomembr. 2006, 1758, 423–428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matthiessen, L.W.; Chalmers, R.L.; Sainsbury, D.C.; Veeramani, S.; Kessell, G.; Humphreys, A.C.; Bond, J.E.; Muir, T.; Gehl, J. Management of cutaneous metastases using electrochemotherapy. Acta Oncol. 2011, 50, 621–629. [Google Scholar] [CrossRef] [PubMed]
- Gehl, J.; Sersa, G.; Matthiessen, L.W.; Muir, T.; Soden, D.; Occhini, A.; Quaglino, P.; Curatolo, P.; Campana, L.G.; Kunte, C.; et al. Updated standard operating procedures for electrochemotherapy of cutaneous tumours and skin metastases. Acta Oncol. 2018, 57, 874–882. [Google Scholar] [CrossRef]
- Miklavcic, D.; Mali, B.; Kos, B.; Heller, R.; Sersa, G. Electrochemotherapy: From the drawing board into medical practice. Biomed. Eng. Online 2014, 13, 29. [Google Scholar] [CrossRef]
- Esmaeili, N.; Friebe, M. Electrochemotherapy: A review of current status, alternative IGP approaches, and future perspectives. J. Healthc. Eng. 2019, 2019, 2784516. [Google Scholar] [CrossRef]
- Cemazar, M.; Miklavcic, D.; Mir, L.M.; Belehradek, J., Jr.; Bonnay, M.; Fourcault, D.; Sersa, G. Electrochemotherapy of tumours resistant to cisplatin: A study in a murine tumour model. Eur. J. Cancer 2001, 37, 1166–1172. [Google Scholar] [CrossRef]
- Barbara, M.; Jarm, T.; Snoj, M.; Serša, G.; Miklavčič, D. Antitumor effectiveness of electrochemotherapy: A systematic review and meta-analysis. Eur. J. Surg. Oncol. 2013, 39, 4–16. [Google Scholar]
- Sersa, G.; Cemazar, M.; Snoj, M. Electrochemotherapy of tumours. Curr. Oncol. 2009, 16, 34–35. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Ghorai, M.K.; Kenney, G.; Stubbe, J. Mechanistic studies on bleomycin-mediated DNA damage: Multiple binding modes can result in double-stranded DNA cleavage. Nucleic Acids Res. 2008, 36, 3781–3790. [Google Scholar] [CrossRef] [PubMed]
- Cemazar, M.; Ambrozic Avgustin, J.; Pavlin, D.; Sersa, G.; Poli, A.; Krhac Levacic, A.; Tesic, N.; Lampreht Tratar, U.; Rak, M.; Tozon, N. Efficacy and safety of electrochemotherapy combined with peritumoral IL-12 gene electrotransfer of canine mast cell tumours. Vet. Comp. Oncol. 2017, 15, 641–654. [Google Scholar] [CrossRef] [PubMed]
- Liu, H. 11-three-dimensional tumor model and t-lymphocytes immunotherapy for cancer. Recent Adv. Cancer Res. Ther. 2012, 245–286. [Google Scholar] [CrossRef]
- Campana, L.G.; Mocellin, S.; Basso, M.; Puccetti, O.; De Salvo, G.L.; Chiarion-Sileni, V.; Vecchiato, A.; Corti, L.; Rossi, C.R.; Nitti, D. Bleomycin-based electrochemotherapy: Clinical outcome from a single institution’s experience with 52 patients. Ann. Surg. Oncol. 2009, 16, 191–199. [Google Scholar] [CrossRef] [PubMed]
- Sersa, G.; Teissie, J.; Cemazar, M.; Signori, E.; Kamensek, U.; Marshall, G.; Miklavcic, D. Electrochemotherapy of tumors as in situ vaccination boosted by immunogene electrotransfer. Cancer Immunol. Immunother. 2015, 64, 1315–1327. [Google Scholar] [CrossRef] [PubMed]
- Mir, L.M.; Orlowski, S.; Poddevin, B.; Belehradek, J., Jr. Electrochemotherapy tumor treatment is improved by interleukin-2 stimulation of the host’s defenses. Eur. Cytokine Netw. 1992, 3, 331–334. [Google Scholar]
- Mir, L.M.; Roth, C.; Orlowski, S.; Quintin-Colonna, F.; Fradelizi, D.; Belehradek, J., Jr.; Kourilsky, P. Systemic antitumor effects of electrochemotherapy combined with histoincompatible cells secreting interleukin-2. J. Immunother. Emphas. Tumor Immunol. 1995, 17, 30–38. [Google Scholar] [CrossRef]
- Andersen, M.H.; Gehl, J.; Reker, S.; Pedersen, L.O.; Becker, J.C.; Geertsen, P.; thor Straten, P. Dynamic changes of specific T cell responses to melanoma correlate with IL-2 administration. Semin. Cancer Biol. 2003, 13, 449–459. [Google Scholar] [CrossRef]
- Torrero, M.N.; Henk, W.G.; Li, S. Regression of high-grade malignancy in mice by bleomycin and interleukin-12 electrochemogenetherapy. Clin. Cancer Res. 2006, 12, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Kos, S.; Blagus, T.; Cemazar, M.; Lampreht Tratar, U.; Stimac, M.; Prosen, L.; Dolinsek, T.; Kamensek, U.; Kranjc, S.; Steinstraesser, L.; et al. Electrotransfer parameters as a tool for controlled and targeted gene expression in skin. Mol. Ther. Nucleic Acids 2016, 5, e356. [Google Scholar] [CrossRef] [PubMed]
- McArthur, H.L.; Page, D.B. Immunotherapy for the treatment of breast cancer: Checkpoint blockade, cancer vaccines, and future directions in combination immunotherapy. Clin. Adv. Hematol. Oncol. 2016, 14, 922–933. [Google Scholar] [PubMed]
- Ventola, C.L. Cancer immunotherapy, Part 3: Challenges and future trends. Pharm. Ther. 2017, 42, 514–521. [Google Scholar]
- Amos, S.M.; Duong, C.P.; Westwood, J.A.; Ritchie, D.S.; Junghans, R.P.; Darcy, P.K.; Kershaw, M.H. Autoimmunity associated with immunotherapy of cancer. Blood 2011, 118, 499–509. [Google Scholar] [CrossRef] [PubMed]
- Kostine, M.; Chiche, L.; Lazaro, E.; Halfon, P.; Charpin, C.; Arniaud, D.; Retornaz, F.; Blanco, P.; Jourde-Chiche, N.; Richez, C.; et al. Opportunistic autoimmunity secondary to cancer immunotherapy (OASI): An emerging challenge. Rev. Med. Interne 2017, 38, 513–525. [Google Scholar] [CrossRef] [PubMed]
- Cha, E.; Daud, A. Plasmid IL-12 electroporation in melanoma. Hum. Vaccines Immunother. 2012, 8, 1734–1738. [Google Scholar] [CrossRef] [Green Version]
- Skrombolas, D.; Frelinger, J.G. Challenges and developing solutions for increasing the benefits of IL-2 treatment in tumor therapy. Expert Rev. Clin. Immunol. 2014, 10, 207–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burkart, C.; Mukhopadhyay, A.; Shirley, S.A.; Connolly, R.J.; Wright, J.H.; Bahrami, A.; Campbell, J.S.; Pierce, R.H.; Canton, D.A. Improving therapeutic efficacy of IL-12 intratumoral gene electrotransfer through novel plasmid design and modified parameters. Gene Ther. 2018, 25, 93–103. [Google Scholar] [CrossRef]
- Schulz-Knappe, P.K.; Lautscham, G. Prevent cancer immunotherapy’s side effects. Genet. Eng. Biotechnol. News 2018, 38, 28–29. [Google Scholar] [CrossRef]
- Guo, M.M.; Wang, Y.; Han, F.H. The study of interfering of endogenous VEGF-C genes and the protein expression gastric cancer cell with siRNA technique. Sichuan Da Xue Xue Bao Yi Xue Ban 2013, 44, 740–743. [Google Scholar] [PubMed]
- Singh, A.; Trivedi, P.; Jain, N.K. Advances in siRNA delivery in cancer therapy. Artif. Cells Nanomed. Biotechnol. 2018, 46, 274–283. [Google Scholar] [CrossRef] [PubMed]
- Bader, A.G.; Brown, D.; Winkler, M. The promise of microRNA replacement therapy. Cancer Res. 2010, 70, 7027–7030. [Google Scholar] [CrossRef] [PubMed]
- Mollaei, H.; Safaralizadeh, R.; Rostami, Z. MicroRNA replacement therapy in cancer. J. Cell. Physiol. 2019, 234, 12369–12384. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Rao, D.D.; Senzer, N.; Nemunaitis, J. RNA interference and cancer therapy. Pharm. Res. 2011, 28, 2983–2995. [Google Scholar] [CrossRef]
- Pavlin, M.; Kandušer, M. New insights into the mechanisms of gene electrotransfer—Experimental and theoretical analysis. Sci. Rep. 2015, 5, 9132. [Google Scholar] [CrossRef] [PubMed]
- Jakstys, B.; Ruzgys, P.; Tamosiunas, M.; Satkauskas, S. Different cell viability assays reveal inconsistent results after bleomycin electrotransfer in vitro. J. Membr. Biol. 2015, 248, 857–863. [Google Scholar] [CrossRef]
- Ruzgys, P.; Jakutaviciute, M.; Chopra, S.; Satkauskas, S. Enhancement of drug electrotransfer by extracellular plasmid DNA. Arch. Biochem. Biophys. 2019, 666, 156–160. [Google Scholar] [CrossRef]
- Hecht, S.M. Bleomycin: New perspectives on the mechanism of action. J. Nat. Prod. 2000, 63, 158–168. [Google Scholar] [CrossRef]
- Bai, H.; Lester, G.M.S.; Petishnok, L.C.; Dean, D.A. Cytoplasmic transport and nuclear import of plasmid DNA. Biosci. Rep. 2017, 37, BSR20160616. [Google Scholar] [CrossRef]
- Dean, D.A.; Strong, D.D.; Zimmer, W.E. Nuclear entry of nonviral vectors. Gene Ther. 2005, 12, 881–890. [Google Scholar] [CrossRef] [Green Version]
- Murray, V.; Chen, J.K.; Chung, L.H. The interaction of the metallo-glycopeptide anti-tumour drug bleomycin with DNA. Int. J. Mol. Sci. 2018, 19, 1372. [Google Scholar] [CrossRef]
- Roy, B.; Hecht, S.M. Hairpin DNA sequences bound strongly by bleomycin exhibit enhanced double-strand cleavage. J. Am. Chem. Soc. 2014, 136, 4382–4393. [Google Scholar] [CrossRef]
- Sankaranarayanan, K.; Sundararajan, R. Electrically-enhanced proliferation control of cancer-stem-cells-like adult human mesenchymal stem cells—A novel modality of treatment. Electroporation Based Ther. Cancer 2014, 127–159. [Google Scholar] [CrossRef]
- Tounekti, O.; Pron, G.; Belehradek, J., Jr.; Mir, L.M. Bleomycin, an apoptosis-mimetic drug that induces two types of cell death depending on the number of molecules internalized. Cancer Res. 1993, 53, 5462–5469. [Google Scholar]
- Muller, W.E.; Zahn, R.K. Determination of the bleomycin-inactivating enzyme in biopsies. GANN 1976, 67, 425–430. [Google Scholar]
- Wu, M.; Yuan, F. Membrane binding of plasmid DNA and endocytic pathways are involved in electrotransfection of mammalian cells. PLoS ONE 2011, 6, e20923. [Google Scholar] [CrossRef]
- Golzio, M.; Teissie, J.; Rols, M.P. Direct visualization at the single-cell level of electrically mediated gene delivery. Proc. Natl. Acad. Sci. USA 2002, 99, 1292–1297. [Google Scholar] [CrossRef] [Green Version]
- Cemazar, M.; Todorovic, V.; Scancar, J.; Lampreht, U.; Stimac, M.; Kamensek, U.; Kranjc, S.; Coer, A.; Sersa, G. Adjuvant TNF-alpha therapy to electrochemotherapy with intravenous cisplatin in murine sarcoma exerts synergistic antitumor effectiveness. Radiol. Oncol. 2015, 49, 32–40. [Google Scholar] [CrossRef]
- Kamensek, U.; Tesic, N.; Sersa, G.; Cemazar, M. Clinically usable interleukin 12 plasmid without an antibiotic resistance gene: Functionality and toxicity study in murine melanoma model. Cancers (Basel) 2018, 10, 60. [Google Scholar] [CrossRef]
- Daud, A.I.; DeConti, R.C.; Andrews, S.; Urbas, P.; Riker, A.I.; Sondak, V.K.; Munster, P.N.; Sullivan, D.M.; Ugen, K.E.; Messina, J.L.; et al. Phase I trial of interleukin-12 plasmid electroporation in patients with metastatic melanoma. J. Clin. Oncol. 2008, 26, 5896–5903. [Google Scholar] [CrossRef]
- Ansell, S.M.; Witzig, T.E.; Kurtin, P.J.; Sloan, J.A.; Jelinek, D.F.; Howell, K.G.; Markovic, S.N.; Habermann, T.M.; Klee, G.G.; Atherton, P.J.; et al. Phase 1 study of interleukin-12 in combination with rituximab in patients with B-cell non-Hodgkin lymphoma. Blood 2002, 99, 67–74. [Google Scholar] [CrossRef] [Green Version]
- Cemazar, M.; Jarm, T.; Sersa, G. Cancer electrogene therapy with interleukin-12. Curr. Gene Ther. 2010, 10, 300–311. [Google Scholar] [CrossRef]
- Pavlin, D.; Cemazar, M.; Sersa, G.; Tozon, N. IL-12 based gene therapy in veterinary medicine. J. Transl. Med. 2012, 10, 234. [Google Scholar] [CrossRef]
- Belardelli, F.; Ferrantini, M.; Proietti, E.; Kirkwood, J.M. Interferon-alpha in tumor immunity and immunotherapy. Cytokine Growth Factor Rev. 2002, 13, 119–134. [Google Scholar] [CrossRef]
- O’Malley, B.W., Jr.; Li, D.; McQuone, S.J.; Ralston, R. Combination nonviral interleukin-2 gene immunotherapy for head and neck cancer: From bench top to bedside. Laryngoscope 2005, 115, 391–404. [Google Scholar] [CrossRef]
- Yao, H.; Ng, S.S.; Huo, L.F.; Chow, B.K.; Shen, Z.; Yang, M.; Sze, J.; Ko, O.; Li, M.; Yue, A.; et al. Effective melanoma immunotherapy with interleukin-2 delivered by a novel polymeric nanoparticle. Mol. Cancer Ther. 2011, 10, 1082–1092. [Google Scholar] [CrossRef]
- Lesueur, L.L.; Mir, L.M.; Andre, F.M. Overcoming the specific toxicity of large plasmids electrotransfer in primary cells in vitro. Mol. Ther. Nucleic Acids 2016, 5, e291. [Google Scholar] [CrossRef]
- Chen, Y.Z.; Yao, X.L.; Tabata, Y.; Nakagawa, S.; Gao, J.Q. Gene carriers and transfection systems used in the recombination of dendritic cells for effective cancer immunotherapy. Clin. Dev. Immunol. 2010, 2010, 565643. [Google Scholar] [CrossRef]
- Shirley, S.A.; Lundberg, C.G.; Li, F.; Burcus, N.; Heller, R. Controlled gene delivery can enhance therapeutic outcome for cancer immune therapy for melanoma. Curr. Gene Ther. 2015, 15, 32–43. [Google Scholar] [CrossRef]
- Sznol, M.; Parkinson, D.R. Clinical applications of IL-2. Oncology 1994, 8, 61–67, 71, 74–75. [Google Scholar] [PubMed]
- Leonard, J.P.; Sherman, M.L.; Fisher, G.L.; Buchanan, L.J.; Larsen, G.; Atkins, M.B.; Sosman, J.A.; Dutcher, J.P.; Vogelzang, N.J.; Ryan, J.L. Effects of single-dose interleukin-12 exposure on interleukin-12-associated toxicity and interferon-gamma production. Blood 1997, 90, 2541–2548. [Google Scholar] [PubMed]
- Jelley-Gibbs, D.M.; Lepak, N.M.; Yen, M.; Swain, S.L. Two distinct stages in the transition from naive CD4 T cells to effectors, early antigen-dependent and late cytokine-driven expansion and differentiation. J. Immunol. 2000, 165, 5017–5026. [Google Scholar] [CrossRef] [PubMed]
- Kishida, T.; Asada, H.; Itokawa, Y.; Yasutomi, K.; Shin-Ya, M.; Gojo, S.; Cui, F.D.; Ueda, Y.; Yamagishi, H.; Imanishi, J.; et al. Electrochemo-gene therapy of cancer: Intratumoral delivery of interleukin-12 gene and bleomycin synergistically induced therapeutic immunity and suppressed subcutaneous and metastatic melanomas in mice. Mol. Ther. 2003, 8, 738–745. [Google Scholar] [CrossRef] [PubMed]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chopra, S.; Ruzgys, P.; Jakutaviciute, M.; Rimgailaite, A.; Navickaitė, D.; Satkauskas, S. A Novel Method for Controlled Gene Expression via Combined Bleomycin and Plasmid DNA Electrotransfer. Int. J. Mol. Sci. 2019, 20, 4047. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20164047
Chopra S, Ruzgys P, Jakutaviciute M, Rimgailaite A, Navickaitė D, Satkauskas S. A Novel Method for Controlled Gene Expression via Combined Bleomycin and Plasmid DNA Electrotransfer. International Journal of Molecular Sciences. 2019; 20(16):4047. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20164047
Chicago/Turabian StyleChopra, Sonam, Paulius Ruzgys, Milda Jakutaviciute, Aiste Rimgailaite, Diana Navickaitė, and Saulius Satkauskas. 2019. "A Novel Method for Controlled Gene Expression via Combined Bleomycin and Plasmid DNA Electrotransfer" International Journal of Molecular Sciences 20, no. 16: 4047. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20164047