Impact of Chronic BDNF Depletion on GABAergic Synaptic Transmission in the Lateral Amygdala



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
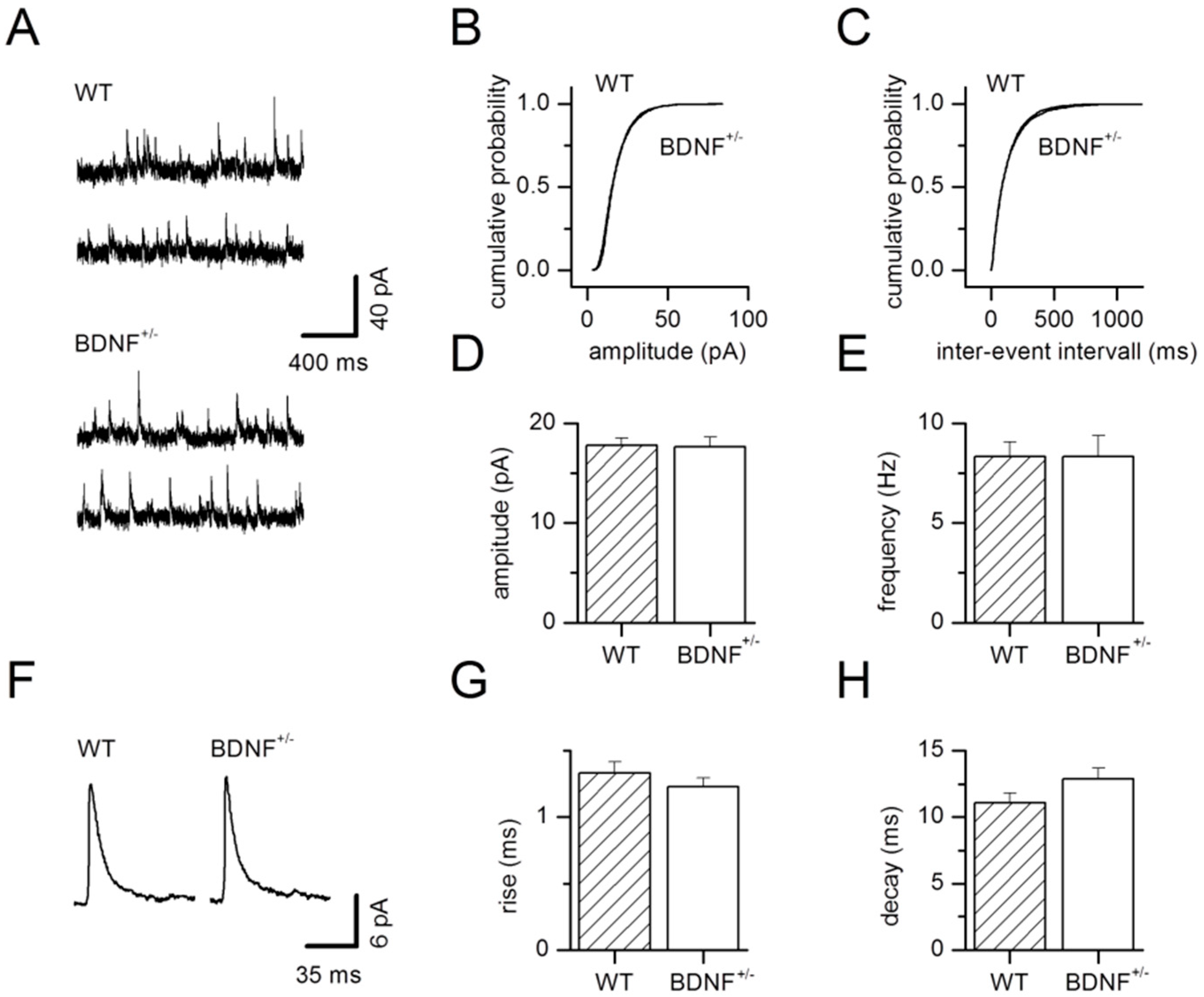
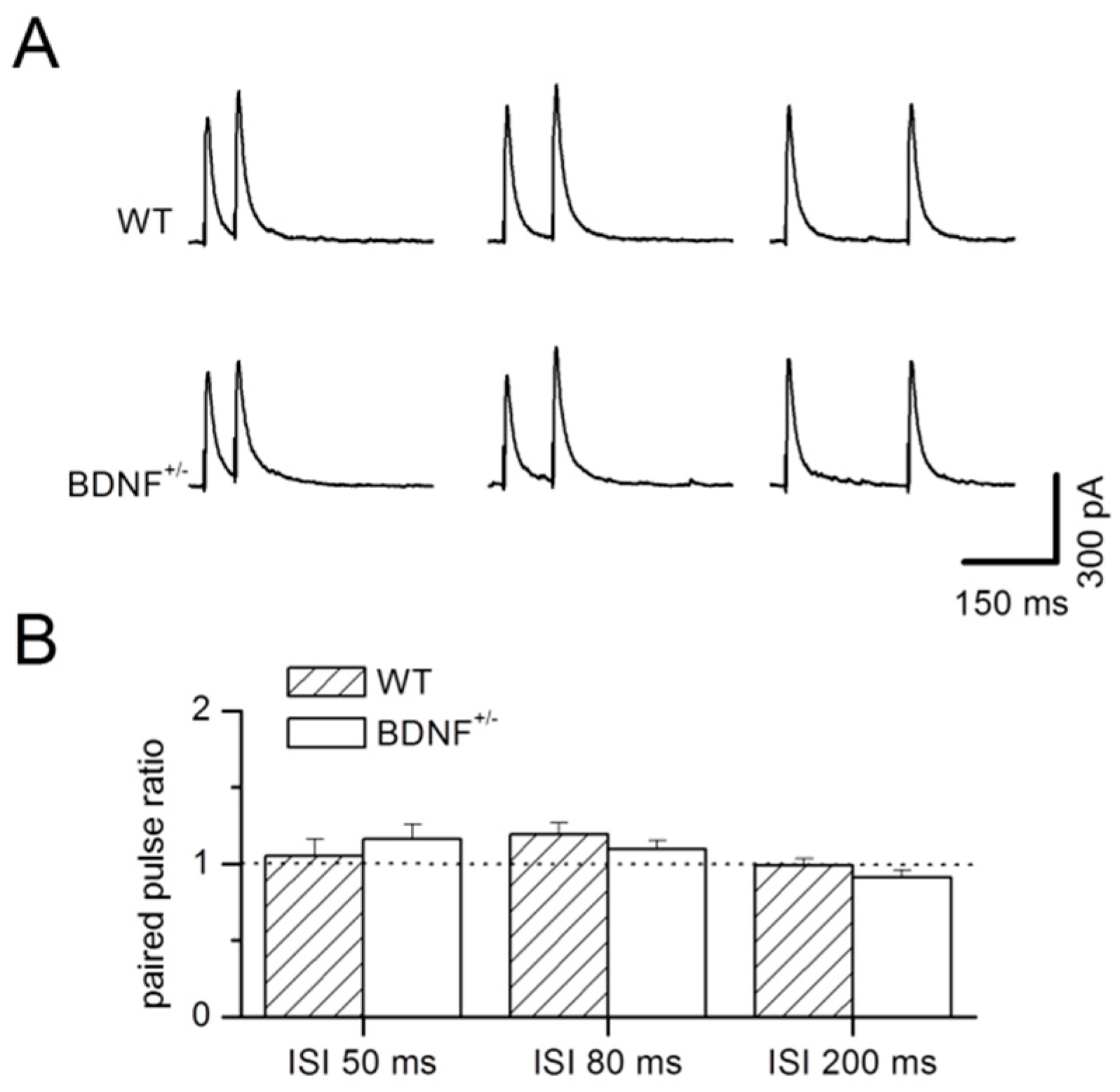
2.1. Basal GABAergic Synaptic Transmission
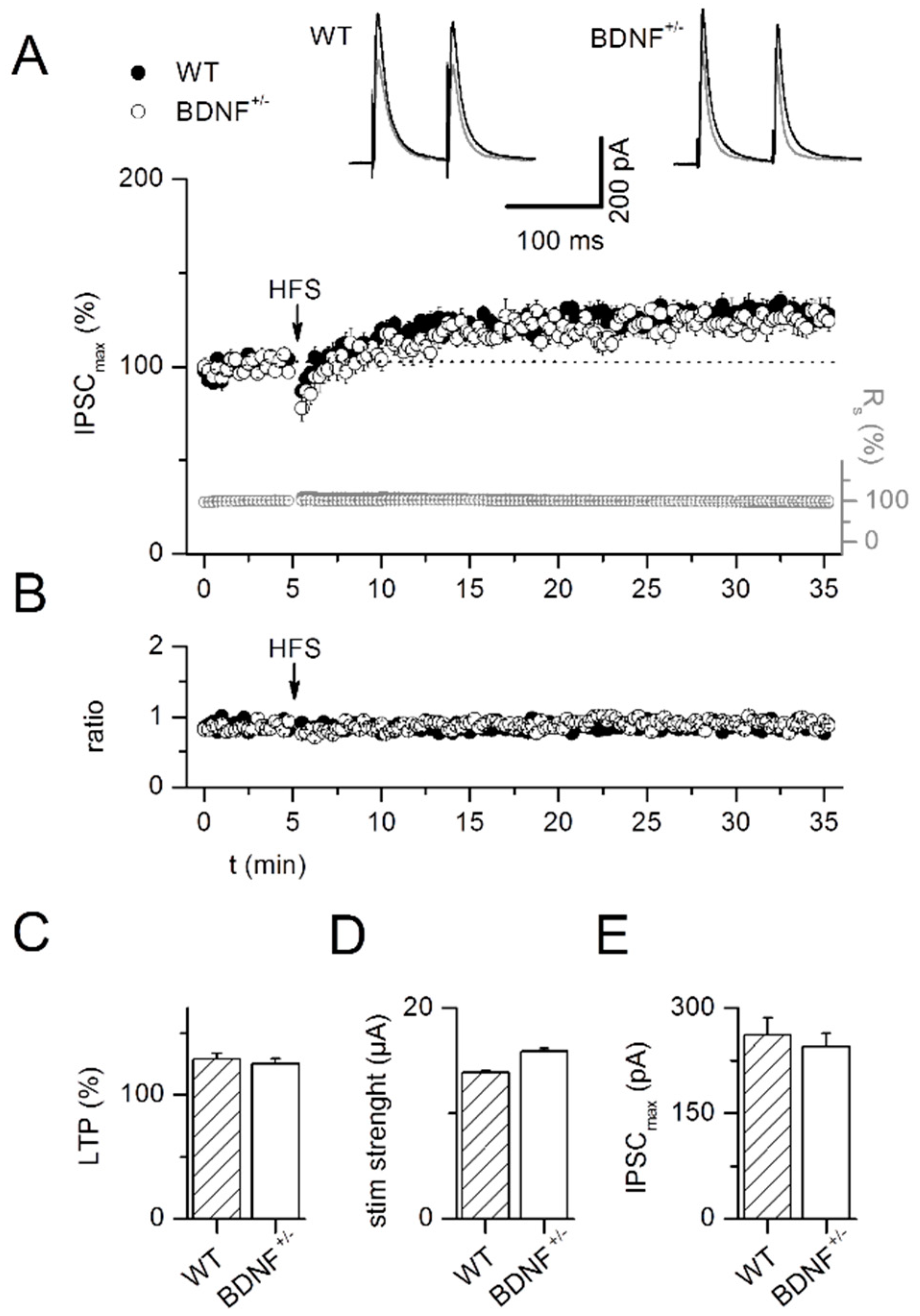
2.2. LTP at GABAergic Synapses (iLTP)
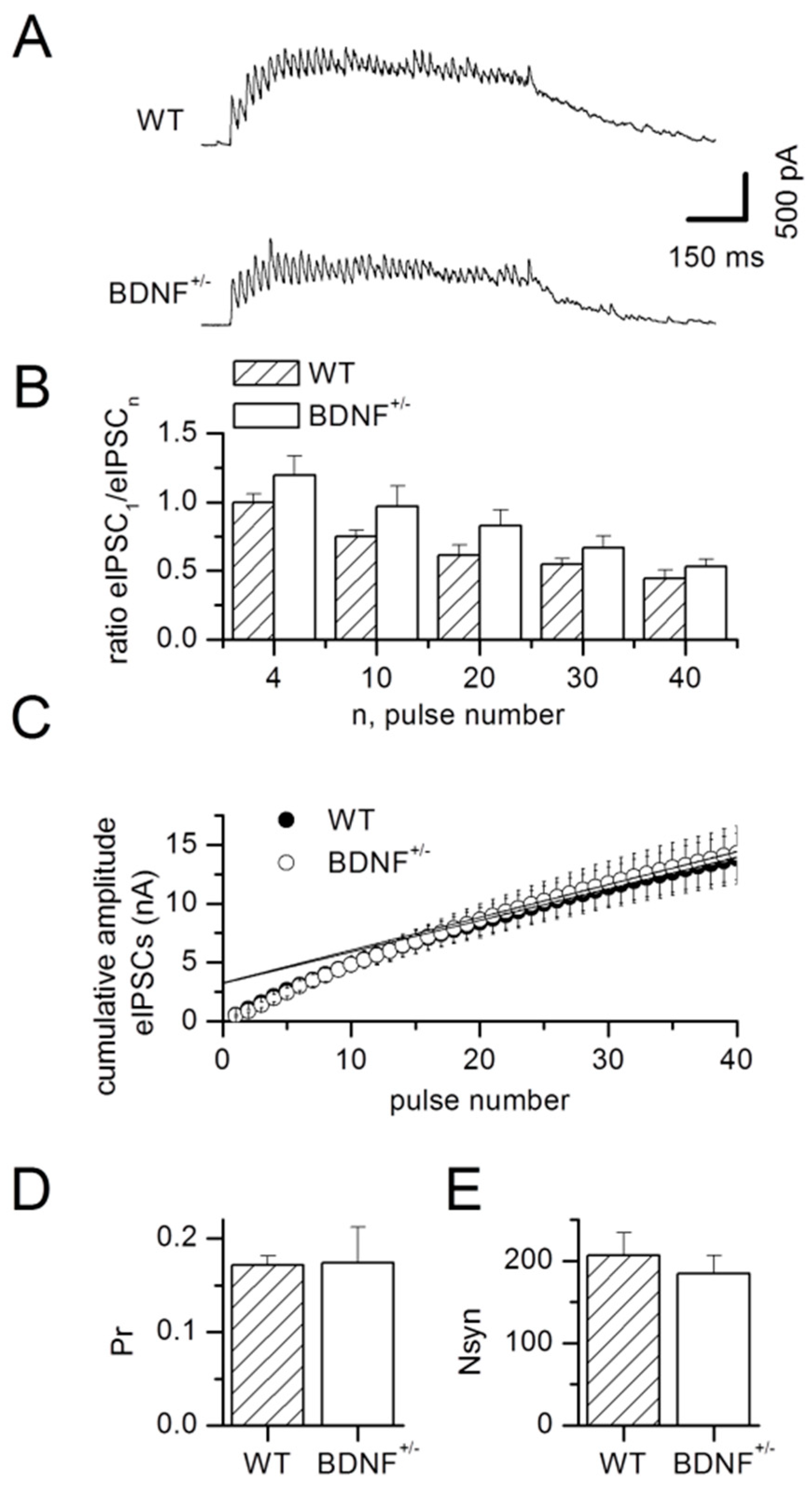
2.3. Glutamatergic Drive onto LA Interneurons
2.4. Modulation of sIPSCs by Norepinephrine (NE)
3. Discussion
3.1. Basal GABAergic Transmission in BDNF+/− Mice
3.2. LTP at GABAergic Synapses in the LA of BDNF+/− Mice
3.3. Norepinephrine
3.4. Functional Implications
4. Materials and Methods
4.1. Animals
4.2. Slice Preparation
4.3. Recording Techniques
4.4. LTP Recordings
4.5. Drugs
4.6. Data Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| BA | Basal amygdala |
| BDNF | Brain-derived neurotrophic factor |
| BDNF+/− | BDNF heterozygous knockout mice |
| LA | Lateral amygdala |
| iLTP | Inhibitory long-term potentiation |
| LTP | Long-term potentiation |
| NE | Norepinephrine |
| TrkB | Tropomyosin-related kinase B |
References
- Gottmann, K.; Mittmann, T.; Lessmann, V. BDNF signaling in the formation, maturation and plasticity of glutamatergic and GABAergic synapses. Exp. Brain Res. 2009, 199, 203–234. [Google Scholar] [CrossRef] [PubMed]
- Cowansage, K.K.; LeDoux, J.E.; Monfils, M.H. Brain-derived neurotrophic factor: A dynamic gatekeeper of neural plasticity. Curr. Mol. Pharmcol. 2010, 3, 12–29. [Google Scholar] [CrossRef]
- Yoshii, A.; Constantine-Paton, M. Postsynaptic BDNF-TrkB signaling in synapse maturation, plasticity, and disease. Dev. Neurobiol. 2010, 70, 304–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, H.; Poo, M.M. Neurotrophin regulation of neural circuit development and function. Nat. Rev. Neurosci. 2013, 14, 7–23. [Google Scholar] [CrossRef] [PubMed]
- Edelmann, E.; Lessmann, V.; Brigadski, T. Pre- and postsynaptic twists in BDNF secretion and action in synaptic plasticity. Neuropharmacology 2014, 76, 610–627. [Google Scholar] [CrossRef] [PubMed]
- Lu, B.; Nagappan, G.; Lu, Y. BDNF and synaptic plasticity, cognitive function, and dysfunction. Handb. Exp. Pharmcol. 2014, 220, 223–250. [Google Scholar]
- Kim, J.; Lee, S.; Kang, S.; Kim, S.H.; Kim, J.C.; Yang, M.; Moon, C. Brain-derived neurotropic factor and GABAergic transmission in neurodegeneration and neuroregeneration. Neural Regen. Res. 2017, 12, 1733–1741. [Google Scholar]
- Brunig, I.; Penschuck, S.; Berninger, B.; Benson, J.; Fritschy, J.M. BDNF reduces miniature inhibitory postsynaptic currents by rapid downregulation of GABA(A) receptor surface expression. Eur. J. Neurosci. 2001, 13, 1320–1328. [Google Scholar] [CrossRef]
- Cheng, Q.; Yeh, H.H. Brain-derived neurotrophic factor attenuates mouse cerebellar granule cell GABA(A) receptor-mediated responses via postsynaptic mechanisms. J. Physiol. 2003, 548, 711–721. [Google Scholar] [CrossRef]
- Hewitt, S.A.; Bains, J.S. Brain-derived neurotrophic factor silences GABA synapses onto hypothalamic neuroendocrine cells through a postsynaptic dynamin-mediated mechanism. J. Neurophysiol. 2006, 95, 2193–2198. [Google Scholar] [CrossRef]
- Lemtiri-Chlieh, F.; Levine, E.S. BDNF evokes release of endogenous cannabinoids at layer 2/3 inhibitory synapses in the neocortex. J. Neurophysiol. 2010, 104, 1923–1932. [Google Scholar] [CrossRef] [PubMed]
- Mizoguchi, Y.; Kanematsu, T.; Hirata, M.; Nabekura, J. A rapid increase in the total number of cell surface functional GABAA receptors induced by brain-derived neurotrophic factor in rat visual cortex. J. Biol. Chem. 2003, 278, 44097–44102. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Ko, H.; Cheung, Z.H.; Yung, K.K.; Yao, T.; Wang, J.J.; Morozov, A.; Ke, Y.; Ip, N.Y.; Yung, W.H. Dual actions of brain-derived neurotrophic factor on GABAergic transmission in cerebellar Purkinje neurons. Exp. Neurol. 2012, 233, 791–798. [Google Scholar] [CrossRef] [PubMed]
- Colino-Oliveira, M.; Rombo, D.M.; Dias, R.B.; Ribeiro, J.A.; Sebastiao, A.M. BDNF-induced presynaptic facilitation of GABAergic transmission in the hippocampus of young adults is dependent of TrkB and adenosine A2A receptors. Purinergic Signal 2016, 12, 283–294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jovanovic, J.N.; Thomas, P.; Kittler, J.T.; Smart, T.G.; Moss, S.J. Brain-derived neurotrophic factor modulates fast synaptic inhibition by regulating GABA(A) receptor phosphorylation, activity, and cell-surface stability. J. Neurosci. Off. J. Soc. Neurosci. 2004, 24, 522–530. [Google Scholar] [CrossRef] [PubMed]
- Olofsdotter, K.; Lindvall, O.; Asztely, F. Increased synaptic inhibition in dentate gyrus of mice with reduced levels of endogenous brain-derived neurotrophic factor. Neuroscience 2000, 101, 531–539. [Google Scholar] [CrossRef]
- Henneberger, C.; Juttner, R.; Rothe, T.; Grantyn, R. Postsynaptic action of BDNF on GABAergic synaptic transmission in the superficial layers of the mouse superior colliculus. J. Neurophysiol. 2002, 88, 595–603. [Google Scholar] [CrossRef]
- Abidin, I.; Eysel, U.T.; Lessmann, V.; Mittmann, T. Impaired GABAergic inhibition in the visual cortex of brain-derived neurotrophic factor heterozygous knockout mice. J. Physiol. 2008, 586, 1885–1901. [Google Scholar] [CrossRef]
- Laudes, T.; Meis, S.; Munsch, T.; Lessmann, V. Impaired transmission at corticothalamic excitatory inputs and intrathalamic GABAergic synapses in the ventrobasal thalamus of heterozygous BDNF knockout mice. Neuroscience 2012, 222, 215–227. [Google Scholar] [CrossRef]
- Ehrlich, I.; Humeau, Y.; Grenier, F.; Ciocchi, S.; Herry, C.; Luthi, A. Amygdala inhibitory circuits and the control of fear memory. Neuron 2009, 62, 757–771. [Google Scholar] [CrossRef]
- Pape, H.C.; Pare, D. Plastic synaptic networks of the amygdala for the acquisition, expression, and extinction of conditioned fear. Physiol. Rev. 2010, 90, 419–463. [Google Scholar] [CrossRef] [PubMed]
- Quirk, G.J.; Gehlert, D.R. Inhibition of the amygdala: Key to pathological states? Ann. N. Y. Acad. Sci. 2003, 985, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Muller, I.; Caliskan, G.; Stork, O. The GAD65 knock out mouse—A model for GABAergic processes in fear- and stress-induced psychopathology. Genes Brain Behav. 2015, 14, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Prager, E.M.; Bergstrom, H.C.; Wynn, G.H.; Braga, M.F. The basolateral amygdala gamma-aminobutyric acidergic system in health and disease. J. Neurosci. Res. 2016, 94, 548–567. [Google Scholar] [CrossRef]
- Aroniadou-Anderjaska, V.; Qashu, F.; Braga, M.F. Mechanisms regulating GABAergic inhibitory transmission in the basolateral amygdala: Implications for epilepsy and anxiety disorders. Amino Acids 2007, 32, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Skelly, M.J.; Ariwodola, O.J.; Weiner, J.L. Fear conditioning selectively disrupts noradrenergic facilitation of GABAergic inhibition in the basolateral amygdala. Neuropharmacology 2017, 113, 231–240. [Google Scholar] [CrossRef] [PubMed]
- Andero, R.; Choi, D.C.; Ressler, K.J. BDNF-TrkB receptor regulation of distributed adult neural plasticity, memory formation, and psychiatric disorders. Prog. Mol. Biol. Transl. Sci. 2014, 122, 169–192. [Google Scholar] [PubMed]
- Ehrlich, D.E.; Josselyn, S.A. Plasticity-related genes in brain development and amygdala-dependent learning. Genes Brain Behav. 2016, 15, 125–143. [Google Scholar] [CrossRef]
- Meis, S.; Endres, T.; Lessmann, V. Postsynaptic BDNF signalling regulates long-term potentiation at thalamo-amygdala afferents. J. Physiol. 2012, 590, 193–208. [Google Scholar] [CrossRef]
- Meis, S.; Endres, T.; Munsch, T.; Lessmann, V. The Relation Between Long-Term Synaptic Plasticity at Glutamatergic Synapses in the Amygdala and Fear Learning in Adult Heterozygous BDNF-Knockout Mice. Cereb. Cortex 2018, 28, 1195–1208. [Google Scholar] [CrossRef]
- Mou, L.; Dias, B.G.; Gosnell, H.; Ressler, K.J. Gephyrin plays a key role in BDNF-dependent regulation of amygdala surface GABAARs. Neuroscience 2013, 255, 33–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mou, L.; Heldt, S.A.; Ressler, K.J. Rapid brain-derived neurotrophic factor-dependent sequestration of amygdala and hippocampal GABA(A) receptors via different tyrosine receptor kinase B-mediated phosphorylation pathways. Neuroscience 2011, 176, 72–85. [Google Scholar] [CrossRef] [PubMed]
- Endres, T.; Lessmann, V. Age-dependent deficits in fear learning in heterozygous BDNF knock-out mice. Learn. Mem. 2012, 19, 561–570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bauer, E.P.; LeDoux, J.E. Heterosynaptic long-term potentiation of inhibitory interneurons in the lateral amygdala. J. Neurosci. Off. J. Soc. Neurosci. 2004, 24, 9507–9512. [Google Scholar] [CrossRef]
- Braga, M.F.; Aroniadou-Anderjaska, V.; Manion, S.T.; Hough, C.J.; Li, H. Stress impairs alpha(1A) adrenoceptor-mediated noradrenergic facilitation of GABAergic transmission in the basolateral amygdala. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 2004, 29, 45–58. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, K.; Tamamaki, N.; Owada, H.; Kakizaki, T.; Kume, N.; Totsuka, M.; Yamamoto, T.; Yawo, H.; Yagi, T.; Obata, K.; et al. Noradrenergic excitation of a subpopulation of GABAergic cells in the basolateral amygdala via both activation of nonselective cationic conductance and suppression of resting K+ conductance: A study using glutamate decarboxylase 67-green fluorescent protein knock-in mice. Neuroscience 2008, 157, 781–797. [Google Scholar] [PubMed]
- Miyajima, M.; Ozaki, M.; Wada, K.; Sekiguchi, M. Noradrenaline-induced spontaneous inhibitory postsynaptic currents in mouse basolateral nucleus of amygdala pyramidal neurons: Comparison with dopamine-induced currents. Neurosci. Lett. 2010, 480, 167–172. [Google Scholar] [CrossRef]
- Meis, S.; Endres, T.; Munsch, T.; Lessmann, V. Presynaptic Regulation of Tonic Inhibition by Neuromodulatory Transmitters in the Basal Amygdala. Mol. Neurobiol. 2018, 55, 8509–8521. [Google Scholar] [CrossRef]
- Holm, M.M.; Nieto-Gonzalez, J.L.; Vardya, I.; Vaegter, C.B.; Nykjaer, A.; Jensen, K. Mature BDNF, but not proBDNF, reduces excitability of fast-spiking interneurons in mouse dentate gyrus. J. Neurosci. Off. J. Soc. Neurosci. 2009, 29, 12412–12418. [Google Scholar] [CrossRef]
- Sakata, K.; Woo, N.H.; Martinowich, K.; Greene, J.S.; Schloesser, R.J.; Shen, L.; Lu, B. Critical role of promoter IV-driven BDNF transcription in GABAergic transmission and synaptic plasticity in the prefrontal cortex. Proc. Natl. Acad. Sci. USA 2009, 106, 5942–5947. [Google Scholar] [CrossRef] [Green Version]
- Szinyei, C.; Narayanan, R.T.; Pape, H.C. Plasticity of inhibitory synaptic network interactions in the lateral amygdala upon fear conditioning in mice. Eur. J. Neurosci. 2007, 25, 1205–1211. [Google Scholar] [CrossRef] [PubMed]
- Pan, B.X.; Dong, Y.; Ito, W.; Yanagawa, Y.; Shigemoto, R.; Morozov, A. Selective gating of glutamatergic inputs to excitatory neurons of amygdala by presynaptic GABAb receptor. Neuron 2009, 61, 917–929. [Google Scholar] [CrossRef] [PubMed]
- Spampanato, J.; Polepalli, J.; Sah, P. Interneurons in the basolateral amygdala. Neuropharmacology 2011, 60, 765–773. [Google Scholar] [CrossRef] [PubMed]
- Lange, M.D.; Doengi, M.; Lesting, J.; Pape, H.C.; Jungling, K. Heterosynaptic long-term potentiation at interneuron-principal neuron synapses in the amygdala requires nitric oxide signalling. J. Physiol. 2012, 590, 131–143. [Google Scholar] [CrossRef] [PubMed]
- Tully, K.; Li, Y.; Tsvetkov, E.; Bolshakov, V.Y. Norepinephrine enables the induction of associative long-term potentiation at thalamo-amygdala synapses. Proc. Natl. Acad. Sci. USA 2007, 104, 14146–14150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lazzaro, S.C.; Hou, M.; Cunha, C.; LeDoux, J.E.; Cain, C.K. Antagonism of lateral amygdala alpha1-adrenergic receptors facilitates fear conditioning and long-term potentiation. Learn. Mem. 2010, 17, 489–493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakata, K.; Duke, S.M. Lack of BDNF expression through promoter IV disturbs expression of monoamine genes in the frontal cortex and hippocampus. Neuroscience 2014, 260, 265–275. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y.; Zhang, Z.; Zhang, C.; Wang, X.; Sakata, K.; Lu, B.; Sun, Q.Q. A key mechanism underlying sensory experience-dependent maturation of neocortical GABAergic circuits in vivo. Proc. Natl. Acad. Sci. USA 2011, 108, 12131–12136. [Google Scholar] [CrossRef] [Green Version]
- Jones, N.C.; Hudson, M.; Foreman, J.; Rind, G.; Hill, R.; Manning, E.E.; van den Buuse, M. Brain-derived neurotrophic factor haploinsufficiency impairs high-frequency cortical oscillations in mice. Eur. J. Neurosci. 2018, 48, 2816–2825. [Google Scholar] [CrossRef]
- Du, X.; Serena, K.; Hwang, W.; Grech, A.M.; Wu, Y.W.C.; Schroeder, A.; Hill, R.A. Prefrontal cortical parvalbumin and somatostatin expression and cell density increase during adolescence and are modified by BDNF and sex. Mol. Cell. Neurosci. 2018, 88, 177–188. [Google Scholar] [CrossRef]
- Glorioso, C.; Sabatini, M.; Unger, T.; Hashimoto, T.; Monteggia, L.M.; Lewis, D.A.; Mirnics, K. Specificity and timing of neocortical transcriptome changes in response to BDNF gene ablation during embryogenesis or adulthood. Mol. Psychiatry 2006, 11, 633–648. [Google Scholar] [CrossRef] [PubMed]
- Guilloux, J.P.; Douillard-Guilloux, G.; Kota, R.; Wang, X.; Gardier, A.M.; Martinowich, K.; Tseng, G.C.; Lewis, D.A.; Sibille, E. Molecular evidence for BDNF- and GABA-related dysfunctions in the amygdala of female subjects with major depression. Mol. Psychiatry 2012, 17, 1130–1142. [Google Scholar] [CrossRef] [PubMed]
- Song, C.; Xu, X.B.; He, Y.; Liu, Z.P.; Wang, M.; Zhang, X.; Li, B.M.; Pan, B.X. Stuttering interneurons generate fast and robust inhibition onto projection neurons with low capacity of short term modulation in mouse lateral amygdala. PLoS ONE 2013, 8, e60154. [Google Scholar]
- Sosulina, L.; Graebenitz, S.; Pape, H.C. GABAergic interneurons in the mouse lateral amygdala: A classification study. J. Neurophysiol. 2010, 104, 617–626. [Google Scholar] [CrossRef] [PubMed]
- Asan, E. The Catecholaminergic Innervation of the Rat Amygdala; Advances in Anatomy, Embryology and Cell Biology Book 142; Springer: Berlin/Heidelberg, Germany, 1998; Volume 142, pp. 1–118. [Google Scholar]
- Farb, C.R.; Chang, W.; Ledoux, J.E. Ultrastructural characterization of noradrenergic axons and Beta-adrenergic receptors in the lateral nucleus of the amygdala. Front. Behav. Neurosci. 2010, 4, 162. [Google Scholar] [CrossRef] [PubMed]
- Galvez, R.; Mesches, M.H.; McGaugh, J.L. Norepinephrine release in the amygdala in response to footshock stimulation. Neurobiol. Learn. Mem. 1996, 66, 253–257. [Google Scholar] [CrossRef] [PubMed]
- Giustino, T.F.; Maren, S. Noradrenergic Modulation of Fear Conditioning and Extinction. Front. Behav. Neurosci. 2018, 12, 43. [Google Scholar] [CrossRef]
- Luscher, B.; Fuchs, T. GABAergic control of depression-related brain states. Adv. Pharmcol. 2015, 73, 97–144. [Google Scholar]
- Korte, M.; Carroll, P.; Wolf, E.; Brem, G.; Thoenen, H.; Bonhoeffer, T. Hippocampal long-term potentiation is impaired in mice lacking brain-derived neurotrophic factor. Proc. Natl. Acad. Sci. USA 1995, 92, 8856–8860. [Google Scholar] [CrossRef]
- Kulisch, C.; Eckers, N.; Albrecht, D. Method of euthanasia affects amygdala plasticity in horizontal brain slices from mice. J. Neurosci. Methods 2011, 201, 340–345. [Google Scholar] [CrossRef]
- Tsvetkov, E.; Carlezon, W.A.; Benes, F.M.; Kandel, E.R.; Bolshakov, V.Y. Fear conditioning occludes LTP-induced presynaptic enhancement of synaptic transmission in the cortical pathway to the lateral amygdala. Neuron 2002, 34, 289–300. [Google Scholar] [CrossRef]
- Kirischuk, S.; Juttner, R.; Grantyn, R. Time-matched pre- and postsynaptic changes of GABAergic synaptic transmission in the developing mouse superior colliculus. J. Physiol. 2005, 563, 795–807. [Google Scholar] [CrossRef] [PubMed]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meis, S.; Endres, T.; Munsch, T.; Lessmann, V. Impact of Chronic BDNF Depletion on GABAergic Synaptic Transmission in the Lateral Amygdala. Int. J. Mol. Sci. 2019, 20, 4310. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20174310
Meis S, Endres T, Munsch T, Lessmann V. Impact of Chronic BDNF Depletion on GABAergic Synaptic Transmission in the Lateral Amygdala. International Journal of Molecular Sciences. 2019; 20(17):4310. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20174310
Chicago/Turabian StyleMeis, Susanne, Thomas Endres, Thomas Munsch, and Volkmar Lessmann. 2019. "Impact of Chronic BDNF Depletion on GABAergic Synaptic Transmission in the Lateral Amygdala" International Journal of Molecular Sciences 20, no. 17: 4310. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20174310