A Fragmenting Protocol with Explicit Hydration for Calculation of Binding Enthalpies of Target-Ligand Complexes at a Quantum Mechanical Level



Abstract
:1. Introduction
2. Results and Discussion
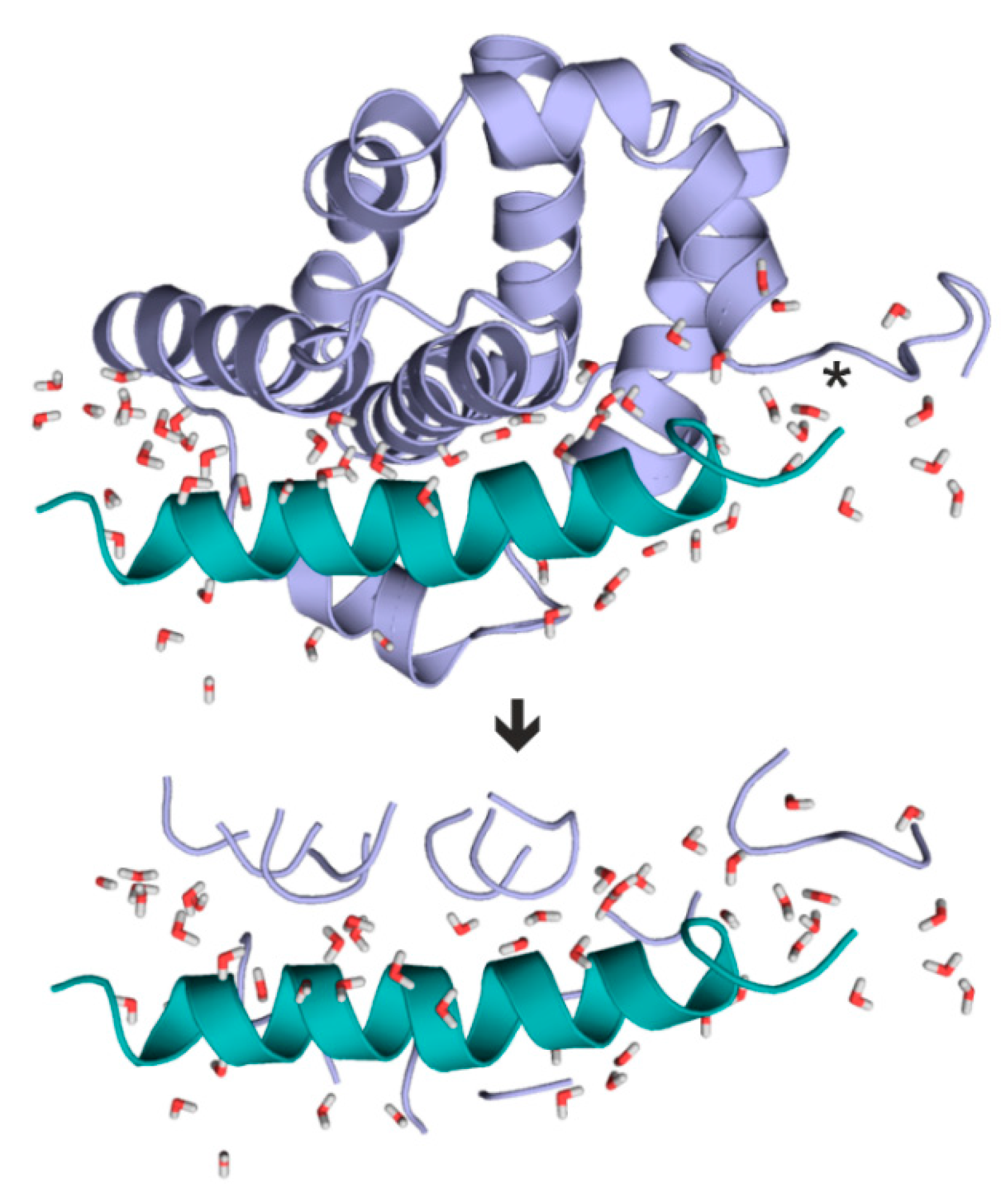
2.1. Fragmenter
2.2. Dry Systems and an Implicit Water Model
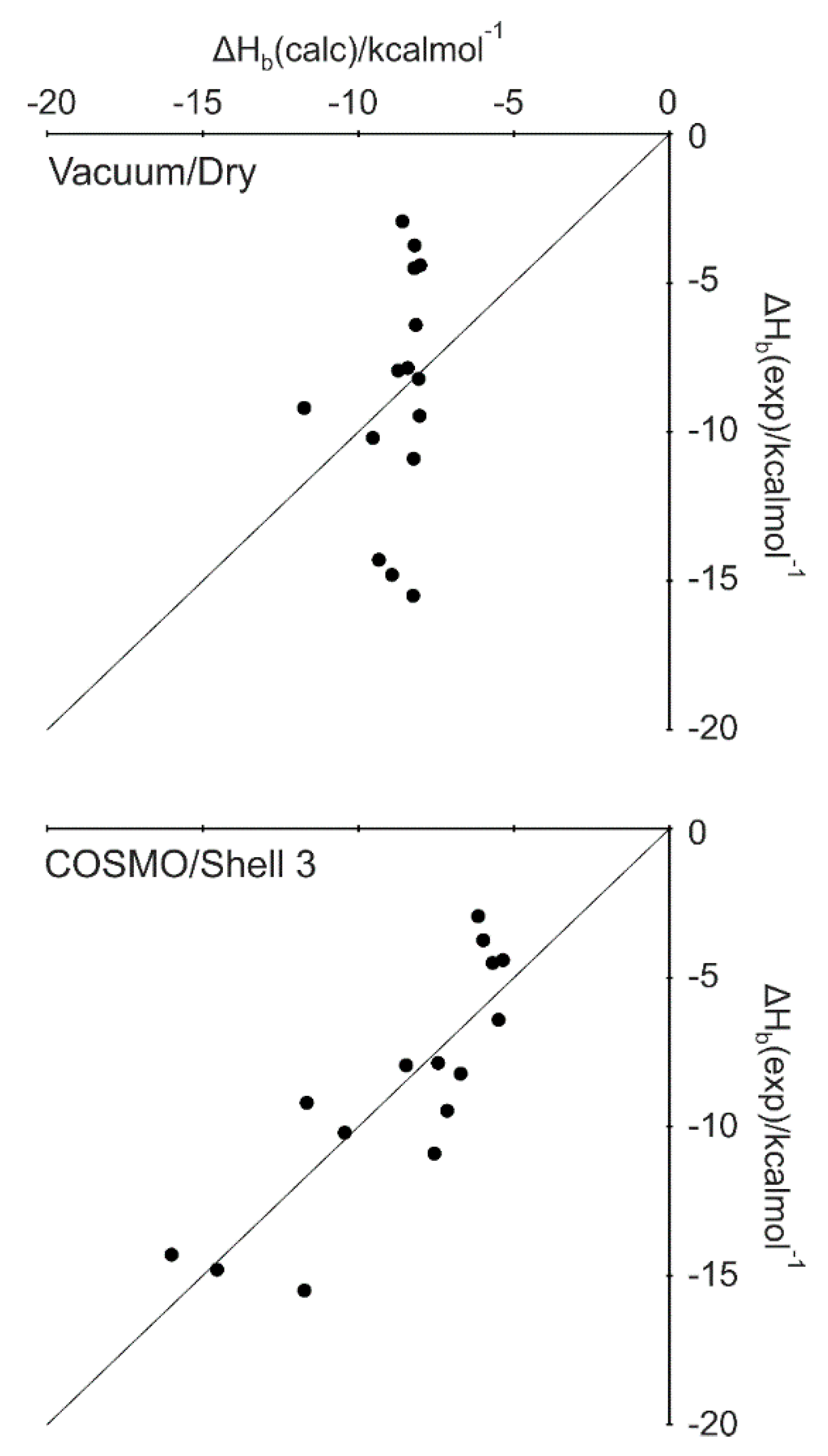
2.3. Explicit Hydration and a Hybrid Model
2.4. Scaling Factor
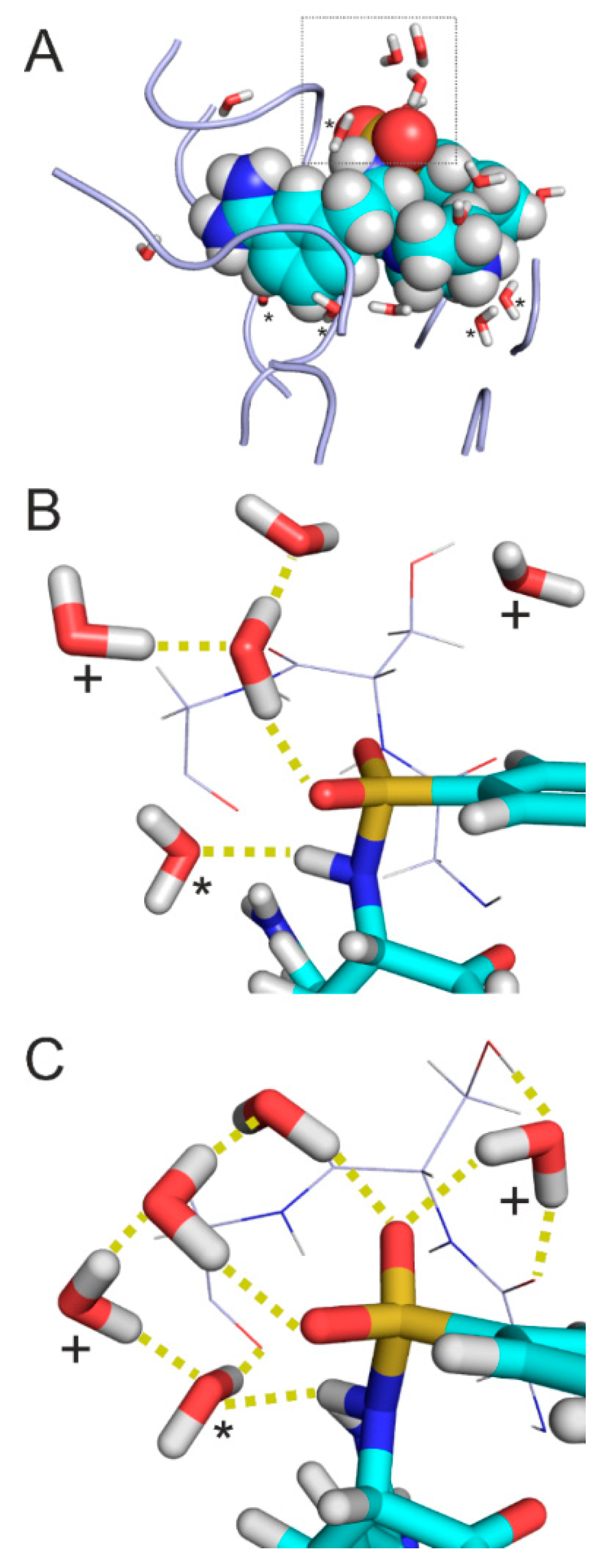
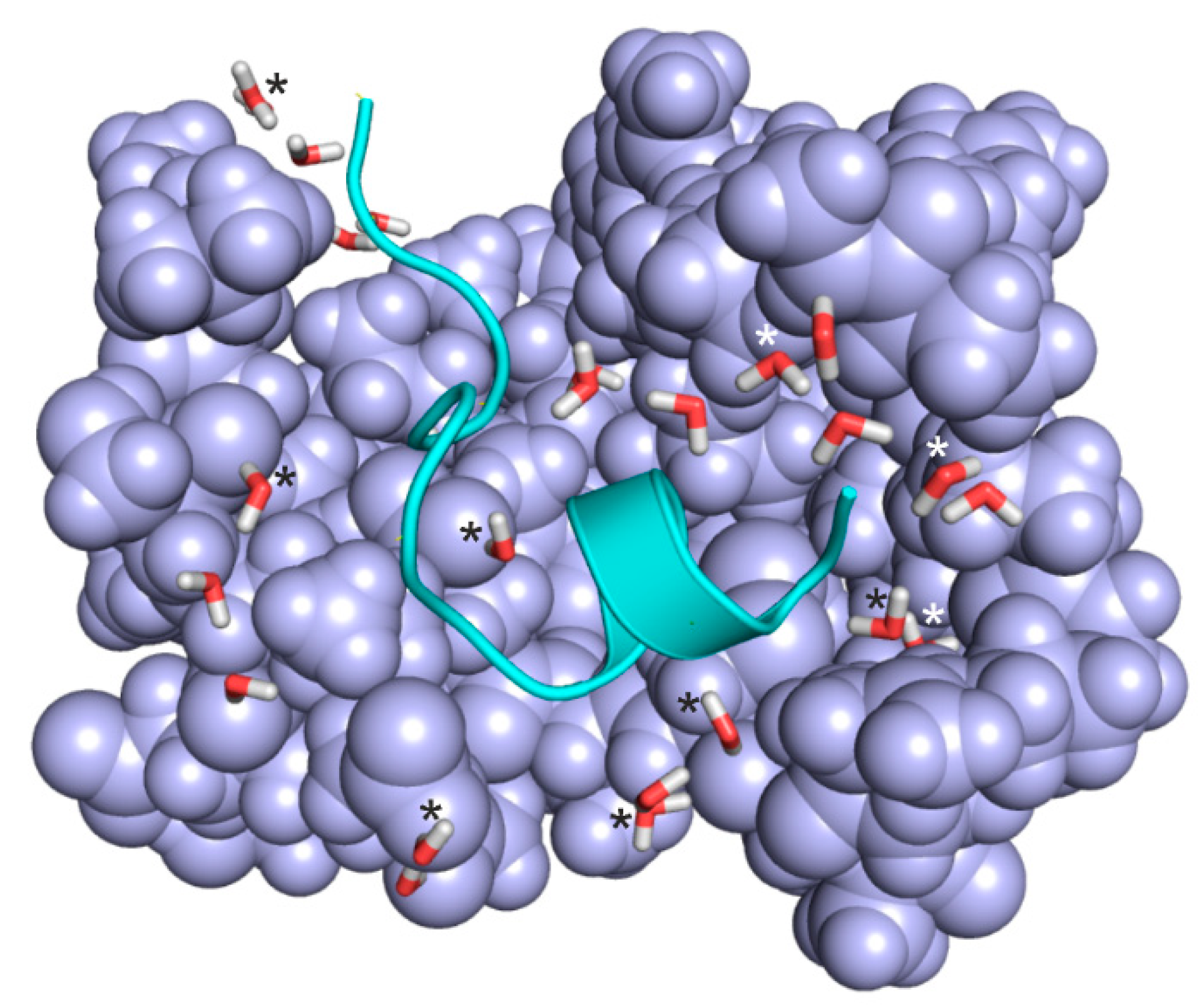
2.5. Case Studies on Hydration Structures
3. Methods
3.1. Preparation of Complexes
3.2. Parameters of Non-Amino Acid Ligands
3.3. Calculation of Interfacial Hydration Structure
3.4. Molecular Mechanics Energy-Minimization during MobyWat Predictions
3.5. Molecular Dynamics of the Protein Target
3.6. Re-Assembly of the Target-Ligand Complex
3.7. Molecular Dynamics of the Target-Ligand Complex
3.8. Production of Interfacial Water Positions
3.9. Molecular Mechanics Energy-Minimization after MobyWat
3.10. Extraction of Target-Ligand Interfaces by Fragmenter
3.10.1. Input
3.10.2. Main Algorithm
3.10.3. Target-Ligand Intermolecular Interaction Energy
3.10.4. Output
3.11. Calculation of Heats of Formation
3.12. Statistics
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| COSMO | Conductor-like screening model |
| Einter | Intermolecular interaction energy |
| ITC | Isothermal titration calorimetry |
| LJ | Lennard-Jones |
| MD | Molecular dynamics |
| MM | Molecular mechanics |
| MW | Molecular weight |
| PDB | Protein Databank |
| PM7 | Parametric method 7 |
| QM | Quantum mechanical |
| RMSE | Root mean square error |
References
- Bajusz, D.; Ferenczy, G.; Keseru, G. Structure-Based Virtual Screening Approaches in Kinase-Directed Drug Discovery. Curr. Top. Med. Chem. 2017, 17, 2235–2259. [Google Scholar] [CrossRef]
- Talhout, R.; Engberts, J.B. Thermodynamic analysis of binding of p-substituted benzamidines to trypsin. Eur. J. Biochem. 2001, 268, 1554–1560. [Google Scholar] [CrossRef]
- Sleigh, S.H.; Seavers, P.R.; Wilkinson, A.J.; Ladbury, J.E.; Tame, J.R.H. Crystallographic and Calorimetric Analysis of Peptide Binding to OppA Protein. J. Mol. Biol. 1999, 291, 393–415. [Google Scholar] [CrossRef]
- Dullweber, F.; Stubbs, M.; Musil, D.; Stürzebecher, J.; Klebe, G. Factorising ligand affinity: A Combined thermodynamic and crystallographic study of trypsin and thrombin inhibition. J. Mol. Biol. 2001, 313, 593–614. [Google Scholar] [CrossRef]
- Palencia, A.; Cobos, E.S.; Mateo, P.L.; Martinez, J.C.; Luque, I. Thermodynamic Dissection of the Binding Energetics of Proline-rich Peptides to the Abl-SH3 Domain: Implications for Rational Ligand Design. J. Mol. Biol. 2004, 336, 527–537. [Google Scholar] [CrossRef]
- McNemar, C.; Snow, M.E.; Windsor, W.T.; Prongay, A.; Mui, P.; Zhang, R.; Durkin, J.; Le, H.V.; Weber, P.C. Thermodynamic and structural analysis of phosphotyrosine polypeptide binding to Grb2-SH2. Biochemistry 1997, 36, 10006–10014. [Google Scholar] [CrossRef]
- Wang, C.; Pawley, N.H.; Nicholson, L.K. The role of backbone motions in ligand binding to the c-Src SH3 domain. J. Mol. Biol. 2001, 313, 873–887. [Google Scholar] [CrossRef] [Green Version]
- Org, T.; Chignola, F.; Chignola, F.; Hetényi, C.; Gaetani, M.; Rebane, A.; Liiv, I.; Maran, U.; Mollica, L.; Bottomley, M.J.; et al. The autoimmune regulator PHD finger binds to non-methylated histone H3K4 to activate gene expression. EMBO Rep. 2008, 9, 370–376. [Google Scholar] [CrossRef]
- Chrencik, J.E.; Brooun, A.; Recht, M.; Kraus, M.L.; Koolpe, M.; Kolatkar, A.; Bruce, R.H.; Martiny-Baron, G.; Widmer, H.; Pasquale, E.B.; et al. Thermodynamic and structural analysis of phosphotyrosine polypeptide binding to Grb2-SH2. Biochemistry 2006, 14, 321–330. [Google Scholar]
- Kozlov, G.; De Crescenzo, G.; Lim, N.S.; Siddiqui, N.; Fantus, D.; Kahvejian, A.; Trempe, J.-F.; Elias, D.; Ekiel, I.; Sonenberg, N.; et al. Structural basis of ligand recognition by PABC, a highly specific peptide-binding domain found in poly(A)-binding protein and a HECT ubiquitin ligase. EMBO J. 2004, 23, 272–281. [Google Scholar] [CrossRef]
- Day, C.L.; Smits, C.; Fan, F.C.; Lee, E.F.; Fairlie, W.D.; Hinds, M.G. Structure of the BH3 domains from the p53-inducible BH3-only proteins Noxa and Puma in complex with Mcl-1. J. Mol. Biol. 2008, 380, 958–971. [Google Scholar] [CrossRef]
- Lu, Z.; Zhang, Y. Interfacing ab initio Quantum Mechanical Method with Classical Drude Osillator Polarizable Model for Molecular Dynamics Simulation of Chemical Reactions. J. Chem. Theory Comput. 2008, 4, 1237–1248. [Google Scholar] [CrossRef]
- Vanommeslaeghe, K.; Guvench, O.; MacKerell, A.D. Molecular Mechanics. Curr. Pharm. Des. 2014, 20, 3281–3292. [Google Scholar] [CrossRef] [Green Version]
- Ganoth, A.; Friedman, R.; Nachliel, E.; Gutman, M. A Molecular Dynamics Study and Free Energy Analysis of Complexes between the Mlc1p Protein and Two IQ Motif Peptides. Biophys. J. 2006, 91, 2436–2450. [Google Scholar] [CrossRef] [Green Version]
- Fenley, A.T.; Muddana, H.S.; Gilson, M.K. Entropy—Enthalpy transduction caused by conformational shifts can obscure the forces driving protein—Ligand binding. Proc. Natl. Acad. Sci. USA 2012, 109, 20006–20011. [Google Scholar] [CrossRef]
- Huggins, D.J. Quantifying the Entropy of Binding for Water Molecules in Protein Cavities by Computing Correlations. Biophys. J. 2015, 108, 928–936. [Google Scholar] [CrossRef] [Green Version]
- Dougherty, D.A. Cation-π Interactions Involving Aromatic Amino Acids. J. Nutr. 2007, 137 (Suppl. S1), 1504–1508. [Google Scholar] [CrossRef]
- Majumdar, S.; Maiti, S.; Ghosh Dastidar, S. Dynamic and Static Water Molecules Complement the TN16 Conformational Heterogeneity Inside the Tubulin Cavity. Biochemistry 2016, 55, 335–347. [Google Scholar] [CrossRef]
- Poole, P.L.; Finney, J.L. Hydration-induced conformational and flexibility changes in lysozyme at low water content. Int. J. Biol. Macromol. 1983, 5, 308–310. [Google Scholar] [CrossRef]
- Jukič, M.; Konc, J.; Gobec, S.; Janežič, D. Identification of Conserved Water Sites in Protein Structures for Drug Design. J. Chem. Inf. Model. 2017, 57, 3094–3103. [Google Scholar] [CrossRef]
- Choi, H.; Kang, H.; Park, H. New solvation free energy function comprising intermolecular solvation and intramolecular self-solvation terms. J. Chem. 2013, 5, 5–8. [Google Scholar] [CrossRef]
- Cui, G.; Swails, J.M.; Manas, E.S. SPAM: A Simple Approach for Profiling Bound Water Molecules. J. Chem. Theory Comput. 2013, 9, 5539–5549. [Google Scholar] [CrossRef]
- García-Sosa, A.T.; Stuart, F.C.; Mancera, R.L. Including Tightly-Bound Water Molecules in de Novo Drug Design. Exemplification through the in Silico Generation of Poly(ADP-ribose)polymerase Ligands. J. Chem. Inf. Model. 2005, 45, 624–633. [Google Scholar] [CrossRef]
- Huggins, D.J.; Tidor, B. Systematic placement of structural water molecules for improved scoring of protein—Ligand interactions. Protein Eng. Des. Sel. 2011, 24, 777–789. [Google Scholar] [CrossRef]
- Ladbury, J.E. Just add water! The effect of water on the specificity of proteinligand binding sites and its potential application to drug design. Cell Chem. Biol. 1996, 3, 973–980. [Google Scholar]
- Lloyd, D.G.; García-Sosa, A.T.; Alberts, I.L.; Todorov, N.P.; Mancera, R.L. The effect of tightly bound water molecules on the structural interpretation of ligand-derived pharmacophore models. J. Comp. Aided Mol. Des. 2004, 18, 89–100. [Google Scholar] [CrossRef]
- Michel, J.; Tirado-Rives, J.; Jorgensen, W.L. Energetics of Displacing Water Molecules from Protein Binding Sites: Consequences for Ligand Optimization. J. Am. Chem. Soc. 2009, 131, 15403–15411. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Z. Computational Modeling of Solvent Effects on Protein-Ligand Interactions Using Fully Polarizable Continuum Model and Rational Drug Design. Commun. Comput. Phys. 2013, 13, 31–60. [Google Scholar] [CrossRef]
- Kunstmann, S.; Gohlke, U.; Broeker, N.K.; Roske, Y.; Heinemann, U.; Santer, M.; Barbirz, S. Solvent Networks Tune Thermodynamics of Oligosaccharide Complex Formation in an Extended Protein Binding Site. J. Am. Chem. Soc. 2018, 140, 10447–10455. [Google Scholar] [CrossRef]
- Brysbaert, G.; Blossey, R.; Lensink, M.F. The Inclusion of Water Molecules in Residue Interaction Networks Identifies Additional Central Residues. Front. Mol. Biosci. 2018, 5, 88. [Google Scholar] [CrossRef]
- Quiocho, F.A.; Wilson, D.K.; Vyas, N.K. Substrate specificity and affinity of a protein modulated by bound water molecules. Nature 1989, 340, 404–407. [Google Scholar] [CrossRef]
- Jeszenői, N.; Bálint, M.; Horváth, I.; van der Spoel, D.; Hetényi, C. Exploration of interfacial hydration networks of target—Ligand complexes. J. Chem. Inf. Model. 2016, 56, 148–158. [Google Scholar] [CrossRef]
- Afonine, P.V.; Grosse-Kunstleve, R.W.; Adams, P.D.; Urzhumstev, A. Bulk-solvent and overall scaling revisited: Faster calculations, improved results. Acta Cryst. D Biol. Cryst. 2013, 69 Pt 4, 625–634. [Google Scholar] [CrossRef]
- Badger, J. Modeling and refinement of water molecules and disordered solvent. Meth. Enzymol. 1997, 277, 344–352. [Google Scholar]
- Finney, J.L. The organization and function of water in protein crystals. Philos. Trans. R. Soc. Lond. B Biol. Sci. 1977, 278, 3–32. [Google Scholar] [CrossRef]
- Halle, B. Protein hydration dynamics in solution: A critical survey. Philos. Trans. R. Soc. Lond. B 2004, 359, 1207–1224. [Google Scholar] [CrossRef]
- Elsässer, S.J.; Huang, H.; Lewis, P.W.; Chin, J.W.; Allis, C.D.; Patel, D.J. DAXX envelops a histone H3.3–H4 dimer for H3.3-specific recognition. Nature 2012, 491, 560–565. [Google Scholar] [CrossRef]
- Abel, R.; Young, T.; Farid, R.; Berne, B.; Friesner, R. Role of the active-site solvent in the thermodynamics of factor Xa ligand binding. J. Am. Chem. Soc. 2008, 130, 2817–2831. [Google Scholar] [CrossRef]
- Pearlstein, R.; Hu, Q.; Zhou, J.; Yowe, D.; Levell, J.; Dale, B.; Kaushik, V.; Daniels, D.; Hanrahan, S.; Sherman, W.; et al. New hypotheses about the structure-function of proprotein convertase subtilisin/kexin type 9: Analysis of the epidermal growth factor-like repeat A docking site using WaterMap. Proteins 2010, 78, 2571–2586. [Google Scholar] [CrossRef]
- Vukovic, S.; Brennan, P.E.; Huggins, D. Exploring the role of water in molecular recognition: Predicting protein ligandability using a combinatorial search of surface hydration sites. J. Phys. Condens. Matter. 2016, 28, 344007. [Google Scholar] [CrossRef]
- Hylsová, M.; Carbain, B.; Fanfrlik, J.; Lepsik, M. Explicit treatment of active-site waters enhances quantum mechanical/implicit solvent scoring: Inhibition of CDK2 by new pyrazolo [1,5-a] pyrimidines. Eur. J. Med. Chem. 2017, 126, 1118–1128. [Google Scholar] [CrossRef]
- García-Sosa, A.T.; Mancera, R.L. Free Energy Calculations of Mutations Involving a Tightly Bound Water Molecule and Ligand Substitutions in a Ligand-Protein Complex. Mol. Inf. 2010, 29, 589–600. [Google Scholar] [CrossRef]
- Lee, S.H.; Rossky, P.J. A comparison of the structure and dynamics of liquid water at hydrophobic and hydrophilic surfaces—A molecular dynamics simulation study. J. Chem. Phys. 1994, 100, 3334–3345. [Google Scholar] [CrossRef]
- Watanabe, G.; Nakajima, D.; Hiroshima, A.; Suzuki, H.; Yoneda, S. Analysis of water channels by molecular dynamics simulation of heterotetrameric sarcosine oxidase. Biophys. Physicobiol. 2015, 12, 131–137. [Google Scholar] [CrossRef]
- Patodia, S.; Bagaria, A.; Chopra, D. Molecular Dynamics Simulation of Proteins: A Brief Overview. J. Phys. Chem. Bioph. 2014, 4, 1. [Google Scholar] [CrossRef]
- Jeszenői, N.; Horváth, I.; Bálint, M.; van der Spoel, D.; Hetényi, C. Mobility-based prediction of hydration structures of protein surfaces. Bioinformatics 2015, 31, 1959–1965. [Google Scholar] [CrossRef]
- Field, M.J.; Bash, P.A.; Karplus, M. A combined quantum mechanical and molecular mechanical potential for molecular dynamics simulations. J. Comp. Chem. 1990, 11, 700–733. [Google Scholar] [CrossRef]
- Hayik, S.A.; Dunbrack, R.; Merz, K.M. A Mixed QM/MM Scoring Function to Predict Protein-Ligand Binding Affinity. J. Chem. Theory Comp. 2010, 6, 3079–3091. [Google Scholar] [CrossRef]
- Menikarachchi, L.C.; Gascón, J.A. QM/MM Approaches in Medicinal Chemistry Research. Curr. Top. Med. Chem. 2010, 10, 46–54. [Google Scholar] [CrossRef]
- Van der Kamp, M.W.; Mulholland, A.J. Combined Quantum Mechanics/Molecular Mechanics (QM/MM) Methods in Computational Enzymology. Biochemistry 2013, 52, 2708–2728. [Google Scholar] [CrossRef]
- Warshel, A.; Levitt, M. Theoretical studies of enzymic reactions: Dielectric, electrostatic and steric stabilization of the carbonium ion in the reaction of lysozyme. J. Mol. Biol. 1976, 103, 227–249. [Google Scholar] [CrossRef]
- Vidossich, P.; Magistrato, A. QM/MM Molecular Dynamics Studies of Metal Binding Proteins. Biomolecules 2014, 4, 616–645. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.W.; Zhang, J.Z.H. Molecular fractionation with conjugate caps for full quantum mechanical calculation of protein—Molecule interaction energy. J. Chem. Phys. 2003, 119, 3599–3605. [Google Scholar] [CrossRef]
- Nikitina, E.; Sulimov, V.; Zayets, V.; Zaitseva, N. Semiempirical calculations of binding enthalpy for protein–ligand complexes. Int. J. Quantum Chem. 2004, 97, 747–763. [Google Scholar] [CrossRef]
- Nikitina, E.; Sulimov, V.; Zayets, V.; Zaitseva, N. Mixed Implicit/Explicit Solvation Models in Quantum Mechanical Calculations of Binding Enthalpy for Protein–Ligand Complexes. Int. J. Quantum Chem. 2006, 106, 1943–1963. [Google Scholar] [CrossRef]
- Dobeš, P.; Otyepka, M.; Strnad, M.; Hobza, P. Interaction Energies for the Purine Inhibitor Roscovitine with Cyclin-Dependent Kinase 2: Correlated Ab Initio Quantum-Chemical, DFT and Empirical Calculations. Chemistry 2006, 12, 4297–4304. [Google Scholar] [CrossRef]
- Freire, E. Do Enthalpy and Entropy Distinguish First in Class from Best in Class? Drug Discov. Today 2008, 13, 869–874. [Google Scholar] [CrossRef]
- Ferenczy, G.G.; Keserű, G.M. Thermodynamics guided lead discovery and optimization. Drug Discov. Today 2010, 15, 919–932. [Google Scholar] [CrossRef]
- Hann, M.M.; Keserü, G.M. Finding the sweet spot: The role of nature and nurture in medicinal chemistry. Nat. Rev. Drug. Discov. 2012, 11, 355–365. [Google Scholar] [CrossRef]
- Zhang, D.W.; Xiang, Y.; Zhang, J.Z.H. New Advance in Computational Chemistry: Full Quantum Mechanical ab Initio Computation of Streptavidin—Biotin Interaction Energy. J. Phys. Chem. B 2003, 107, 12039–12041. [Google Scholar] [CrossRef]
- Zhang, D.W.; Xiang, Y.; Gao, A.M.; Zhang, J.Z.H. Quantum mechanical map for protein-ligand binding with application to β-trypsin/benzamidine complex. J. Chem. Phys. 2004, 120, 1145–1148. [Google Scholar] [CrossRef]
- Brown, S.; Shirts, M.; Mobley, D. Free-energy calculations in structure-based drug design. Drug Des. Struct. Ligand Based Approaches 2010, 2010, 61–86. [Google Scholar]
- Klamt, A.; Schüürmann, G. COSMO: A New Approach to Dielectric Screening in Solvents with Explicit Expressions for the Screening Energy and its Gradient. J. Chem. Soc. Perk. Trans. 1993, 2, 799–805. [Google Scholar] [CrossRef]
- Weichenberger, C.X.; Afonine, P.V.; Kantardjieff, K.; Rupp, B. The solvent component of macromolecular crystals. Acta Cryst. D Biol. Cryst. 2015, 71, 1023–1038. [Google Scholar] [CrossRef] [Green Version]
- Halle, B. Biomolecular cryocrystallography: Structural changes during flash-cooling. Proc. Natl. Acad. Sci. USA 2004, 101, 4793–4798. [Google Scholar] [CrossRef] [Green Version]
- Schmidtke, P.; Barril, X.; Luque, F.J.; Murray, J.B. Shielded hydrogen bonds as structural determinants of binding kinetics: Application in drug design. J. Am. Chem. Soc. 2011, 133, 18903–18910. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Guex, N.; Peitsch, M.C. SWISS-MODEL and the Swiss-PdbViewer: An environment for comparative protein modeling. Electrophoresis 1997, 18, 2714–2723. [Google Scholar] [CrossRef]
- Schrödinger Release 2019-3: Maestro; Schrödinger, LLC: New York, NY, USA, 2019.
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general Amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Gille, A.L.; Dutmer, B.C.; Gilbert, T.M. PCMODEL 9.2. J. Am. Chem. Soc. 2009, 131, 5714. [Google Scholar] [CrossRef]
- Halgren, T. Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94. J. Comput. Chem. 1996, 17, 490–519. [Google Scholar] [CrossRef]
- Stewart, J.J.P. MOPAC2009, 2009; Steward Computational Chemistry: Colorado Springs, CO, USA, 2008. [Google Scholar]
- Stewart, J.J.P. Optimization of parameters for semiempirical methods V: Modification of NDDO approximations and application to 70 elements. J. Mol. Model. 2007, 13, 1173–1213. [Google Scholar] [CrossRef] [Green Version]
- Vanquelef, E.; Simon, S.; Marquant, G.; Garcia, E.; Klimerak, G.; Delepine, J.C.; Cieplak, P.; Dupradeau, F.Y. R.E.D. Server: A web service for deriving RESP and ESP charges and building force field libraries for new molecules and molecular fragments. Nucleic Acids Res. 2011, 39, W511–W517. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Krishnan, R.; Binkley, J.S.; Seeger, R.; Pople, J.A. Self-consistent molecular-orbital methods. XX. Basis set for correlated wave-functions. J. Chem. Phys. 1980, 72, 650–654. [Google Scholar] [CrossRef]
- Schmidt, M.W.; Baldridge, K.K.; Boatz, J.A.; Elbert, S.T.; Gordon, M.S.; Jensen, J.H.; Koseki, S.; Matsunaga, N.; Nguyen, K.A.; Su, S.J.; et al. General atomic and molecular electronic-structure system. J. Comp. Chem. 1993, 14, 1347–1363. [Google Scholar] [CrossRef]
- Case, D.; Darden, T.; Cheatham Iii, T.; Simmerling, C.; Wang, J.; Duke, R.; Luo, R.; Walker, R.; Zhang, W.; Merz, K. AmberTools, 15; Amber; University of California: San Francisco, CA, USA, 2015. [Google Scholar]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef]
- Pronk, S.; Pall, S.; Schulz, R.; Larsson, P.; Bjelkmar, P.; Apostolov, R.; Shirts, M.R.; Smith, J.C.; Kasson, P.M.; van der Spoel, D.; et al. GROMACS 4.5: A high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 2013, 29, 845–854. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Lindorff-Larsen, K.; Piana, S.; Palmo, K.; Maragakis, P.; Klepeis, J.L.; Dror, R.O.; Shaw, D.E. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins 2010, 78, 1950–1958. [Google Scholar] [CrossRef] [Green Version]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef] [Green Version]
- Darden, T.; York, D.; Pedersen, L. Particle Mesh Ewald—An N.log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Nose, S.; Klein, M.L. Constant pressure molecular-dynamics for molecular systems. Mol. Phys. 1983, 50, 1055–1076. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single-crystals—A new molecular-dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef]
- Wang, J.; Cieplak, P.; Li, J.; Cai, Q.; Hsieh, M.J.; Luo, R.; Duan, Y. Development of polarizable models for molecular mechanical calculations. 4. van der Waals parametrization. J. Phys. Chem. B 2012, 116, 7088–7101. [Google Scholar] [CrossRef]
- Hanson, R.M.; Prilusky, J.; Renjian, Z.; Nakane, T.; Sussman, J.L. JSmol and the Next-Generation Web-Based Representation of 3D Molecular Structure as Applied to Proteopedia. Isr. J. Chem. 2013, 53, 207–216. [Google Scholar] [CrossRef]
- Stewart, J.J.P. Openmopac Online Manual. 2016. Available online: http://www.openmopac.net (accessed on 1 March 2013).
- Stewart, J.J.P. Optimization of parameters for semiempirical methods VI: More modifications to the NDDO approximations and re-optimization of parameters. J. Mol. Model. 2013, 19, 1–32. [Google Scholar] [CrossRef]
- Stewart, J.J.P. Application of localized molecular orbitals to the solution of semiempirical self-consistent field equations. Int. J. Quant. Chem. 1996, 58, 133–146. [Google Scholar] [CrossRef]
- Baker, J. An Algorithm for the Location of Transition States. J. Comp. Chem. 1986, 7, 385. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| System a | Res b (Å) | Target | Ligand | Water Count | ΔHb(exp) d | |||
|---|---|---|---|---|---|---|---|---|
| Name | MW c | Shell 1 | Shell 2 | Shell 3 | kcal mol−1 | |||
| 3ptb_ben | 1.7 | beta-trypsin | benzamidine | 121.2 | 1 | 6 | 7 | −4.507 [2] |
| 3ptb_pme | 1.7 | beta-trypsin | p-methylbenzamidine | 135.2 | 1 | 5 | 6 | −4.412 [2] |
| 3ptb_pam | 1.7 | beta-trypsin | p-aminobenzamidine | 136.2 | 3 | 4 | 7 | −6.417 [2] |
| 3ptb_pmo | 1.7 | beta-trypsin | p-methoxybenzamidine | 151.2 | 1 | 6 | 7 | −3.742 [2] |
| 3ptb_pad | 1.7 | beta-trypsin | p-amidinobenzamidine | 164.2 | 2 | 8 | 10 | −2.935 [2] |
| 1k1l | 2.5 | bovine trypsin | NAPe-piperazine | 467.6 | 5 | 10 | 15 | −7.863 [4] |
| 1k1m | 2.2 | bovine trypsin | NAP e−4-acetyl-piperazine | 508.6 | 4 | 12 | 16 | −8.222 [4] |
| 1k1i | 2.2 | bovine trypsin | NAP e-D-pipecolinic acid | 508.6 | 2 | 13 | 15 | −10.899 [4] |
| 1k1j | 2.2 | bovine trypsin | NAP e-isopipecolinic acid methyl ester | 523.6 | 3 | 13 | 16 | −9.465 [4] |
| 1jyr | 1.55 | Grb2 SH2 domain | APS-PTR e-VNVQN | 1069.0 | 1 | 14 | 15 | −7.94 [6] |
| 1rlq | NA | C-src tyrosine kinase SH3 domain | RALPPLPRY | 1084.3 | 2 | 25 | 27 | −10.2 [7] |
| 2ke1 | NA | autoimmune regulator | ARTKQTARKS | 1150.3 | 12 | 15 | 27 | −9.2 [8] |
| 2bba | 1.65 | EphB4 receptor | NYLFSPNGPIARAW | 1606.8 | 12 | 15 | 27 | −15.5 [9] |
| 1jgn | NA | human poly(A)-binding protein | VVKSNLNPNAKEFVPGVKYGNI | 2389.8 | 14 | 34 | 48 | −14.8 [10] |
| 2roc | NA | induced myeloid leukemia cell differentiation protein homolog | EEEWAREIGAQLRRIADDLNAQYERRM | 3317.6 | 14 | 38 | 52 | −14.3 [11] |
| System | Vacuum | COSMO | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Dry | Shell 1 | Shell 2 | Shell 3 | Dry | Shell 1 | Shell 2 | Shell 3 | Shell 3 b | |
| |ε| a | |||||||||
| 3ptb_ben | 3.70 | 3.18 | 2.90 | 1.93 | 3.73 | 2.33 | 2.45 | 1.18 | 0.85 |
| 3ptb_pme | 3.60 | 3.09 | 3.05 | 2.03 | 0.53 | 1.12 | 2.48 | 0.95 | 1.22 |
| 3ptb_pam | 1.74 | 1.29 | 0.88 | 0.03 | 0.01 | 0.02 | 0.44 | 0.92 | 3.03 |
| 3ptb_pmo | 4.45 | 3.91 | 3.71 | 2.64 | 3.59 | 3.25 | 3.32 | 2.24 | 0.33 |
| 3ptb_pad | 5.65 | 5.11 | 5.04 | 4.25 | 3.03 | 3.12 | 4.32 | 3.22 | 1.37 |
| 1k1l | 0.56 | 0.39 | 0.10 | 0.10 | 2.59 | 1.05 | 0.56 | 0.43 | 1.74 |
| 1k1m | 0.16 | 0.56 | 0.36 | 0.79 | 2.71 | 1.17 | 0.75 | 1.50 | 3.12 |
| 1k1i | 2.67 | 3.19 | 2.95 | 3.51 | 2.80 | 3.62 | 2.88 | 3.34 | 4.60 |
| 1k1j | 1.43 | 1.81 | 1.60 | 2.10 | 0.57 | 2.60 | 1.47 | 2.32 | 3.75 |
| 1jyr | 0.78 | 0.28 | 0.53 | 0.09 | 1.73 | 0.36 | 0.40 | 0.53 | 0.36 |
| 1rlq | 0.67 | 0.64 | 0.27 | 0.33 | 0.37 | 2.13 | 0.61 | 0.24 | 0.16 |
| 2ke1 | 2.54 | 4.67 | 4.66 | 5.28 | 4.38 | 4.21 | 5.73 | 2.46 | 2.89 |
| 2bba | 7.25 | 6.38 | 7.21 | 6.23 | 4.34 | 2.13 | 6.58 | 3.77 | 3.31 |
| 1jgn | 5.88 | 5.24 | 4.75 | 2.56 | 1.61 | 0.27 | 3.25 | 0.25 | 1.36 |
| 2roc | 4.96 | 4.11 | 3.74 | 1.26 | 2.76 | 1.24 | 3.05 | 1.71 | 3.92 |
| R2 | 0.06 | 0.18 | 0.19 | 0.44 | 0.51 | 0.65 | 0.33 | 0.73 | 0.93 |
| R2(cv) c | 0.00 | 0.01 | 0.02 | 0.22 | 0.34 | 0.54 | 0.07 | 0.65 | 0.91 |
| F | 0.81 | 2.77 | 3.14 | 10.20 | 13.46 | 24.28 | 6.36 | 34.55 | 179.66 |
| RMSE a | 4.02 | 3.76 | 3.72 | 3.10 | 2.90 | 2.45 | 3.40 | 2.17 | 2.65 |
| tα | 0.90 | 1.66 | 1.77 | 3.19 | 3.67 | 4.93 | 2.52 | 5.88 | 13.40 |
| tβ | −5.56 | −5.04 | −4.68 | −3.90 | −2.18 | −3.99 | −4.24 | −2.81 | - |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Horváth, I.; Jeszenői, N.; Bálint, M.; Paragi, G.; Hetényi, C. A Fragmenting Protocol with Explicit Hydration for Calculation of Binding Enthalpies of Target-Ligand Complexes at a Quantum Mechanical Level. Int. J. Mol. Sci. 2019, 20, 4384. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20184384
Horváth I, Jeszenői N, Bálint M, Paragi G, Hetényi C. A Fragmenting Protocol with Explicit Hydration for Calculation of Binding Enthalpies of Target-Ligand Complexes at a Quantum Mechanical Level. International Journal of Molecular Sciences. 2019; 20(18):4384. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20184384
Chicago/Turabian StyleHorváth, István, Norbert Jeszenői, Mónika Bálint, Gábor Paragi, and Csaba Hetényi. 2019. "A Fragmenting Protocol with Explicit Hydration for Calculation of Binding Enthalpies of Target-Ligand Complexes at a Quantum Mechanical Level" International Journal of Molecular Sciences 20, no. 18: 4384. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20184384