Genetic Factors of Cerebral Small Vessel Disease and Their Potential Clinical Outcome


Abstract
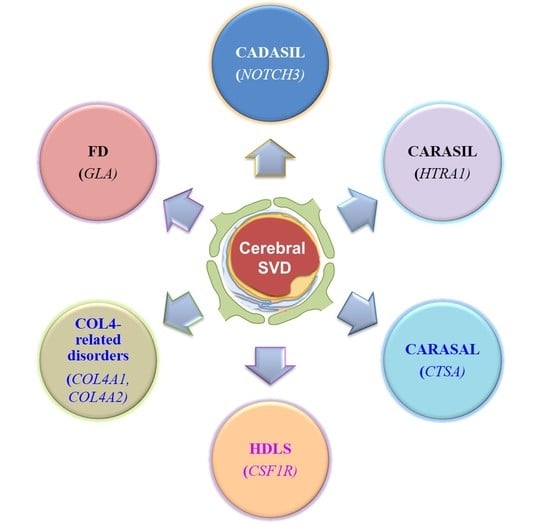
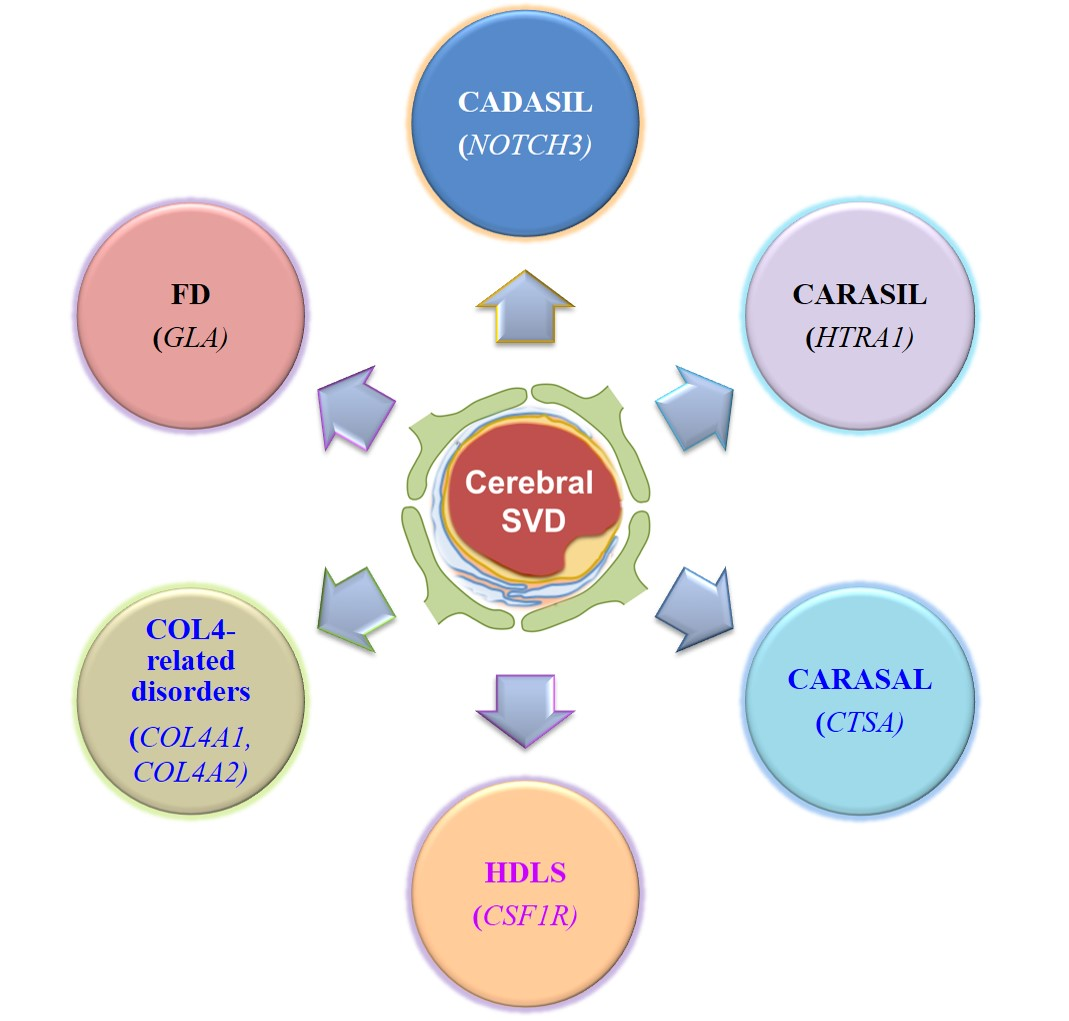
:
1. Introduction
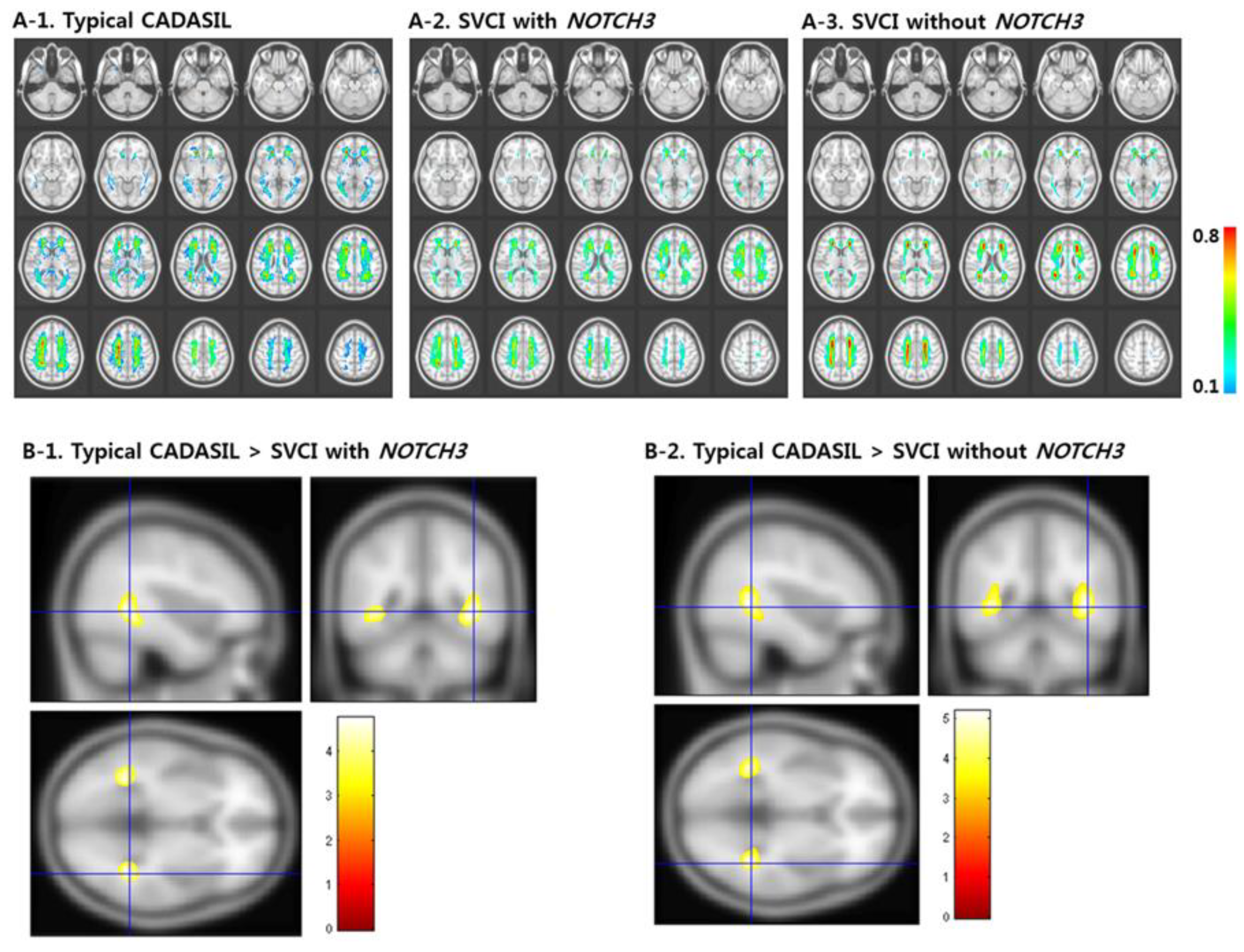
2. Cerebral Autosomal-Dominant Arteriopathy with Subcortical Infarcts and Leukoencephalopathy (CADASIL)
3. Cerebral Autosomal Recessive Arteriopathy with Subcortical Infarcts and Leukoencephalopathy (CARASIL)
4. Cathepsin A–Related Arteriopathy with Strokes and Leukoencephalopathy (CARASAL)
5. Hereditary Diffuse Leukoencephalopathy with Spheroids (HDLS)
6. COL4A1/2-Related Brain Small-Vessel Disease
7. Fabry Disease
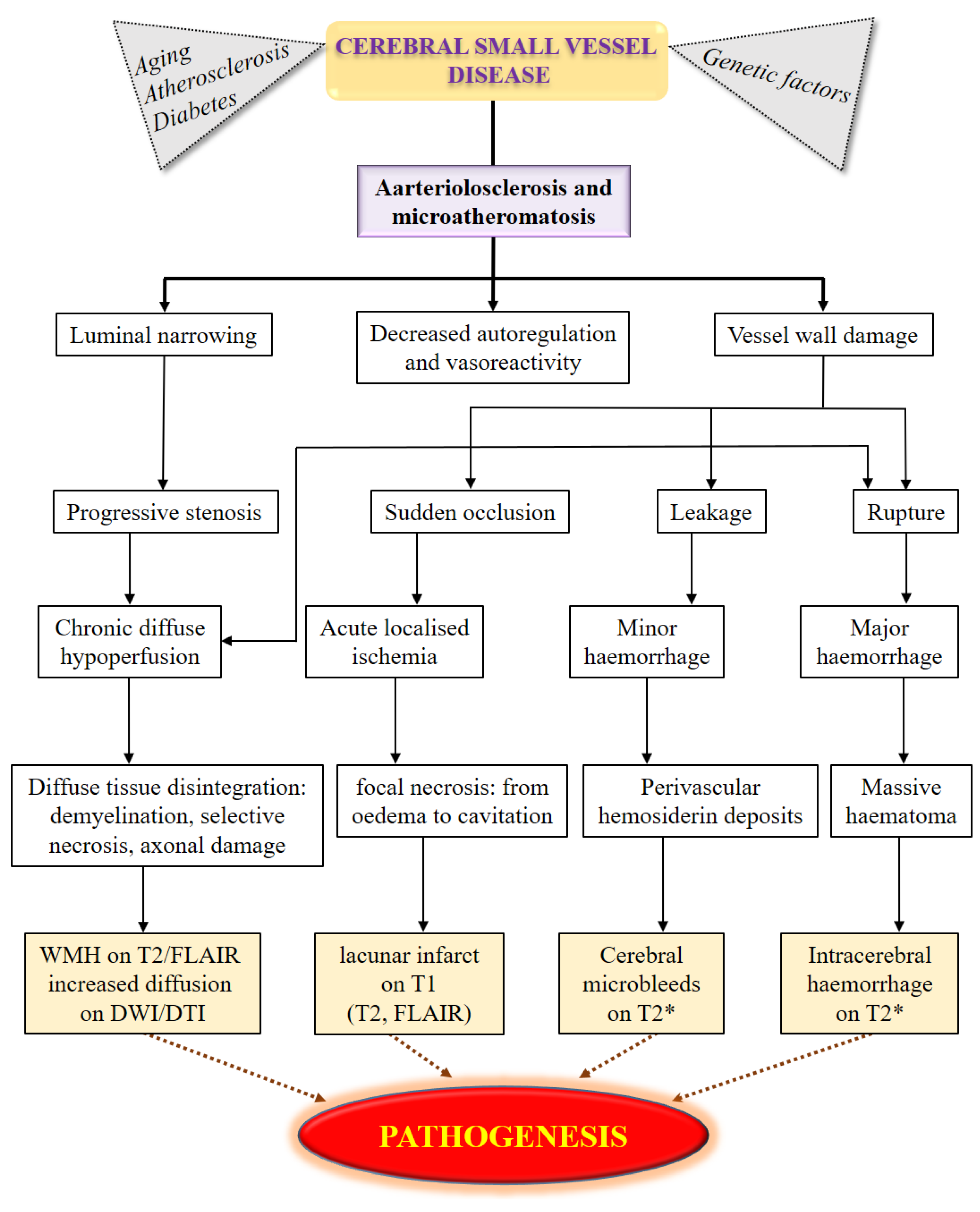
8. Possible Mechanisms of Cerebral Small Vessel Diseases Association Genetics
9. Conclusions and Future Perspective
Author Contributions
Funding
Conflicts of Interest
References
- Choi, J.C. Genetics of Cerebral Small Vessel Disease. J. Stroke 2015, 17, 7–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giau, V.V.; Bagyinszky, E.; An, S.S.A.; Kim, S. Clinical genetic strategies for early onset neurodegenerative diseases. Mol. Cell. Toxicol. 2018, 14, 123–142. [Google Scholar] [CrossRef]
- van Giau, V.; An, S.S.A.; Bagyinszky, E.; Kim, S. Gene panels and primers for next generation sequencing studies on neurodegenerative disorders. Mol. Cell. Toxicol. 2015, 11, 89–143. [Google Scholar] [CrossRef]
- White, H.; Boden-Albala, B.; Wang, C.; Elkind, M.S.; Rundek, T.; Wright, C.B.; Sacco, R.L. Ischemic stroke subtype incidence among whites, blacks, and Hispanics: The Northern Manhattan Study. Circulation 2005, 111, 1327–1331. [Google Scholar] [CrossRef] [PubMed]
- Sacco, S.; Marini, C.; Totaro, R.; Russo, T.; Cerone, D.; Carolei, A. A population-based study of the incidence and prognosis of lacunar stroke. Neurology 2006, 66, 1335–1338. [Google Scholar] [CrossRef] [PubMed]
- Chamorro, A.; Sacco, R.L.; Mohr, J.P.; Foulkes, M.A.; Kase, C.S.; Tatemichi, T.K.; Wolf, P.A.; Price, T.R.; Hier, D.B. Clinical-computed tomographic correlations of lacunar infarction in the Stroke Data Bank. Stroke 1991, 22, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Petty, G.W.; Brown, R.D., Jr.; Whisnant, J.P.; Sicks, J.D.; O’Fallon, W.M.; Wiebers, D.O. Ischemic stroke subtypes: A population-based study of incidence and risk factors. Stroke 1999, 30, 2513–2516. [Google Scholar] [CrossRef]
- Jung, K.H.; Lee, S.H.; Kim, B.J.; Yu, K.H.; Hong, K.S.; Lee, B.C.; Roh, J.K. Secular trends in ischemic stroke characteristics in a rapidly developed country: Results from the Korean Stroke Registry Study (secular trends in Korean stroke). Circ. Cardiovasc. Qual. Outcomes 2012, 5, 327–334. [Google Scholar] [CrossRef]
- Syed, N.A.; Khealani, B.A.; Ali, S.; Hasan, A.; Akhtar, N.; Brohi, H.; Mozaffar, T.; Ahmed, N.; Hameed, A.; Baig, S.M.; et al. Ischemic stroke subtypes in Pakistan: The Aga Khan University Stroke Data Bank. JPMA 2003, 53, 584–588. [Google Scholar]
- Tsai, C.F.; Thomas, B.; Sudlow, C.L. Epidemiology of stroke and its subtypes in Chinese vs. white populations: A systematic review. Neurology 2013, 81, 264–272. [Google Scholar] [CrossRef]
- Turin, T.C.; Kita, Y.; Rumana, N.; Nakamura, Y.; Takashima, N.; Ichikawa, M.; Sugihara, H.; Morita, Y.; Hirose, K.; Okayama, A.; et al. Ischemic stroke subtypes in a Japanese population: Takashima Stroke Registry, 1988–2004. Stroke 2010, 41, 1871–1876. [Google Scholar] [CrossRef] [PubMed]
- Kubo, M.; Hata, J.; Doi, Y.; Tanizaki, Y.; Iida, M.; Kiyohara, Y. Secular trends in the incidence of and risk factors for ischemic stroke and its subtypes in Japanese population. Circulation 2008, 118, 2672–2678. [Google Scholar] [CrossRef] [PubMed]
- Wolma, J.; Nederkoorn, P.J.; Goossens, A.; Vergouwen, M.D.; van Schaik, I.N.; Vermeulen, M. Ethnicity a risk factor? The relation between ethnicity and large- and small-vessel disease in White people, Black people, and Asians within a hospital-based population. Eur. J. Neurol. 2009, 16, 522–527. [Google Scholar] [CrossRef] [PubMed]
- Hilal, S.; Mok, V.; Youn, Y.C.; Wong, A.; Ikram, M.K.; Chen, C.L. Prevalence, risk factors and consequences of cerebral small vessel diseases: Data from three Asian countries. J. Neurol. Neurosurg. Psychiatry 2017, 88, 669–674. [Google Scholar] [CrossRef] [PubMed]
- Wardlaw, J.M.; Smith, E.E.; Biessels, G.J.; Cordonnier, C.; Fazekas, F.; Frayne, R.; Lindley, R.I.; O’Brien, J.T.; Barkhof, F.; Benavente, O.R.; et al. Neuroimaging standards for research into small vessel disease and its contribution to ageing and neurodegeneration. Lancet Neurol. 2013, 12, 822–838. [Google Scholar] [CrossRef] [Green Version]
- Moran, C.; Phan, T.G.; Srikanth, V.K. Cerebral small vessel disease: A review of clinical, radiological, and histopathological phenotypes. Int. J. Stroke 2012, 7, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Rost, N.S.; Fitzpatrick, K.; Biffi, A.; Kanakis, A.; Devan, W.; Anderson, C.D.; Cortellini, L.; Furie, K.L.; Rosand, J. White matter hyperintensity burden and susceptibility to cerebral ischemia. Stroke 2010, 41, 2807–2811. [Google Scholar] [CrossRef]
- Rost, N.S.; Rahman, R.M.; Biffi, A.; Smith, E.E.; Kanakis, A.; Fitzpatrick, K.; Lima, F.; Worrall, B.B.; Meschia, J.F.; Brown, R.D.; et al. White matter hyperintensity volume is increased in small vessel stroke subtypes. Neurology 2010, 75, 1670–1677. [Google Scholar] [CrossRef]
- Cheung, N.; Liew, G.; Lindley, R.I.; Liu, E.Y.; Wang, J.J.; Hand, P.; Baker, M.; Mitchell, P.; Wong, T.Y. Retinal fractals and acute lacunar stroke. Ann. Neurol. 2010, 68, 107–111. [Google Scholar] [CrossRef]
- Toyoda, K. Cerebral Small Vessel Disease and Chronic Kidney Disease. J. Stroke 2015, 17, 31–37. [Google Scholar] [CrossRef] [Green Version]
- Martini, S.; Williams, S.; Moretti, P.; Woo, D.; Worrall, B. A molecular/genetic approach to cerebral small-vessel disease: Beyond aging and hypertension. Brain Circ. 2015, 1, 79–87. [Google Scholar] [CrossRef]
- Tan, R.; Traylor, M.; Rutten-Jacobs, L.; Markus, H. New insights into mechanisms of small vessel disease stroke from genetics. Clin. Sci. (London, England 1979) 2017, 131, 515–531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giau, V.V.; Bagyinszky, E.; Yang, Y.S.; Youn, Y.C.; An, S.S.A.; Kim, S.Y. Genetic analyses of early-onset Alzheimer’s disease using next generation sequencing. Sci. Rep. 2019, 9, 8368. [Google Scholar] [CrossRef] [PubMed]
- Giau, V.V.; Senanarong, V.; Bagyinszky, E.; An, S.S.A.; Kim, S. Analysis of 50 Neurodegenerative Genes in Clinically Diagnosed Early-Onset Alzheimer’s Disease. Int. J. Mol. Sci. 2019, 20, 1514. [Google Scholar] [CrossRef] [PubMed]
- Dichgans, M. Genetics of ischaemic stroke. Lancet Neurol. 2007, 6, 149–161. [Google Scholar] [CrossRef]
- Craggs, L.J.; Yamamoto, Y.; Deramecourt, V.; Kalaria, R.N. Microvascular pathology and morphometrics of sporadic and hereditary small vessel diseases of the brain. Brain Pathol. (Zur. Switz.) 2014, 24, 495–509. [Google Scholar] [CrossRef] [PubMed]
- Joutel, A.; Corpechot, C.; Ducros, A.; Vahedi, K.; Chabriat, H.; Mouton, P.; Alamowitch, S.; Domenga, V.; Cecillion, M.; Marechal, E.; et al. Notch3 mutations in CADASIL, a hereditary adult-onset condition causing stroke and dementia. Nature 1996, 383, 707–710. [Google Scholar] [CrossRef] [PubMed]
- Hara, K.; Shiga, A.; Fukutake, T.; Nozaki, H.; Miyashita, A.; Yokoseki, A.; Kawata, H.; Koyama, A.; Arima, K.; Takahashi, T.; et al. Association of HTRA1 mutations and familial ischemic cerebral small-vessel disease. N. Engl. J. Med. 2009, 360, 1729–1739. [Google Scholar] [CrossRef] [PubMed]
- Verdura, E.; Herve, D.; Scharrer, E.; Amador Mdel, M.; Guyant-Marechal, L.; Philippi, A.; Corlobe, A.; Bergametti, F.; Gazal, S.; Prieto-Morin, C.; et al. Heterozygous HTRA1 mutations are associated with autosomal dominant cerebral small vessel disease. Brain 2015, 138, 2347–2358. [Google Scholar] [CrossRef]
- Bugiani, M.; Kevelam, S.H.; Bakels, H.S.; Waisfisz, Q.; Ceuterick-de Groote, C.; Niessen, H.W.; Abbink, T.E.; Lesnik Oberstein, S.A.; van der Knaap, M.S. Cathepsin A-related arteriopathy with strokes and leukoencephalopathy (CARASAL). Neurology 2016, 87, 1777–1786. [Google Scholar] [CrossRef]
- Rademakers, R.; Baker, M.; Nicholson, A.M.; Rutherford, N.J.; Finch, N.; Soto-Ortolaza, A.; Lash, J.; Wider, C.; Wojtas, A.; DeJesus-Hernandez, M.; et al. Mutations in the colony stimulating factor 1 receptor (CSF1R) gene cause hereditary diffuse leukoencephalopathy with spheroids. Nat. Genet. 2011, 44, 200–205. [Google Scholar] [CrossRef] [PubMed]
- Gould, D.B.; Phalan, F.C.; van Mil, S.E.; Sundberg, J.P.; Vahedi, K.; Massin, P.; Bousser, M.G.; Heutink, P.; Miner, J.H.; Tournier-Lasserve, E.; et al. Role of COL4A1 in small-vessel disease and hemorrhagic stroke. N. Engl. J. Med. 2006, 354, 1489–1496. [Google Scholar] [CrossRef] [PubMed]
- Revesz, T.; Holton, J.L.; Lashley, T.; Plant, G.; Frangione, B.; Rostagno, A.; Ghiso, J. Genetics and molecular pathogenesis of sporadic and hereditary cerebral amyloid angiopathies. Acta Neuropathol. 2009, 118, 115–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Silva, T.M.; Miller, A.A. Cerebral Small Vessel Disease: Targeting Oxidative Stress as a Novel Therapeutic Strategy? Front. Pharmacol. 2016, 7, 61. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, Y.; Craggs, L.; Baumann, M.; Kalimo, H.; Kalaria, R.N. Review: Molecular genetics and pathology of hereditary small vessel diseases of the brain. Neuropathol. Appl. Neurobiol. 2011, 37, 94–113. [Google Scholar] [CrossRef] [PubMed]
- Desmond, D.W.; Moroney, J.T.; Lynch, T.; Chan, S.; Chin, S.S.; Mohr, J.P. The natural history of CADASIL: A pooled analysis of previously published cases. Stroke 1999, 30, 1230–1233. [Google Scholar] [CrossRef] [PubMed]
- Fattapposta, F.; Restuccia, R.; Pirro, C.; Malandrini, A.; Locuratolo, N.; Amabile, G.; Bianco, F. Early diagnosis in cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL): The role of MRI. Funct. Neurol. 2004, 19, 239–242. [Google Scholar] [PubMed]
- Malandrini, A.; Gaudiano, C.; Gambelli, S.; Berti, G.; Serni, G.; Bianchi, S.; Federico, A.; Dotti, M.T. Diagnostic value of ultrastructural skin biopsy studies in CADASIL. Neurology 2007, 68, 1430–1432. [Google Scholar] [CrossRef] [PubMed]
- Stojanov, D.; Aracki-Trenkic, A.; Vojinovic, S.; Ljubisavljevic, S.; Benedeto-Stojanov, D.; Tasic, A.; Vujnovic, S. Imaging characteristics of cerebral autosomal dominant arteriopathy with subcortical infarcts and leucoencephalopathy (CADASIL). Bosn. J. Basic Med. Sci. 2015, 15, 1–8. [Google Scholar] [CrossRef]
- Kim, K.W.; Kwon, H.; Kim, Y.-E.; Yoon, C.W.; Kim, Y.J.; Kim, Y.B.; Lee, J.M.; Yoon, W.T.; Kim, H.J.; Lee, J.S.; et al. Multimodal imaging analyses in patients with genetic and sporadic forms of small vessel disease. Sci. Rep. 2019, 9, 787. [Google Scholar] [CrossRef]
- Davous, P. CADASIL: A review with proposed diagnostic criteria. Eur. J. Neurol. 1998, 5, 219–233. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.E.; Beaudin, A.E. New insights into cerebral small vessel disease and vascular cognitive impairment from MRI. Curr. Opin. Neurol. 2018, 31, 36–43. [Google Scholar] [CrossRef] [PubMed]
- ter Telgte, A.; van Leijsen, E.M.C.; Wiegertjes, K.; Klijn, C.J.M.; Tuladhar, A.M.; de Leeuw, F.-E. Cerebral small vessel disease: From a focal to a global perspective. Nat. Rev. Neurol. 2018, 14, 387–398. [Google Scholar] [CrossRef] [PubMed]
- Rutten, J.W.; Haan, J.; Terwindt, G.M.; van Duinen, S.G.; Boon, E.M.; Lesnik Oberstein, S.A. Interpretation of NOTCH3 mutations in the diagnosis of CADASIL. Expert Rev. Mol. Diagn. 2014, 14, 593–603. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, S.; Dotti, M.T.; Federico, A. Physiology and pathology of notch signalling system. J. Cell. Physiol. 2006, 207, 300–308. [Google Scholar] [CrossRef] [PubMed]
- Duering, M.; Karpinska, A.; Rosner, S.; Hopfner, F.; Zechmeister, M.; Peters, N.; Kremmer, E.; Haffner, C.; Giese, A.; Dichgans, M.; et al. Co-aggregate formation of CADASIL-mutant NOTCH3: A single-particle analysis. Hum. Mol. Genet. 2011, 20, 3256–3265. [Google Scholar] [CrossRef]
- Joutel, A.; Andreux, F.; Gaulis, S.; Domenga, V.; Cecillon, M.; Battail, N.; Piga, N.; Chapon, F.; Godfrain, C.; Tournier-Lasserve, E. The ectodomain of the Notch3 receptor accumulates within the cerebrovasculature of CADASIL patients. J. Clin. Investig. 2000, 105, 597–605. [Google Scholar] [CrossRef] [PubMed]
- Monet-Lepretre, M.; Haddad, I.; Baron-Menguy, C.; Fouillot-Panchal, M.; Riani, M.; Domenga-Denier, V.; Dussaule, C.; Cognat, E.; Vinh, J.; Joutel, A. Abnormal recruitment of extracellular matrix proteins by excess Notch3 ECD: A new pathomechanism in CADASIL. Brain 2013, 136, 1830–1845. [Google Scholar] [CrossRef] [PubMed]
- Ruchoux, M.M.; Guerouaou, D.; Vandenhaute, B.; Pruvo, J.P.; Vermersch, P.; Leys, D. Systemic vascular smooth muscle cell impairment in cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy. Acta Neuropathol. 1995, 89, 500–512. [Google Scholar] [CrossRef] [PubMed]
- Tikka, S.; Mykkanen, K.; Ruchoux, M.M.; Bergholm, R.; Junna, M.; Poyhonen, M.; Yki-Jarvinen, H.; Joutel, A.; Viitanen, M.; Baumann, M.; et al. Congruence between NOTCH3 mutations and GOM in 131 CADASIL patients. Brain 2009, 132, 933–939. [Google Scholar] [CrossRef] [Green Version]
- Federico, A.; Bianchi, S.; Dotti, M.T. The spectrum of mutations for CADASIL diagnosis. Neurol. Sci. 2005, 26, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Mazzei, R.; Conforti, F.L.; Lanza, P.L.; Sprovieri, T.; Lupo, M.R.; Gallo, O.; Patitucci, A.; Magariello, A.; Caracciolo, M.; Gabriele, A.L.; et al. A novel Notch3 gene mutation not involving a cysteine residue in an Italian family with CADASIL. Neurology 2004, 63, 561–564. [Google Scholar] [CrossRef] [PubMed]
- Coupland, K.; Lendahl, U.; Karlström, H. Role of NOTCH3 Mutations in the Cerebral Small Vessel Disease Cerebral Autosomal Dominant Arteriopathy With Subcortical Infarcts and Leukoencephalopathy. Stroke 2018, 49, 2793–2800. [Google Scholar] [CrossRef] [PubMed]
- Wollenweber, F.A.; Hanecker, P.; Bayer-Karpinska, A.; Malik, R.; Bazner, H.; Moreton, F.; Muir, K.W.; Muller, S.; Giese, A.; Opherk, C.; et al. Cysteine-sparing CADASIL mutations in NOTCH3 show proaggregatory properties in vitro. Stroke 2015, 46, 786–792. [Google Scholar] [CrossRef] [PubMed]
- Tuominen, S.; Juvonen, V.; Amberla, K.; Jolma, T.; Rinne, J.O.; Tuisku, S.; Kurki, T.; Marttila, R.; Poyhonen, M.; Savontaus, M.L.; et al. Phenotype of a homozygous CADASIL patient in comparison to 9 age-matched heterozygous patients with the same R133C Notch3 mutation. Stroke 2001, 32, 1767–1774. [Google Scholar] [CrossRef] [PubMed]
- Liem, M.K.; Lesnik Oberstein, S.A.; Vollebregt, M.J.; Middelkoop, H.A.; van der Grond, J.; van den Helderman Enden, A.T. Homozygosity for a NOTCH3 mutation in a 65-year-old CADASIL patient with mild symptoms: A family report. J. Neurol. 2008, 255, 1978–1980. [Google Scholar] [CrossRef]
- Ragno, M.; Pianese, L.; Morroni, M.; Cacchio, G.; Manca, A.; Di Marzio, F.; Silvestri, S.; Miceli, C.; Scarcella, M.; Onofrj, M.; et al. “CADASIL coma” in an Italian homozygous CADASIL patient: Comparison with clinical and MRI findings in age-matched heterozygous patients with the same G528C NOTCH3 mutation. Neurol. Sci. 2013, 34, 1947–1953. [Google Scholar] [CrossRef]
- Soong, B.W.; Liao, Y.C.; Tu, P.H.; Tsai, P.C.; Lee, I.H.; Chung, C.P.; Lee, Y.C. A homozygous NOTCH3 mutation p.R544C and a heterozygous TREX1 variant p.C99MfsX3 in a family with hereditary small vessel disease of the brain. J. Chin. Med. Assoc. 2013, 76, 319–324. [Google Scholar] [CrossRef] [Green Version]
- Vinciguerra, C.; Rufa, A.; Bianchi, S.; Sperduto, A.; De Santis, M.; Malandrini, A.; Dotti, M.T.; Federico, A. Homozygosity and severity of phenotypic presentation in a CADASIL family. Neurol. Sci. 2014, 35, 91–93. [Google Scholar] [CrossRef]
- Coto, E.; Menendez, M.; Navarro, R.; Garcia-Castro, M.; Alvarez, V. A new de novo Notch3 mutation causing CADASIL. Eur. J. Neurol. 2006, 13, 628–631. [Google Scholar] [CrossRef]
- Stojanov, D.; Grozdanovic, D.; Petrovic, S.; Benedeto-Stojanov, D.; Stefanovic, I.; Stojanovic, N.; Ilic, D.N. De novo mutation in the NOTCH3 gene causing CADASIL. Bosn. J. Basic Med. Sci. 2014, 14, 48–50. [Google Scholar] [CrossRef] [PubMed]
- Dichgans, M.; Ludwig, H.; Muller-Hocker, J.; Messerschmidt, A.; Gasser, T. Small in-frame deletions and missense mutations in CADASIL: 3D models predict misfolding of Notch3 EGF-like repeat domains. Eur. J. Hum. Genet. 2000, 8, 280–285. [Google Scholar] [CrossRef] [PubMed]
- Dotti, M.T.; De Stefano, N.; Bianchi, S.; Malandrini, A.; Battisti, C.; Cardaioli, E.; Federico, A. A novel NOTCH3 frameshift deletion and mitochondrial abnormalities in a patient with CADASIL. Arch. Neurol. 2004, 61, 942–945. [Google Scholar] [CrossRef] [PubMed]
- Dichgans, M.; Herzog, J.; Gasser, T. NOTCH3 mutation involving three cysteine residues in a family with typical CADASIL. Neurology 2001, 57, 1714–1717. [Google Scholar] [CrossRef] [PubMed]
- Muiño, E.; Gallego-Fabrega, C.; Cullell, N.; Carrera, C.; Torres, N.; Krupinski, J.; Roquer, J.; Montaner, J.; Fernández-Cadenas, I. Systematic Review of Cysteine-Sparing NOTCH3 Missense Mutations in Patients with Clinical Suspicion of CADASIL. Int. J. Mol. Sci. 2017, 18, 1964. [Google Scholar] [CrossRef] [PubMed]
- Brass, S.D.; Smith, E.E.; Arboleda-Velasquez, J.F.; Copen, W.A.; Frosch, M.P. Case records of the Massachusetts General Hospital. Case 12-2009. A 46-year-old man with migraine, aphasia, and hemiparesis and similarly affected family members. N. Engl. J. Med. 2009, 360, 1656–1665. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Choi, E.J.; Choi, C.G.; Kim, G.; Choi, J.H.; Yoo, H.W.; Kim, J.S. Characteristics of CADASIL in Korea: A novel cysteine-sparing Notch3 mutation. Neurology 2006, 66, 1511–1516. [Google Scholar] [CrossRef]
- Ungaro, C.; Mazzei, R.; Conforti, F.L.; Sprovieri, T.; Servillo, P.; Liguori, M.; Citrigno, L.; Gabriele, A.L.; Magariello, A.; Patitucci, A.; et al. CADASIL: Extended polymorphisms and mutational analysis of the NOTCH3 gene. J. Neurosci. Res. 2009, 87, 1162–1167. [Google Scholar] [CrossRef]
- Ge, W.; Kuang, H.; Wei, B.; Bo, L.; Xu, Z.; Xu, X.; Geng, D.; Sun, M. A novel cysteine-sparing NOTCH3 mutation in a Chinese family with CADASIL. PLoS ONE 2014, 9, e104533. [Google Scholar] [CrossRef]
- Ampuero, I.; Alegre-Abarrategui, J.; Rodal, I.; Espana, A.; Ros, R.; Sendon, J.L.; Galloway, E.G.; Cervello, A.; Caminero, A.B.; Zabala, A.; et al. On the diagnosis of CADASIL. J. Alzheimer’s Dis. 2009, 17, 787–794. [Google Scholar] [CrossRef]
- Roy, B.; Maksemous, N.; Smith, R.A.; Menon, S.; Davies, G.; Griffiths, L.R. Two novel mutations and a previously unreported intronic polymorphism in the NOTCH3 gene. Mutat. Res. 2012, 732, 3–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santa, Y.; Uyama, E.; Chui, D.H.; Arima, M.; Kotorii, S.; Takahashi, K.; Tabira, T. Genetic, clinical and pathological studies of CADASIL in Japan: A partial contribution of Notch3 mutations and implications of smooth muscle cell degeneration for the pathogenesis. J. Neurol. Sci. 2003, 212, 79–84. [Google Scholar] [CrossRef]
- Abramycheva, N.; Stepanova, M.; Kalashnikova, L.; Zakharova, M.; Maximova, M.; Tanashyan, M.; Lagoda, O.; Fedotova, E.; Klyushnikov, S.; Konovalov, R.; et al. New mutations in the Notch3 gene in patients with cerebral autosomal dominant arteriopathy with subcortical infarcts and leucoencephalopathy (CADASIL). J. Neurol. Sci. 2015, 349, 196–201. [Google Scholar] [CrossRef] [PubMed]
- Homer, V.; George, P.M.; du Toit, S.; Davidson, J.S.; Wilson, C.J. Novel human pathological mutations. Gene symbol: APOB. Disease: Normotriglyceridemic hypobetalipoproteinemia. Hum. Genet. 2007, 121, 645–646. [Google Scholar] [PubMed]
- Fouillade, C.; Chabriat, H.; Riant, F.; Mine, M.; Arnoud, M.; Magy, L.; Bousser, M.G.; Tournier-Lasserve, E.; Joutel, A. Activating NOTCH3 mutation in a patient with small-vessel-disease of the brain. Hum. Mutat. 2008, 29, 452. [Google Scholar] [CrossRef] [PubMed]
- Nishimoto, Y.; Shibata, M.; Nihonmatsu, M.; Nozaki, H.; Shiga, A.; Shirata, A.; Yamane, K.; Kosakai, A.; Takahashi, K.; Nishizawa, M.; et al. A novel mutation in the HTRA1 gene causes CARASIL without alopecia. Neurology 2011, 76, 1353–1355. [Google Scholar] [CrossRef]
- Bersano, A.; Ranieri, M.; Ciammola, A.; Cinnante, C.; Lanfranconi, S.; Dotti, M.T.; Candelise, L.; Baschirotto, C.; Ghione, I.; Ballabio, E.; et al. Considerations on a mutation in the NOTCH3 gene sparing a cysteine residue: A rare polymorphism rather than a CADASIL variant. Funct. Neurol. 2012, 27, 247–252. [Google Scholar] [PubMed]
- Chavoshi Tarzjani, S.P.; Shahzadeh Fazeli, S.A.; Sanati, M.H.; Mirzayee, Z. Genetic study of the NOTCH3 gene in CADASIL patients. Egypt. J. Med. Hum. Genet. 2018, 19, 425–427. [Google Scholar] [CrossRef]
- Wang, Z.; Yuan, Y.; Zhang, W.; Lv, H.; Hong, D.; Chen, B.; Liu, Y.; Luan, X.; Xie, S.; Wu, S. NOTCH3 mutations and clinical features in 33 mainland Chinese families with CADASIL. J. Neurol. Neurosurg. Psychiatry 2011, 82, 534–539. [Google Scholar] [CrossRef]
- Mizuno, T.; Muranishi, M.; Torugun, T.; Tango, H.; Nagakane, Y.; Kudeken, T.; Kawase, Y.; Kawabe, K.; Oshima, F.; Yaoi, T.; et al. Two Japanese CADASIL families exhibiting Notch3 mutation R75P not involving cysteine residue. Intern. Med. (Tokyo Jpn.) 2008, 47, 2067–2072. [Google Scholar] [CrossRef]
- Kotorii, S.; Takahashi, K.; Kamimura, K.; Nishio, T.; Arima, K.; Yamada, H.; Uyama, E.; Uchino, M.; Suenaga, A.; Matsumoto, M.; et al. Mutations of the notch3 gene in non-caucasian patients with suspected CADASIL syndrome. Dement. Geriatr. Cogn. Disord. 2001, 12, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Scheid, R.; Heinritz, W.; Leyhe, T.; Thal, D.R.; Schober, R.; Strenge, S.; von Cramon, D.Y.; Froster, U.G. Cysteine-sparing notch3 mutations: Cadasil or cadasil variants? Neurology 2008, 71, 774–776. [Google Scholar] [CrossRef] [PubMed]
- Mazzei, R.; Guidetti, D.; Ungaro, C.; Conforti, F.L.; Muglia, M.; Cenacchi, G.; Lanza, P.L.; Patitucci, A.; Sprovieri, T.; Riguzzi, P.; et al. First evidence of a pathogenic insertion in the NOTCH3 gene causing CADASIL. J. Neurol. Neurosurg. Psychiatry 2008, 79, 108–110. [Google Scholar] [CrossRef] [PubMed]
- Kilarski, L.L.; Rutten-Jacobs, L.C.; Bevan, S.; Baker, R.; Hassan, A.; Hughes, D.A.; Markus, H.S. Prevalence of CADASIL and Fabry Disease in a Cohort of MRI Defined Younger Onset Lacunar Stroke. PLoS ONE 2015, 10, e0136352. [Google Scholar] [CrossRef] [PubMed]
- Cai, B.; Zeng, J.; Lin, Y.; Lin, Y.; Lin, W.; Lin, W.; Li, Z.; Wang, N. A frameshift mutation in HTRA1 expands CARASIL syndrome and peripheral small arterial disease to the Chinese population. Neurol. Sci. 2015, 36, 1387–1391. [Google Scholar] [CrossRef] [PubMed]
- Fukutake, T.; Hirayama, K. Familial young-adult-onset arteriosclerotic leukoencephalopathy with alopecia and lumbago without arterial hypertension. Eur. Neurol. 1995, 35, 69–79. [Google Scholar] [CrossRef]
- Oide, T.; Nakayama, H.; Yanagawa, S.; Ito, N.; Ikeda, S.; Arima, K. Extensive loss of arterial medial smooth muscle cells and mural extracellular matrix in cerebral autosomal recessive arteriopathy with subcortical infarcts and leukoencephalopathy (CARASIL). Neuropathology 2008, 28, 132–142. [Google Scholar] [CrossRef]
- Arima, K.; Yanagawa, S.; Ito, N.; Ikeda, S. Cerebral arterial pathology of CADASIL and CARASIL (Maeda syndrome). Neuropathology 2003, 23, 327–334. [Google Scholar] [CrossRef]
- Fukutake, T. Cerebral autosomal recessive arteriopathy with subcortical infarcts and leukoencephalopathy (CARASIL): From discovery to gene identification. J. Stroke Cerebrovasc. Dis. 2011, 20, 85–93. [Google Scholar] [CrossRef]
- Okeda, R.; Murayama, S.; Sawabe, M.; Kuroiwa, T. Pathology of the cerebral artery in Binswanger’s disease in the aged: Observation by serial sections and morphometry of the cerebral arteries. Neuropathology 2004, 24, 21–29. [Google Scholar] [CrossRef]
- Chen, Y.; He, Z.; Meng, S.; Li, L.; Yang, H.; Zhang, X. A novel mutation of the high-temperature requirement A serine peptidase 1 (HTRA1) gene in a Chinese family with cerebral autosomal recessive arteriopathy with subcortical infarcts and leukoencephalopathy (CARASIL). J. Int. Med. Res. 2013, 41, 1445–1455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.L.; Li, C.F.; Guo, H.W.; Cao, B.Z. A novel mutation in the HTRA1 gene identified in Chinese CARASIL pedigree. CNS Neurosci. Ther. 2012, 18, 867–869. [Google Scholar] [CrossRef] [PubMed]
- Di Donato, I.; Bianchi, S.; Gallus, G.N.; Cerase, A.; Taglia, I.; Pescini, F.; Nannucci, S.; Battisti, C.; Inzitari, D.; Pantoni, L.; et al. Heterozygous mutations of HTRA1 gene in patients with familial cerebral small vessel disease. CNS Neurosci. Ther. 2017, 23, 759–765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendioroz, M.; Fernandez-Cadenas, I.; Del Rio-Espinola, A.; Rovira, A.; Sole, E.; Fernandez-Figueras, M.T.; Garcia-Patos, V.; Sastre-Garriga, J.; Domingues-Montanari, S.; Alvarez-Sabin, J.; et al. A missense HTRA1 mutation expands CARASIL syndrome to the Caucasian population. Neurology 2010, 75, 2033–2035. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, S.; Di Palma, C.; Gallus, G.N.; Taglia, I.; Poggiani, A.; Rosini, F.; Rufa, A.; Muresanu, D.F.; Cerase, A.; Dotti, M.T.; et al. Two novel HTRA1 mutations in a European CARASIL patient. Neurology 2014, 82, 898–900. [Google Scholar] [CrossRef] [PubMed]
- Bayrakli, F.; Balaban, H.; Gurelik, M.; Hizmetli, S.; Topaktas, S. Mutation in the HTRA1 gene in a patient with degenerated spine as a component of CARASIL syndrome. Turk. Neurosurg. 2014, 24, 67–69. [Google Scholar] [CrossRef] [PubMed]
- Ibrahimi, M.; Nozaki, H.; Lee, A.; Onodera, O.; Reichwein, R.; Wicklund, M.; El-Ghanem, M. A CARASIL Patient from Americas with Novel Mutation and Atypical Features: Case Presentation and Literature Review. Cerebrovasc. Dis. (Basel Switz.) 2017, 44, 135–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bougea, A.; Velonakis, G.; Spantideas, N.; Anagnostou, E.; Paraskevas, G.; Kapaki, E.; Kararizou, E. The first Greek case of heterozygous cerebral autosomal recessive arteriopathy with subcortical infarcts and leukoencephalopathy: An atypical clinico-radiological presentation. Neuroradiol. J. 2017, 30, 583–585. [Google Scholar] [CrossRef]
- Nozaki, H.; Kato, T.; Nihonmatsu, M.; Saito, Y.; Mizuta, I.; Noda, T.; Koike, R.; Miyazaki, K.; Kaito, M.; Ito, S.; et al. Distinct molecular mechanisms of HTRA1 mutants in manifesting heterozygotes with CARASIL. Neurology 2016, 86, 1964–1974. [Google Scholar] [CrossRef]
- Menezes Cordeiro, I.; Nzwalo, H.; Sa, F.; Ferreira, R.B.; Alonso, I.; Afonso, L.; Basilio, C. Shifting the CARASIL paradigm: Report of a non-Asian family and literature review. Stroke 2015, 46, 1110–1112. [Google Scholar] [CrossRef]
- Nozaki, H.; Nishizawa, M.; Onodera, O. Features of cerebral autosomal recessive arteriopathy with subcortical infarcts and leukoencephalopathy. Stroke 2014, 45, 3447–3453. [Google Scholar] [CrossRef] [PubMed]
- Herve, D.; Chabriat, H.; Rigal, M.; Dalloz, M.A.; Kawkabani Marchini, A.; De Lepeleire, J.; Fontaine, B.; Ceuterick-de Groote, C.; Alili, N.; Mine, M.; et al. A novel hereditary extensive vascular leukoencephalopathy mapping to chromosome 20q13. Neurology 2012, 79, 2283–2287. [Google Scholar] [CrossRef] [PubMed]
- d’Azzo, A.; Bonten, E. Molecular Mechanisms of Pathogenesis in a Glycosphigolipid and a Glycoprotein Storage Disease. Biochem. Soc. Trans. 2010, 38, 1453–1457. [Google Scholar] [CrossRef] [PubMed]
- Strisciuglio, P.; Parenti, G.; Giudice, C.; Lijoi, S.; Hoogeveen, A.T.; d’Azzo, A. The presence of a reduced amount of 32-kd “protective” protein is a distinct biochemical finding in late infantile galactosialidosis. Hum. Genet. 1988, 80, 304–306. [Google Scholar] [CrossRef] [PubMed]
- Hammond, T.R.; Gadea, A.; Dupree, J.; Kerninon, C.; Nait-Oumesmar, B.; Aguirre, A.; Gallo, V. Astrocyte-derived endothelin-1 inhibits remyelination through notch activation. Neuron 2014, 81, 588–602. [Google Scholar] [CrossRef] [PubMed]
- Baba, Y.; Ghetti, B.; Baker, M.C.; Uitti, R.J.; Hutton, M.L.; Yamaguchi, K.; Bird, T.; Lin, W.; DeLucia, M.W.; Dickson, D.W.; et al. Hereditary diffuse leukoencephalopathy with spheroids: Clinical, pathologic and genetic studies of a new kindred. Acta Neuropathol. 2006, 111, 300–311. [Google Scholar] [CrossRef]
- Konno, T.; Yoshida, K.; Mizuno, T.; Kawarai, T.; Tada, M.; Nozaki, H.; Ikeda, S.I.; Nishizawa, M.; Onodera, O.; Wszolek, Z.K.; et al. Clinical and genetic characterization of adult-onset leukoencephalopathy with axonal spheroids and pigmented glia associated with CSF1R mutation. Eur. J. Neurol. 2017, 24, 37–45. [Google Scholar] [CrossRef]
- Sundal, C.; Ekholm, S.; Andersen, O. White matter disorders with autosomal dominant heredity: A review with personal clinical case studies and their MRI findings. Acta Neurol. Scand. 2010, 121, 328–337. [Google Scholar] [CrossRef]
- Sundal, C.; Jonsson, L.; Ljungberg, M.; Zhong, J.; Tian, W.; Zhu, T.; Linden, T.; Borjesson-Hanson, A.; Andersen, O.; Ekholm, S. Different stages of white matter changes in the original HDLS family revealed by advanced MRI techniques. J. Neuroimaging 2014, 24, 444–452. [Google Scholar] [CrossRef]
- Axelsson, R.; Roytta, M.; Sourander, P.; Akesson, H.O.; Andersen, O. Hereditary diffuse leucoencephalopathy with spheroids. Acta Psychiatr. Scand. Suppl. 1984, 314, 1–65. [Google Scholar]
- Prinz, M.; Priller, J. Microglia and brain macrophages in the molecular age: From origin to neuropsychiatric disease. Nat. Rev. Neurosci. 2014, 15, 300–312. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, A. Microglia and brain macrophages: An update. Neuropathology 2017, 37, 452–464. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, A.M.; Baker, M.C.; Finch, N.A.; Rutherford, N.J.; Wider, C.; Graff-Radford, N.R.; Nelson, P.T.; Clark, H.B.; Wszolek, Z.K.; Dickson, D.W.; et al. CSF1R mutations link POLD and HDLS as a single disease entity. Neurology 2013, 80, 1033–1040. [Google Scholar] [CrossRef] [PubMed]
- Locascio, L.E.; Donoghue, D.J. KIDs rule: Regulatory phosphorylation of RTKs. Trends Biochem. Sci. 2013, 38, 75–84. [Google Scholar] [CrossRef]
- Kraya, T.; Quandt, D.; Pfirrmann, T.; Kindermann, A.; Lampe, L.; Schroeter, M.L.; Kohlhase, J.; Stoevesandt, D.; Hoffmann, K.; Villavicencio-Lorini, P. Functional characterization of a novel CSF1R mutation causing hereditary diffuse leukoencephalopathy with spheroids. Mol. Genet. Genom. Med. 2019, 7, e00595. [Google Scholar] [CrossRef] [PubMed]
- Vahedi, K.; Mascart, F.; Mary, J.Y.; Laberenne, J.E.; Bouhnik, Y.; Morin, M.C.; Ocmant, A.; Velly, C.; Colombel, J.F.; Matuchansky, C. Reliability of antitransglutaminase antibodies as predictors of gluten-free diet compliance in adult celiac disease. Am. J. Gastroenterol. 2003, 98, 1079–1087. [Google Scholar] [CrossRef]
- Shah, S.; Ellard, S.; Kneen, R.; Lim, M.; Osborne, N.; Rankin, J.; Stoodley, N.; van der Knaap, M.; Whitney, A.; Jardine, P. Childhood presentation of COL4A1 mutations. Dev. Med. Child Neurol. 2012, 54, 569–574. [Google Scholar] [CrossRef] [Green Version]
- Lanfranconi, S.; Markus, H.S. COL4A1 mutations as a monogenic cause of cerebral small vessel disease: A systematic review. Stroke 2010, 41, e513–e518. [Google Scholar] [CrossRef]
- Verbeek, E.; Meuwissen, M.E.; Verheijen, F.W.; Govaert, P.P.; Licht, D.J.; Kuo, D.S.; Poulton, C.J.; Schot, R.; Lequin, M.H.; Dudink, J.; et al. COL4A2 mutation associated with familial porencephaly and small-vessel disease. Eur. J. Hum. Genet. 2012, 20, 844–851. [Google Scholar] [CrossRef] [Green Version]
- Khoshnoodi, J.; Pedchenko, V.; Hudson, B.G. Mammalian collagen IV. Microsc. Res. Tech. 2008, 71, 357–370. [Google Scholar] [CrossRef] [Green Version]
- Gould, D.B.; Phalan, F.C.; Breedveld, G.J.; van Mil, S.E.; Smith, R.S.; Schimenti, J.C.; Aguglia, U.; van der Knaap, M.S.; Heutink, P.; John, S.W. Mutations in Col4a1 cause perinatal cerebral hemorrhage and porencephaly. Science 2005, 308, 1167–1171. [Google Scholar] [CrossRef] [PubMed]
- Plaisier, E.; Gribouval, O.; Alamowitch, S.; Mougenot, B.; Prost, C.; Verpont, M.C.; Marro, B.; Desmettre, T.; Cohen, S.Y.; Roullet, E.; et al. COL4A1 mutations and hereditary angiopathy, nephropathy, aneurysms, and muscle cramps. N. Engl. J. Med. 2007, 357, 2687–2695. [Google Scholar] [CrossRef] [PubMed]
- Kuo, D.S.; Labelle-Dumais, C.; Gould, D.B. COL4A1 and COL4A2 mutations and disease: Insights into pathogenic mechanisms and potential therapeutic targets. Hum. Mol. Genet. 2012, 21, R97–R110. [Google Scholar] [CrossRef] [PubMed]
- Meuwissen, M.E.; Halley, D.J.; Smit, L.S.; Lequin, M.H.; Cobben, J.M.; de Coo, R.; van Harssel, J.; Sallevelt, S.; Woldringh, G.; van der Knaap, M.S.; et al. The expanding phenotype of COL4A1 and COL4A2 mutations: Clinical data on 13 newly identified families and a review of the literature. Genet. Med. 2015, 17, 843–853. [Google Scholar] [CrossRef] [PubMed]
- Kato, M.; Wang, L.; Putta, S.; Wang, M.; Yuan, H.; Sun, G.; Lanting, L.; Todorov, I.; Rossi, J.J.; Natarajan, R. Post-transcriptional up-regulation of Tsc-22 by Ybx1, a target of miR-216a, mediates TGF-{beta}-induced collagen expression in kidney cells. J. Biol. Chem. 2010, 285, 34004–34015. [Google Scholar] [CrossRef] [PubMed]
- Chung, A.C.; Huang, X.R.; Meng, X.; Lan, H.Y. miR-192 mediates TGF-beta/Smad3-driven renal fibrosis. J. Am. Soc. Nephrol. 2010, 21, 1317–1325. [Google Scholar] [CrossRef] [PubMed]
- Jeanne, M.; Labelle-Dumais, C.; Jorgensen, J.; Kauffman, W.B.; Mancini, G.M.; Favor, J.; Valant, V.; Greenberg, S.M.; Rosand, J.; Gould, D.B. COL4A2 Mutations Impair COL4A1 and COL4A2 Secretion and Cause Hemorrhagic Stroke. Am. J. Hum. Genet. 2012, 90, 91–101. [Google Scholar] [CrossRef] [Green Version]
- Yoneda, Y.; Haginoya, K.; Arai, H.; Yamaoka, S.; Tsurusaki, Y.; Doi, H.; Miyake, N.; Yokochi, K.; Osaka, H.; Kato, M.; et al. De novo and inherited mutations in COL4A2, encoding the type IV collagen alpha2 chain cause porencephaly. Am. J. Hum. Genet. 2012, 90, 86–90. [Google Scholar] [CrossRef]
- Alavi, M.V.; Mao, M.; Pawlikowski, B.T.; Kvezereli, M.; Duncan, J.L.; Libby, R.T.; John, S.W.M.; Gould, D.B. Col4a1 mutations cause progressive retinal neovascular defects and retinopathy. Sci. Rep. 2016, 6, 18602. [Google Scholar] [CrossRef]
- Schmidt, H.; Zeginigg, M.; Wiltgen, M.; Freudenberger, P.; Petrovic, K.; Cavalieri, M.; Gider, P.; Enzinger, C.; Fornage, M.; Debette, S.; et al. Genetic variants of the NOTCH3 gene in the elderly and magnetic resonance imaging correlates of age-related cerebral small vessel disease. Brain 2011, 134, 3384–3397. [Google Scholar] [CrossRef] [Green Version]
- Weng, Y.C.; Sonni, A.; Labelle-Dumais, C.; de Leau, M.; Kauffman, W.B.; Jeanne, M.; Biffi, A.; Greenberg, S.M.; Rosand, J.; Gould, D.B. COL4A1 mutations in patients with sporadic late-onset intracerebral hemorrhage. Ann. Neurol. 2012, 71, 470–477. [Google Scholar] [CrossRef] [PubMed]
- Weng, J.; Li, Y.; Xu, W.; Shi, L.; Zhang, Q.; Zhu, D.; Hu, Y.; Zhou, Z.; Yan, X.; Tian, H.; et al. Effect of intensive insulin therapy on beta-cell function and glycaemic control in patients with newly diagnosed type 2 diabetes: A multicentre randomised parallel-group trial. Lancet (Lond. Engl.) 2008, 371, 1753–1760. [Google Scholar] [CrossRef]
- Robin, N.H.; Taylor, C.J.; McDonald-McGinn, D.M.; Zackai, E.H.; Bingham, P.; Collins, K.J.; Earl, D.; Gill, D.; Granata, T.; Guerrini, R.; et al. Polymicrogyria and deletion 22q11.2 syndrome: Window to the etiology of a common cortical malformation. Am. J. Med. Genet. A 2006, 140, 2416–2425. [Google Scholar] [CrossRef] [PubMed]
- Brady, R.O.; Gal, A.E.; Bradley, R.M.; Martensson, E.; Warshaw, A.L.; Laster, L. Enzymatic defect in Fabry’s disease. Ceramidetrihexosidase deficiency. N. Engl. J. Med. 1967, 276, 1163–1167. [Google Scholar] [CrossRef] [PubMed]
- Guffon, N. Clinical presentation in female patients with Fabry disease. J. Med. Genet. 2003, 40, e38. [Google Scholar] [CrossRef] [PubMed]
- Arends, M.; Wanner, C.; Hughes, D.; Mehta, A.; Oder, D.; Watkinson, O.T.; Elliott, P.M.; Linthorst, G.E.; Wijburg, F.A.; Biegstraaten, M.; et al. Characterization of Classical and Nonclassical Fabry Disease: A Multicenter Study. J. Am. Soc. Nephrol. 2017, 28, 1631–1641. [Google Scholar] [CrossRef] [PubMed]
- Buechner, S.; Moretti, M.; Burlina, A.P.; Cei, G.; Manara, R.; Ricci, R.; Mignani, R.; Parini, R.; Di Vito, R.; Giordano, G.P.; et al. Central nervous system involvement in Anderson-Fabry disease: A clinical and MRI retrospective study. J. Neurol. Neurosurg. Psychiatry 2008, 79, 1249–1254. [Google Scholar] [CrossRef] [PubMed]
- Hopkin, R.J.; Jefferies, J.L.; Laney, D.A.; Lawson, V.H.; Mauer, M.; Taylor, M.R.; Wilcox, W.R. The management and treatment of children with Fabry disease: A United States-based perspective. Mol. Genet. Metab. 2016, 117, 104–113. [Google Scholar] [CrossRef] [PubMed]
- Gal, A. Molecular Genetics of Fabry Disease and Genotype—Phenotype Correlation. In Fabry Disease; Elstein, D., Altarescu, G., Beck, M., Eds.; Springer: Dordrecht, The Netherlands, 2010; pp. 3–19. [Google Scholar]
- Verovnik, F.; Benko, D.; Vujkovac, B.; Linthorst, G.E. Remarkable variability in renal disease in a large Slovenian family with Fabry disease. Eur. J. Hum. Genet. 2004, 12, 678–681. [Google Scholar] [CrossRef] [PubMed]
- Froissart, R.; Guffon, N.; Vanier, M.T.; Desnick, R.J.; Maire, I. Fabry disease: D313Y is an alpha-galactosidase A sequence variant that causes pseudodeficient activity in plasma. Mol. Genet. Metab. 2003, 80, 307–314. [Google Scholar] [CrossRef]
- Shi, Q.; Chen, J.; Pongmoragot, J.; Lanthier, S.; Saposnik, G. Prevalence of Fabry disease in stroke patients—A systematic review and meta-analysis. J. Stroke Cerebrovasc. Dis. 2014, 23, 985–992. [Google Scholar] [CrossRef] [PubMed]
- Meikle, P.J.; Hopwood, J.J.; Clague, A.E.; Carey, W.F. Prevalence of lysosomal storage disorders. JAMA 1999, 281, 249–254. [Google Scholar] [CrossRef] [PubMed]
- Houge, G.; Skarbovik, A.J. Fabry disease—A diagnostic and therapeutic challenge. Tidsskr. Nor. Laegeforen. 2005, 125, 1004–1006. [Google Scholar] [PubMed]
- Yasuda, M.; Shabbeer, J.; Benson, S.D.; Maire, I.; Burnett, R.M.; Desnick, R.J. Fabry disease: Characterization of alpha-galactosidase A double mutations and the D313Y plasma enzyme pseudodeficiency allele. Hum. Mutat. 2003, 22, 486–492. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, S.; Ortiz, A.; Germain, D.P.; Viana-Baptista, M.; Caldeira-Gomes, A.; Camprecios, M.; Fenollar-Cortes, M.; Gallegos-Villalobos, A.; Garcia, D.; Garcia-Robles, J.A.; et al. The alpha-galactosidase A p.Arg118Cys variant does not cause a Fabry disease phenotype: Data from individual patients and family studies. Mol. Genet. Metab. 2015, 114, 248–258. [Google Scholar] [CrossRef]
- Meng, Y.; Zhang, W.M.; Shi, H.P.; Wei, M.; Huang, S.Z. Clinical manifestations and mutation study in 16 Chinese patients with Fabry disease. Zhonghua Yi Xue Za Zhi 2010, 90, 551–554. [Google Scholar] [PubMed]
- Mukdsi, J.H.; Gutiérrez, S.; Barrón, B.; Novoa, P.; Fernández, S.; de Diller, A.B.; Torres, A.I.; Formica, R.N., Jr.; Orías, M. A renal variant of Fabry disease: A case with a novel Gal A hemizygote mutation. J. Nephropathol. 2012, 1, 194–197. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Niño, M.D.; Ortiz, A. Is it or is it not a pathogenic mutation? Is it or is it not the podocyte? J. Nephropathol. 2012, 1, 152–154. [Google Scholar] [CrossRef] [Green Version]
- Baptista, M.V.; Ferreira, S.; Pinho, E.M.T.; Carvalho, M.; Cruz, V.T.; Carmona, C.; Silva, F.A.; Tuna, A.; Rodrigues, M.; Ferreira, C.; et al. Mutations of the GLA gene in young patients with stroke: The PORTYSTROKE study--screening genetic conditions in Portuguese young stroke patients. Stroke 2010, 41, 431–436. [Google Scholar] [CrossRef]
- Gaspar, P.; Herrera, J.; Rodrigues, D.; Cerezo, S.; Delgado, R.; Andrade, C.F.; Forascepi, R.; Macias, J.; del Pino, M.D.; Prados, M.D.; et al. Frequency of Fabry disease in male and female haemodialysis patients in Spain. BMC Med. Genet. 2010, 11, 19. [Google Scholar] [CrossRef]
- Elliott, P.; Baker, R.; Pasquale, F.; Quarta, G.; Ebrahim, H.; Mehta, A.B.; Hughes, D.A. Prevalence of Anderson-Fabry disease in patients with hypertrophic cardiomyopathy: The European Anderson-Fabry Disease survey. Heart (Br. Card. Soc.) 2011, 97, 1957–1960. [Google Scholar] [CrossRef] [PubMed]
- Rolfs, A.; Fazekas, F.; Grittner, U.; Dichgans, M.; Martus, P.; Holzhausen, M.; Bottcher, T.; Heuschmann, P.U.; Tatlisumak, T.; Tanislav, C.; et al. Acute cerebrovascular disease in the young: The Stroke in Young Fabry Patients study. Stroke 2013, 44, 340–349. [Google Scholar] [CrossRef] [PubMed]
- Turaca, L.T.; Pessoa, J.G.; Motta, F.L.; Munoz Rojas, M.V.; Muller, K.B.; Lourenco, C.M.; Junior Marques, W.; D’Almeida, V.; Martins, A.M.; Pesquero, J.B. New mutations in the GLA gene in Brazilian families with Fabry disease. J. Hum. Genet. 2012, 57, 347–351. [Google Scholar] [CrossRef] [PubMed]
- Spada, M.; Pagliardini, S.; Yasuda, M.; Tukel, T.; Thiagarajan, G.; Sakuraba, H.; Ponzone, A.; Desnick, R.J. High Incidence of Later-Onset Fabry Disease Revealed by Newborn Screening. Am. J. Hum. Genet. 2006, 79, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Hsu, T.R.; Niu, D.M. Fabry disease: Review and experience during newborn screening. Trends Cardiovasc. Med. 2018, 28, 274–281. [Google Scholar] [CrossRef] [PubMed]
- Parfenov, V.A.; Ostroumova, O.D.; Ostroumova, T.M.; Kochetkov, A.I.; Fateeva, V.V.; Khacheva, K.K.; Khakimova, G.R.; Epstein, O.I. Vascular cognitive impairment: Pathophysiological mechanisms, insights into structural basis, and perspectives in specific treatments. Neuropsychiatr. Dis. Treat. 2019, 15, 1381–1402. [Google Scholar] [CrossRef] [PubMed]
- Giau, V.V.; Bagyinszky, E.; An, S.S.A.; Kim, S.Y. Role of apolipoprotein E in neurodegenerative diseases. Neuropsychiatr. Dis. Treat. 2015, 11, 1723–1737. [Google Scholar] [CrossRef] [PubMed]
- Giau, V.V.; Lee, H.; Shim, K.H.; Bagyinszky, E.; An, S.S.A. Genome-editing applications of CRISPR-Cas9 to promote in vitro studies of Alzheimer’s disease. Clin. Interv. Aging 2018, 13, 221–233. [Google Scholar] [CrossRef]
- Shen, L.; An, S.S.A.; Bagyinszky, E.; Van Giau, V.; Choi, S.H.; Kim, S.Y. Novel GRN mutations in Koreans with Alzheimer’s disease. Mol. Cell. Toxicol. 2019, 15, 345–352. [Google Scholar] [CrossRef]
- Beaufort, N.; Scharrer, E.; Kremmer, E.; Lux, V.; Ehrmann, M.; Huber, R.; Houlden, H.; Werring, D.; Haffner, C.; Dichgans, M. Cerebral small vessel disease-related protease HtrA1 processes latent TGF-beta binding protein 1 and facilitates TGF-beta signaling. Proc. Natl. Acad. Sci. USA 2014, 111, 16496–16501. [Google Scholar] [CrossRef]
- Richards, A.; van den Maagdenberg, A.M.; Jen, J.C.; Kavanagh, D.; Bertram, P.; Spitzer, D.; Liszewski, M.K.; Barilla-Labarca, M.L.; Terwindt, G.M.; Kasai, Y.; et al. C-terminal truncations in human 3’–5’ DNA exonuclease TREX1 cause autosomal dominant retinal vasculopathy with cerebral leukodystrophy. Nat. Genet. 2007, 39, 1068–1070. [Google Scholar] [CrossRef] [PubMed]
- French, C.R.; Seshadri, S.; Destefano, A.L.; Fornage, M.; Arnold, C.R.; Gage, P.J.; Skarie, J.M.; Dobyns, W.B.; Millen, K.J.; Liu, T.; et al. Mutation of FOXC1 and PITX2 induces cerebral small-vessel disease. J. Clin. Investig. 2014, 124, 4877–4881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sondergaard, C.B.; Nielsen, J.E.; Hansen, C.K.; Christensen, H. Hereditary cerebral small vessel disease and stroke. Clin. Neurol. Neurosurg. 2017, 155, 45–57. [Google Scholar] [CrossRef] [PubMed]
- Giau, V.V.; Bagyinszky, E.; An, S.S.A. Potential Fluid Biomarkers for the Diagnosis of Mild Cognitive Impairment. Int. J. Mol. Sci. 2019, 20, 4149. [Google Scholar] [CrossRef] [PubMed]
- Fleischer, V.; Radetz, A.; Ciolac, D.; Muthuraman, M.; Gonzalez-Escamilla, G.; Zipp, F.; Groppa, S. Graph Theoretical Framework of Brain Networks in Multiple Sclerosis: A Review of Concepts. Neuroscience 2017, 403, 35–53. [Google Scholar] [CrossRef] [PubMed]
- Giau, V.V.; An, S.S.A.; Hulme, J.J. Mitochondrial therapeutic interventions in Alzheimer’s disease. Neurol. Sci. 2018, 395, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Van Giau, V.; An, S.S. Emergence of exosomal miRNAs as a diagnostic biomarker for Alzheimer’s disease. J. Neurol. Sci. 2016, 360, 141–152. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
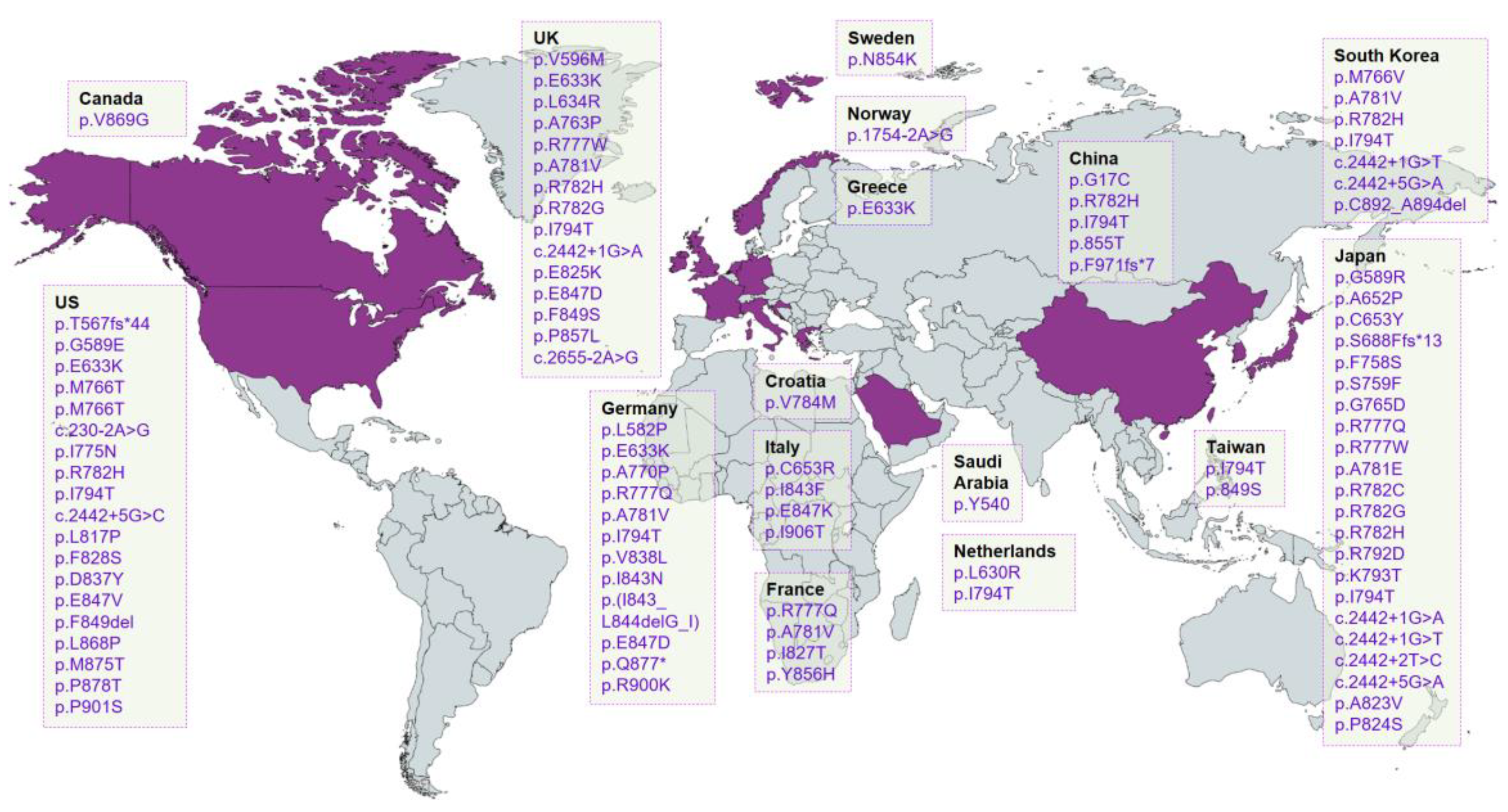{kind=link}
| Diseases | Inheritance | Chromosome Locus | Gene | Mutation Findings | Protein | Cerebral Features | Neuroimaging Finding | Pathological Findings | References |
|---|---|---|---|---|---|---|---|---|---|
| CADASIL | Autosomal dominant | 19q12 | NOTCH3 | Over 256 missense mutations or rare deletions and insertions were reported | Transmembrane receptor | Migraine with & aura, recurrent was chemic strokes, mood disturbance, cognitive decline, disability > death 65–70 years WMH, lacunar infarcts, dilated PVS, micro bleeds, brain atrophy | Typical of cerebral SVD plus in anterior temporal lobe and external capsular | Granular osmiophilic material found in the walls of affected arterioles | [27] |
| CARASIL | Autosomal recessive | 10q25 | HTRA1 | At least 17 mutations were identified in 25 families worldwide | HtrA serine peptidase/protease 1 | Recurrent is chemic strokes, Cognitive decline, disability > death | Similar to CADASIL, diffuse WM lesions and small infarcts in basal ganglia | Arteriosclerotic changes, WM changes. No GOM deposition. Hyaline degeneration and thickening and splitting of internal lamina | [28,29] |
| CARASAL | Autosomal dominant | 20q13.12 | CTSA | Many galactosialidosis patients related to CTSA gene point mutations were reported | Cathepsin A (CathA) | Therapy-resistant hypertension, strokes, and slow and late cognitive deterioration | A diffuse, progressive leukoencephalopathy preceding the onset of strokes and disproportionate to the degree of clinical severity | Endothelin-1 overexpression coincides with increased numbers of premyelinating OPCs, decreased MBP amounts, abundance of axons without myelin, and features of remyelination failure | [30] |
| HDLS | Autosomal dominant | 5q32 | CSF1R | Approximately 60 pathogenic variants have been reported in patients with HDLS | CSF-1 receptor | Stroke episodes with pyramidal, bulbar and | Diffuse WM lesions, lacunar strokes and atrophy | Diffuse gliosis, moderate loss of axons and many axonal spheroids | [31] |
| COL4-related disorders | Autosomal dominant | 13q34 | COL4A, COL4A2 | Over 50 types of mutations have been reported to dat | Collagen type IV, alpha chains | Infantileemiparesis, intracerebralhemorrhage (perinatal, youngoradult) porencephaliccysts, microbleeds, WMH, intracranial aneurysms (HANAC) | Typical of cerebral SVD | Defects in the basement membrane | [32] |
| FD | X-linked | Xq22 | GLA | Around 585 pathogenic mutations have been reported in the GLA gene to date | Lysosomal α-galactosidase A | Typically, stroke is considered a manifestation of end stage | Multifocal WMH lesions, intracranial arterial dolichectasia | Lysomal storage materials in vascular endothelial cells and smooth muscle cells | [33,34] |
| Mutation | Exon | Age of Onset | Gender | Population | Clinical Phenotypes | References |
|---|---|---|---|---|---|---|
| p.Arg61Trp | 2 | 46 | NA | American | Migraine, aphasia, hemiparesis, probable familial | [66] |
| p.Arg75Pro | 3 | 53 | M | Korean | Typical CADASIL symptoms, granular osmiophilic granules on skin biopsy in a patient. Probable positive family history | [67] |
| p.Arg75Pro | 47 | F | ||||
| p.Arg75Pro | 65 | M | ||||
| p.Arg75Pro | 40–50s | F | Japanese | Proband: depression, akinetic mutism, pseudobalbar palsy, and tetraplegia; MRI: mild frontal and temporal lobe atrophy multiple cerebral infarctions at the bilateral basal ganglia other family members: stiffness, headache, vertigo, dizziness; MRI: e infarctions at the bilateral basal ganglia | [80] | |
| p.Arg75Pro | 64 | F | right thalamic hemorrhage, akinetic mutism, pseudobalbar palsy, repeated cerebral infarctions MRI: diffuse cerebral atrophy Family members were affected with dementia | |||
| p.Arg75Pro | 34 | M | Chinese | Cerebral infarction, transient ischaemic attack with tinnitus, irritable; MRI: small infract in different brain areas (deep white matter, basal ganglia) | [79] | |
| p.Asp80Gly | 50s | F | German | Imaging and clinical symptoms: typical CADASIL phenotype. Mutation may be segregated with diseases | [54] | |
| p.Arg107Trp | NA | NA | German | CADASIL, no clinical phenotype | [68] | |
| 63 | M | UK | Hypertension, Mother was affected with stroke | [84] | ||
| p.Pro109Thr | 57 | F | Iranian | CADASIL, no detailed clinical phenotype. Mutation co-existed with NOTCH3 Pro203His mutation. | [78] | |
| p.Gly149Val | 4 | 39 | F | Chinese | Initial symptoms: progressive dizziness, memory dysfunctions. MRI: leukoencephalopathy and confluent signal abnormalities; Family history probable positive | [69] |
| p.Gln151Glu | NA | NA | Italian | CADASIL, white matter hyperintensities, migraine, no detailed clinical phenotype | [68] | |
| p.Gln151Glu | NA | NA | Spanish | CADASIL, white matter hyperintensities, stroke, no detailed clinical phenotype | [70] | |
| p.His170Arg | NA | NA | Spanish | CADASIL, white matter hyperintensities not specified, no detailed clinical phenotype | [70] | |
| p.His170Arg | NA | NA | Oceanian | CADASIL, not specified white matter hyperintensities, stroke, no clinical phenotype | [71] | |
| p.Ala198Thr | NA | NA | Italian | CADASIL, white matter hyperintensities, migraine, no detailed clinical phenotype | [68] | |
| p.Ala202Val | NA | F | Oceanian | CADASIL, not specified white matter hyperintensities, stroke, no clinical phenotype | [70] | |
| p.Pro203His | 57 | F | Iranian | CADASIL, no detailed clinical phenotype. Mutation co-existed with NOTCH3 Pro109Thr mutation. | [78] | |
| p.Arg207His | NA | NA | Italian | CADASIL, white matter hyperintensities, migraine no detailed clinical phenotype | [68] | |
| p.Arg213Lys | 36 | M | Japanese | CADASIL, white matter hyperintensities, migraine, dementia, stroke, positive family history | [71] | |
| p.Arg213Lys | 10 | M | Japanese | CADASIL, white matter hyperintensities, dementia migraine, stroke, positive family history | [72] | |
| p.Val237Met | 5 | 71 | F | Japanese | CADASIL, white matter hyperintensities, stroke, gait disturbances, dementia, positive family history | [81] |
| p.Val252Met | 63 | M | Russian | strokes and/or transient ischemic attacks, pseudobulbar palsy, pyramidal signs. MRI: hyperintensities of the external capsules, temporal lobes, lacunar infarcts in the cerebellum and/or brainstem, cerebral hemispheres | [73] | |
| p.Glu309Lys | 6 | NA | NA | Italian | CADASIL, white matter hyperintensities, migraine, dementia, family history positive | [68] |
| p.Ser497Lys | 9 | NA | NA | Russian | CADASIL, non-specified white matter hyperintensities, appeared in unaffected individuals | [73] |
| p.Thr577Ala | 11 | NA | NA | Portuguese | Unclear phenotype, non-specified white matter hyperintensities | [74] |
| p.Arg592Ser | 11 | NA | NA | Italian | CADASIL, white matter hyperintensities, migraine, dementia, probable positive family history | [68] |
| p.Val644Asp | 12 | NA | NA | Italian | CADASIL, white matter hyperintensities, migraine, dementia, probable positive family history | [68] |
| p.Ser978Arg | 18 | NA | F | Portuguese | CADASIL, white matter hyperintensities, stroke seizure, psychiatric symptoms | [74] |
| p.Ala1020Pro | 19 | Adolescence | F | German | CADASIL, white matter hyperintensities, hypertension, migraine, dementia, positive family history | [82] |
| p.Ala1020Pro | 19 | NA | F | German | CADASIL, white matter hyperintensities, hypertension, psychiatric disturbance, positive family history | |
| p.Thr1098Ser | 20 | 39 | M | Chinese | CADASIL, white matter hyperintensities, stroke, psychiatric disturbances, dementia, positive family history | [79] |
| p.His1133Gln | 21 | Unknown | Unknown | Russian | CADASIL, non-specified white matter hyperintensities, | [85] |
| p.His1235Lys | 22 | Unknown | Unknown | Italian | CADASIL, white matter hyperintensities | [68] |
| p.Lys1515Pro | 25 | 35 | F | French | CADASIL, white matter hyperintensities, migraine, dementia, positive family history | [75] |
| p.Val1762Met | 29 | Childhood | F | Italian | CADASIL, white matter hyperintensities, psychiatric dysfunctions, positive family history | [77] |
| Mutation | Exon | Gender | AOO | Clinical Phenotypes | Mutation Type | Ethnicity | References |
|---|---|---|---|---|---|---|---|
| Asian patients | |||||||
| p.Ala252Thr | 3 | F | Teenage | Lumbago in her teens, leukoaraiosis in brain, stroke, gait disturbance, pseudobulbar palsy, pyramidal signs | Homozygous missense | Japanese | [28] |
| p.Val297Met | 4 | M/F | 14–16 | Alopecia in teens, leukoaraiosis in brain, spondylosis, dementia, gait disturbance, pseudobulbar palsy, pyramidal signs | |||
| p.Arg302X | 4 | F/M | 14/16 | Alopecia in teens, spondylosis, dementia, gait disturbance, possible stroke, pseudobulbar palsy, pyramidal signs | |||
| p.Arg370X | 6 | F | 18 | Alopecia in teens, spondylosis, dementia, gait disturbance, possible stroke, pseudobulbar palsy, pyramidal signs | |||
| p.Arg274Gln | 4 | F | 14 | Lumbago in her teens, lumbar and cervical spondylosis in her 30s, later subcortical ischemic lesions and spastic paraparesis and intellectual dysfunctions | [76] | ||
| p.Gly283Glu | 4 | M | 49 | Cognitive impairment, gait disturbance, spondylosis, pseudobulbar palsy, hyperreflexia in limbs | Heterozygous missense | [99] | |
| p.Pro285Leu | 4 | M | 20 | Alopecia in his 20s, gait distubances in his 30s, cognitive dysfunction in his 50s. Spondylosis, hyperreflexia in limbs, Babinski reflexes | |||
| M | 51 | Stroke, cognitive impairment and gait disturbance in his 50s. Spondylosis, pseudobulbar palsy, hyperreflexia in limbs | |||||
| p.Arg302Gln | 4 | M | 20-63 | Alopecia possible in their 20–40s. Spondylosis, pseudobulbar palsy (not all patients), hyperreflexia in limbs, Babinski reflex | |||
| p.Thr319Ile | 4 | M | 53 | Spondylosis, pseudobulbar palsy, hyperreflexia in limbs, Babinski reflex | |||
| p.Pro285Leu | 4 | F | 24 | Progressive alopecia from birth, right limb disability, lumbar pain, lethargy, memory dysfunctions | Homozygous missense | Chinese | [91] |
| p.Leu364Pro | 6 | F | 25 | right foot and lumbar pain, prolapse of lumbar intervertebral disk, mild alopecia, intellectual dysfunction, spastic gait Babinski signs | Homozygous missense | [92] | |
| p.Gly56Alafs*160 | 1 | M | 28 | recurrent stroke, hair loss and low back pain, lower limb weakness, alopecia, pyramidal signs | Homozygous/Frameshift | [85] | |
| Caucasian patients | |||||||
| p.Gly295Arg | 4 | M | 34 | Alopecia, dysarthria, dysphagia, emotional instability, and spastic gait, Babinski signLater, cognitive impairment and upper limb weakness, pseudobulbar syndrome | Homozygous missense | Spanish | [94] |
| p.Arg370X | 6 | F | 29 | Alopecia, back and neck pain, right-sided weakness, difficulty in walking | Homozygous nonsense | Turkish | [96] |
| p.Glu42fs | 1 | F | 29 | chronic lumbar and cervical pain from the age of 14, ischemic strokes with left hemiparesis and dysarthria, without alopecia and cognitive dysfunctions | compound heterozygous deletion & missense | Romanian | [95] |
| p.Ala321Thr | 4 | ||||||
| p.Arg166Cys | 3 | M | 33 | Alopecia, transient ischemic attacks, lacunar strokes, cervical, and lumbar pain, acute psychosis, cognitive impairment, dysarthria, spastic paraparesis, hyperreflexia, bilateral Babinski’s sign. | Homozygous missense | Portuguese | [100] |
| p.Gly206Arg | 3 | M | 24 | Alopecia, chronic back pain presented with recurrent ischemic strokes, hearing impairment | homozygous missense | American | [97] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giau, V.V.; Bagyinszky, E.; Youn, Y.C.; An, S.S.A.; Kim, S.Y. Genetic Factors of Cerebral Small Vessel Disease and Their Potential Clinical Outcome. Int. J. Mol. Sci. 2019, 20, 4298. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20174298
Giau VV, Bagyinszky E, Youn YC, An SSA, Kim SY. Genetic Factors of Cerebral Small Vessel Disease and Their Potential Clinical Outcome. International Journal of Molecular Sciences. 2019; 20(17):4298. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20174298
Chicago/Turabian StyleGiau, Vo Van, Eva Bagyinszky, Young Chul Youn, Seong Soo A. An, and Sang Yun Kim. 2019. "Genetic Factors of Cerebral Small Vessel Disease and Their Potential Clinical Outcome" International Journal of Molecular Sciences 20, no. 17: 4298. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20174298