High Concentration of C5a-Induced Mitochondria-Dependent Apoptosis in Murine Kidney Endothelial Cells

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
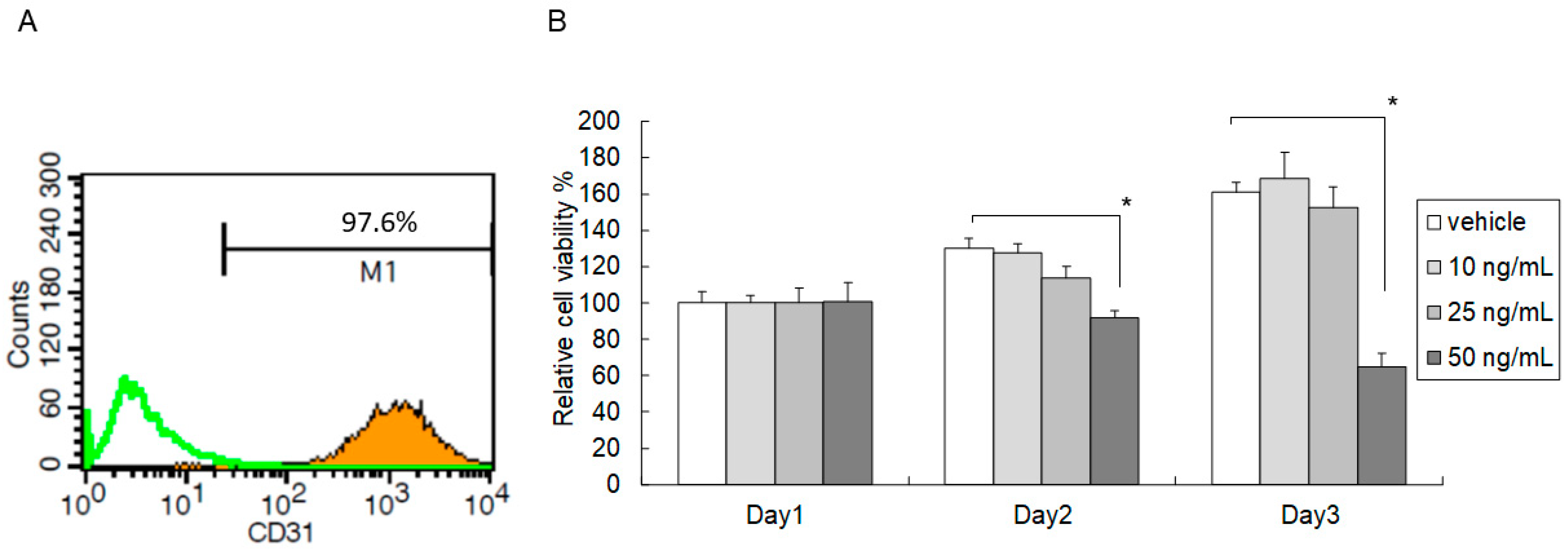
2.1. High-Dose C5a Treatment Reduced Mouse KEC Growth
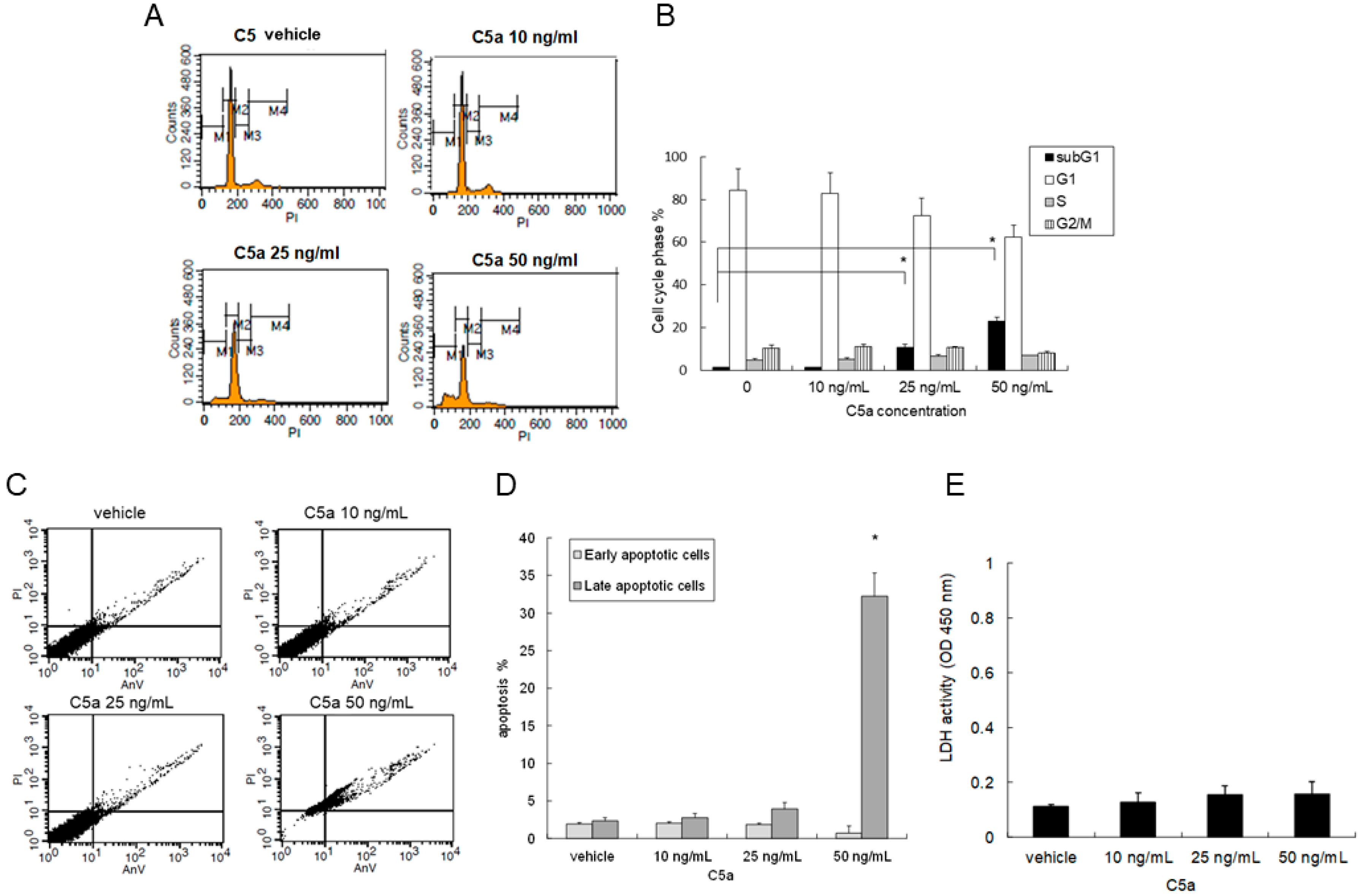
2.2. High-Dose C5a Treatment Induced Apoptosis of Mouse KECs
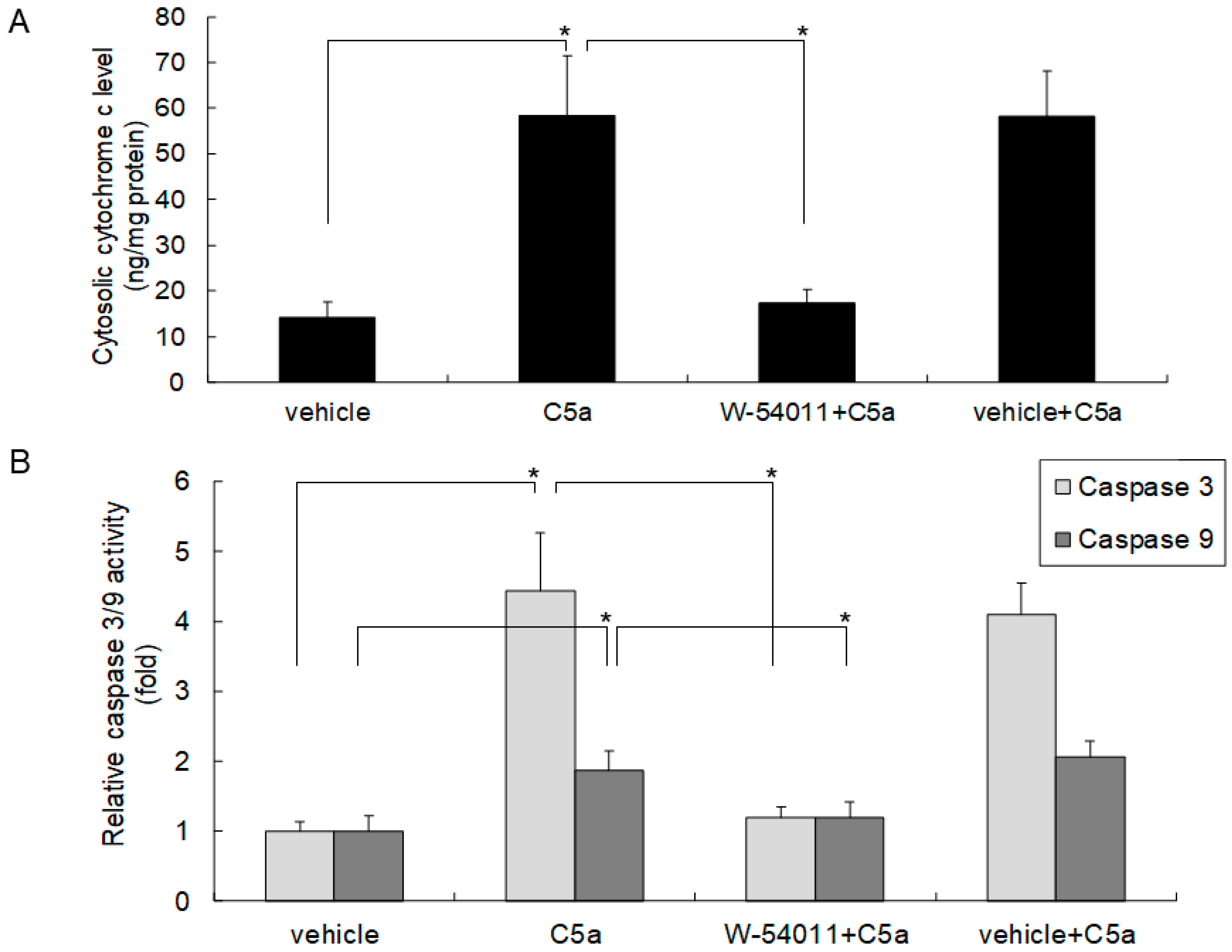
2.3. High-Dose C5a Treatment Induced Cytochrome c and Caspase 3/9 Activities through C5aR in Mouse KECs
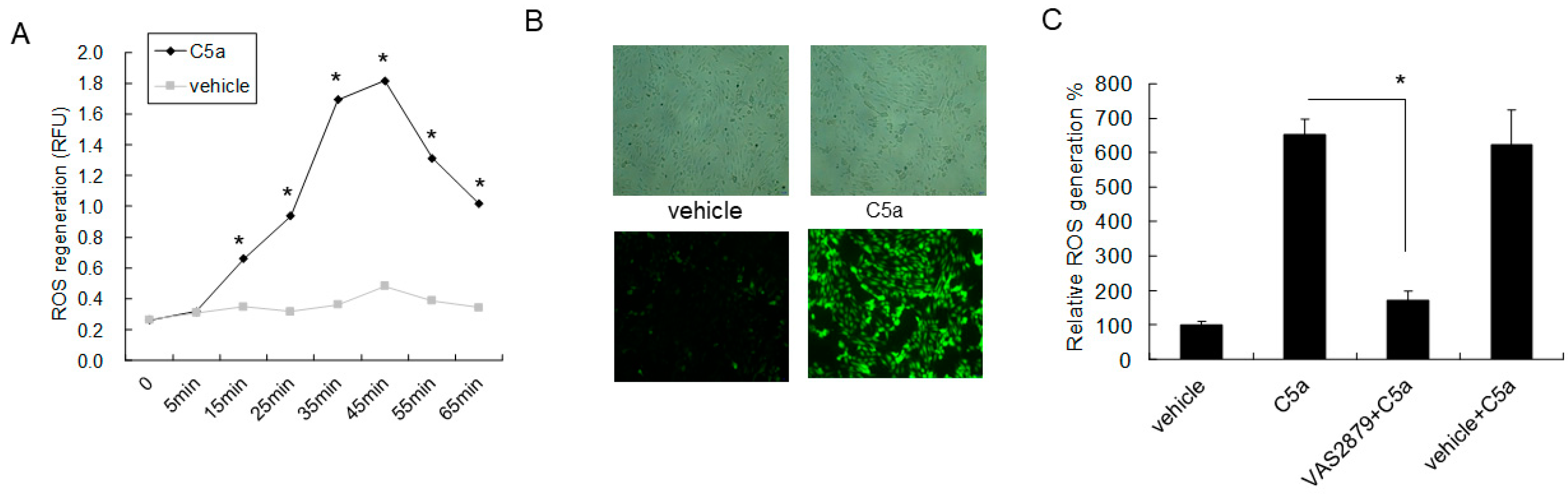
2.4. High-Dose C5a Treatment Induced Oxidative Stress via NOXs-Dependent ROS Generation in Mouse KECs
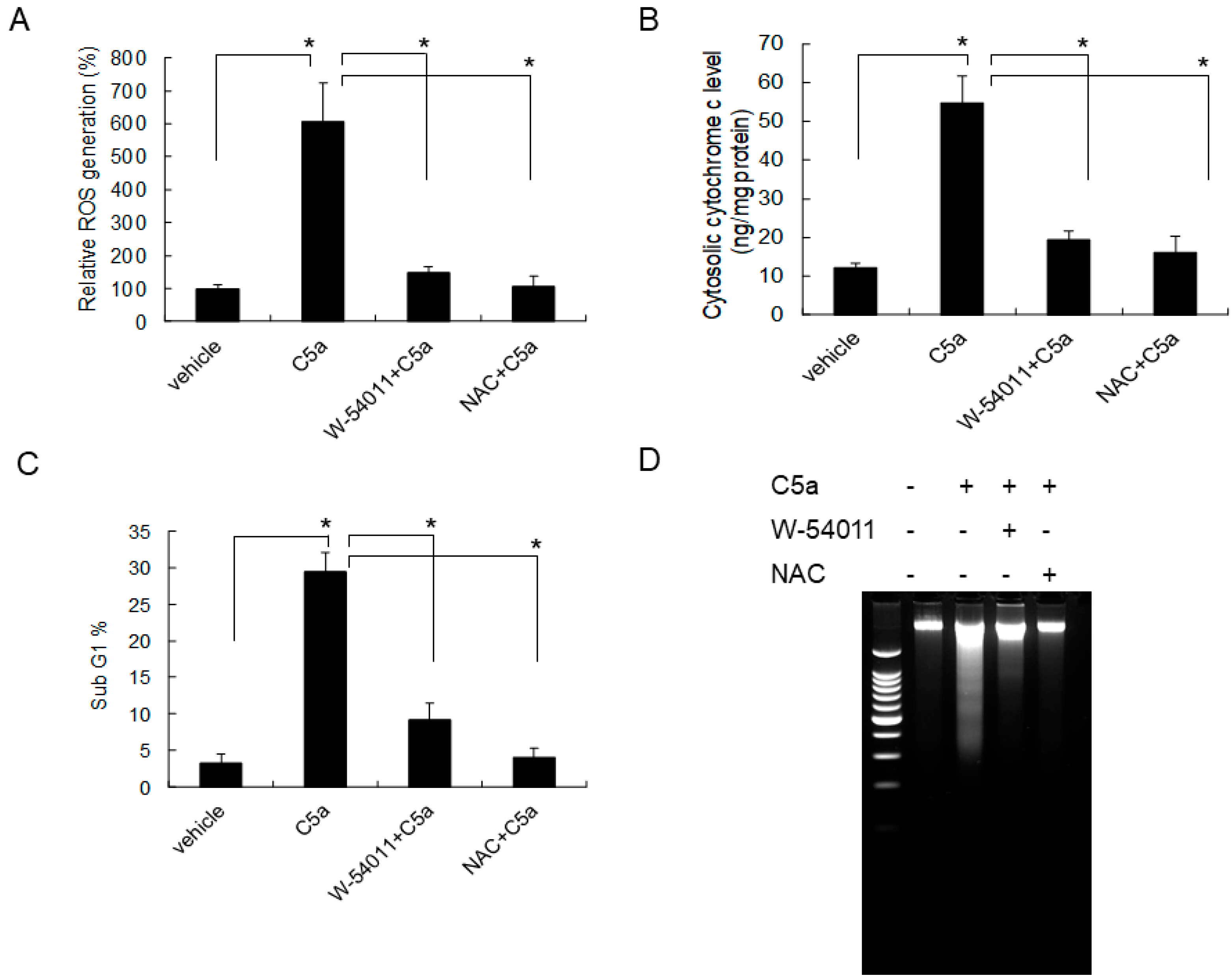
2.5. C5aR Inhibitors (W-54011) or NAC Rescued High-Dose C5a Treatment Induced ROS Formation and Apoptosis in Mouse KECs
3. Discussion
4. Materials and Methods
4.1. Mouse Kidney Endothelial Cell (KEC) Preparation
4.2. Recombinant Protein and Chemical Reagents
4.3. Cell Viability Determination
4.4. Cell Cycle and Apoptosis Analysis Determined by Flow Cytometry
4.5. Lactate Dehydrogenase (LDH) Assay
4.6. Cytochrome c Release and Caspase 3/9 Activity Determination
4.7. ROS Formation Determination by DCF-DA
4.8. Visualization of DNA Fragmentation with Gel Electrophoresis
4.9. Statistical Analysis
Author Contributions
Funding
Conflicts of Interest
References
- Schulman, E.S.; Post, T.J.; Henson, P.M.; Giclas, P.C. Differential effects of the complement peptides, C5a and C5a des Arg on human basophil and lung mast cell histamine release. J. Clin. Investig. 1988, 81, 918–923. [Google Scholar] [CrossRef] [PubMed]
- Becker, E.L. The relationship of the chemotactic behavior of the complement-derived factors, C3a, C5a, and C567, and a bacterial chemotactic factor to their ability to activate the proesterase 1 of rabbit polymorphonuclear leukocytes. J. Exp. Med. 1972, 135, 376–387. [Google Scholar] [CrossRef] [PubMed]
- Wenderfer, S.E.; Ke, B.; Hollmann, T.J.; Wetsel, R.A.; Lan, H.Y.; Braun, M.C. C5a receptor deficiency attenuates T cell function and renal disease in MRLlpr mice. J. Am. Soc. Nephrol. 2005, 16, 3572–3582. [Google Scholar] [CrossRef] [PubMed]
- Gueler, F.; Rong, S.; Gwinner, W.; Mengel, M.; Bröcker, V.; Schön, S.; Greten, T.F.; Hawlisch, H.; Polakowski, T.; Schnatbaum, K.; et al. Complement 5a receptor inhibition improves renal allograft survival. J. Am. Soc. Nephrol. 2008, 19, 2302–2312. [Google Scholar] [CrossRef] [PubMed]
- Arumugam, T.V.; Shiels, I.A.; Strachan, A.J.; Abbenante, G.; Fairlie, D.P.; Taylor, S.M. A small molecule C5a receptor antagonist protects kidneys from ischemia/reperfusion injury in rats. Kidney Int. 2003, 63, 134–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Vries, B.; Kohl, J.; Leclercq, W.K.; Wolfs, T.G.; van Bijnen, A.A.; Heeringa, P.; Buurman, W.A. Complement factor C5a mediates renal ischemia-reperfusion injury independent from neutrophils. J. Immunol. 2003, 170, 3883–3889. [Google Scholar] [CrossRef] [PubMed]
- Branten, A.J.; van den Born, J.; Jansen, J.L.; Assmann, K.J.; Wetzels, J.F. Familial nephropathy differing from minimal change nephropathy and focal glomerulosclerosis. Kidney Int. 2001, 59, 693–701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pagtalunan, M.E.; Miller, P.L.; Jumping-Eagle, S.; Nelson, R.G.; Myers, B.D.; Rennke, H.G.; Coplon, N.S.; Sun, L.; Meyer, T.W. Podocyte loss and progressive glomerular injury in type II diabetes. J. Clin. Investig. 1997, 99, 342–348. [Google Scholar] [CrossRef] [PubMed]
- Stillman, I.E.; Karumanchi, S.A. The glomerular injury of preeclampsia. J. Am. Soc. Nephrol. 2007, 18, 2281–2284. [Google Scholar] [CrossRef] [PubMed]
- Eremina, V.; Sood, M.; Haigh, J.; Nagy, A.; Lajoie, G.; Ferrara, N.; Gerber, H.P.; Kikkawa, Y.; Miner, J.H.; Quaggin, S.E. Glomerular-specific alterations of VEGF-A expression lead to distinct congenital and acquired renal diseases. J. Clin. Investig. 2003, 111, 707–716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, I.J.; Chou, C.H.; Yang, Y.H.; Lin, W.C.; Lin, Y.H.; Chow, L.P.; Lee, H.H.; Kao, P.G.; Liau, W.T.; Jou, T.S.; et al. Inhibition of Rho-associated kinase relieves C5a-induced proteinuria in murine nephrotic syndrome. Cell. Mol. Life Sci. 2015, 72, 3157–3171. [Google Scholar] [CrossRef] [PubMed]
- Back, M. Inflammatory signaling through leukotriene receptors in atherosclerosis. Curr. Atheroscler. Rep. 2008, 10, 244–251. [Google Scholar] [CrossRef] [PubMed]
- Riedemann, N.C.; Guo, R.F.; Neff, T.A.; Laudes, I.J.; Keller, K.A.; Sarma, V.J.; Markiewski, M.M.; Mastellos, D.; Strey, C.W.; Pierson, C.L.; et al. Increased C5a receptor expression in sepsis. J. Clin. Investig. 2002, 110, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, S.R.; Radhakrishnamurthy, B.; Vijayagopal, P.; Berenson, G.S. Proteoglycans, lipoproteins, and atherosclerosis. Adv. Exp. Med. Biol. 1991, 285, 373–381. [Google Scholar] [PubMed]
- Wesche, D.E.; Lomas-Neira, J.L.; Perl, M.; Chung, C.S.; Ayala, A. Leukocyte apoptosis and its significance in sepsis and shock. J. Leukoc. Biol. 2005, 78, 325–337. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Peng, Q.; Xing, G.; Li, K.; Wang, N.; Farrar, C.A.; Meader, L.; Sacks, S.H.; Zhou, W. Deficiency of C5aR prolongs renal allograft survival. J. Am. Soc. Nephrol. 2010, 21, 1344–1353. [Google Scholar] [CrossRef] [PubMed]
- Foreman, K.E.; Vaporciyan, A.A.; Bonish, B.K.; Jones, M.L.; Johnson, K.J.; Glovsky, M.M.; Eddy, S.M.; Ward, P.A. C5a-induced expression of P-selectin in endothelial cells. J. Clin. Investig. 1994, 94, 1147–1155. [Google Scholar] [CrossRef] [PubMed]
- Gasque, P.; Singhrao, S.K.; Neal, J.W.; Gotze, O.; Morgan, B.P. Expression of the receptor for complement C5a (CD88) is up-regulated on reactive astrocytes, microglia, and endothelial cells in the inflamed human central nervous system. Am. J. Pathol. 1997, 150, 31–41. [Google Scholar] [PubMed]
- Guo, R.F.; Sun, L.; Gao, H.; Shi, K.X.; Rittirsch, D.; Sarma, V.J.; Zetoune, F.S.; Ward, P.A. In vivo regulation of neutrophil apoptosis by C5a during sepsis. J. Leukoc. Biol. 2006, 80, 1575–1583. [Google Scholar] [CrossRef] [PubMed]
- Monsinjon, T.; Gasque, P.; Chan, P.; Ischenko, A.; Brady, J.J.; Fontaine, M.C. Regulation by complement C3a and C5a anaphylatoxins of cytokine production in human umbilical vein endothelial cells. FASEB J. 2003, 17, 1003–1014. [Google Scholar] [CrossRef]
- Mollnes, T.E.; Brekke, O.L.; Fung, M.; Fure, H.; Christiansen, D.; Bergseth, G.; Videm, V.; Lappegard, K.T.; Kohl, J.; Lambris, J.D. Essential role of the C5a receptor in E coli-induced oxidative burst and phagocytosis revealed by a novel lepirudin-based human whole blood model of inflammation. Blood 2002, 100, 1869–1877. [Google Scholar] [PubMed]
- Riedemann, N.C.; Guo, R.F.; Bernacki, K.D.; Reuben, J.S.; Laudes, I.J.; Neff, T.A.; Gao, H.; Speyer, C.; Sarma, V.J.; Zetoune, F.S.; et al. Regulation by C5a of neutrophil activation during sepsis. Immunity 2003, 19, 193–202. [Google Scholar] [CrossRef]
- Krug, N.; Tschernig, T.; Erpenbeck, V.J.; Hohlfeld, J.M.; Kohl, J. Complement factors C3a and C5a are increased in bronchoalveolar lavage fluid after segmental allergen provocation in subjects with asthma. Am. J. Respir. Crit. Care Med. 2001, 164, 1841–1843. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.A.; Maasch, C.; Vater, A.; Klussmann, S.; Morser, J.; Leung, L.L.; Atkinson, C.; Tomlinson, S.; Heeger, P.S.; Nicolls, M.R. Targeting complement component 5a promotes vascular integrity and limits airway remodeling. Proc. Natl. Acad. Sci. USA 2013, 110, 6061–6066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lechner, J.; Chen, M.; Hogg, R.E.; Toth, L.; Silvestri, G.; Chakravarthy, U.; Xu, H. Higher plasma levels of complement C3a, C4a and C5a increase the risk of subretinal fibrosis in neovascular age-related macular degeneration: Complement activation in AMD. Immun. Ageing 2016, 13, 4. [Google Scholar] [CrossRef] [PubMed]
- Solomkin, J.S.; Jenkins, M.K.; Nelson, R.D.; Chenoweth, D.; Simmons, R.L. Neutrophil dysfunction in sepsis. II. Evidence for the role of complement activation products in cellular deactivation. Surgery 1981, 90, 319–327. [Google Scholar]
- Wood, A.J.T.; Vassallo, A.; Summers, C.; Chilvers, E.R.; Conway-Morris, A. C5a anaphylatoxin and its role in critical illness-induced organ dysfunction. Eur. J. Clin. Investig. 2018, 48, e13028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oppermann, M.; Gotze, O. Plasma clearance of the human C5a anaphylatoxin by binding to leucocyte C5a receptors. Immunology 1994, 82, 516–521. [Google Scholar] [PubMed]
- Boor, P.; Konieczny, A.; Villa, L.; Schult, A.-L.; Bücher, E.; Rong, S.; Kunter, U.; van Roeyen, C.R.C.; Polakowski, T.; Hawlisch, H.; et al. Complement C5 Mediates Experimental Tubulointerstitial Fibrosis. J. Am. Soc. Nephrol. 2007, 18, 1508–1515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kestilä, M.; Lenkkeri, U.; Männikkö, M.; Lamerdin, J.; McCready, P.; Putaala, H.; Ruotsalainen, V.; Morita, T.; Nissinen, M.; Herva, R.; et al. Positionally cloned gene for a novel glomerular protein—nephrin—is mutated in congenital nephrotic syndrome. Mol. Cell. 1998, 1, 575–582. [Google Scholar] [CrossRef]
- Tryggvason, K.; Patrakka, J.; Wartiovaara, J. Hereditary proteinuria syndromes and mechanisms of proteinuria. N. Engl. J. Med. 2006, 354, 1387–1401. [Google Scholar] [CrossRef] [PubMed]
- Yuen, D.A.; Stead, B.E.; Zhang, Y.; White, K.E.; Kabir, M.G.; Thai, K.; Advani, S.L.; Connelly, K.A.; Takano, T.; Zhu, L.; et al. eNOS deficiency predisposes podocytes to injury in diabetes. J. Am. Soc. Nephrol. 2012, 23, 1810–1823. [Google Scholar] [CrossRef] [PubMed]
- Davis, B.; Dei Cas, A.; Long, D.A.; White, K.E.; Hayward, A.; Ku, C.H.; Woolf, A.S.; Bilous, R.; Viberti, G.; Gnudi, L. Podocyte-specific expression of angiopoietin-2 causes proteinuria and apoptosis of glomerular endothelia. J. Am. Soc. Nephrol. 2007, 18, 2320–2329. [Google Scholar] [CrossRef] [PubMed]
- Klos, A.; Tenner, A.J.; Johswich, K.O.; Ager, R.R.; Reis, E.S.; Kohl, J. The role of the anaphylatoxins in health and disease. Mol. Immunol. 2009, 46, 2753–2766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, R.; Coulthard, L.G.; Wu, M.C.; Taylor, S.M.; Woodruff, T.M. C5L2: A controversial receptor of complement anaphylatoxin, C5a. FASEB J. 2013, 27, 855–864. [Google Scholar] [CrossRef] [PubMed]
- Gerard, C.; Gerard, N.P. C5A anaphylatoxin and its seven transmembrane-segment receptor. Annu. Rev. Immunol. 1994, 12, 775–808. [Google Scholar] [CrossRef]
- Haviland, D.L.; McCoy, R.L.; Whitehead, W.T.; Akama, H.; Molmenti, E.P.; Brown, A.; Haviland, J.C.; Parks, W.C.; Perlmutter, D.H.; Wetsel, R.A. Cellular expression of the C5a anaphylatoxin receptor (C5aR): Demonstration of C5aR on nonmyeloid cells of the liver and lung. J. Immunol. 1995, 154, 1861–1869. [Google Scholar]
- Fayyazi, A.; Scheel, O.; Werfel, T.; Schweyer, S.; Oppermann, M.; Gotze, O.; Radzun, H.J.; Zwirner, J. The C5a receptor is expressed in normal renal proximal tubular but not in normal pulmonary or hepatic epithelial cells. Immunology 2000, 99, 38–45. [Google Scholar] [CrossRef]
- Schraufstatter, I.U.; Trieu, K.; Sikora, L.; Sriramarao, P.; DiScipio, R. Complement c3a and c5a induce different signal transduction cascades in endothelial cells. J. Immunol. 2002, 169, 2102–2110. [Google Scholar] [CrossRef]
- Ward, P.A. The dark side of C5a in sepsis. Nat. Rev. Immunol. 2004, 4, 133–142. [Google Scholar] [CrossRef]
- Albrecht, E.A.; Chinnaiyan, A.M.; Varambally, S.; Kumar-Sinha, C.; Barrette, T.R.; Sarma, J.V.; Ward, P.A. C5a-induced gene expression in human umbilical vein endothelial cells. Am. J. Pathol. 2004, 164, 849–859. [Google Scholar] [CrossRef]
- Perianayagam, M.C.; Balakrishnan, V.S.; King, A.J.; Pereira, B.J.; Jaber, B.L. C5a delays apoptosis of human neutrophils by a phosphatidylinositol 3-kinase-signaling pathway. Kidney Int. 2002, 61, 456–463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perianayagam, M.C.; Balakrishnan, V.S.; Pereira, B.J.; Jaber, B.L. C5a delays apoptosis of human neutrophils via an extracellular signal-regulated kinase and Bad-mediated signalling pathway. Eur. J. Clin. Investig. 2004, 34, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Mahajan, S.D.; Parikh, N.U.; Woodruff, T.M.; Jarvis, J.N.; Lopez, M.; Hennon, T.; Cunningham, P.; Quigg, R.J.; Schwartz, S.A.; Alexander, J.J. C5a alters blood-brain barrier integrity in a human in vitro model of systemic lupus erythematosus. Immunology 2015, 146, 130–143. [Google Scholar] [CrossRef] [PubMed]
- Jacob, A.; Hack, B.; Bai, T.; Brorson, J.R.; Quigg, R.J.; Alexander, J.J. Inhibition of C5a receptor alleviates experimental CNS lupus. J. Neuroimmunol. 2010, 221, 46–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahajan, S.D.; Tutino, V.M.; Redae, Y.; Meng, H.; Siddiqui, A.; Woodruff, T.M.; Jarvis, J.N.; Hennon, T.; Schwartz, S.; Quigg, R.J.; et al. C5a induces caspase-dependent apoptosis in brain vascular endothelial cells in experimental lupus. Immunology 2016, 148, 407–419. [Google Scholar] [CrossRef] [PubMed]
- Drummond, G.R.; Sobey, C.G. Endothelial NADPH oxidases: Which NOX to target in vascular disease? Trends Endocrinol. Metab. 2014, 25, 452–463. [Google Scholar] [CrossRef] [PubMed]
- Green, D.R.; Kroemer, G. Pharmacological manipulation of cell death: Clinical applications in sight? J. Clin. Investig. 2005, 115, 2610–2617. [Google Scholar] [CrossRef]
- Riedl, S.J.; Salvesen, G.S. The apoptosome: Signalling platform of cell death. Nat. Rev. Mol. Cell Biol. 2007, 8, 405–413. [Google Scholar] [CrossRef]
- Sanz, A.B.; Santamaría, B.; Ruiz-Ortega, M.; Egido, J.; Ortiz, A. Mechanisms of renal apoptosis in health and disease. J. Am. Soc. Nephrol. 2008, 19, 1634–1642. [Google Scholar] [CrossRef]
- Olsen, T.S.; Olsen, H.S.; Hansen, H.E. Tubular ultrastructure in acute renal failure in man: Epithelial necrosis and regeneration. Vichows Arch. A Pathol. Anat. 1985, 406, 75–89. [Google Scholar] [CrossRef]
- Shankland, S.J. The podocyte’s response to injury: Role in proteinuria and glomerulosclerosis. Kidney Int. 2006, 69, 2131–2147. [Google Scholar] [CrossRef]
- Hughes, J.; Savill, J.S. Apoptosis in glomerulonephritis. Curr. Opin. Nephrol. Hypertens. 2005, 14, 389–395. [Google Scholar] [CrossRef] [PubMed]
- Ferri, K.F.; Kroemer, G. Organelle-specific initiation of cell death pathways. Nat. Cell Biol. 2001, 3, E255–E263. [Google Scholar] [CrossRef] [PubMed]
- Ravagnan, L.; Roumier, T.; Kroemer, G. Mitochondria, the killer organelles and their weapons. J. Cell. Physiol. 2002, 192, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Justo, P.; Lorz, C.; Sanz, A.; Egido, J.; Ortiz, A. Intracellular Mechanisms of Cyclosporin A–Induced Tubular Cell Apoptosis. J. Am. Soc. Nephrol. 2003, 14, 3072–3080. [Google Scholar] [CrossRef]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsai, I.-J.; Lin, W.-C.; Yang, Y.-H.; Tseng, Y.-L.; Lin, Y.-H.; Chou, C.-H.; Tsau, Y.-K. High Concentration of C5a-Induced Mitochondria-Dependent Apoptosis in Murine Kidney Endothelial Cells. Int. J. Mol. Sci. 2019, 20, 4465. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20184465
Tsai I-J, Lin W-C, Yang Y-H, Tseng Y-L, Lin Y-H, Chou C-H, Tsau Y-K. High Concentration of C5a-Induced Mitochondria-Dependent Apoptosis in Murine Kidney Endothelial Cells. International Journal of Molecular Sciences. 2019; 20(18):4465. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20184465
Chicago/Turabian StyleTsai, I-Jung, Wei-Chou Lin, Yao-Hsu Yang, Yu-Lin Tseng, Yen-Hung Lin, Chia-Hung Chou, and Yong-Kwei Tsau. 2019. "High Concentration of C5a-Induced Mitochondria-Dependent Apoptosis in Murine Kidney Endothelial Cells" International Journal of Molecular Sciences 20, no. 18: 4465. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20184465