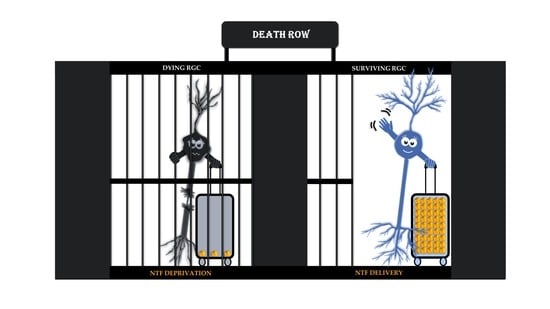
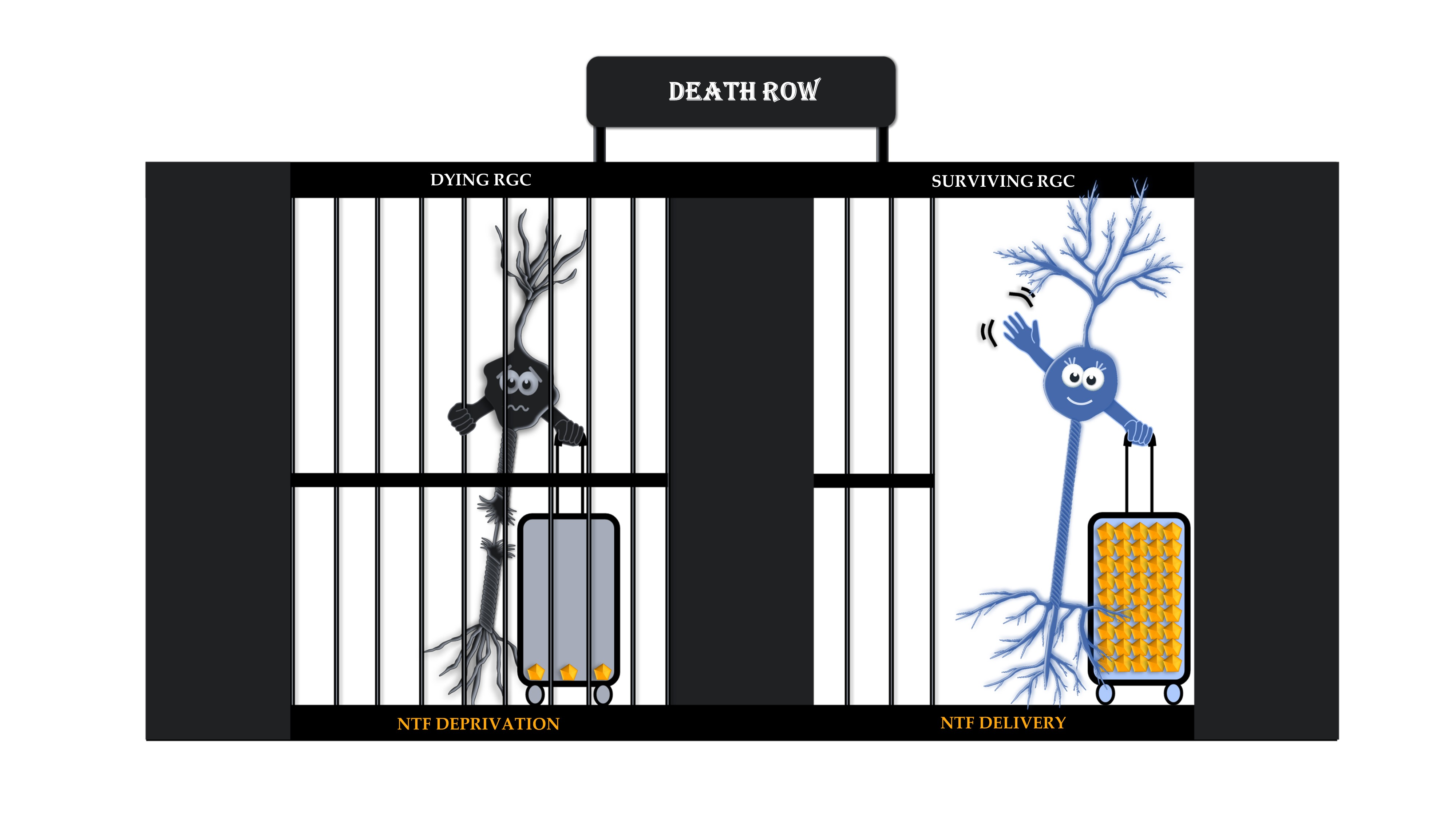
Target-Derived Neurotrophic Factor Deprivation Puts Retinal Ganglion Cells on Death Row: Cold Hard Evidence and Caveats



Abstract
:
{kind=link}
{kind=link}
{kind=link}
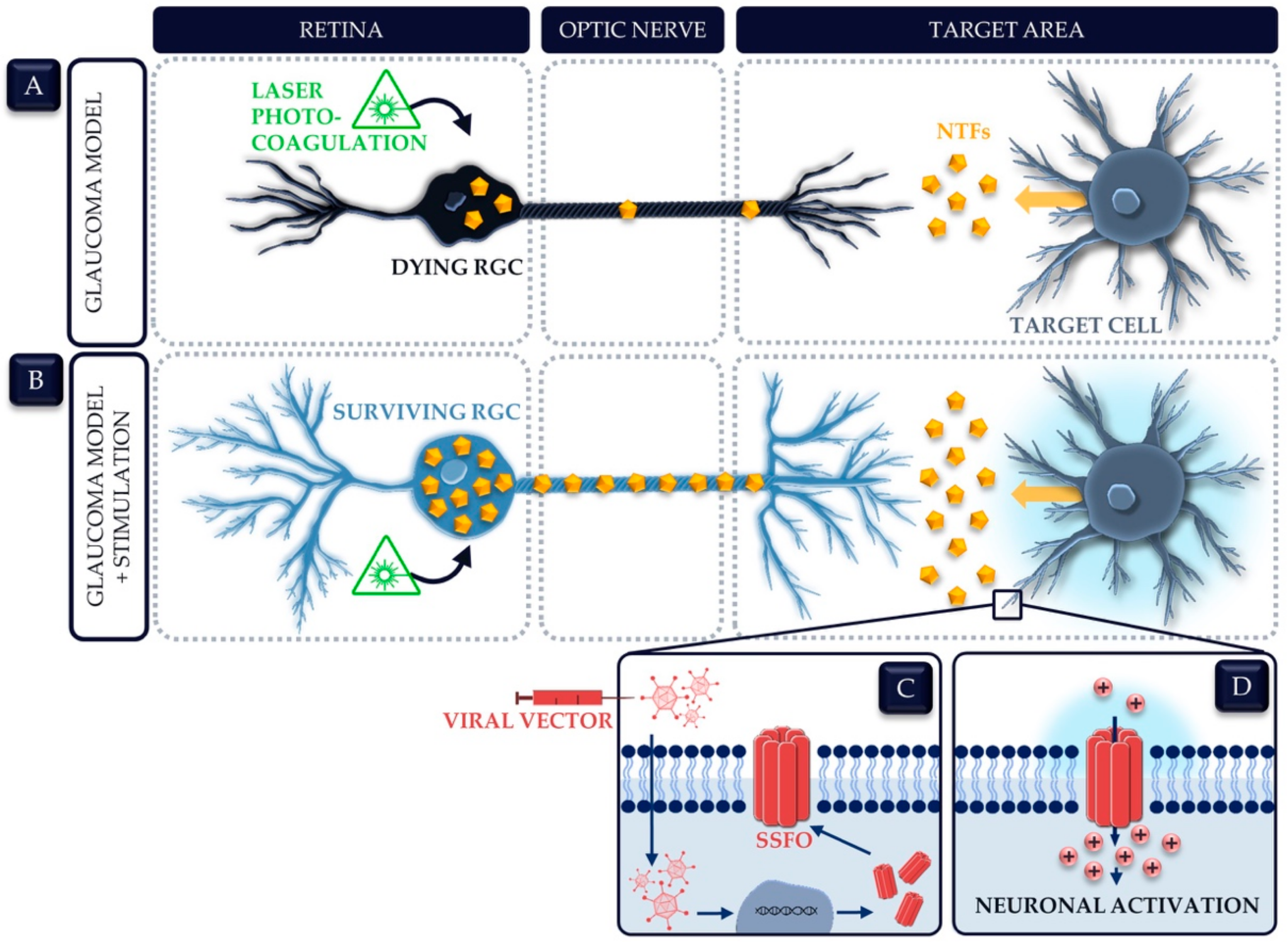{kind=link}
1. Introduction
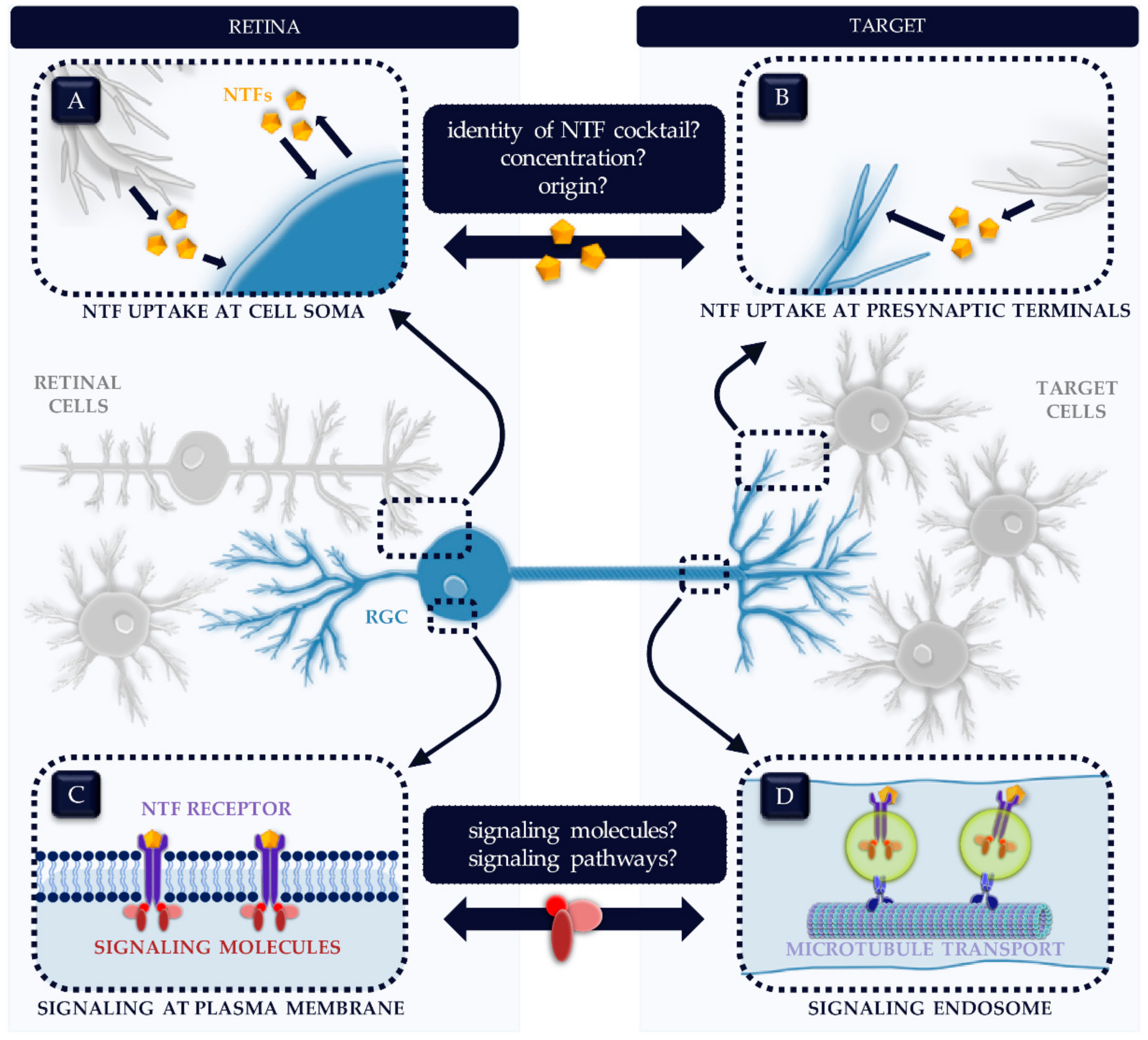
2. Which NTFs and Signaling Pathways Are Involved?
3. NTFs in the Developing and Adult Nervous System
4. Which Tangible Evidence Corroborates the NTF Deprivation Theory in Glaucoma?
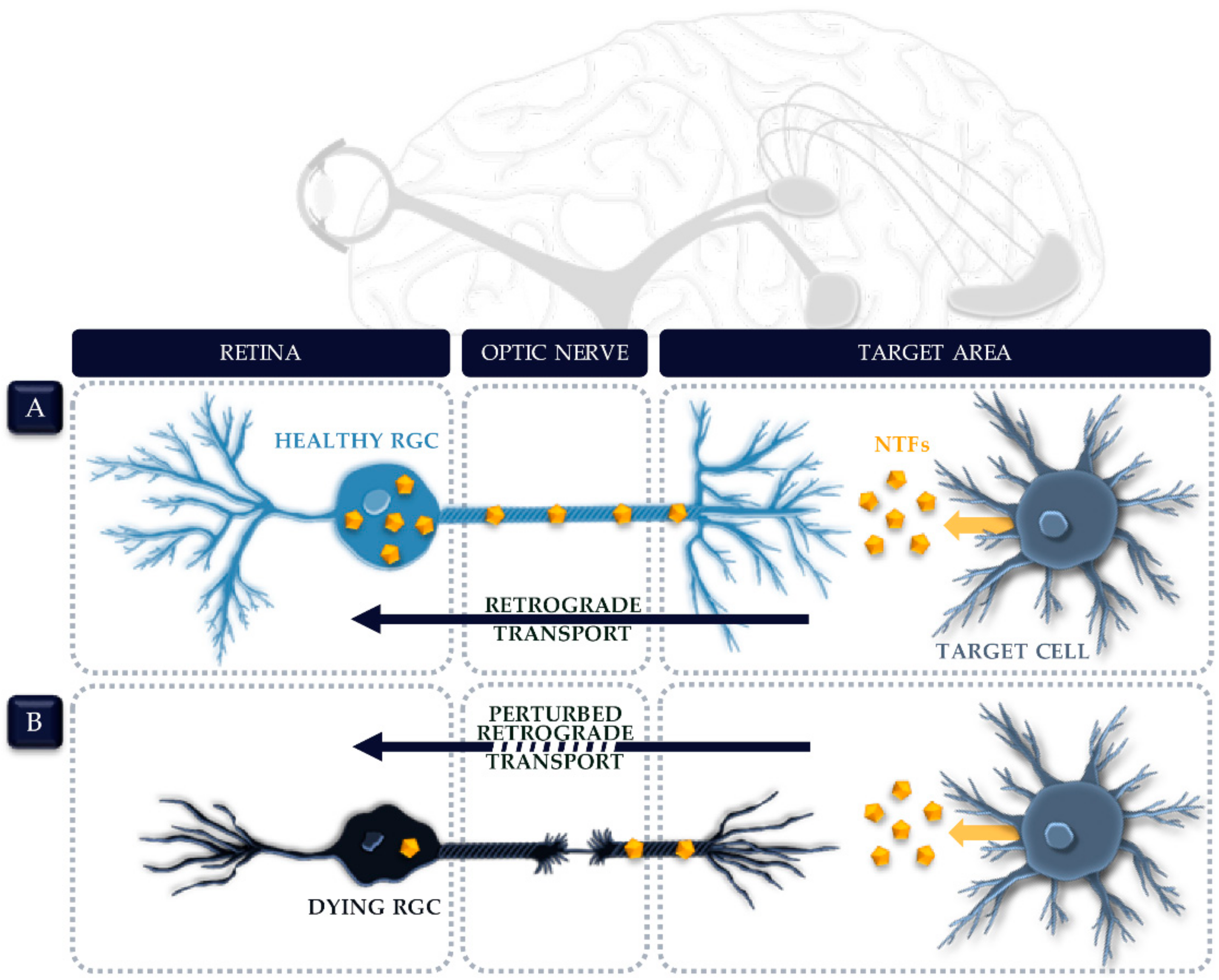
4.1. Axonal Transport Deficits in Glaucoma
4.2. NTF Transport along the Optic Nerve
4.3. NTF Deprivation in the Glaucomatous Retina
5. NTFs as Neuroprotective Therapy in Glaucoma: What Information Are We Missing?
5.1. First Shortcoming of NTF Therapy Efforts: The Transient Effect
5.2. Second Shortcoming of NTF Therapy Efforts: The Focus on Monotherapy
5.3. Third Shortcoming of NTF Therapy Efforts: Superfluous Supplementation
5.4. Fourth Shortcoming of NTF Therapy Efforts: A Possible Difference between Local and Target-Derived NTFs
5.5. Tackling the Shortcomings of NTF Therapy Efforts
6. Fishing for Interspecies Differences in the NTF Deprivation Theory
7. Concluding Remarks and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| BDNF | Brain-derived neurotrophic factor |
| bFGF | Basic fibroblast growth factor |
| CNTF | Ciliary nerve trophic factor |
| dLGN | Dorsal lateral geniculate nucleus |
| ES | Electrical stimulation |
| GDNF | Glial cell-derived neurotrophic factor |
| Hsp70 | Heat shock protein 70 |
| IGF-1 | Insulin-like growth factor |
| JAK/STAT | Janus kinase/signal transducers and activators of transcription |
| JNK | c-Jun N-terminal kinase |
| MAPK/ERK | Mitogen-activated protein kinase/extracellular-signal-regulated kinase |
| NF-κB | Nuclear factor kappa-light-chain-enhancer of activated B |
| NGF | Nerve growth factor |
| NT-3 | Neurotrophin-3 |
| NT-4/5 | Neurotrophin-4/5 |
| NT-6 | Neurotrophin-6 |
| NT-7 | Neurotrophin-7 |
| NTF | Neurotrophic factor |
| ONI | Optic nerve injury |
| p75NTR | p75 neurotrophin |
| PI3K/AKT | Phosphoinositol-3 kinase/protein kinase B |
| PLC-γ | Phospholipase C- γ |
| RGC | Retinal ganglion cell |
| RT-PCR | Reverse transcription polymerase chain reaction |
| SC | Superior colliculus |
| SSFO | Stabilized step function opsin |
| TGF-β | Transforming growth factor β |
| Trk | Tropomyosin related kinase |
References
- Foster, P.J. The definition and classification of glaucoma in prevalence surveys. Br. J. Ophthalmol. 2002, 86, 238–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chua, B.; Goldberg, I. Neuroprotective agents in glaucoma therapy: Recent developments and future directions. Expert Rev. Ophthalmol. 2010, 5, 627–636. [Google Scholar] [CrossRef]
- Almasieh, M.; Wilson, A.M.; Morquette, B.; Cueva Vargas, J.L.; Di Polo, A. The molecular basis of retinal ganglion cell death in glaucoma. Prog. Retin. Eye Res. 2012, 31, 152–181. [Google Scholar] [CrossRef] [PubMed]
- Foldvari, M.; Chen, D.W. The intricacies of neurotrophic factor therapy for retinal ganglion cell rescue in glaucoma: A case for gene therapy. Neural Regen. Res. 2016, 11, 875–877. [Google Scholar] [CrossRef]
- Nickells, R.W. From ocular hypertension to ganglion cell death: A theoretical sequence of events leading to glaucoma. Can. J. Ophthalmol. 2007, 42, 278–287. [Google Scholar] [CrossRef]
- Nickells, R.W. The Cell and Molecular Biology of Glaucoma: Mechanisms of Retinal Ganglion Cell Death. Investig. Opthalmol. Vis. Sci. 2012, 53, 2476–2481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vrabec, J.P.; Levin, L.A. The neurobiology of cell death in glaucoma. Eye 2007, 21, S11–S14. [Google Scholar] [CrossRef] [PubMed]
- Johnson, E.C.; Guo, Y.; Cepurna, W.O.; Morrison, J.C. Neurotrophin roles in retinal ganglion cell survival: Lessons from rat glaucoma models. Exp. Eye Res. 2009, 88, 808–815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knox, D.L. Optic Nerve Hydropic Axonal Degeneration and Blocked Retrograde Axoplasmic Transport. Arch. Ophthalmol. 2007, 125, 347–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burgoyne, C.F. A biomechanical paradigm for axonal insult within the optic nerve head in aging and glaucoma. Exp. Eye Res. 2011, 93, 120–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quigley, H.A.; Davis, E.B.; Anderson, D.R. Descending optic nerve degeneration in primates. Invest. Ophthalmol. Vis. Sci. 1977, 16, 841–849. [Google Scholar] [PubMed]
- Ceni, C.; Unsain, N.; Zeinieh, M.P.; Barker, P.A. Neurotrophins in the Regulation of Cellular Survival and Death. In Neurotrophic Factors; Lewin, G.R., Carter, B.D., Eds.; Springer: Berlin, Germany, 2014; Volume 220, pp. 193–221. [Google Scholar]
- Fahy, E.T.; Chrysostomou, V.; Crowston, J.G. Mini-Review: Impaired Axonal Transport and Glaucoma. Curr. Eye Res. 2016, 41, 273–283. [Google Scholar] [PubMed]
- Millecamps, S.; Julien, J.-P. Axonal transport deficits and neurodegenerative diseases. Nat. Rev. Neurosci. 2013, 14, 161–176. [Google Scholar] [CrossRef] [PubMed]
- Bartus, R.T.; Johnson, E.M. Clinical tests of neurotrophic factors for human neurodegenerative diseases, part 1: Where have we been and what have we learned? Neurobiol. Dis. 2017, 97, 156–168. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Hopkins, J.J.; Heier, J.S.; Birch, D.G.; Halperin, L.S.; Albini, T.A.; Brown, D.M.; Jaffe, G.J.; Taoj, W.; Williams, G.A. Ciliary neurotrophic factor delivered by encapsulated cell intraocular implants for treatment of geographic atrophy in age-related macular degeneration. Proc. Natl. Acad. Sci. USA 2011. [Google Scholar] [CrossRef] [PubMed]
- Lanni, C.; Stanga, S.; Racchi, M.; Govoni, S. The Expanding Universe of Neurotrophic Factors: Therapeutic Potential in Aging and Age-Associated Disorders. Curr. Pharm. Des. 2010, 16, 698–717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levy, Y.S.; Gilgun-Sherki, Y.; Melamed, E.; Offen, D. Therapeutic Potential of Neurotrophic Factors in Neurodegenerative Diseases. BioDrugs 2005, 19, 97–127. [Google Scholar] [CrossRef]
- Caminos, E.; Becker, E.; Martín-Zanca, D.; Vecino, E. Neurotrophins and their receptors in the tench retina during optic nerve regeneration. J. Comp. Neurol. 1999, 404, 321–331. [Google Scholar] [CrossRef]
- McMahon, S.; Murinson, B. Therapeutic Potential of Neurotrophic Factors. BioDrugs 2005, 19, 97–127. [Google Scholar]
- Nilsson, A.S.; Fainzilber, M.; Falck, P.; Ibáñez, C.F. Neurotrophin-7: A novel member of the neurotrophin family from the zebrafish. FEBS Lett. 1998, 424, 285–290. [Google Scholar] [CrossRef]
- Park, B.-C.; Tibudan, M.; Samaraweera, M.; Shen, X.; Yue, B.Y.J.T. Interaction between two glaucoma genes, optineurin and myocilin. Genes to Cells 2007, 12, 969–979. [Google Scholar] [CrossRef] [PubMed]
- Sippl, C.; Bosserhoff, A.K.; Fischer, D.; Tamm, E.R. Depletion of optineurin in RGC-5 cells derived from retinal neurons causes apoptosis and reduces the secretion of neurotrophins. Exp. Eye Res. 2011, 93, 669–680. [Google Scholar] [CrossRef] [PubMed]
- Nowak, A.; Majsterek, I.; Przybyłowska-Sygut, K.; Pytel, D.; Szymanek, K.; Szaflik, J.; Szaflik, J.P. Analysis of the Expression and Polymorphism of APOE, HSP, BDNF, and GRIN2B Genes Associated with the Neurodegeneration Process in the Pathogenesis of Primary Open Angle Glaucoma. Biomed. Res. Int. 2015, 2015, 1–14. [Google Scholar]
- Pasutto, F.; Matsumoto, T.; Mardin, C.Y.; Sticht, H.; Brandstätter, J.H.; Michels-Rautenstrauss, K.; Weisschuh, N.; Gramer, E.; Ramdas, W.D.; van Koolwijk, L.M.E.; et al. Heterozygous NTF4 Mutations Impairing Neurotrophin-4 Signaling in Patients with Primary Open-Angle Glaucoma. Am. J. Hum. Genet. 2009, 85, 447–456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nowak, A.; Szaflik, J.P.; Gacek, M.; Przybylowska-Sygut, K.; Kamińska, A.; Szaflik, J.; Majsterek, I. BDNF and HSP gene polymorphisms and their influence on the progression of primary open-angle glaucoma in a Polish population. Arch. Med. Sci. 2014, 6, 1206–1213. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, W.; Crooks, K.; Schmidt, S.; Allingham, R.R.; Hauser, M.A. No Evidence of Association of Heterozygous NTF4 Mutations in Patients with Primary Open-Angle Glaucoma. Am. J. Hum. Genet. 2010, 86, 498–499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, K.N.; Kaur, I.; Parikh, R.S.; Mandal, A.K.; Chandrasekhar, G.; Thomas, R.; Chakrabarti, S. Variations in NTF4, VAV2, and VAV3 Genes Are Not Involved with Primary Open-Angle and Primary Angle-Closure Glaucomas in an Indian Population. Investig. Opthalmol. Vis. Sci. 2010, 51, 4937. [Google Scholar]
- Chowdary, P.D.; Che, D.L.; Cui, B. Neurotrophin Signaling via Long-Distance Axonal Transport. Annu. Rev. Phys. Chem. 2012, 63, 571–594. [Google Scholar] [CrossRef]
- Chao, M.V. Neurotrophins and their receptors: A convergence point for many signalling pathways. Nat. Rev. Neurosci. 2003, 4, 299–309. [Google Scholar]
- Levkovitch-Verbin, H. Retinal Ganglion Cell Apoptotic Pathway in Glaucoma. In New Trends in Basic and Clinical Research of Glaucoma: A Neurodegenerative Disease of the Visual System, Part A; Bagetta, G., Nucci, C., Eds.; Elsevier: Amsterdam, The Netherlands, 2015; Volume 220, pp. 37–57. [Google Scholar]
- Huang, E.J.; Reichardt, L.F. Neurotrophins: Roles in Neuronal Development and Function. Annu. Rev. Neurosci. 2001, 24, 677–736. [Google Scholar] [Green Version]
- Nafissi, N.; Foldvari, M. Neuroprotective therapies in glaucoma: I. Neurotrophic factor delivery. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2016, 8, 240–254. [Google Scholar] [CrossRef] [PubMed]
- Skaper, S.D. The neurotrophin family of neurotrophic factors: An overview. Methods Mol. Biol. 2012, 846, 1–12. [Google Scholar] [PubMed]
- Lu, B.; Pang, P.T.; Woo, N.H. The yin and yang of neurotrophin action. Nat. Rev. Neurosci. 2005, 6, 603–614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drinkut, A.; Tillack, K.; Meka, D.P.; Schulz, J.B.; Kügler, S.; Kramer, E.R. Ret is essential to mediate GDNF’s neuroprotective and neuroregenerative effect in a Parkinson disease mouse model. Cell Death Dis. 2016, 7, e2359. [Google Scholar] [CrossRef] [PubMed]
- Huang, E.J.; Reichardt, L.F. Trk Receptors: Roles in Neuronal Signal Transduction. Annu. Rev. Biochem. 2003, 72, 609–642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, K.; Enomoto, H. Retrograde transport of neurotrophic factor signaling: Implications in neuronal development and pathogenesis. J. Biochem. 2016, 160, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Oppenheim, R. Cell Death During Development Of The Nervous System. Annu. Rev. Neurosci. 1991, 14, 453–501. [Google Scholar] [CrossRef] [PubMed]
- Conradt, B. Genetic Control of Programmed Cell Death During Animal Development. Annu. Rev. Genet. 2009, 43, 493–523. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.K.; Chew, K.S.; McNeill, D.S.; Keeley, P.W.; Ecker, J.L.; Mao, B.Q.; Pahlberg, J.; Kim, B.; Lee, S.C.S.; Fox, M.A.; et al. Apoptosis Regulates ipRGC Spacing Necessary for Rods and Cones to Drive Circadian Photoentrainment. Neuron 2013, 77, 503–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oppenheim, R.W. The neurotrophic theory and naturally occurring motoneuron death. Trends Neurosci. 1989, 12, 252–255. [Google Scholar] [CrossRef]
- Zweifel, L.S.; Kuruvilla, R.; Ginty, D.D. Functions and mechanisms of retrograde neurotrophin signalling. Nat. Rev. Neurosci. 2005, 6, 615–625. [Google Scholar] [CrossRef] [PubMed]
- Quigley, H.A.; Nickells, R.W.; Kerrigan, L.A.; Pease, M.E.; Thibault, D.J.; Zack, D.J. Retinal ganglion cell death in experimental glaucoma and after axotomy occurs by apoptosis. Invest. Ophthalmol. Vis. Sci. 1995, 36, 774–786. [Google Scholar] [PubMed]
- Yamaguchi, Y.; Miura, M. Programmed cell death in neurodevelopment. Dev. Cell 2015, 32, 478–490. [Google Scholar] [CrossRef] [PubMed]
- Spalding, K.L.; Cui, Q.; Harvey, A.R. The effects of central administration of neurotrophins or transplants of fetal tectal tissue on retinal ganglion cell survival following removal of the superior colliculus in neonatal rats. Dev. Brain Res. 1998, 107, 133–142. [Google Scholar] [CrossRef]
- Bear, M.F.; Connors, B.W.; Paradiso, M.A. The Central Visual System. In Neuroscience: Exploring the Brain, 3rd ed.; Lupash, E., Connolly, E., Dilernia, B., Williams, P.C., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2007; pp. 313–315. [Google Scholar]
- Martersteck, E.M.; Hirokawa, K.E.; Evarts, M.; Bernard, A.; Duan, X.; Li, Y.; Ng, L.; Oh, S.W.; Ouellette, B.; Royall, J.J.; et al. Diverse Central Projection Patterns of Retinal Ganglion Cells. Cell Rep. 2017, 18, 2058–2072. [Google Scholar] [CrossRef] [PubMed]
- Whitmore, A.V.; Libby, R.T.; John, S.W.M. Glaucoma: Thinking in new ways—a rôle for autonomous axonal self-destruction and other compartmentalised processes? Prog. Retin. Eye Res. 2005, 24, 639–662. [Google Scholar] [CrossRef] [PubMed]
- Harvey, A.R.; Robertson, D. Time-course and extent of retinal ganglion cell death following ablation of the superior colliculus in neonatal rats. J. Comp. Neurol. 1992, 325, 83–94. [Google Scholar] [CrossRef]
- Carpenter, P.; Sefton, A.J.; Dreher, B.; Lim, W.-L. Role of target tissue in regulating the development of retinal ganglion cells in the albino rat: Effects of kainate lesions in the superior colliculus. J. Comp. Neurol. 1986, 251, 240–259. [Google Scholar] [CrossRef]
- Perry, V.H.; Cowey, A. A sensitive period for ganglion cell degeneration and the formation of aberrant retino-fugal connections following tectal lesions in rats. Neuroscience 1982, 7, 583–594. [Google Scholar] [CrossRef]
- Yang, X.; Chou, T.H.; Ruggeri, M.; Porciatti, V. A new mouse model of inducible, chronic retinal ganglion cell dysfunction not associated with cell death. Investig. Ophthalmol. Vis. Sci. 2013, 54, 1898–1904. [Google Scholar] [CrossRef]
- Pearson, H.E.; Thompson, T.P. Atrophy and Degeneration of Ganglion Cells in Central Retina Following Loss of Postsynaptic Target Neurons in the Dorsal Lateral Geniculate Nucleus of the Adult Cat. Exp. Neurol. 1993, 119, 113–119. [Google Scholar] [CrossRef]
- Ellis, E.M.; Gauvain, G.; Sivyer, B.; Murphy, G.J. Shared and distinct retinal input to the mouse superior colliculus and dorsal lateral geniculate nucleus. J. Neurophysiol. 2016, 116, 602–610. [Google Scholar] [CrossRef] [PubMed]
- De Groef, L.; Dekeyster, E.; Geeraerts, E.; Lefevere, E.; Stalmans, I.; Salinas-Navarro, M.; Moons, L. Differential visual system organization and susceptibility to experimental models of optic neuropathies in three commonly used mouse strains. Exp. Eye Res. 2016, 145, 235–247. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.H.; Park, K.K.; Luo, X.; Porciatti, V. Retrograde signaling in the optic nerve is necessary for electrical responsiveness of retinal ganglion cells. Investig. Ophthalmol. Vis. Sci. 2013, 54, 1236–1243. [Google Scholar] [CrossRef] [PubMed]
- Salinas-Navarro, M.; Alarcón-Martínez, L.; Valiente-Soriano, F.J.; Jiménez-López, M.; Mayor-Torroglosa, S.; Avilés-Trigueros, M.; Villegas-Pérez, M.P.; Vidal-Sanz, M. Ocular hypertension impairs optic nerve axonal transport leading to progressive retinal ganglion cell degeneration. Exp. Eye Res. 2010, 90, 168–183. [Google Scholar] [CrossRef] [PubMed]
- Johansson, J.-O. Inhibition and recovery of retrograde axoplasmic transport in rat optic nerve during and after elevated IOP in vivo. Exp. Eye Res. 1988, 46, 223–227. [Google Scholar] [CrossRef]
- Jakobs, T.C.; Libby, R.T.; Ben, Y.; John, S.W.M.; Masland, R.H. Retinal ganglion cell degeneration is topological but not cell type specific in DBA/2J mice. J. Cell Biol. 2005, 171, 313–325. [Google Scholar] [CrossRef]
- Minckler, D.S.; Bunt, A.H.; Johanson, G.W. Orthograde and retrograde axoplasmic transport during acute ocular hypertension in the monkey. Invest. Ophthalmol. Vis. Sci. 1977, 16, 426–441. [Google Scholar]
- Valiente-Soriano, F.J.; Salinas-Navarro, M.; Jiménez-López, M.; Alarcón-Martínez, L.; Ortín-Martínez, A.; Bernal-Garro, J.M.; Avilés-Trigueros, M.; Agudo-Barriuso, M.; Villegas-Pérez, M.P.; Vidal-Sanz, M. Effects of Ocular Hypertension in the Visual System of Pigmented Mice. PLoS ONE 2015, 10, e0121134. [Google Scholar] [CrossRef]
- Kim, D.H.; Kim, H.S.; Ahn, M.D.; Chun, M.H. Ganglion Cell Death in Rat Retina by Persistent Intraocular Pressure Elevation. Korean J. Ophthalmol. 2004, 18, 15–22. [Google Scholar] [CrossRef] [Green Version]
- Buckingham, B.P.; Inman, D.M.; Lambert, W.; Oglesby, E.; Calkins, D.J.; Steele, M.R.; Vetter, M.L.; Marsh-Armstrong, N.; Horner, P.J. Progressive Ganglion Cell Degeneration Precedes Neuronal Loss in a Mouse Model of Glaucoma. J. Neurosci. 2008, 28, 2735–2744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, K.R.G.; Quigley, H.A.; Valenta, D.; Kielczewski, J.; Pease, M.E. Optic nerve dynein motor protein distribution changes with intraocular pressure elevation in a rat model of glaucoma. Exp. Eye Res. 2006, 83, 255–262. [Google Scholar] [CrossRef] [PubMed]
- Radius, R.L.; Anderson, D.R. Rapid axonal transport in primate optic nerve: Distribution of Pressure-Induced Interruption. Arch. Ophthalmol. 1981, 99, 650–654. [Google Scholar] [PubMed]
- Howell, G.R.; Libby, R.T.; Jakobs, T.C.; Smith, R.S.; Phalan, F.C.; Barter, J.W.; Barbay, J.M.; Marchant, J.K.; Mahesh, N.; Porciatti, V.; et al. Axons of retinal ganglion cells are insulted in the optic nerve early in DBA/2J glaucoma. J. Cell Biol. 2007, 179, 1523–1537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dengler-Crish, C.M.; Smith, M.A.; Inman, D.M.; Wilson, G.N.; Young, J.W.; Crish, S.D. Anterograde transport blockade precedes deficits in retrograde transport in the visual projection of the DBA/2J mouse model of glaucoma. Front. Neurosci. 2014, 8, 290. [Google Scholar] [PubMed] [Green Version]
- Quigley, H.A.; Addicks, E.M.; Green, W.R.; Maumenee, A.E. Optic nerve damage in human glaucoma: Ii. The Site of Injury and Susceptibility to Damage. Arch. Ophthalmol. 1981, 99, 635–649. [Google Scholar] [CrossRef] [PubMed]
- Anderson, D.R.; Hendrickson, A. Effect of intraocular pressure on rapid axoplasmic transport in monkey optic nerve. Invest. Ophthalmol. 1974, 13, 771–783. [Google Scholar] [PubMed]
- Crish, S.D.; Sappington, R.M.; Inman, D.M.; Horner, P.J.; Calkins, D.J. Distal axonopathy with structural persistence in glaucomatous neurodegeneration. Proc. Natl. Acad. Sci. USA 2010, 107, 5196–5201. [Google Scholar] [CrossRef] [Green Version]
- Crish, S.D.; Dapper, J.D.; MacNamee, S.E.; Balaram, P.; Sidorova, T.N.; Lambert, W.S.; Calkins, D.J. Failure of axonal transport induces a spatially coincident increase in astrocyte BDNF prior to synapse loss in a central target. Neuroscience 2013, 229, 55–70. [Google Scholar] [CrossRef]
- Lambert, W.S.; Ruiz, L.; Crish, S.D.; Wheeler, L.A.; Calkins, D.J. Brimonidine prevents axonal and somatic degeneration of retinal ganglion cell neurons. Mol. Neurodegener. 2011, 6, 4. [Google Scholar] [CrossRef]
- Guo, Y.; Johnson, E.; Cepurna, W.; Jia, L.; Dyck, J.; Morrison, J.C. Does elevated intraocular pressure reduce retinal TRKB-mediated survival signaling in experimental glaucoma? Exp. Eye Res. 2009, 89, 921–933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scott-Solomon, E.; Kuruvilla, R. Mechanisms of neurotrophin trafficking via Trk receptors. Mol. Cell. Neurosci. 2018, 91, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Schlamp, C.L.; Li, Y.; Dietz, J.A.; Janssen, K.T.; Nickells, R.W. Progressive ganglion cell loss and optic nerve degeneration in DBA/2J mice is variable and asymmetric. BMC Neurosci. 2006, 7, 66. [Google Scholar] [CrossRef]
- Agarwal, N.; Agarwal, R.; Kumar, D.M.; Ondricek, A.; Clark, A.F.; Wordinger, R.J.; Pang, I.-H. Comparison of expression profile of neurotrophins and their receptors in primary and transformed rat retinal ganglion cells. Mol. Vis. 2007, 13, 1311–1318. [Google Scholar]
- Wu, Q.; Zhang, M.; Song, B.W.; Lu, B.; Hu, P. Expression of ciliary neurotrophic factor after induction of ocular hypertension in the retina of rats. Chin. Med. J. (Engl.) 2007, 120, 1825–1829. [Google Scholar] [CrossRef]
- Kimura, A.; Namekata, K.; Guo, X.; Harada, C.; Harada, T. Neuroprotection, growth factors and BDNF-TRKB signalling in retinal degeneration. Int. J. Mol. Sci. 2016, 17, 1584. [Google Scholar]
- Beltran, W.A.; Zhang, Q.; Kijas, J.W.; Gu, D.; Rohrer, H.; Jordan, J.A.; Aguirre, G.D. Cloning, mapping, and retinal expression of the canine ciliary neurotrophic factor receptor α (CNTFRα). Investig. Ophthalmol. Vis. Sci. 2003, 44, 3642–3649. [Google Scholar] [CrossRef]
- Koeberle, P.D.; Ball, A.K. Neurturin enhances the survival of axotomized retinal ganglion cells in vivo: Combined effects with glial cell line-derived neurotrophic factor and brain-derived neurotrophic factor. Neuroscience 2002, 110, 555–567. [Google Scholar] [PubMed]
- Miotke, J.A.; MacLennan, A.J.; Meyer, R.L. Immunohistochemical localization of CNTFRα in adult mouse retina and optic nerve following intraorbital nerve crush: Evidence for the axonal loss of a trophic factor receptor after injury. J. Comp. Neurol. 2007, 500, 384–400. [Google Scholar] [CrossRef]
- Parmhans, N.; Sajgo, S.; Niu, J.; Luo, W.; Badea, T.C. Characterization of retinal ganglion cell, horizontal cell, and amacrine cell types expressing the neurotrophic receptor tyrosine kinase Ret. J. Comp. Neurol. 2018, 526, 742–766. [Google Scholar] [CrossRef]
- Hofer, M.; Pagliusi, S.R.; Hohn, A.; Leibrock, J.; Barde, Y.A. Regional distribution of brain-derived neurotrophic factor mRNA in the adult mouse brain. EMBO J. 1990, 9, 2459–2464. [Google Scholar] [CrossRef] [PubMed]
- Wetmore, C.; Ernfors, P.; Persson, H.; Olson, L. Localization of brain-derived neurotrophic factor mRNA to neurons in the brain by in situ hybridization. Exp. Neurol. 1990, 109, 141–152. [Google Scholar] [CrossRef]
- Dekeyster, E.; Geeraerts, E.; Buyens, T.; Van Den Haute, C.; Baekelandt, V.; De Groef, L.; Salinas-Navarro, M.; Moons, L. Tackling glaucoma from within the brain: An unfortunate interplay of BDNF and TrkB. PLoS ONE 2015, 10, e0142067. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, H.; Ito, Y.; Nakamura, S.; Shimazawa, M.; Hara, H. Involvement of brain-derived neurotrophic factor in time-dependent neurodegeneration in the murine superior colliculus after intravitreal injection of N-methyl-D-aspartate. Mol. Vis. 2009, 15, 662–669. [Google Scholar] [PubMed]
- Ito, Y.; Shimazawa, M.; Inokuchi, Y.; Fukumitsu, H.; Furukawa, S.; Araie, M.; Hara, H. Degenerative alterations in the visual pathway after NMDA-induced retinal damage in mice. Brain Res. 2008, 1212, 89–101. [Google Scholar] [PubMed]
- Pöyhönen, S.; Er, S.; Domanskyi, A.; Airavaara, M. Effects of Neurotrophic Factors in Glial Cells in the Central Nervous System: Expression and Properties in Neurodegeneration and Injury. Front. Physiol. 2019, 10, 486. [Google Scholar] [PubMed]
- Pease, M.E.; McKinnon, S.J.; Quigley, H.A.; Kerrigan-Baumrind, L.A.; Zack, D.J. Obstructed axonal transport of BDNF and its receptor TrkB in experimental glaucoma. Invest. Ophthalmol. Vis. Sci. 2000, 41, 764–774. [Google Scholar] [PubMed]
- Quigley, H.A.; McKinnon, S.J.; Zack, D.J.; Pease, M.E.; Kerrigan-Baumrind, L.A.; Kerrigan, D.F.; Mitchell, R.S. Retrograde axonal transport of BDNF in retinal ganglion cells is blocked by acute IOP elevation in rats. Invest. Ophthalmol. Vis. Sci. 2000, 41, 3460–3466. [Google Scholar]
- Yan, Q.; Wang, J.; Matheson, C.R.; Urich, J.L. Glial cell line-derived neurotrophic factor (GDNF) promotes the survival of axotomized retinal ganglion cells in adult rats: Comparison to and combination with brain-derived neurotrophic factor (BDNF). J. Neurobiol. 1999, 38, 382–390. [Google Scholar]
- Takihara, Y.; Inatani, M.; Hayashi, H.; Adachi, N.; Iwao, K.; Inoue, T.; Iwao, M.; Tanihara, H. Dynamic imaging of axonal transport in living retinal ganglion cells in vitro. Investig. Ophthalmol. Vis. Sci. 2011, 52, 3039–3045. [Google Scholar]
- Ghaffariyeh, A.; Honarpisheh, N.; Shakiba, Y.; Puyan, S.; Chamacham, T.; Zahedi, F.; Zarrineghbal, M. Brain-derived neurotrophic factor in patients with normal-tension glaucoma. Optom. J. Am. Optom. Assoc. 2009, 80, 635–638. [Google Scholar]
- Ghaffariyeh, A.; Honarpisheh, N.; Heidari, M.H.; Puyan, S.; Abasov, F. Brain-Derived Neurotrophic Factor as a Biomarker in Primary Open-Angle Glaucoma. Optom. Vis. Sci. 2011, 88, 80–85. [Google Scholar] [CrossRef] [PubMed]
- Shpak, A.A.; Guekht, A.B.; Druzhkova, T.A.; Kozlova, K.I.; Gulyaeva, N.V. Brain-Derived Neurotrophic Factor in Patients with Primary Open-Angle Glaucoma and Age-related Cataract. Curr. Eye Res. 2018, 43, 224–231. [Google Scholar] [CrossRef] [PubMed]
- Pietrucha-Dutczak, M.; Amadio, M.; Govoni, S.; Lewin-Kowalik, J.; Smedowski, A. The role of endogenous neuroprotective mechanisms in the prevention of retinal ganglion cells degeneration. Front. Neurosci. 2018, 12, 834. [Google Scholar] [PubMed]
- Perez, M.-T.R.; Caminos, E. Expression of brain-derived neurotrophic factor and of its functional receptor in neonatal and adult rat retina. Neurosci. Lett. 1995, 183, 96–99. [Google Scholar] [CrossRef]
- Lambert, W.; Agarwal, R.; Howe, W.; Clark, A.F.; Wordinger, R.J. Neurotrophin and neurotrophin receptor expression by cells of the human lamina cribrosa. Invest. Ophthalmol. Vis. Sci. 2001, 42, 2315–2323. [Google Scholar] [PubMed]
- Vecino, E.; Garía-Grespo, D.; Garía, M.; Martinez-Millán, L.; Sharma, S.C.; Carrascal, E. Rat retinal ganglion cells co-express brain derived neurotrophic factor (BDNF) and its receptor TrkB. Vis. Res. 2002, 42, 151–157. [Google Scholar] [CrossRef] [Green Version]
- Johnson, E.C.; Deppmeier, L.M.H.; Wentzien, S.K.F.; Hsu, I.; Morrison, J.C. Chronology of optic nerve head and retinal responses to elevated intraocular pressure. Investig. Ophthalmol. Vis. Sci. 2000, 41, 431–442. [Google Scholar]
- Herzog, K.-H.; von Bartheld, C.S. Contributions of the Optic Tectum and the Retina as Sources of Brain-Derived Neurotrophic Factor for Retinal Ganglion Cells in the Chick Embryo. J. Neurosci. 1998, 18, 2891–2906. [Google Scholar] [CrossRef]
- Seki, M.; Tanaka, T.; Sakai, Y.; Fukuchi, T.; Abe, H.; Nawa, H.; Takei, N. Müller Cells as a Source of Brain-derived Neurotrophic Factor in the Retina: Noradrenaline Upregulates Brain-derived Neurotrophic Factor Levels in Cultured Rat Müller Cells. Neurochem. Res. 2005, 30, 1163–1170. [Google Scholar] [CrossRef]
- García, M.; Forster, V.; Hicks, D.; Vecino, E. In vivo expression of neurotrophins and neurotrophin receptors is conserved in adult porcine retina in vitro. Investig. Ophthalmol. Vis. Sci. 2003, 44, 4532–4541. [Google Scholar]
- Chong, R.S.; Martin, K.R. Glial cell interactions and glaucoma. Curr. Opin. Ophthalmol. 2015, 26, 73–77. [Google Scholar] [PubMed] [Green Version]
- Galindo-Romero, C.; Valiente-Soriano, F.J.; Jiménez-López, M.; García-Ayuso, D.; Villegas-Pérez, M.P.; Vidal-Sanz, M.; Agudo-Barriuso, M. Effect of brain-derived neurotrophic factor on mouse axotomized retinal ganglion cells and phagocytic microglia. Investig. Ophthalmol. Vis. Sci. 2013, 54, 974–985. [Google Scholar] [CrossRef] [PubMed]
- Harada, T.; Harada, C.; Kohsaka, S.; Wada, E.; Yoshida, K.; Ohno, S.; Mamada, H.; Tanaka, K.; Parada, L.F.; Wada, K. Microglia-Müller glia cell interactions control neurotrophic factor production during light-induced retinal degeneration. J. Neurosci. 2002, 22, 9228–9236. [Google Scholar] [CrossRef] [PubMed]
- Langmann, T. Microglia activation in retinal degeneration. J. Leukoc. Biol. 2007, 81, 1345–1351. [Google Scholar] [PubMed]
- Inanc Tekin, M.; Sekeroglu, M.A.; Demirtas, C.; Tekin, K.; Doguizi, S.; Bayraktar, S.; Yilmazbas, P. Brain-Derived Neurotrophic Factor in Patients With Age-Related Macular Degeneration and Its Correlation With Retinal Layer Thicknesses. Investig. Opthalmol. Vis. Sci. 2018, 59, 2833. [Google Scholar] [CrossRef] [PubMed]
- Chitranshi, N.; Dheer, Y.; Abbasi, M.; You, Y.; Graham, S.L.; Gupta, V. Glaucoma Pathogenesis and Neurotrophins: Focus on the Molecular and Genetic Basis for Therapeutic Prospects. Curr. Neuropharmacol. 2018, 16, 1018–1035. [Google Scholar] [PubMed]
- Di Polo, A.; Aigner, L.J.; Dunn, R.J.; Bray, G.M.; Aguayo, A.J. Prolonged delivery of brain-derived neurotrophic factor by adenovirus-infected Muller cells temporarily rescues injured retinal ganglion cells. Proc. Natl. Acad. Sci. USA 1998, 95, 3978–3983. [Google Scholar] [CrossRef] [PubMed]
- Clarke, D.B.; Bray, G.M.; Aguayo, A.J. Prolonged administration of NT-4/5 fails to rescue most axotomized retinal ganglion cells in adult rats. Vis. Res. 1998, 38, 1517–1524. [Google Scholar] [CrossRef] [Green Version]
- Frank, L.; Ventimiglia, R.; Anderson, K.; Lindsay, R.M.; Rudge, J.S. BDN F Down-regulates Neurotrophin Responsiveness, TrkB Protein and TrkB mRNA Levels in Cultured Rat Hippocampal Neurons. Eur. J. Neurosci. 1996, 8, 1220–1230. [Google Scholar]
- Chen, H.; Weber, A.J. Brain-derived neurotrophic factor reduces TrkB protein and mRNA in the normal retina and following optic nerve crush in adult rats. Brain Res. 2004, 1011, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Ratican, S.E.; Osborne, A.; Martin, K.R. Progress in Gene Therapy to Prevent Retinal Ganglion Cell Loss in Glaucoma and Leber’s Hereditary Optic Neuropathy. Neural Plast. 2018, 2018, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Proenca, C.C.; Song, M.; Lee, F.S. Differential effects of BDNF and neurotrophin 4 (NT4) on endocytic sorting of TrkB receptors. J. Neurochem. 2016, 138, 397–406. [Google Scholar] [CrossRef] [PubMed]
- Osborne, A.; Wang, A.X.Z.; Tassoni, A.; Widdowson, P.S.; Martin, K.R. Design of a Novel Gene Therapy Construct to Achieve Sustained Brain-Derived Neurotrophic Factor Signaling in Neurons. Hum. Gene Ther. 2018, 29, 828–841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, L.; Sapieha, P.; Kittlerova, P.; Hauswirth, W.W.; Di Polo, A. TrkB gene transfer protects retinal ganglion cells from axotomy-induced death in vivo. J. Neurosci. 2002, 22, 3977–3986. [Google Scholar] [CrossRef] [PubMed]
- Nuzzi, R.; Tridico, F. Glaucoma: Biological trabecular and neuroretinal pathology with perspectives of therapy innovation and preventive diagnosis. Front. Neurosci. 2017, 11, 494. [Google Scholar] [CrossRef] [PubMed]
- Johnson, T.V.; Bull, N.D.; Martin, K.R. Neurotrophic factor delivery as a protective treatment for glaucoma. Exp. Eye Res. 2011, 93, 196–203. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Pernet, V.; Hauswirth, W.W.; Di Polo, A. Activation of the Extracellular Signal-Regulated Kinase 1/2 Pathway by AAV Gene Transfer Protects Retinal Ganglion Cells in Glaucoma. Mol. Ther. 2005, 12, 402–412. [Google Scholar] [CrossRef]
- Malik, J.M.I.; Shevtsova, Z.; Bähr, M.; Kügler, S. Long-term in vivo inhibition of CNS neurodegeneration by Bcl-XL gene transfer. Mol. Ther. 2005, 11, 373–381. [Google Scholar] [CrossRef]
- Planchamp, V.; Bermel, C.; Tönges, L.; Ostendorf, T.; Kügler, S.; Reed, J.C.; Kermer, P.; Bähr, M.; Lingor, P. BAG1 promotes axonal outgrowth and regeneration in vivo via Raf-1 and reduction of ROCK activity. Brain 2008, 131, 2606–2619. [Google Scholar] [CrossRef] [Green Version]
- McKinnon, S.J.; Lehman, D.M.; Tahzib, N.G.; Ransom, N.L.; Reitsamer, H.A.; Liston, P.; LaCasse, E.; Li, Q.; Korneluk, R.G.; Hauswirth, W.W. Baculoviral IAP repeat-containing-4 protects optic nerve axons in a rat glaucoma model. Mol. Ther. 2002, 5, 780–787. [Google Scholar] [PubMed]
- Kügler, S.; Klöcker, N.; Kermer, P.; Isenmann, S.; Bähr, M. Transduction of axotomized retinal ganglion cells by adenoviral vector administration at the optic nerve stump: An in vivo model system for the inhibition of neuronal apoptotic cell death. Gene Ther. 1999, 6, 1759–1767. [Google Scholar] [PubMed]
- Kermer, P.; Klöcker, N.; Labes, M.; Bähr, M. Inhibition of CPP32-Like Proteases Rescues Axotomized Retinal Ganglion Cells from Secondary Cell Death In Vivo. J. Neurosci. 1998, 18, 4656–4662. [Google Scholar] [CrossRef] [PubMed]
- Thoenen, H.; Sendtner, M. Neurotrophins: From enthusiastic expectations through sobering experiences to rational therapeutic approaches. Nat. Neurosci. 2002, 5, 1046–1050. [Google Scholar] [PubMed]
- Barker, P.A. p75NTR: A study in contrasts. Cell Death Differ. 1998, 5, 346–356. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Xu, J.; Brahimi, F.; Zhuo, Y.; Sarunic, M.V.; Uri Saragovi, H. An agonistic TrKb mAb causes sustained TrkB activation, delays RGC death, and protects the retinal structure in optic nerve axotomy and in glaucoma. Investig. Ophthalmol. Vis. Sci. 2010, 51, 4722–4731. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Dergham, P.; Nedev, H.; Xu, J.; Galan, A.; Rivera, J.C.; ZhiHua, S.; Mehta, H.M.; Woo, S.B.; Sarunic, M.V.; et al. Chronic and Acute Models of Retinal Neurodegeneration TrkA Activity Are Neuroprotective whereas p75 NTR Activity Is Neurotoxic through a Paracrine Mechanism. J. Biol. Chem. 2010, 285, 39392–39400. [Google Scholar]
- Gupta, V.K.; You, Y.; Li, J.C.; Klistorner, A.; Graham, S.L. Protective Effects of 7,8-Dihydroxyflavone on Retinal Ganglion and RGC-5 Cells Against Excitotoxic and Oxidative Stress. J. Mol. Neurosci. 2013, 49, 96–104. [Google Scholar] [CrossRef]
- Rheaume, B.A.; Jereen, A.; Bolisetty, M.; Sajid, M.S.; Yang, Y.; Renna, K.; Sun, L.; Robson, P.; Trakhtenberg, E.F. Single cell transcriptome profiling of retinal ganglion cells identifies cellular subtypes. Nat. Commun. 2018, 9, 2759. [Google Scholar] [CrossRef]
- Logan, A.; Ahmed, Z.; Baird, A.; Gonzalez, A.M.; Berry, M. Neurotrophic factor synergy is required for neuronal survival and disinhibited axon regeneration after CNS injury. Brain 2006, 129, 490–502. [Google Scholar] [CrossRef]
- Flachsbarth, K.; Jankowiak, W.; Kruszewski, K.; Helbing, S.; Bartsch, S.; Bartsch, U. Pronounced synergistic neuroprotective effect of GDNF and CNTF on axotomized retinal ganglion cells in the adult mouse. Exp. Eye Res. 2018, 176, 258–265. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, Y.; Bikbova, G.; Baba, T.; Yamamoto, S.; Oshitari, T. In vivo effects of single or combined topical neuroprotective and regenerative agents on degeneration of retinal ganglion cells in rat optic nerve crush model. Sci. Rep. 2019, 9, 101. [Google Scholar] [CrossRef] [PubMed]
- Pease, M.E.; Zack, D.J.; Berlinicke, C.; Bloom, K.; Cone, F.; Wang, Y.; Klein, R.L.; Hauswirth, W.W.; Quigley, H.A. Effect of cntf on retinal ganglion cell survival in experimental glaucoma. Investig. Ophthalmol. Vis. Sci. 2009, 50, 2194–2200. [Google Scholar]
- Klöcker, N.; Kermer, P.; Weishaupt, J.H.; Labes, M.; Ankerhold, R.; Bähr, M. Brain-derived neurotrophic factor-mediated neuroprotection of adult rat retinal ganglion cells in vivo does not exclusively depend on phosphatidyl-inositol-3′-kinase/protein kinase B signaling. J. Neurosci. 2000, 20, 6962–6967. [Google Scholar] [CrossRef] [PubMed]
- West, A.E.; Pruunsild, P.; Timmusk, T. Neurotrophins: Transcription and translation. Handb. Exp. Pharmacol. 2014, 220, 67–100. [Google Scholar] [PubMed]
- Corredor, R.G.; Goldberg, J.L. Electrical activity enhances neuronal survival and regeneration. J. Neural Eng. 2009, 6, 055001. [Google Scholar] [CrossRef] [PubMed]
- Kolarow, R.; Kuhlmann, C.R.W.; Munsch, T.; Zehendner, C.; Brigadski, T.; Luhmann, H.J.; Lessmann, V. BDNF-induced nitric oxide signals in cultured rat hippocampal neurons: Time course, mechanism of generation, and effect on neurotrophin secretion. Front. Cell. Neurosci. 2014, 8, 323. [Google Scholar] [CrossRef]
- Sehic, A.; Guo, S.; Cho, K.-S.; Corraya, R.M.; Chen, D.F.; Utheim, T.P. Electrical Stimulation as a Means for Improving Vision. Am. J. Pathol. 2016, 186, 2783–2797. [Google Scholar] [CrossRef] [Green Version]
- Manthey, A.L.; Liu, W.; Jiang, Z.X.; Lee, M.H.K.; Ji, J.; So, K.-F.; Lai, J.S.M.; Lee, V.W.H.; Chiu, K. Using Electrical Stimulation to Enhance the Efficacy of Cell Transplantation Therapies for Neurodegenerative Retinal Diseases: Concepts, Challenges, and Future Perspectives. Cell Transplant. 2017, 26, 949–965. [Google Scholar] [Green Version]
- Sato, T.; Fujikado, T.; Lee, T.-S.; Tano, Y. Direct Effect of Electrical Stimulation on Induction of Brain-Derived Neurotrophic Factor from Cultured Retinal Müller Cells. Investig. Opthalmol. Vis. Sci. 2008, 49, 4641. [Google Scholar] [CrossRef]
- Zhou, W.T.; Ni, Y.Q.; Jin, Z.B.; Zhang, M.; Wu, J.H.; Zhu, Y.; Xu, G.Z.; Gan, D.K. Electrical stimulation ameliorates light-induced photoreceptor degeneration in vitro via suppressing the proinflammatory effect of microglia and enhancing the neurotrophic potential of Müller cells. Exp. Neurol. 2012, 238, 192–208. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Fujikado, T.; Morimoto, T.; Matsushita, K.; Harada, T.; Tano, Y. Effect of electrical stimulation on IGF-1 transcription by L-type calcium channels in cultured retinal Müller cells. Jpn. J. Ophthalmol. 2008, 52, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Ni, Y.; Gan, D.; Xu, H.; Xu, G.; Da, C. Neuroprotective effect of transcorneal electrical stimulation on light-induced photoreceptor degeneration. Exp. Neurol. 2009, 219, 439–452. [Google Scholar]
- Ciavatta, V.T.; Kim, M.; Wong, P.; Nickerson, J.M.; Shuler, R.K.; Mclean, G.Y.; Pardue, M.T. Retinal expression of Fgf2 in RCS rats with subretinal microphotodiode array. Investig. Ophthalmol. Vis. Sci. 2009, 50, 4523–4530. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, T.; Miyoshi, T.; Matsuda, S.; Tano, Y.; Fujikado, T.; Fukuda, Y. Transcorneal electrical stimulation rescues axotomized retinal ganglion cells by activating endogenous retinal IGF-1 system. Investig. Ophthalmol. Vis. Sci. 2005, 46, 2147–2155. [Google Scholar] [CrossRef] [PubMed]
- Willmann, G.; Schäferhoff, K.; Fischer, M.D.; Arango-Gonzalez, B.; Bolz, S.; Naycheva, L.; Röck, T.; Bonin, M.; Bartz-Schmidt, K.U.; Zrenner, E.; et al. Gene expression profiling of the retina after transcorneal electrical stimulation in wild-type Brown Norway rats. Investig. Ophthalmol. Vis. Sci. 2011, 52, 7529–7537. [Google Scholar]
- Henrich-Noack, P.; Lazik, S.; Sergeeva, E.; Wagner, S.; Voigt, N.; Prilloff, S.; Fedorov, A.; Sabel, B.A. Transcorneal alternating current stimulation after severe axon damage in rats results in “long-term silent survivor” neurons. Brain Res. Bull. 2013, 95, 7–14. [Google Scholar] [CrossRef]
- Osako, T.; Chuman, H.; Maekubo, T.; Ishiai, M.; Kawano, N.; Nao-i, N. Effects of steroid administration and transcorneal electrical stimulation on the anatomic and electrophysiologic deterioration of nonarteritic ischemic optic neuropathy in a rodent model. Jpn. J. Ophthalmol. 2013, 57, 410–415. [Google Scholar] [CrossRef] [PubMed]
- Fujikado, T.; Morimoto, T.; Matsushita, K.; Shimojo, H.; Okawa, Y.; Tano, Y. Effect of Transcorneal Electrical Stimulation in Patients with Nonarteritic Ischemic Optic Neuropathy or Traumatic Optic Neuropathy. Jpn. J. Ophthalmol. 2006, 50, 266–273. [Google Scholar] [CrossRef] [PubMed]
- Gall, C.; Fedorov, A.B.; Ernst, L.; Borrmann, A.; Sabel, B.A. Repetitive transorbital alternating current stimulation in optic neuropathy. NeuroRehabilitation 2010, 27, 335–341. [Google Scholar] [PubMed]
- Gall, C.; Sgorzaly, S.; Schmidt, S.; Brandt, S.; Fedorov, A.; Sabel, B.A. Noninvasive transorbital alternating current stimulation improves subjective visual functioning and vision-related quality of life in optic neuropathy. Brain Stimul. 2011, 4, 175–188. [Google Scholar] [CrossRef] [PubMed]
- Fedorov, A.; Jobke, S.; Bersnev, V.; Chibisova, A.; Chibisova, Y.; Gall, C.; Sabel, B.A. Restoration of vision after optic nerve lesions with noninvasive transorbital alternating current stimulation: A clinical observational study. Brain Stimul. 2011, 4, 189–201. [Google Scholar] [CrossRef] [PubMed]
- Gall, C.; Schmidt, S.; Schittkowski, M.P.; Antal, A.; Ambrus, G.G.; Paulus, W.; Dannhauer, M.; Michalik, R.; Mante, A.; Bola, M.; et al. Alternating current stimulation for vision restoration after optic nerve damage: A randomized clinical trial. PLoS ONE 2016, 11, e0156134. [Google Scholar] [CrossRef] [PubMed]
- Weber, A.J.; Viswanáthan, S.; Ramanathan, C.; Harman, C.D. Combined application of BDNF to the eye and brain enhances ganglion cell survival and function in the cat after optic nerve injury. Investig. Ophthalmol. Vis. Sci. 2010, 51, 327–334. [Google Scholar]
- Segal, R.A. Selectivity in neurotrophin signaling: Theme and variations. Annu. Rev. Neurosci. 2003, 26, 299–330. [Google Scholar] [PubMed]
- Quigley, H.A.; Addicks, E.M. Chronic experimental glaucoma in primates. II. Effect of extended intraocular pressure elevation on optic nerve head and axonal transport. Invest. Ophthalmol. Vis. Sci. 1980, 19, 137–152. [Google Scholar] [PubMed]
- Cosker, K.E.; Courchesne, S.L.; Segal, R.A. Action in the axon: Generation and transport of signaling endosomes. Curr. Opin. Neurobiol. 2008, 18, 270–275. [Google Scholar]
- Howe, C.L.; Valletta, J.S.; Rusnak, A.S.; Mobley, W.C. NGF Signaling from Clathrin-Coated Vesicles. Neuron 2001, 32, 801–814. [Google Scholar] [CrossRef]
- Delcroix, J.-D.; Valletta, J.S.; Wu, C.; Hunt, S.J.; Kowal, A.S.; Mobley, W.C. NGF Signaling in Sensory Neurons. Neuron 2003, 39, 69–84. [Google Scholar] [CrossRef]
- Zheng, J.; Shen, W.H.; Lu, T.J.; Zhou, Y.; Chen, Q.; Wang, Z.; Xiang, T.; Zhu, Y.C.; Zhang, C.; Duan, S.; et al. Clathrin-dependent endocytosis is required for TrkB-dependent Akt-mediated neuronal protection and dendritic growth. J. Biol. Chem. 2008, 283, 13280–13288. [Google Scholar] [CrossRef]
- Qu, J.; Wang, D.; Grosskreutz, C.L. Mechanisms of retinal ganglion cell injury and defense in glaucoma. Exp. Eye Res. 2010, 91, 48–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ibáñez, C.F. Message in a bottle: Long-range retrograde signaling in the nervous system. Trends Cell Biol. 2007, 17, 519–528. [Google Scholar] [PubMed]
- Harrington, A.W.; Ginty, D.D. Long-distance retrograde neurotrophic factor signalling in neurons. Nat. Rev. Neurosci. 2013, 14, 177–187. [Google Scholar] [CrossRef] [PubMed]
- Ye, M.; Lehigh, K.M.; Ginty, D.D. Multivesicular bodies mediate long-range retrograde NGF-TrkA signaling. Elife 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Cosker, K.E.; Segal, R.A. Neuronal signaling through endocytosis. Cold Spring Harb. Perspect. Biol. 2014, 6, a020669. [Google Scholar] [CrossRef]
- Lom, B.; Cogen, J.; Sanchez, A.L.; Vu, T.; Cohen-Cory, S. Local and Target-Derived Brain-Derived Neurotrophic Factor Exert Opposing Effects on the Dendritic Arborization of Retinal Ganglion Cells In Vivo. J. Neurosci. 2018, 22, 7639–7649. [Google Scholar] [CrossRef]
- Watson, F.L.; Heerssen, H.M.; Bhattacharyya, A.; Klesse, L.; Lin, M.Z.; Segal, R.A. Neurotrophins use the Erk5 pathway to mediate a retrograde survival response. Nat. Neurosci. 2001, 4, 981–988. [Google Scholar] [CrossRef]
- Van Oterendorp, C.; Sgouris, S.; Schallner, N.; Biermann, J.; Lagrèze, W.A. Retrograde neurotrophic signaling in rat retinal ganglion cells is transmitted via the ERK5 but not the ERK1/2 pathway. Investig. Ophthalmol. Vis. Sci. 2014, 55, 658–665. [Google Scholar] [CrossRef]
- Geeraerts, E.; Claes, M.; Dekeyster, E.; Salinas-Navarro, M.; De Groef, L.; Van den Haute, C.; Scheyltjens, I.; Baekelandt, V.; Arckens, L.; Moons, L. Optogenetic Stimulation of the Superior Colliculus Confers Retinal Neuroprotection in a Mouse Glaucoma Model. J. Neurosci. 2019, 39, 2313–2325. [Google Scholar] [CrossRef] [Green Version]
- Hasel, P.; Dando, O.; Jiwaji, Z.; Baxter, P.; Todd, A.C.; Heron, S.; Márkus, N.M.; McQueen, J.; Hampton, D.W.; Torvell, M.; et al. Neurons and neuronal activity control gene expression in astrocytes to regulate their development and metabolism. Nat. Commun. 2017, 8, 15132. [Google Scholar] [CrossRef]
- Galindo-Romero, C.; Avilés-Trigueros, M.; Jiménez-López, M.; Valiente-Soriano, F.J.; Salinas-Navarro, M.; Nadal-Nicolás, F.; Villegas-Pérez, M.P.; Vidal-Sanz, M.; Agudo-Barriuso, M. Axotomy-induced retinal ganglion cell death in adult mice: Quantitative and topographic time course analyses. Exp. Eye Res. 2011, 92, 377–387. [Google Scholar] [CrossRef] [PubMed]
- Heiduschka, P.; Thanos, S. Restoration of the retinofugal pathway. Prog. Retin. Eye Res. 2000, 19, 577–606. [Google Scholar] [CrossRef]
- Intrinsic survival mechanisms for retinal ganglion cells. Eur. J. Ophthalmol. 1999, 9, S12–S16. [CrossRef]
- Humphrey, M.F.; Beazley, L.D. Retinal ganglion cell death during optic nerve regeneration in the froghyla moorei. J. Comp. Neurol. 1985, 236, 382–402. [Google Scholar] [CrossRef] [PubMed]
- Beazley, L.D.; Darbyy, J.E.; Perry, V.H. Cell death in the retinal ganglion cell layer during optic nerve regeneration for the frog Rana pipiens. Vis. Res. 1986, 26, 543–556. [Google Scholar] [CrossRef]
- WATANABE, M.; INUKAI, N.; FUKUDA, Y. Survival of retinal ganglion cells after transection of the optic nerve in adult cats: A quantitative study within two weeks. Vis. Neurosci. 2001, 18, 137–145. [Google Scholar] [CrossRef]
- Muchnick, N.; Hibbard, E. Avian retinal ganglion cells resistant to degeneration after optic nerve lesion. Exp. Neurol. 1980, 68, 205–216. [Google Scholar] [CrossRef]
- Soto, I.; Marie, B.; Baro, D.J.; Blanco, R.E. FGF-2 modulates expression and distribution of GAP-43 in frog retinal ganglion cells after optic nerve injury. J. Neurosci. Res. 2003, 73, 507–517. [Google Scholar] [CrossRef]
- Duprey-Díaz, M.V.; Blagburn, J.M.; Blanco, R.E. Exogenous Modulation of Retinoic Acid Signaling Affects Adult RGC Survival in the Frog Visual System after Optic Nerve Injury. PLoS ONE 2016, 11, e0162626. [Google Scholar] [CrossRef]
- Bollaerts, I.; Veys, L.; Geeraerts, E.; Andries, L.; De Groef, L.; Buyens, T.; Salinas-Navarro, M.; Moons, L.; Van Hove, I. Complementary research models and methods to study axonal regeneration in the vertebrate retinofugal system. Brain Struct. Funct. 2018, 223, 545–567. [Google Scholar] [CrossRef]
- Scalia, F.; Arango, V.; Singman, E.L. Loss and displacement of ganglion cells after optic nerve regeneration in adultRana pipiens. Brain Res. 1985, 344, 267–280. [Google Scholar] [CrossRef]
- Williams, D.L. Regenerating reptile retinas: A comparative approach to restoring retinal ganglion cell function. Eye 2017, 31, 167–172. [Google Scholar] [CrossRef] [PubMed]
- Ramón, Y.; Cajal, S.; DeFelipe, J.; Jones, E.G.; May, R.M. Cajal’s Degeneration and Regeneration of the Nervous System; DeFelipe, J., Jones, E.G., Eds.; Oxford University Press: Oxford, UK, 2012; ISBN 9780199847242. [Google Scholar]
- Germain, F.; Calvo, M.; de la Villa, P. Rabbit retinal ganglion cell survival after optic nerve section and its effect on the inner plexiform layer. Exp. Eye Res. 2004, 78, 95–102. [Google Scholar] [PubMed]
- Berkelaar, M.; Clarke, D.; Wang, Y.; Bray, G.; Aguayo, A. Axotomy results in delayed death and apoptosis of retinal ganglion cells in adult rats. J. Neurosci. 1994, 14, 4368–4374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magharious, M.M.; D’Onofrio, P.M.; Koeberle, P.D. Optic Nerve Transection: A Model of Adult Neuron Apoptosis in the Central Nervous System. J. Vis. Exp. 2011, 12, 2241. [Google Scholar] [CrossRef] [PubMed]
- Grafstein, B.; Ingoglia, N.A. Intracranial transection of the optic nerve in adult mice: Preliminary observations. Exp. Neurol. 1982, 76, 318–330. [Google Scholar] [PubMed]
- Allcutt, D.; Berry, M.; Sievers, J. A qualitative comparison of the reactions of retinal ganglion cell axons to optic nerve crush in neonatal and adult mice. Dev. Brain Res. 1984, 16, 231–240. [Google Scholar] [CrossRef]
- Misantone, L.J.; Gershenbaum, M.; Murray, M. Viability of retinal ganglion cells after optic nerve crush in adult rats. J. Neurocytol. 1984, 13, 449–465. [Google Scholar] [CrossRef] [PubMed]
- Agudo, M.; Pérez-Marín, M.C.; Lönngren, U.; Sobrado, P.; Conesa, A.; Cánovas, I.; Salinas-Navarro, M.; Miralles-Imperial, J.; Hallböök, F.; Vidal-Sanz, M. Time course profiling of the retinal transcriptome after optic nerve transection and optic nerve crush. Mol. Vis. 2008, 14, 1050–1063. [Google Scholar] [PubMed]
- Li, H.-J.; Sun, Z.-L.; Yang, X.-T.; Zhu, L.; Feng, D.-F. Exploring Optic Nerve Axon Regeneration. Curr. Neuropharmacol. 2017, 15, 861–873. [Google Scholar] [PubMed] [Green Version]
- Villegas-Pérez, M.-P.; Vidal-Sanz, M.; Rasminsky, M.; Bray, G.M.; Aguayo, A.J. Rapid and protracted phases of retinal ganglion cell loss follow axotomy in the optic nerve of adult rats. J. Neurobiol. 1993, 24, 23–36. [Google Scholar] [CrossRef] [PubMed]
- Peinado-Ramón, P.; Salvador, M.; Villegas-Pérez, M.P.; Vidal-Sanz, M. Effects of axotomy and intraocular administration of NT-4, NT-3, and brain-derived neurotrophic factor on the survival of adult rat retinal ganglion cells. A quantitative in vivo study. Invest. Ophthalmol. Vis. Sci. 1996, 37, 489–500. [Google Scholar] [PubMed]
- Bähr, M. Live or let die—Retinal ganglion cell death and survival during development and in the lesioned adult CNS. Trends Neurosci. 2000, 23, 483–490. [Google Scholar] [CrossRef]
- Isenmann, S.; Wahl, C.; Krajewski, S.; Reed, J.C.; Bähr, M. Up-regulation of Bax protein in degenerating retinal ganglion cells precedes apoptotic cell death after optic nerve lesion in the rat. Eur. J. Neurosci. 1997, 9, 1763–1772. [Google Scholar] [CrossRef] [PubMed]
- Leaver, S.G.; Cui, Q.; Plant, G.W.; Arulpragasam, A.; Hisheh, S.; Verhaagen, J.; Harvey, A.R. AAV-mediated expression of CNTF promotes long-term survival and regeneration of adult rat retinal ganglion cells. Gene Ther. 2006, 13, 1328–1341. [Google Scholar] [Green Version]
- Murray, M.; Grafstein, B. Changes in the morphology and amino acid incorporation of regenerating goldfish optic neurons. Exp. Neurol. 1969, 23, 544–560. [Google Scholar] [CrossRef]
- Kato, S.; Matsukawa, T.; Koriyama, Y.; Sugitani, K.; Ogai, K. A molecular mechanism of optic nerve regeneration in fish: The retinoid signaling pathway. Prog. Retin. Eye Res. 2013, 37, 13–30. [Google Scholar] [CrossRef]
- Zou, S.; Tian, C.; Ge, S.; Hu, B. Neurogenesis of Retinal Ganglion Cells Is Not Essential to Visual Functional Recovery after Optic Nerve Injury in Adult Zebrafish. PLoS ONE 2013, 8, e57280. [Google Scholar] [CrossRef]
- Zou, S.; Yin, W.; Huang, Y.; Tian, C.; Ge, S.; Hu, B. Functional Regeneration and Remyelination in the Zebrafish Optic Nerve. In Neural Regeneration; Elsevier: Amsterdam, The Netherlands, 2015; pp. 21–41. ISBN 9780128018347. [Google Scholar]
- Rodger, J.; Dunlop, S.A. Central Nerve Regeneration in Reptiles. In Neural Regeneration; Elsevier: Amsterdam, The Netherlands, 2015; pp. 43–55. ISBN 9780128018347. [Google Scholar]
- Beckers, A.; Van Dyck, A.; Bollaerts, I.; Van houcke, J.; Lefevere, E.; Andries, L.; Agostinone, J.; Van Hove, I.; Di Polo, A.; Lemmens, K.; et al. An Antagonistic Axon-Dendrite Interplay Enables Efficient Neuronal Repair in the Adult Zebrafish Central Nervous System. Mol. Neurobiol. 2019, 56, 3175–3192. [Google Scholar] [CrossRef]
- Wyatt, C.; Ebert, A.; Reimer, M.M.; Rasband, K.; Hardy, M.; Chien, C.-B.; Becker, T.; Becker, C.G. Analysis of the astray/robo2 Zebrafish Mutant Reveals that Degenerating Tracts Do Not Provide Strong Guidance Cues for Regenerating Optic Axons. J. Neurosci. 2010, 30, 13838–13849. [Google Scholar] [CrossRef]
- McCurley, A.T.; Callard, G.V.G.V. Time course analysis of gene expression patterns in zebrafish eye during optic nerve regeneration. J. Exp. Neurosci. 2010, 4, 17–33. [Google Scholar]
- Dunlop, S.A.; Tennant, M.; Beazley, L.D. Extent of retinal ganglion cell death in the frogLitoria moorei after optic nerve regeneration induced by lesions of different sizes. J. Comp. Neurol. 2002, 446, 276–287. [Google Scholar] [PubMed]
- Zhao, Y.; Szaro, B.G. The return of phosphorylated and nonphosphorylated epitopes of neurofilament proteins to the regenerating optic nerve ofXenopus laevis. J. Comp. Neurol. 1994, 343, 158–172. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Yu, H.; Deaton, S.K.; Szaro, B.G. Heterogeneous Nuclear Ribonucleoprotein K, an RNA-Binding Protein, Is Required for Optic Axon Regeneration in Xenopus laevis. J. Neurosci. 2012, 32, 3563–3574. [Google Scholar] [PubMed] [Green Version]
- Stelzner, D.J.; Bohn, R.C.; Strauss, J.A. Regeneration of the frog optic nerve. Neurochem. Pathol. 1986, 5, 255–288. [Google Scholar] [CrossRef] [PubMed]
- Dunlop, S.A. Axonal sprouting in the optic nerve is not a prerequisite for successful regeneration. J. Comp. Neurol. 2003, 465, 319–334. [Google Scholar] [CrossRef] [PubMed]
- Matsukawa, T.; Arai, K.; Koriyama, Y.; Liu, Z.; Kato, S. Axonal Regeneration of Fish Optic Nerve after Injury. Biol. Pharm. Bull. 2004, 27, 445–451. [Google Scholar] [Green Version]
- Koriyama, Y.; Homma, K.; Sugitani, K.; Higuchi, Y.; Matsukawa, T.; Murayama, D.; Kato, S. Upregulation of IGF-I in the goldfish retinal ganglion cells during the early stage of optic nerve regeneration. Neurochem. Int. 2007, 50, 749–756. [Google Scholar] [Green Version]
- Homma, K.; Koriyama, Y.; Mawatari, K.; Higuchi, Y.; Kosaka, J.; Kato, S. Early downregulation of IGF-I decides the fate of rat retinal ganglion cells after optic nerve injury. Neurochem. Int. 2007, 50, 741–748. [Google Scholar] [CrossRef]
- Lenkowski, J.R.; Raymond, P.A. Müller glia: Stem cells for generation and regeneration of retinal neurons in teleost fish. Prog. Retin. Eye Res. 2014, 40, 94–123. [Google Scholar] [CrossRef]
- Cudeiro, J.; Rivadulla, C. Sight and insight – on the physiological role of nitric oxide in the visual system. Trends Neurosci. 1999, 22, 109–116. [Google Scholar] [CrossRef]
- Koriyama, Y.; Yasuda, R.; Homma, K.; Mawatari, K.; Nagashima, M.; Sugitani, K.; Matsukawa, T.; Kato, S. Nitric oxide-cGMP signaling regulates axonal elongation during optic nerve regeneration in the goldfish in vitro and in vivo. J. Neurochem. 2009, 110, 890–901. [Google Scholar] [PubMed]
- Lee, E.J.; Kim, K.Y.; Gu, T.H.; Moon, J.I.; Kim, I.B.; Lee, M.Y.; Oh, S.J.; Chun, M.H. Neuronal nitric oxide synthase is expressed in the axotomized ganglion cells of the rat retina. Brain Res. 2003, 986, 174–180. [Google Scholar] [CrossRef]
- Koeberle, P.D.; Ball, A.K. Nitric Oxide Synthase Inhibition Delays Axonal Degeneration and Promotes the Survival of Axotomized Retinal Ganglion Cells. Exp. Neurol. 1999, 158, 366–381. [Google Scholar] [CrossRef] [PubMed]
- Canossa, M.; Giordano, E.; Cappello, S.; Guarnieri, C.; Ferri, S. Nitric oxide down-regulates brain-derived neurotrophic factor secretion in cultured hippocampal neurons. Proc. Natl. Acad. Sci. USA 2002, 99, 3282–3287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chidlow, G.; Wood, J.P.M.; Casson, R.J. Expression of inducible heat shock proteins Hsp27 and Hsp70 in the visual pathway of rats subjected to various models of retinal ganglion cell injury. PLoS ONE 2014, 9, e114838. [Google Scholar]
- Kamioka, Y.; Fujikawa, C.; Ogai, K.; Sugitani, K.; Watanabe, S.; Kato, S.; Wakasugi, K. Functional characterization of fish neuroglobin: Zebrafish neuroglobin is highly expressed in amacrine cells after optic nerve injury and can translocate into ZF4 cells. Biochim. Biophys. Acta Proteins Proteom. 2013, 1834, 1779–1788. [Google Scholar] [CrossRef]
- Sugitani, K.; Koriyama, Y.; Ogai, K.; Wakasugi, K.; Kato, S. A Possible Role of Neuroglobin in the Retina After Optic Nerve Injury: A Comparative Study of Zebrafish and Mouse Retina. Adv. Exp. Med. Biol. 2016, 854, 671–675. [Google Scholar]
- Rosenzweig, S.; Raz-Prag, D.; Nitzan, A.; Galron, R.; Paz, M.; Jeserich, G.; Neufeld, G.; Barzilai, A.; Solomon, A.S. Sema-3A indirectly disrupts the regeneration process of goldfish optic nerve after controlled injury. Graefes Arch. Clin. Exp. Ophthalmol. 2010, 248, 1423–1435. [Google Scholar]
- Shirvan, A.; Kimron, M.; Holdengreber, V.; Ziv, I.; Ben-Shaul, Y.; Melamed, S.; Melamed, E.; Barzilai, A.; Solomon, A.S. Anti-semaphorin 3A Antibodies Rescue Retinal Ganglion Cells from Cell Death following Optic Nerve Axotomy. J. Biol. Chem. 2002, 277, 49799–49807. [Google Scholar] [CrossRef] [Green Version]
- Ogai, K.; Nishitani, M.; Kuwana, A.; Mawatari, K.; Koriyama, Y.; Sugitani, K.; Nakashima, H.; Kato, S. Regeneration-associated genes on optic nerve regeneration in fish retina. Adv. Exp. Med. Biol. 2014, 801, 441–446. [Google Scholar] [PubMed]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Claes, M.; De Groef, L.; Moons, L. Target-Derived Neurotrophic Factor Deprivation Puts Retinal Ganglion Cells on Death Row: Cold Hard Evidence and Caveats. Int. J. Mol. Sci. 2019, 20, 4314. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20174314
Claes M, De Groef L, Moons L. Target-Derived Neurotrophic Factor Deprivation Puts Retinal Ganglion Cells on Death Row: Cold Hard Evidence and Caveats. International Journal of Molecular Sciences. 2019; 20(17):4314. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20174314
Chicago/Turabian StyleClaes, Marie, Lies De Groef, and Lieve Moons. 2019. "Target-Derived Neurotrophic Factor Deprivation Puts Retinal Ganglion Cells on Death Row: Cold Hard Evidence and Caveats" International Journal of Molecular Sciences 20, no. 17: 4314. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20174314