Interplay Between Mitochondrial Peroxiredoxins and ROS in Cancer Development and Progression


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Mitochondrial Prdxs
3. Characteristics of mitochondrial Prdxs
4. ROS and Mitochondrial Prdxs
5. ROS and Mitochondrial Antioxidants in Oncogenic Signaling
6. Prdx3 and Carcinogenesis
7. Prdx5 and Carcinogenesis
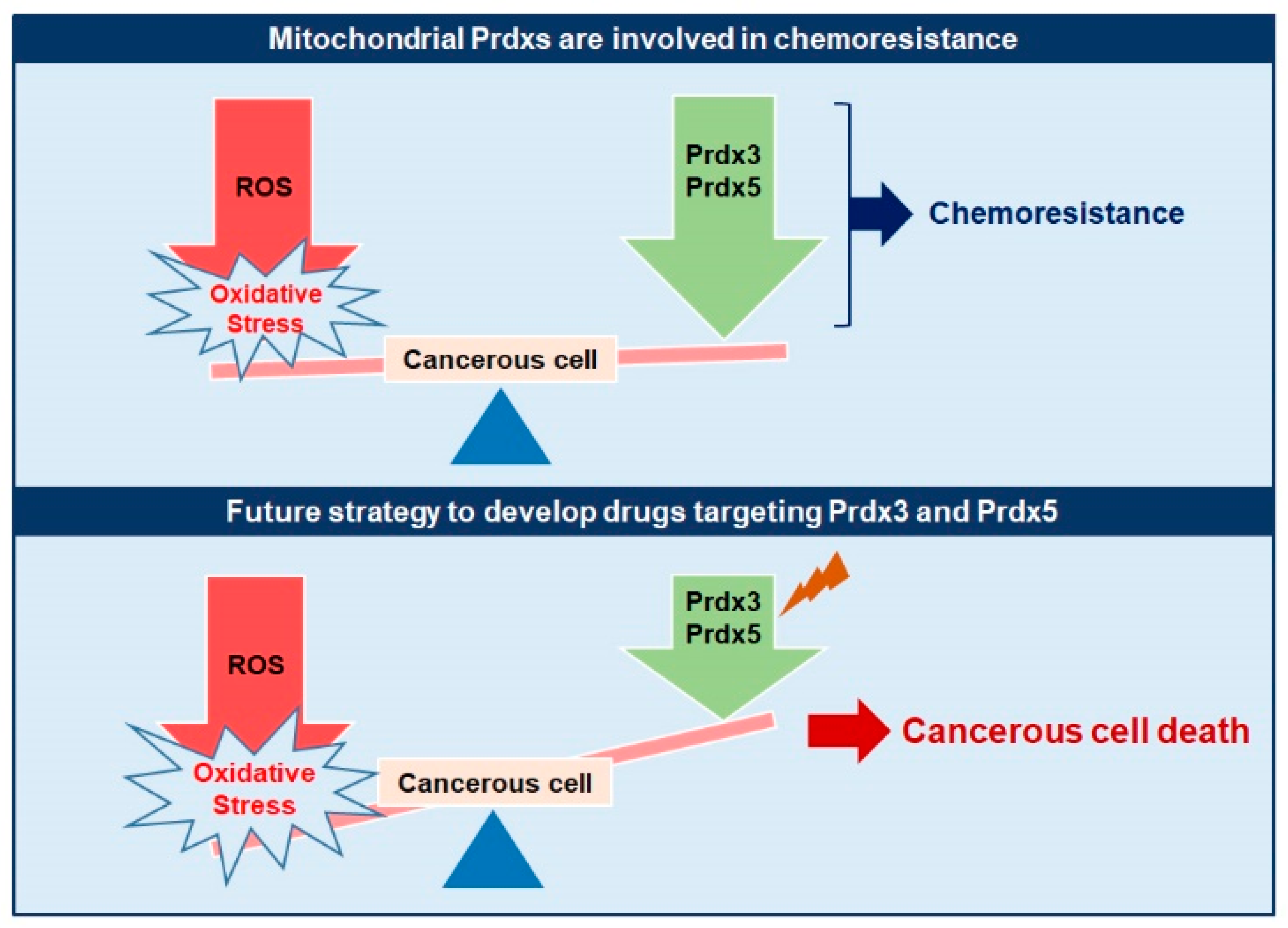
8. Mitochondrial Prdxs and Chemoresistance
9. Concluding Remarks
10. Future Directions
- Most of the studies reporting the upregulation of mitochondrial Prdxs in human cancers are cell-line specific, it will be more advantageous to design in-vivo studies to explore the interaction of these Prdxs in cellular environment.
- Mitochondrial Prdxs have the ability to function as molecular chaperons, enzyme activators, and can be involved in protein-protein interactions beyond their enzymatic peroxidase functions. Accordingly, the transgenic animal (mouse) models should be used to demonstrate the role of mitochondrial Prdxs in oncogenic signaling. It will be beneficial to understand the exact mechanism of action of these Prdxs in cancer and to design effective drugs targeting a particular pathway associated with cancer survival and progression.
- Although a plethora of studies describe the regulation of mitochondrial Prdxs by different transcription factors, oncogenes, and microRNAs in different types of cancer, but the exact mechanism of mitochondrial Prdxs in different types of cancers and their upstream and downstream regulators is lacking. Incorporation of variety of omics techniques i.e., transcriptomics, proteomics, and metabolomics into the in vitro and in vivo studies of mitochondrial Prdxs in cancer development can help to elucidate signaling mechanism in future studies.
- More clinical investigations are needed to evaluate the differences in the expression of Prdx3 and Prdx5 between normal and diseased state. In addition, the expression of mitochondrial Prdxs during the early and late stage of cancer should be analyzed to demonstrate their role as anti-oncogenic or pro-oncogenic in different cellular context.
- The upregulated expression of Prdx3 and Prdx5 is associated with the development of chemoresistance in different cancers and selective targeting of these mitochondrial Prdxs can lead to sensitization of cancer cells to chemotherapy. This fact should be investigated in detail to unveil the underlying mechanisms.
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ROS | Reactive oxygen species |
| Prdxs | Peroxiredoxins |
| ATP | Adenosine triphosphate |
| SOD | Superoxide dismutase |
| Cys | Cysteine |
| Cp | Peroxidatic cysteine |
| Cr | Resolving cysteine |
| ERK | Extracellular signal related kinase |
| JNK | Jun N-terminal kinase |
| MAPK | Mitogen activated protein kinase |
| PTP | Protein tyrosine phosphatases |
| PTEN | Phosphatase and tensin homologues |
| CSCs | Cancerous stem cells |
| NRF2 | Nuclear factor erythroid 2 related factor |
| KEAP1 | Kelch like ECH associated proteins |
| AREs | Antioxidant responsive elements |
| HIFs | Hypoxia inducible factors |
| PHDs | Prolyl hydroxylases |
| pVHL | von Hippel-Laundau protein |
| CCRCC | Clear cell renal cell carcinoma |
| FOXM1 | Forkhead box protein 1 |
| LNCaP | Lymph node carcinoma of prostate |
| TIMP1 | TIMP metallopeptidase inhibitor 1 |
| ECM | Extracellular matrix |
| HepG2 | Human hepatocellular carcinoma/hepatoma cell lines |
| MCF-7 | Mitichigan cancer foundation-7 cancerous cell lines |
| HER2 | Human epithelial growth factor receptor 2 |
| Trx2 | Thioredoxin 2 |
| TrxR2 | Thioredoxin reductase 2 |
| Srx | Sulfiredoxin |
| MM | Malignant mesothelioma |
| NF-κB | Nuclear factor κB |
| ARE | Antioxidant responsive element |
| IRE | Insulin responsive element |
| GRE | Glucocorticoid responsive element |
| Ets ½ | E twenty six transcription factor 1 and 2 |
| HMGB1 | High mobility group protein B1 |
References
- Panchal, K.; Tiwari, A.K. Mitochondrial dynamics, a key executioner in neurodegenerative diseases. Mitochondrion 2018. [Google Scholar] [CrossRef] [PubMed]
- Austad, S.N. The Comparative Biology of Mitochondrial Function and the Rate of Aging. Integr. Comp. Biol. 2018, 58, 559–566. [Google Scholar] [CrossRef] [PubMed]
- Moreira, O.C.; Estebanez, B.; Martinez-Florez, S.; de Paz, J.A.; Cuevas, M.J.; Gonzalez-Gallego, J. Mitochondrial Function and Mitophagy in the Elderly: Effects of Exercise. Oxid. Med. Cell. Longev. 2017, 2017, 2012798. [Google Scholar] [CrossRef] [PubMed]
- Scorrano, L. Keeping mitochondria in shape: A matter of life and death. Eur. J. Clin. Investig. 2013, 43, 886–893. [Google Scholar] [CrossRef] [PubMed]
- Horbay, R.; Bilyy, R. Mitochondrial dynamics during cell cycling. Apoptosis 2016, 21, 1327–1335. [Google Scholar] [CrossRef] [PubMed]
- Jeong, S.Y.; Seol, D.W. The role of mitochondria in apoptosis. BMB Rep. 2008, 41, 11–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Figueiredo-Pereira, C.; Oudot, C.; Vieira, H.L.; Brenner, C. Mitochondrion: A Common Organelle for Distinct Cell Deaths? Int. Rev. Cell Mol. Biol. 2017, 331, 245–287. [Google Scholar] [PubMed]
- Parsons, M.J.; Green, D.R. Mitochondria in cell death. Essays Biochem. 2010, 47, 99–114. [Google Scholar] [CrossRef] [Green Version]
- Fulda, S. Mitochondria, redox signaling and cell death in cancer. Biol. Chem. 2016, 397, 583. [Google Scholar] [CrossRef]
- Chandel, N.S. Mitochondria as signaling organelles. BMC Biol. 2014, 12, 34. [Google Scholar] [CrossRef]
- Chandel, N.S. Evolution of Mitochondria as Signaling Organelles. Cell Metab. 2015, 22, 204–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andreyev, A.Y.; Kushnareva, Y.E.; Starkov, A.A. Mitochondrial metabolism of reactive oxygen species. Biochemistry (Moscow) 2005, 70, 200–214. [Google Scholar] [CrossRef] [PubMed]
- Turrens, J.F. Mitochondrial formation of reactive oxygen species. J. Physiol. 2003, 552 Pt 2, 335–344. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, X.; Vikash, V.; Ye, Q.; Wu, D.; Liu, Y.; Dong, W. ROS and ROS-Mediated Cellular Signaling. Oxid. Med. Cell. Longev. 2016, 2016, 4350965. [Google Scholar] [CrossRef] [PubMed]
- Brieger, K.; Schiavone, S.; Miller, F.J., Jr.; Krause, K.H. Reactive oxygen species: From health to disease. Swiss Med. Wkly. 2012, 142, w13659. [Google Scholar] [CrossRef] [PubMed]
- Roy, J.; Galano, J.M.; Durand, T.; Le Guennec, J.Y.; Lee, J.C. Physiological role of reactive oxygen species as promoters of natural defenses. FASEB J. 2017, 31, 3729–3745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fridovich, I. The biology of oxygen radicals. Science 1978, 201, 875–880. [Google Scholar] [CrossRef] [PubMed]
- Grivennikova, V.G.; Vinogradov, A.D. Mitochondrial production of reactive oxygen species. Biochemistry (Moscow) 2013, 78, 1490–1511. [Google Scholar] [CrossRef]
- Wong, H.S.; Dighe, P.A.; Mezera, V.; Monternier, P.A.; Brand, M.D. Production of superoxide and hydrogen peroxide from specific mitochondrial sites under different bioenergetic conditions. J. Biol. Chem. 2017, 292, 16804–16809. [Google Scholar] [CrossRef] [Green Version]
- Holzerova, E.; Prokisch, H. Mitochondria: Much ado about nothing? How dangerous is reactive oxygen species production? Int. J. Biochem. Cell Biol. 2015, 63, 16–20. [Google Scholar] [CrossRef] [Green Version]
- Teppo, H.R.; Soini, Y.; Karihtala, P. Reactive Oxygen Species-Mediated Mechanisms of Action of Targeted Cancer Therapy. Oxid. Med. Cell. Longev. 2017, 2017, 1485283. [Google Scholar] [CrossRef] [PubMed]
- Hybertson, B.M.; Gao, B.; Bose, S.K.; McCord, J.M. Oxidative stress in health and disease: The therapeutic potential of Nrf2 activation. Mol. Asp. Med. 2011, 32, 234–246. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B. Free radicals and antioxidants: Updating a personal view. Nutr. Rev. 2012, 70, 257–265. [Google Scholar] [CrossRef] [PubMed]
- Schieber, M.; Chandel, N.S. ROS function in redox signaling and oxidative stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P. Antioxidants as therapies: Can we improve on nature? Free Radic. Biol. Med. 2014, 66, 20–23. [Google Scholar] [CrossRef] [PubMed]
- Dandekar, A.; Mendez, R.; Zhang, K. Cross talk between ER stress, oxidative stress, and inflammation in health and disease. Methods Mol. Biol. 2015, 1292, 205–214. [Google Scholar] [PubMed]
- Kattoor, A.J.; Pothineni, N.V.K.; Palagiri, D.; Mehta, J.L. Oxidative Stress in Atherosclerosis. Curr. Atheroscler. Rep. 2017, 19, 42. [Google Scholar] [CrossRef]
- Oyewole, A.O.; Birch-Machin, M.A. Mitochondria-targeted antioxidants. FASEB J. 2015, 29, 4766–4771. [Google Scholar] [CrossRef]
- Bjorklund, G.; Chirumbolo, S. Role of oxidative stress and antioxidants in daily nutrition and human health. Nutrition 2017, 33, 311–321. [Google Scholar] [CrossRef]
- Cortassa, S.; O’Rourke, B.; Aon, M.A. Redox-optimized ROS balance and the relationship between mitochondrial respiration and ROS. Biochim. Biophys. Acta 2014, 1837, 287–295. [Google Scholar] [CrossRef]
- Ji, Y.; Chae, S.; Lee, H.K.; Park, I.; Kim, C.; Ismail, T.; Kim, Y.; Park, J.W.; Kwon, O.S.; Kang, B.S.; et al. Peroxiredoxin5 Controls Vertebrate Ciliogenesis by Modulating Mitochondrial Reactive Oxygen Species. Antioxid. Redox Signal. 2019, 30, 1731–1745. [Google Scholar] [CrossRef] [PubMed]
- Sies, H. Oxidative stress: A concept in redox biology and medicine. Redox Biol. 2015, 4, 180–183. [Google Scholar] [CrossRef] [PubMed]
- Rhee, S.G. Overview on Peroxiredoxin. Mol. Cells 2016, 39, 5. [Google Scholar]
- Rhee, S.G.; Kil, I.S. Multiple Functions and Regulation of Mammalian Peroxiredoxins. Annu. Rev. Biochem. 2017, 86, 749–775. [Google Scholar] [CrossRef] [PubMed]
- Chae, S.; Lee, H.K.; Kim, Y.K.; Sim, H.J.; Ji, Y.; Kim, C.; Ismail, T.; Park, J.W.; Kwon, O.S.; Kang, B.S.; et al. Peroxiredoxin1, a novel regulator of pronephros development, influences retinoic acid and Wnt signaling by controlling ROS levels. Sci. Rep. 2017, 7, 8874. [Google Scholar] [CrossRef] [PubMed]
- Karplus, P.A.; Hall, A. Structural survey of the peroxiredoxins. Subcell. Biochem. 2007, 44, 41–60. [Google Scholar] [PubMed]
- Wood, Z.A.; Schroder, E.; Robin Harris, J.; Poole, L.B. Structure, mechanism and regulation of peroxiredoxins. Trends Biochem. Sci. 2003, 28, 32–40. [Google Scholar] [CrossRef]
- Hall, A.; Nelson, K.; Poole, L.B.; Karplus, P.A. Structure-based insights into the catalytic power and conformational dexterity of peroxiredoxins. Antioxid. Redox Signal. 2011, 15, 795–815. [Google Scholar] [CrossRef]
- Schroder, E.; Littlechild, J.A.; Lebedev, A.A.; Errington, N.; Vagin, A.A.; Isupov, M.N. Crystal structure of decameric 2-Cys peroxiredoxin from human erythrocytes at 1.7 A resolution. Structure 2000, 8, 605–615. [Google Scholar] [CrossRef]
- Barranco-Medina, S.; Lazaro, J.J.; Dietz, K.J. The oligomeric conformation of peroxiredoxins links redox state to function. FEBS Lett. 2009, 583, 1809–1816. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.H.; Lee, W.; Kim, E.E. Crystal structures of human peroxiredoxin 6 in different oxidation states. Biochem. Biophys. Res. Commun. 2016, 477, 717–722. [Google Scholar] [CrossRef] [PubMed]
- Poole, L.B.; Nelson, K.J. Distribution and Features of the Six Classes of Peroxiredoxins. Mol. Cells 2016, 39, 53–59. [Google Scholar] [PubMed]
- Cao, Z.; Lindsay, J.G.; Isaacs, N.W. Mitochondrial peroxiredoxins. Subcell. Biochem. 2007, 44, 295–315. [Google Scholar] [PubMed]
- Yamamoto, T.; Matsui, Y.; Natori, S.; Obinata, M. Cloning of a housekeeping-type gene (MER5) preferentially expressed in murine erythroleukemia cells. Gene 1989, 80, 337–343. [Google Scholar] [PubMed]
- Nemoto, Y.; Yamamoto, T.; Takada, S.; Matsui, Y.; Obinata, M. Antisense RNA of the latent period gene (MER5) inhibits the differentiation of murine erythroleukemia cells. Gene 1990, 91, 261–265. [Google Scholar] [CrossRef]
- Yang, H.Y.; Jeong, D.K.; Kim, S.H.; Chung, K.J.; Cho, E.J.; Yang, U.; Lee, S.R.; Lee, T.H. The role of peroxiredoxin III on late stage of proerythrocyte differentiation. Biochem. Biophys. Res. Commun. 2007, 359, 1030–1036. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, K.; Copeland, N.G.; Jenkins, N.A.; Obinata, M. Mammalian antioxidant protein complements alkylhydroperoxide reductase (ahpC) mutation in Escherichia coli. Biochem. J. 1995, 307 Pt 2, 377–381. [Google Scholar] [CrossRef]
- Watabe, S.; Hiroi, T.; Yamamoto, Y.; Fujioka, Y.; Hasegawa, H.; Yago, N.; Takahashi, S.Y. SP-22 is a thioredoxin-dependent peroxide reductase in mitochondria. Eur. J. Biochem. 1997, 249, 52–60. [Google Scholar] [CrossRef]
- Garrard, L.J.; Goodman, J.M. Two genes encode the major membrane-associated protein of methanol-induced peroxisomes from Candida boidinii. J. Biol. Chem. 1989, 264, 13929–13937. [Google Scholar]
- Yamashita, H.; Avraham, S.; Jiang, S.; London, R.; Van Veldhoven, P.P.; Subramani, S.; Rogers, R.A.; Avraham, H. Characterization of human and murine PMP20 peroxisomal proteins that exhibit antioxidant activity in vitro. J. Biol. Chem. 1999, 274, 29897–29904. [Google Scholar] [CrossRef]
- Knoops, B.; Clippe, A.; Bogard, C.; Arsalane, K.; Wattiez, R.; Hermans, C.; Duconseille, E.; Falmagne, P.; Bernard, A. Cloning and characterization of AOEB166, a novel mammalian antioxidant enzyme of the peroxiredoxin family. J. Biol. Chem. 1999, 274, 30451–30458. [Google Scholar] [CrossRef] [PubMed]
- Seo, M.S.; Kang, S.W.; Kim, K.; Baines, I.C.; Lee, T.H.; Rhee, S.G. Identification of a new type of mammalian peroxiredoxin that forms an intramolecular disulfide as a reaction intermediate. J. Biol. Chem. 2000, 275, 20346–20354. [Google Scholar] [CrossRef] [PubMed]
- Kropotov, A.; Usmanova, N.; Serikov, V.; Zhivotovsky, B.; Tomilin, N. Mitochondrial targeting of human peroxiredoxin V protein and regulation of PRDX5 gene expression by nuclear transcription factors controlling biogenesis of mitochondria. FEBS J. 2007, 274, 5804–5814. [Google Scholar] [CrossRef] [PubMed]
- Knoops, B.; Goemaere, J.; Van der Eecken, V.; Declercq, J.P. Peroxiredoxin 5: Structure, mechanism, and function of the mammalian atypical 2-Cys peroxiredoxin. Antioxid. Redox Signal. 2011, 15, 817–829. [Google Scholar] [CrossRef] [PubMed]
- Trujillo, M.; Ferrer-Sueta, G.; Thomson, L.; Flohe, L.; Radi, R. Kinetics of peroxiredoxins and their role in the decomposition of peroxynitrite. Subcell. Biochem. 2007, 44, 83–113. [Google Scholar] [PubMed]
- Lambert, A.J.; Brand, M.D. Reactive oxygen species production by mitochondria. Methods Mol. Biol. 2009, 554, 165–181. [Google Scholar] [PubMed]
- Kimura, S.; Waszczak, C.; Hunter, K.; Wrzaczek, M. Bound by Fate: The Role of Reactive Oxygen Species in Receptor-Like Kinase Signaling. Plant Cell 2017, 29, 638–654. [Google Scholar] [CrossRef] [Green Version]
- Choudhury, F.K.; Rivero, R.M.; Blumwald, E.; Mittler, R. Reactive oxygen species, abiotic stress and stress combination. Plant J. 2017, 90, 856–867. [Google Scholar] [CrossRef]
- Li, Z.; Xu, X.; Leng, X.; He, M.; Wang, J.; Cheng, S.; Wu, H. Roles of reactive oxygen species in cell signaling pathways and immune responses to viral infections. Arch. Virol. 2017, 162, 603–610. [Google Scholar] [CrossRef]
- Finkel, T. From sulfenylation to sulfhydration: What a thiolate needs to tolerate. Sci. Signal. 2012, 5, pe10. [Google Scholar] [CrossRef]
- Messens, J.; Collet, J.F. Thiol-disulfide exchange in signaling: Disulfide bonds as a switch. Antioxid. Redox Signal. 2013, 18, 1594–1596. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.A.; Zhang, Q.; Wang, Y.; Ge, W.; Guo, D. Prediction of redox-sensitive cysteines using sequential distance and other sequence-based features. BMC Bioinform. 2016, 17, 316. [Google Scholar] [CrossRef] [PubMed]
- Deponte, M.; Lillig, C.H. Enzymatic control of cysteinyl thiol switches in proteins. Biol. Chem. 2015, 396, 401–413. [Google Scholar] [CrossRef] [PubMed]
- Kil, I.S.; Ryu, K.W.; Lee, S.K.; Kim, J.Y.; Chu, S.Y.; Kim, J.H.; Park, S.; Rhee, S.G. Circadian Oscillation of Sulfiredoxin in the Mitochondria. Mol. Cell 2015, 59, 651–663. [Google Scholar] [CrossRef] [PubMed]
- Mishanina, T.V.; Libiad, M.; Banerjee, R. Biogenesis of reactive sulfur species for signaling by hydrogen sulfide oxidation pathways. Nat. Chem. Biol. 2015, 11, 457–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, M.P. Mitochondrial thiols in antioxidant protection and redox signaling: Distinct roles for glutathionylation and other thiol modifications. Antioxid. Redox Signal. 2012, 16, 476–495. [Google Scholar] [CrossRef] [PubMed]
- Andreyev, A.Y.; Kushnareva, Y.E.; Murphy, A.N.; Starkov, A.A. Mitochondrial ROS Metabolism: 10 Years Later. Biochemistry (Moscow) 2015, 80, 517–531. [Google Scholar] [CrossRef]
- Brand, M.D. Mitochondrial generation of superoxide and hydrogen peroxide as the source of mitochondrial redox signaling. Free Radic. Biol. Med. 2016, 100, 14–31. [Google Scholar] [CrossRef]
- Winterbourn, C.C.; Hampton, M.B. Thiol chemistry and specificity in redox signaling. Free Radic. Biol. Med. 2008, 45, 549–561. [Google Scholar] [CrossRef]
- Sharapov, M.G.; Ravin, V.K.; Novoselov, V.I. Peroxyredoxins as multifunctional enzymes. Mol. Biol. (Mosk) 2014, 48, 600–628. [Google Scholar] [CrossRef]
- Kudryavtseva, A.V.; Krasnov, G.S.; Dmitriev, A.A.; Alekseev, B.Y.; Kardymon, O.L.; Sadritdinova, A.F.; Fedorova, M.S.; Pokrovsky, A.V.; Melnikova, N.V.; Kaprin, A.D.; et al. Mitochondrial dysfunction and oxidative stress in aging and cancer. Oncotarget 2016, 7, 44879–44905. [Google Scholar] [CrossRef] [PubMed]
- Moloney, J.N.; Cotter, T.G. ROS signalling in the biology of cancer. Semin. Cell Dev. Biol. 2018, 80, 50–64. [Google Scholar] [CrossRef] [PubMed]
- Gill, J.G.; Piskounova, E.; Morrison, S.J. Cancer, Oxidative Stress, and Metastasis. Cold Spring Harb. Symp. Quant. Biol. 2016, 81, 163–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Hu, X.; Liu, Y.; Dong, S.; Wen, Z.; He, W.; Zhang, S.; Huang, Q.; Shi, M. ROS signaling under metabolic stress: Cross-talk between AMPK and AKT pathway. Mol Cancer 2017, 16, 79. [Google Scholar] [CrossRef] [PubMed]
- Chio, I.I.C.; Tuveson, D.A. ROS in Cancer: The Burning Question. Trends Mol. Med. 2017, 23, 411–429. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Nagarajan, A.; Malvi, P.; Wajapeyee, N. Oncogene-directed alterations in cancer cell metabolism. Trends Cancer 2016, 2, 365–377. [Google Scholar] [CrossRef]
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [Green Version]
- Szatrowski, T.P.; Nathan, C.F. Production of large amounts of hydrogen peroxide by human tumor cells. Cancer Res. 1991, 51, 794–798. [Google Scholar]
- Yuan, D.; Huang, S.; Berger, E.; Liu, L.; Gross, N.; Heinzmann, F.; Ringelhan, M.; Connor, T.O.; Stadler, M.; Meister, M.; et al. Kupffer Cell-Derived Tnf Triggers Cholangiocellular Tumorigenesis through JNK due to Chronic Mitochondrial Dysfunction and ROS. Cancer Cell 2017, 31, 771–789 e6. [Google Scholar] [CrossRef]
- Weinberg, F.; Hamanaka, R.; Wheaton, W.W.; Weinberg, S.; Joseph, J.; Lopez, M.; Kalyanaraman, B.; Mutlu, G.M.; Budinger, G.R.; Chandel, N.S. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc. Natl. Acad. Sci. USA 2010, 107, 8788–8793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhillon, A.S.; Hagan, S.; Rath, O.; Kolch, W. MAP kinase signalling pathways in cancer. Oncogene 2007, 26, 3279–3290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faes, S.; Dormond, O. PI3K and AKT: Unfaithful Partners in Cancer. Int. J. Mol. Sci. 2015, 16, 21138–21152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Xu, Y.; Zhou, Q.; Chen, M.; Zhang, Y.; Liang, H.; Zhao, J.; Zhong, W.; Wang, M. PI3K in cancer: Its structure, activation modes and role in shaping tumor microenvironment. Future Oncol. 2018, 14, 665–674. [Google Scholar] [CrossRef] [PubMed]
- Dolcet, X.; Llobet, D.; Pallares, J.; Matias-Guiu, X. NF-kB in development and progression of human cancer. Virchows Arch. 2005, 446, 475–482. [Google Scholar] [CrossRef] [PubMed]
- Tilborghs, S.; Corthouts, J.; Verhoeven, Y.; Arias, D.; Rolfo, C.; Trinh, X.B.; van Dam, P.A. The role of Nuclear Factor-kappa B signaling in human cervical cancer. Crit. Rev. Oncol. Hematol. 2017, 120, 141–150. [Google Scholar] [CrossRef] [PubMed]
- De Sa Junior, P.L.; Camara, D.A.D.; Porcacchia, A.S.; Fonseca, P.M.M.; Jorge, S.D.; Araldi, R.P.; Ferreira, A.K. The Roles of ROS in Cancer Heterogeneity and Therapy. Oxid. Med. Cell. Longev. 2017, 2017, 2467940. [Google Scholar] [CrossRef] [PubMed]
- Martindale, J.L.; Holbrook, N.J. Cellular response to oxidative stress: Signaling for suicide and survival. J. Cell. Physiol. 2002, 192, 1–15. [Google Scholar] [CrossRef]
- Ranjan, P.; Anathy, V.; Burch, P.M.; Weirather, K.; Lambeth, J.D.; Heintz, N.H. Redox-dependent expression of cyclin D1 and cell proliferation by Nox1 in mouse lung epithelial cells. Antioxid. Redox Signal. 2006, 8, 1447–1459. [Google Scholar] [CrossRef]
- Shimura, T.; Sasatani, M.; Kamiya, K.; Kawai, H.; Inaba, Y.; Kunugita, N. Mitochondrial reactive oxygen species perturb AKT/cyclin D1 cell cycle signaling via oxidative inactivation of PP2A in lowdose irradiated human fibroblasts. Oncotarget 2016, 7, 3559–3570. [Google Scholar] [CrossRef]
- Wang, P.; Zeng, Y.; Liu, T.; Zhang, C.; Yu, P.W.; Hao, Y.X.; Luo, H.X.; Liu, G. Chloride intracellular channel 1 regulates colon cancer cell migration and invasion through ROS/ERK pathway. World J. Gastroenterol. 2014, 20, 2071–2078. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Nikulenkov, F.; Zawacka-Pankau, J.; Li, H.; Gabdoulline, R.; Xu, J.; Eriksson, S.; Hedstrom, E.; Issaeva, N.; Kel, A.; et al. ROS-dependent activation of JNK converts p53 into an efficient inhibitor of oncogenes leading to robust apoptosis. Cell Death Differ. 2014, 21, 612–623. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Lu, M.; Zhang, Q. Chloride intracellular channel 1 regulates migration and invasion in gastric cancer by triggering the ROS-mediated p38 MAPK signaling pathway. Mol. Med. Rep. 2015, 12, 8041–8047. [Google Scholar] [CrossRef] [PubMed]
- Prasad, S.; Gupta, S.C.; Tyagi, A.K. Reactive oxygen species (ROS) and cancer: Role of antioxidative nutraceuticals. Cancer Lett. 2017, 387, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Leslie, N.R.; Bennett, D.; Lindsay, Y.E.; Stewart, H.; Gray, A.; Downes, C.P. Redox regulation of PI 3-kinase signalling via inactivation of PTEN. EMBO J. 2003, 22, 5501–5510. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Rovira, I.I.; Finkel, T. Oxidants painting the cysteine chapel: Redox regulation of PTPs. Dev. Cell 2002, 2, 251–252. [Google Scholar] [CrossRef]
- Harris, I.S.; Blaser, H.; Moreno, J.; Treloar, A.E.; Gorrini, C.; Sasaki, M.; Mason, J.M.; Knobbe, C.B.; Rufini, A.; Halle, M.; et al. PTPN12 promotes resistance to oxidative stress and supports tumorigenesis by regulating FOXO signaling. Oncogene 2014, 33, 1047–1054. [Google Scholar] [CrossRef]
- Qian, X.; Nie, X.; Yao, W.; Klinghammer, K.; Sudhoff, H.; Kaufmann, A.M.; Albers, A.E. Reactive oxygen species in cancer stem cells of head and neck squamous cancer. Semin. Cancer Biol. 2018, 53, 248–257. [Google Scholar] [CrossRef]
- Ding, S.; Li, C.; Cheng, N.; Cui, X.; Xu, X.; Zhou, G. Redox Regulation in Cancer Stem Cells. Oxid. Med. Cell. Longev. 2015, 2015, 750798. [Google Scholar] [CrossRef]
- Hamai, A.; Caneque, T.; Muller, S.; Mai, T.T.; Hienzsch, A.; Ginestier, C.; Charafe-Jauffret, E.; Codogno, P.; Mehrpour, M.; Rodriguez, R. An iron hand over cancer stem cells. Autophagy 2017, 13, 1465–1466. [Google Scholar] [CrossRef]
- Zhang, B.B.; Wang, D.G.; Guo, F.F.; Xuan, C. Mitochondrial membrane potential and reactive oxygen species in cancer stem cells. Fam. Cancer 2015, 14, 19–23. [Google Scholar] [CrossRef] [PubMed]
- Watson, J. Oxidants, antioxidants and the current incurability of metastatic cancers. Open Biol. 2013, 3, 120144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Negrini, S.; Gorgoulis, V.G.; Halazonetis, T.D. Genomic instability—An evolving hallmark of cancer. Nat. Rev. Mol. Cell Biol. 2010, 11, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Yeo, C.Q.X.; Alexander, I.; Lin, Z.; Lim, S.; Aning, O.A.; Kumar, R.; Sangthongpitag, K.; Pendharkar, V.; Ho, V.H.B.; Cheok, C.F. p53 Maintains Genomic Stability by Preventing Interference between Transcription and Replication. Cell Rep. 2016, 15, 132–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carroll, P.A.; Freie, B.W.; Mathsyaraja, H.; Eisenman, R.N. The MYC transcription factor network: Balancing metabolism, proliferation and oncogenesis. Front. Med. 2018, 12, 412–425. [Google Scholar] [CrossRef] [PubMed]
- Gabay, M.; Li, Y.; Felsher, D.W. MYC activation is a hallmark of cancer initiation and maintenance. Cold Spring Harb. Perspect. Med. 2014, 4, a014241. [Google Scholar] [CrossRef] [PubMed]
- Liou, G.Y.; Storz, P. Reactive oxygen species in cancer. Free Radic. Res. 2010, 44, 479–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galadari, S.; Rahman, A.; Pallichankandy, S.; Thayyullathil, F. Reactive oxygen species and cancer paradox: To promote or to suppress? Free Radic. Biol. Med. 2017, 104, 144–164. [Google Scholar] [CrossRef] [PubMed]
- Calaf, G.M.; Aguayo, F.; Sergi, C.M.; Juarranz, A.; Roy, D. Antioxidants and Cancer: Theories, Techniques, and Trials in Preventing Cancer. Oxid. Med. Cell. Longev. 2018, 2018, 5363064. [Google Scholar] [CrossRef]
- Mates, J.M.; Perez-Gomez, C.; Nunez de Castro, I. Antioxidant enzymes and human diseases. Clin. Biochem. 1999, 32, 595–603. [Google Scholar] [CrossRef]
- Bazhin, A.V.; Philippov, P.P.; Karakhanova, S. Reactive Oxygen Species in Cancer Biology and Anticancer Therapy. Oxid. Med. Cell. Longev. 2016, 2016, 4197815. [Google Scholar] [CrossRef] [PubMed]
- Schafer, Z.T.; Grassian, A.R.; Song, L.; Jiang, Z.; Gerhart-Hines, Z.; Irie, H.Y.; Gao, S.; Puigserver, P.; Brugge, J.S. Antioxidant and oncogene rescue of metabolic defects caused by loss of matrix attachment. Nature 2009, 461, 109–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dayem, A.A.; Choi, H.Y.; Kim, J.H.; Cho, S.G. Role of oxidative stress in stem, cancer, and cancer stem cells. Cancers (Basel) 2010, 2, 859–884. [Google Scholar] [CrossRef] [PubMed]
- Tong, L.; Chuang, C.C.; Wu, S.; Zuo, L. Reactive oxygen species in redox cancer therapy. Cancer Lett. 2015, 367, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Karakhanova, S.; Hartwig, W.; D’Haese, J.G.; Philippov, P.P.; Werner, J.; Bazhin, A.V. Mitochondria and Mitochondrial ROS in Cancer: Novel Targets for Anticancer Therapy. J. Cell. Physiol. 2016, 231, 2570–2581. [Google Scholar] [CrossRef] [PubMed]
- Krajka-Kuzniak, V.; Paluszczak, J.; Baer-Dubowska, W. The Nrf2-ARE signaling pathway: An update on its regulation and possible role in cancer prevention and treatment. Pharm. Rep. 2017, 69, 393–402. [Google Scholar] [CrossRef] [PubMed]
- Deshmukh, P.; Unni, S.; Krishnappa, G.; Padmanabhan, B. The Keap1-Nrf2 pathway: Promising therapeutic target to counteract ROS-mediated damage in cancers and neurodegenerative diseases. Biophys. Rev. 2017, 9, 41–56. [Google Scholar] [CrossRef] [PubMed]
- Pandey, P.; Singh, A.K.; Singh, M.; Tewari, M.; Shukla, H.S.; Gambhir, I.S. The see-saw of Keap1-Nrf2 pathway in cancer. Crit. Rev. Oncol. Hematol. 2017, 116, 89–98. [Google Scholar] [CrossRef]
- Itoh, K.; Wakabayashi, N.; Katoh, Y.; Ishii, T.; Igarashi, K.; Engel, J.D.; Yamamoto, M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999, 13, 76–86. [Google Scholar] [CrossRef] [Green Version]
- Raghunath, A.; Nagarajan, R.; Sundarraj, K.; Panneerselvam, L.; Perumal, E. Genome-wide identification and analysis of Nrf2 binding sites—Antioxidant response elements in zebrafish. Toxicol. Appl. Pharm. 2018, 360, 236–248. [Google Scholar] [CrossRef]
- Rojo de la Vega, M.; Chapman, E.; Zhang, D.D. NRF2 and the Hallmarks of Cancer. Cancer Cell 2018, 34, 21–43. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, K.; Yamamoto, M. The KEAP1-NRF2 System in Cancer. Front. Oncol. 2017, 7, 85. [Google Scholar] [CrossRef] [PubMed]
- Nault, J.C.; Rebouissou, S.; Zucman Rossi, J. NRF2/KEAP1 and Wnt/beta-catenin in the multistep process of liver carcinogenesis in humans and rats. Hepatology 2015, 62, 677–679. [Google Scholar] [CrossRef] [PubMed]
- Jeong, Y.; Hoang, N.T.; Lovejoy, A.; Stehr, H.; Newman, A.M.; Gentles, A.J.; Kong, W.; Truong, D.; Martin, S.; Chaudhuri, A.; et al. Role of KEAP1/NRF2 and TP53 Mutations in Lung Squamous Cell Carcinoma Development and Radiation Resistance. Cancer Discov. 2017, 7, 86–101. [Google Scholar] [CrossRef] [PubMed]
- Budanov, A.V. The role of tumor suppressor p53 in the antioxidant defense and metabolism. Subcell. Biochem. 2014, 85, 337–358. [Google Scholar] [PubMed]
- Bakalova, R.; Zhelev, Z.; Shibata, S.; Nikolova, B.; Aoki, I.; Higashi, T. Impressive Suppression of Colon Cancer Growth by Triple Combination SN38/EF24/Melatonin: “Oncogenic” Versus “Onco-Suppressive” Reactive Oxygen Species. Anticancer Res. 2017, 37, 5449–5458. [Google Scholar] [PubMed]
- Ladelfa, M.F.; Toledo, M.F.; Laiseca, J.E.; Monte, M. Interaction of p53 with tumor suppressive and oncogenic signaling pathways to control cellular reactive oxygen species production. Antioxid. Redox Signal. 2011, 15, 1749–1761. [Google Scholar] [CrossRef] [PubMed]
- Leonova, K.I.; Shneyder, J.; Antoch, M.P.; Toshkov, I.A.; Novototskaya, L.R.; Komarov, P.G.; Komarova, E.A.; Gudkov, A.V. A small molecule inhibitor of p53 stimulates amplification of hematopoietic stem cells but does not promote tumor development in mice. Cell Cycle 2010, 9, 1434–1443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aubrey, B.J.; Kelly, G.L.; Janic, A.; Herold, M.J.; Strasser, A. How does p53 induce apoptosis and how does this relate to p53-mediated tumour suppression? Cell Death Differ. 2018, 25, 104–113. [Google Scholar] [CrossRef] [PubMed]
- Bensaad, K.; Tsuruta, A.; Selak, M.A.; Vidal, M.N.; Nakano, K.; Bartrons, R.; Gottlieb, E.; Vousden, K.H. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell 2006, 126, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Ros, S.; Floter, J.; Kaymak, I.; Da Costa, C.; Houddane, A.; Dubuis, S.; Griffiths, B.; Mitter, R.; Walz, S.; Blake, S.; et al. 6-Phosphofructo-2-kinase/fructose-2,6-biphosphatase 4 is essential for p53-null cancer cells. Oncogene 2017, 36, 3287–3299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.H.; Choi, S.I.; Won, K.Y.; Lim, S.J. Distinctive interrelation of p53 with SCO2, COX, and TIGAR in human gastric cancer. Pathol. Res. Pr. 2016, 212, 904–910. [Google Scholar] [CrossRef] [PubMed]
- Coomans de Brachene, A.; Demoulin, J.B. FOXO transcription factors in cancer development and therapy. Cell. Mol. Life Sci. 2016, 73, 1159–1172. [Google Scholar] [CrossRef] [PubMed]
- Link, W.; Fernandez-Marcos, P.J. FOXO transcription factors at the interface of metabolism and cancer. Int. J. Cancer 2017, 141, 2379–2391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neuzillet, C.; Tijeras-Raballand, A.; de Mestier, L.; Cros, J.; Faivre, S.; Raymond, E. MEK in cancer and cancer therapy. Pharm. Ther. 2014, 141, 160–171. [Google Scholar] [CrossRef] [PubMed]
- Zou, X.; Blank, M. Targeting p38 MAP kinase signaling in cancer through post-translational modifications. Cancer Lett. 2017, 384, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Chiu, D.K.; Tse, A.P.; Xu, I.M.; Cui, J.D.; Lai, R.K.; Li, L.L.; Koh, H.Y.; Tsang, F.H.; Wei, L.L.; Wong, C.M.; et al. Hypoxia inducible factor HIF-1 promotes myeloid-derived suppressor cells accumulation through ENTPD2/CD39L1 in hepatocellular carcinoma. Nat. Commun. 2017, 8, 517. [Google Scholar] [CrossRef]
- Huang, Y.; Lin, D.; Taniguchi, C.M. Hypoxia inducible factor (HIF) in the tumor microenvironment: Friend or foe? Sci. China Life Sci. 2017, 60, 1114–1124. [Google Scholar] [CrossRef]
- Pugh, C.W.; Ratcliffe, P.J. The von Hippel-Lindau tumor suppressor, hypoxia-inducible factor-1 (HIF-1) degradation, and cancer pathogenesis. Semin. Cancer Biol. 2003, 13, 83–89. [Google Scholar] [CrossRef]
- Yang, M.; Su, H.; Soga, T.; Kranc, K.R.; Pollard, P.J. Prolyl hydroxylase domain enzymes: Important regulators of cancer metabolism. Hypoxia 2014, 2, 127–142. [Google Scholar]
- Berra, E.; Benizri, E.; Ginouves, A.; Volmat, V.; Roux, D.; Pouyssegur, J. HIF prolyl-hydroxylase 2 is the key oxygen sensor setting low steady-state levels of HIF-1alpha in normoxia. EMBO J. 2003, 22, 4082–4090. [Google Scholar] [CrossRef] [PubMed]
- Palazon, A.; Tyrakis, P.A.; Macias, D.; Velica, P.; Rundqvist, H.; Fitzpatrick, S.; Vojnovic, N.; Phan, A.T.; Loman, N.; Hedenfalk, I.; et al. An HIF-1alpha/VEGF-A Axis in Cytotoxic T Cells Regulates Tumor Progression. Cancer Cell 2017, 32, 669–683 e5. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.Y.; Zhu, P.; Jin, X.P. Association between the expression of HIF-1alpha and VEGF and prognostic implications in primary liver cancer. Genet. Mol. Res. 2016, 15. [Google Scholar] [CrossRef]
- Cui, Y.; Qin, L.; Wu, J.; Qu, X.; Hou, C.; Sun, W.; Li, S.; Vaughan, A.T.; Li, J.J.; Liu, J. SIRT3 Enhances Glycolysis and Proliferation in SIRT3-Expressing Gastric Cancer Cells. PLoS ONE 2015, 10, e0129834. [Google Scholar] [CrossRef] [PubMed]
- Torrens-Mas, M.; Oliver, J.; Roca, P.; Sastre-Serra, J. SIRT3: Oncogene and Tumor Suppressor in Cancer. Cancers (Basel) 2017, 9, 90. [Google Scholar] [CrossRef]
- Athreya, K.; Xavier, M.F. Antioxidants in the Treatment of Cancer. Nutr. Cancer 2017, 69, 1099–1104. [Google Scholar] [CrossRef]
- Bonner, M.Y.; Arbiser, J.L. The antioxidant paradox: What are antioxidants and how should they be used in a therapeutic context for cancer. Future Med. Chem. 2014, 6, 1413–1422. [Google Scholar] [CrossRef]
- Hampton, M.B.; Vick, K.A.; Skoko, J.J.; Neumann, C.A. Peroxiredoxin Involvement in the Initiation and Progression of Human Cancer. Antioxid. Redox Signal. 2018, 28, 591–608. [Google Scholar] [CrossRef]
- Olmos, Y.; Sanchez-Gomez, F.J.; Wild, B.; Garcia-Quintans, N.; Cabezudo, S.; Lamas, S.; Monsalve, M. SirT1 regulation of antioxidant genes is dependent on the formation of a FoxO3a/PGC-1alpha complex. Antioxid. Redox Signal. 2013, 19, 1507–1521. [Google Scholar] [CrossRef]
- Song, I.S.; Jeong, Y.J.; Jeong, S.H.; Heo, H.J.; Kim, H.K.; Bae, K.B.; Park, Y.H.; Kim, S.U.; Kim, J.M.; Kim, N.; et al. FOXM1-Induced PRX3 Regulates Stemness and Survival of Colon Cancer Cells via Maintenance of Mitochondrial Function. Gastroenterology 2015, 149, 1006–1016 e9. [Google Scholar] [CrossRef]
- Song, I.S.; Jeong, Y.J.; Seo, Y.J.; Byun, J.M.; Kim, Y.N.; Jeong, D.H.; Han, J.; Kim, K.T.; Jang, S.W. Peroxiredoxin 3 maintains the survival of endometrial cancer stem cells by regulating oxidative stress. Oncotarget 2017, 8, 92788–92800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ummanni, R.; Barreto, F.; Venz, S.; Scharf, C.; Barett, C.; Mannsperger, H.A.; Brase, J.C.; Kuner, R.; Schlomm, T.; Sauter, G.; et al. Peroxiredoxins 3 and 4 are overexpressed in prostate cancer tissue and affect the proliferation of prostate cancer cells in vitro. J. Proteome Res. 2012, 11, 2452–2466. [Google Scholar] [CrossRef] [PubMed]
- Whitaker, H.C.; Patel, D.; Howat, W.J.; Warren, A.Y.; Kay, J.D.; Sangan, T.; Marioni, J.C.; Mitchell, J.; Aldridge, S.; Luxton, H.J.; et al. Peroxiredoxin-3 is overexpressed in prostate cancer and promotes cancer cell survival by protecting cells from oxidative stress. Br. J. Cancer 2013, 109, 983–993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
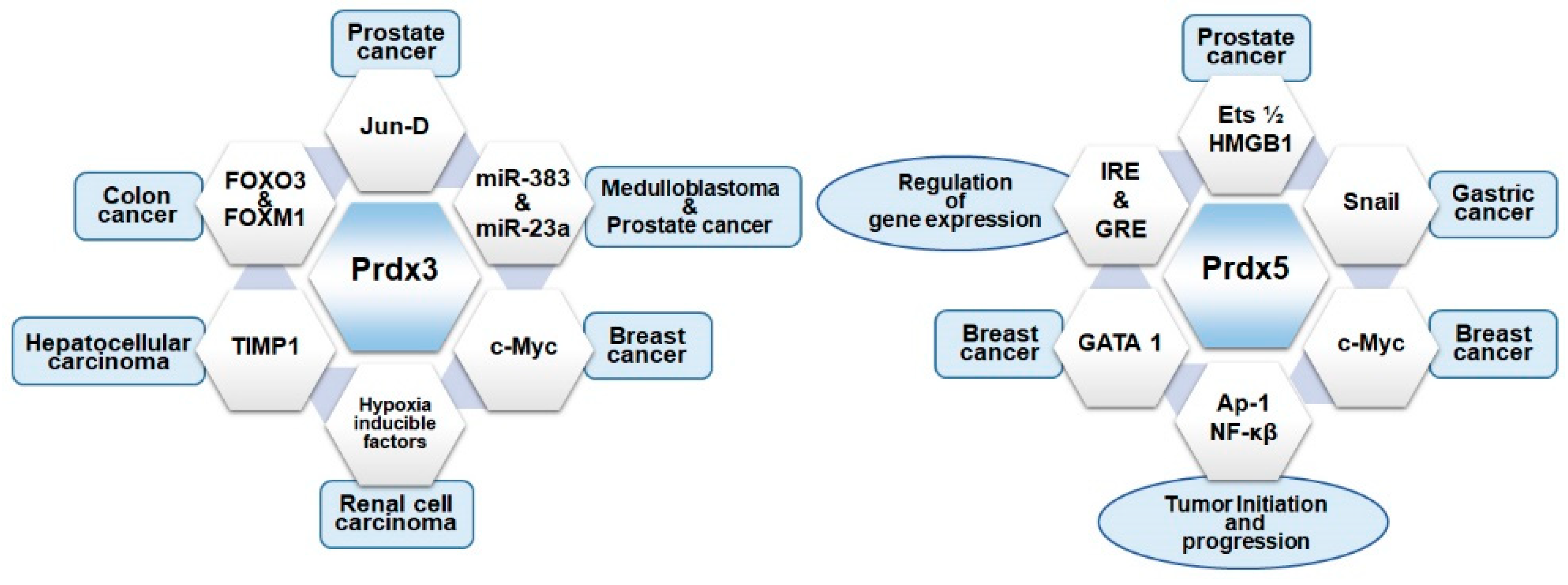
- Li, K.K.; Pang, J.C.; Lau, K.M.; Zhou, L.; Mao, Y.; Wang, Y.; Poon, W.S.; Ng, H.K. MiR-383 is downregulated in medulloblastoma and targets peroxiredoxin 3 (PRDX3). Brain Pathol. 2013, 23, 413–425. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.M.; Zhang, S.F.; Cheng, Z.Q.; Peng, Q.Z.; Hu, J.T.; Gao, L.K.; Xu, J.; Jin, H.T.; Liu, H.Y. MicroRNA383 regulates expression of PRDX3 in human medulloblastomas. Zhonghua Bing Li Xue Za Zhi 2012, 41, 547–552. [Google Scholar] [PubMed]
- He, H.C.; Zhu, J.G.; Chen, X.B.; Chen, S.M.; Han, Z.D.; Dai, Q.S.; Ling, X.H.; Fu, X.; Lin, Z.Y.; Deng, Y.H.; et al. MicroRNA-23b downregulates peroxiredoxin III in human prostate cancer. FEBS Lett. 2012, 586, 2451–2458. [Google Scholar] [CrossRef] [PubMed]
- Elliott, B.; Millena, A.C.; Matyunina, L.; Zhang, M.; Zou, J.; Wang, G.; Zhang, Q.; Bowen, N.; Eaton, V.; Webb, G.; et al. Essential role of JunD in cell proliferation is mediated via MYC signaling in prostate cancer cells. Cancer Lett. 2019, 448, 155–167. [Google Scholar] [CrossRef]
- Xi, H.; Gao, Y.H.; Han, D.Y.; Li, Q.Y.; Feng, L.J.; Zhang, W.; Ji, G.; Xiao, J.C.; Zhang, H.Z.; Wei, Q. Hypoxia inducible factor-1alpha suppresses Peroxiredoxin 3 expression to promote proliferation of CCRCC cells. FEBS Lett. 2014, 588, 3390–3394. [Google Scholar] [CrossRef] [PubMed]
- Ismail, S.; Mayah, W.; Battia, H.E.; Gaballah, H.; Jiman-Fatani, A.; Hamouda, H.; Afifi, M.A.; Elmashad, N.; Saadany, S.E. Plasma nuclear factor kappa B and serum peroxiredoxin 3 in early diagnosis of hepatocellular carcinoma. Asian Pac. J. Cancer Prev. 2015, 16, 1657–1663. [Google Scholar] [CrossRef]
- Liu, Z.; Hu, Y.; Liang, H.; Sun, Z.; Feng, S.; Deng, H. Silencing PRDX3 Inhibits Growth and Promotes Invasion and Extracellular Matrix Degradation in Hepatocellular Carcinoma Cells. J. Proteome Res. 2016, 15, 1506–1514. [Google Scholar] [CrossRef]
- Shi, L.; Wu, L.L.; Yang, J.R.; Chen, X.F.; Zhang, Y.; Chen, Z.Q.; Liu, C.L.; Chi, S.Y.; Zheng, J.Y.; Huang, H.X.; et al. Serum peroxiredoxin3 is a useful biomarker for early diagnosis and assessemnt of prognosis of hepatocellular carcinoma in Chinese patients. Asian Pac. J. Cancer Prev. 2014, 15, 2979–2986. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Feng, R.; Du, L. The role of enoyl-CoA hydratase short chain 1 and peroxiredoxin 3 in PP2-induced apoptosis in human breast cancer MCF-7 cells. FEBS Lett. 2010, 584, 3185–3192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pendharkar, N.; Gajbhiye, A.; Taunk, K.; RoyChoudhury, S.; Dhali, S.; Seal, S.; Mane, A.; Abhang, S.; Santra, M.K.; Chaudhury, K.; et al. Quantitative tissue proteomic investigation of invasive ductal carcinoma of breast with luminal B HER2 positive and HER2 enriched subtypes towards potential diagnostic and therapeutic biomarkers. J. Proteom. 2016, 132, 112–130. [Google Scholar] [CrossRef] [PubMed]
- Kalinina, E.V.; Berezov, T.T.; Shtil, A.A.; Chernov, N.N.; Glazunova, V.A.; Novichkova, M.D.; Nurmuradov, N.K. Expression of peroxiredoxin 1, 2, 3, and 6 genes in cancer cells during drug resistance formation. Bull. Exp. Biol. Med. 2012, 153, 878–881. [Google Scholar] [CrossRef]
- McDonald, C.; Muhlbauer, J.; Perlmutter, G.; Taparra, K.; Phelan, S.A. Peroxiredoxin proteins protect MCF-7 breast cancer cells from doxorubicin-induced toxicity. Int. J. Oncol. 2014, 45, 219–226. [Google Scholar] [CrossRef]
- Safaeian, M.; Hildesheim, A.; Gonzalez, P.; Yu, K.; Porras, C.; Li, Q.; Rodriguez, A.C.; Sherman, M.E.; Schiffman, M.; Wacholder, S.; et al. Single nucleotide polymorphisms in the PRDX3 and RPS19 and risk of HPV persistence and cervical precancer/cancer. PLoS ONE 2012, 7, e33619. [Google Scholar] [CrossRef]
- Byun, J.M.; Kim, S.S.; Kim, K.T.; Kang, M.S.; Jeong, D.H.; Lee, D.S.; Jung, E.J.; Kim, Y.N.; Han, J.; Song, I.S.; et al. Overexpression of peroxiredoxin-3 and -5 is a potential biomarker for prognosis in endometrial cancer. Oncol. Lett. 2018, 15, 5111–5118. [Google Scholar] [CrossRef] [Green Version]
- Han, S.; Shen, H.; Jung, M.; Hahn, B.S.; Jin, B.K.; Kang, I.; Ha, J.; Choe, W. Expression and prognostic significance of human peroxiredoxin isoforms in endometrial cancer. Oncol. Lett. 2012, 3, 1275–1279. [Google Scholar] [CrossRef]
- Kinnula, V.L.; Lehtonen, S.; Sormunen, R.; Kaarteenaho-Wiik, R.; Kang, S.W.; Rhee, S.G.; Soini, Y. Overexpression of peroxiredoxins I, II, III, V, and VI in malignant mesothelioma. J. Pathol. 2002, 196, 316–323. [Google Scholar] [CrossRef]
- Cunniff, B.; Wozniak, A.N.; Sweeney, P.; DeCosta, K.; Heintz, N.H. Peroxiredoxin 3 levels regulate a mitochondrial redox setpoint in malignant mesothelioma cells. Redox Biol. 2014, 3, 79–87. [Google Scholar] [CrossRef] [Green Version]
- Forshaw, T.E.; Holmila, R.; Nelson, K.J.; Lewis, J.E.; Kemp, M.L.; Tsang, A.W.; Poole, L.B.; Lowther, W.T.; Furdui, C.M. Peroxiredoxins in Cancer and Response to Radiation Therapies. Antioxidants (Basel) 2019, 8, 11. [Google Scholar] [CrossRef] [PubMed]
- Idelchik, M.; Begley, U.; Begley, T.J.; Melendez, J.A. Mitochondrial ROS control of cancer. Semin. Cancer Biol. 2017, 47, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Nguyen-Nhu, N.T.; Berck, J.; Clippe, A.; Duconseille, E.; Cherif, H.; Boone, C.; Van der Eecken, V.; Bernard, A.; Banmeyer, I.; Knoops, B. Human peroxiredoxin 5 gene organization, initial characterization of its promoter and identification of alternative forms of mRNA. Biochim. Biophys. Acta 2007, 1769, 472–483. [Google Scholar] [CrossRef] [PubMed]
- Usmanova, N.; Tomilin, N.; Zhivotovsky, B.; Kropotov, A. Transcription factor GABP/NRF-2 controlling biogenesis of mitochondria regulates basal expression of peroxiredoxin V but the mitochondrial function of peroxiredoxin V is dispensable in the dog. Biochimie 2011, 93, 306–313. [Google Scholar] [CrossRef] [PubMed]
- Graves, J.A.; Metukuri, M.; Scott, D.; Rothermund, K.; Prochownik, E.V. Regulation of reactive oxygen species homeostasis by peroxiredoxins and c-Myc. J. Biol. Chem. 2009, 284, 6520–6529. [Google Scholar] [CrossRef]
- Shiota, M.; Izumi, H.; Miyamoto, N.; Onitsuka, T.; Kashiwagi, E.; Kidani, A.; Hirano, G.; Takahashi, M.; Ono, M.; Kuwano, M.; et al. Ets regulates peroxiredoxin1 and 5 expressions through their interaction with the high-mobility group protein B1. Cancer Sci. 2008, 99, 1950–1959. [Google Scholar] [CrossRef]
- Liu, F.; Zhang, Y.; Men, T.; Jiang, X.; Yang, C.; Li, H.; Wei, X.; Yan, D.; Feng, G.; Yang, J.; et al. Quantitative proteomic analysis of gastric cancer tissue reveals novel proteins in platelet-derived growth factor b signaling pathway. Oncotarget 2017, 8, 22059–22075. [Google Scholar] [CrossRef]
- Bur, H.; Haapasaari, K.M.; Turpeenniemi-Hujanen, T.; Kuittinen, O.; Auvinen, P.; Marin, K.; Koivunen, P.; Sormunen, R.; Soini, Y.; Karihtala, P. Oxidative stress markers and mitochondrial antioxidant enzyme expression are increased in aggressive Hodgkin lymphomas. Histopathology 2014, 65, 319–327. [Google Scholar] [CrossRef]
- Seo, M.J.; Liu, X.; Chang, M.; Park, J.H. GATA-binding protein 1 is a novel transcription regulator of peroxiredoxin 5 in human breast cancer cells. Int. J. Oncol. 2012, 40, 655–664. [Google Scholar]
- Li, S.; Hu, X.; Ye, M.; Zhu, X. The prognostic values of the peroxiredoxins family in ovarian cancer. Biosci. Rep. 2018, 38. [Google Scholar] [CrossRef] [Green Version]
- Sienko, J.; Gaj, P.; Czajkowski, K.; Nowis, D. Peroxiredoxin-5 is a negative survival predictor in ovarian cancer. Ginekol. Polska 2019, 90, 6. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Zhou, Q.; Tao, X.; Shen, Z.; Luo, H.; Zhu, X. Expression and function of peroxiredoxins in gynecological malignancies. Front. Biosci. (Landmark Ed.) 2016, 21, 986–997. [Google Scholar] [PubMed] [Green Version]
- Fernandez-Ranvier, G.G.; Weng, J.; Yeh, R.F.; Shibru, D.; Khafnashar, E.; Chung, K.W.; Hwang, J.; Duh, Q.Y.; Clark, O.H.; Kebebew, E. Candidate diagnostic markers and tumor suppressor genes for adrenocortical carcinoma by expression profile of genes on chromosome 11q13. World J. Surg. 2008, 32, 873–881. [Google Scholar] [CrossRef] [PubMed]
- Chung, Y.M.; Yoo, Y.D.; Park, J.K.; Kim, Y.T.; Kim, H.J. Increased expression of peroxiredoxin II confers resistance to cisplatin. Anticancer Res. 2001, 21, 1129–1133. [Google Scholar] [PubMed]
- Kwee, J.K. A paradoxical chemoresistance and tumor suppressive role of antioxidant in solid cancer cells: A strange case of Dr. Jekyll and Mr. Hyde. Biomed. Res. Int. 2014, 2014, 209845. [Google Scholar] [CrossRef] [PubMed]
- Poschmann, G.; Grzendowski, M.; Stefanski, A.; Bruns, E.; Meyer, H.E.; Stuhler, K. Redox proteomics reveal stress responsive proteins linking peroxiredoxin-1 status in glioma to chemosensitivity and oxidative stress. Biochim. Biophys. Acta 2015, 1854, 624–631. [Google Scholar] [CrossRef] [PubMed]
- Roininen, N.; Haapasaari, K.M.; Karihtala, P. The Role of Redox-Regulating Enzymes in Inoperable Breast Cancers Treated with Neoadjuvant Chemotherapy. Oxid. Med. Cell. Longev. 2017, 2017, 2908039. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Diaz, A.J.; Yen, Y. The role of peroxiredoxin II in chemoresistance of breast cancer cells. Breast Cancer (Dove Med. Press) 2014, 6, 73–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Yu, A.Q. The functional role of peroxiredoxin 3 in reactive oxygen species, apoptosis, and chemoresistance of cancer cells. J. Cancer Res. Clin. Oncol. 2015, 141, 2071–2077. [Google Scholar] [CrossRef] [PubMed]
- Song, I.S.; Kim, H.K.; Jeong, S.H.; Lee, S.R.; Kim, N.; Rhee, B.D.; Ko, K.S.; Han, J. Mitochondrial peroxiredoxin III is a potential target for cancer therapy. Int. J. Mol. Sci. 2011, 12, 7163–7185. [Google Scholar] [CrossRef] [PubMed]
- Nicolussi, A.; D’Inzeo, S.; Capalbo, C.; Giannini, G.; Coppa, A. The role of peroxiredoxins in cancer. Mol. Clin. Oncol. 2017, 6, 139–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kropotov, A.; Gogvadze, V.; Shupliakov, O.; Tomilin, N.; Serikov, V.B.; Tomilin, N.V.; Zhivotovsky, B. Peroxiredoxin V is essential for protection against apoptosis in human lung carcinoma cells. Exp. Cell Res. 2006, 312, 2806–2815. [Google Scholar] [CrossRef] [PubMed]
- Yeldag, G.; Rice, A.; Del Rio Hernandez, A. Chemoresistance and the Self-Maintaining Tumor Microenvironment. Cancers (Basel) 2018, 10, 471. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J. Cancer stem cells and chemoresistance: The smartest survives the raid. Pharm. Ther. 2016, 160, 145–158. [Google Scholar] [CrossRef] [PubMed]
- Gorrini, C.; Harris, I.S.; Mak, T.W. Modulation of oxidative stress as an anticancer strategy. Nat. Rev. Drug Discov. 2013, 12, 931–947. [Google Scholar] [CrossRef] [PubMed]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ismail, T.; Kim, Y.; Lee, H.; Lee, D.-S.; Lee, H.-S. Interplay Between Mitochondrial Peroxiredoxins and ROS in Cancer Development and Progression. Int. J. Mol. Sci. 2019, 20, 4407. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20184407
Ismail T, Kim Y, Lee H, Lee D-S, Lee H-S. Interplay Between Mitochondrial Peroxiredoxins and ROS in Cancer Development and Progression. International Journal of Molecular Sciences. 2019; 20(18):4407. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20184407
Chicago/Turabian StyleIsmail, Tayaba, Youni Kim, Hongchan Lee, Dong-Seok Lee, and Hyun-Shik Lee. 2019. "Interplay Between Mitochondrial Peroxiredoxins and ROS in Cancer Development and Progression" International Journal of Molecular Sciences 20, no. 18: 4407. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20184407