Deficiency of Mitochondrial Aspartate-Glutamate Carrier 1 Leads to Oligodendrocyte Precursor Cell Proliferation Defects Both In Vitro and In Vivo

 , , , ,
, , , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
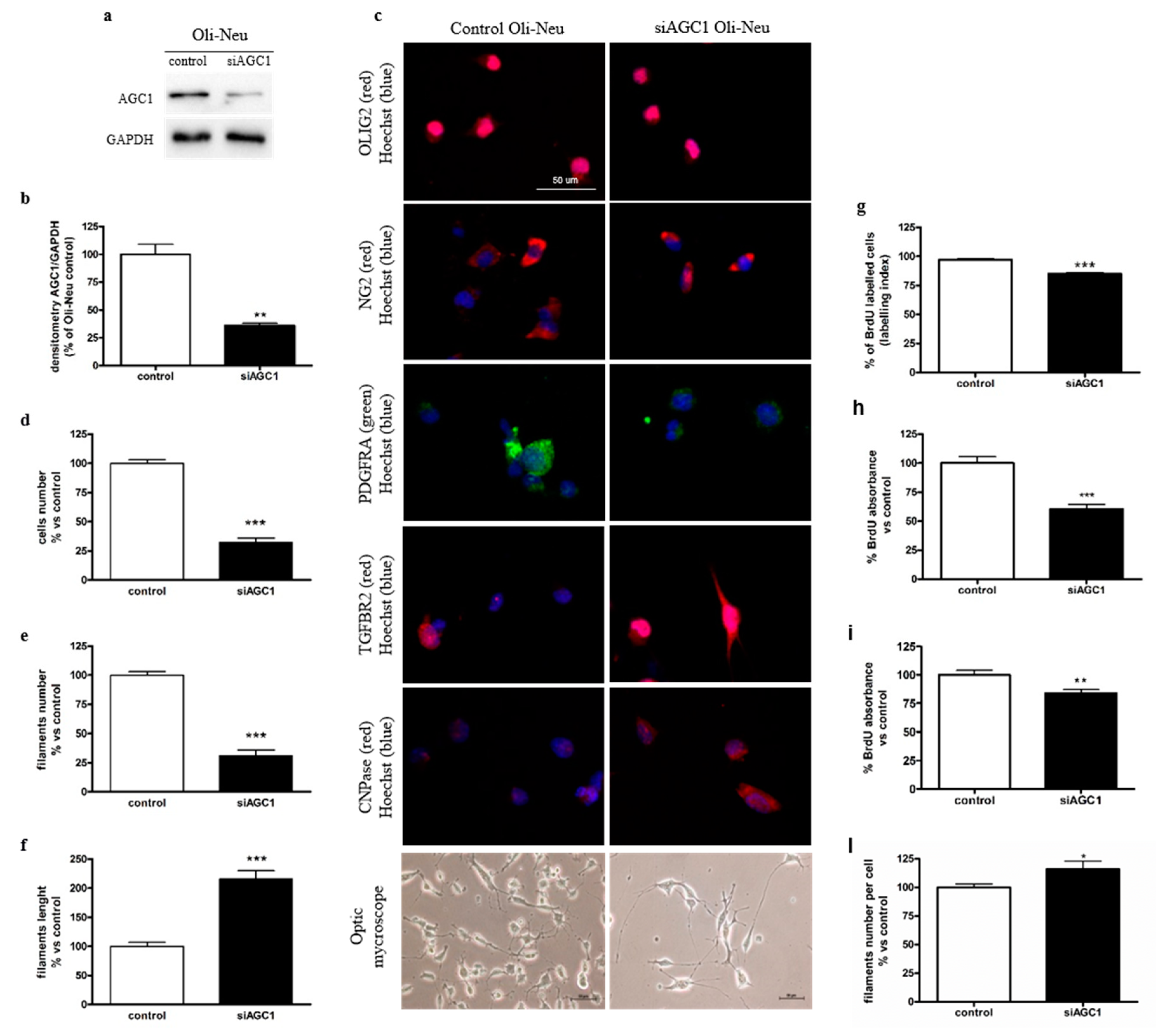
2.1. Effect of AGC1 Silencing on Oli-Neu Cell Differentiation and Proliferation
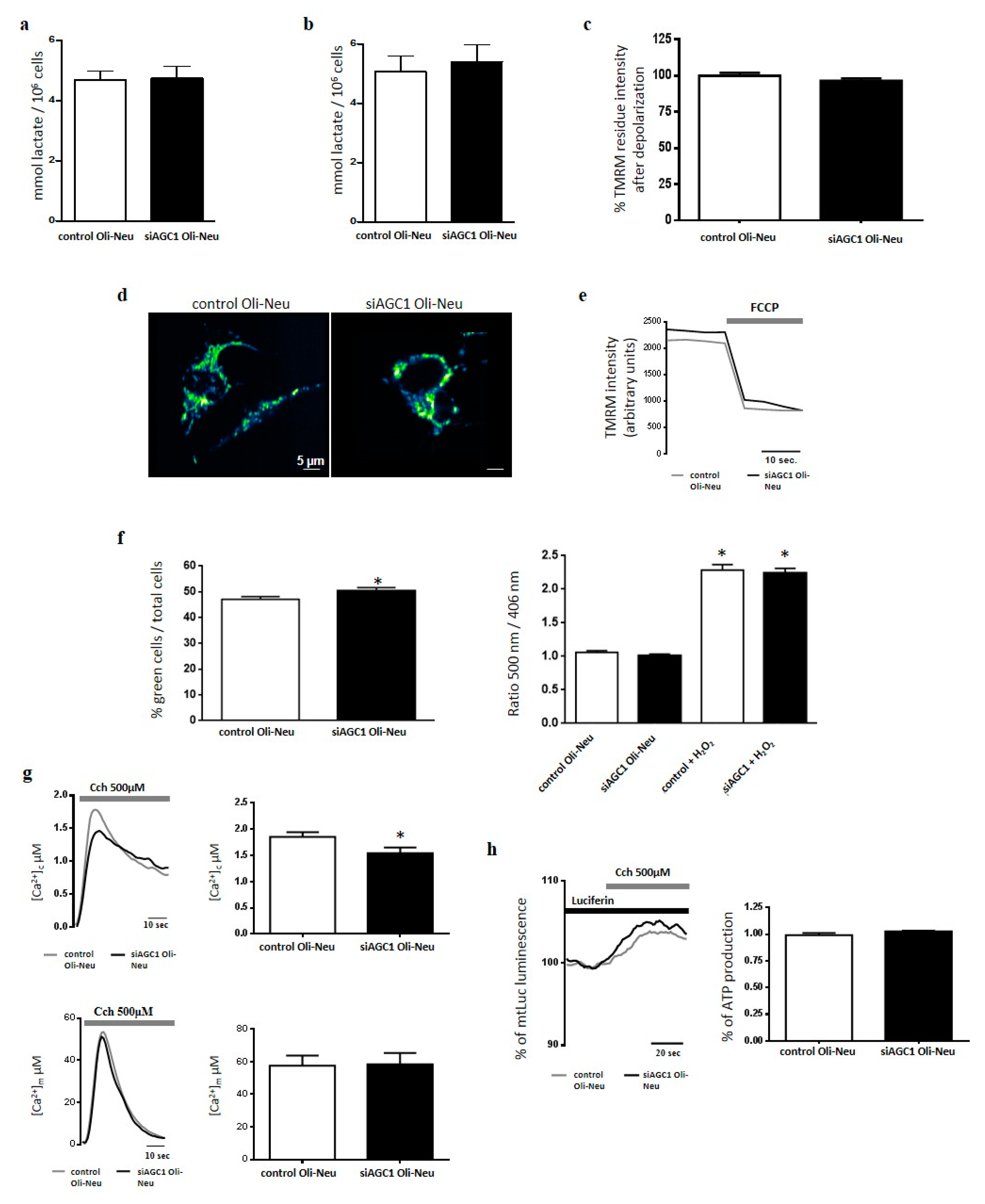
2.2. Effect of AGC1 Silencing on Oli-Neu Cell Lactate Production, Ros, [Ca2+], [Atp] and Mitochondrial Membrane Potential
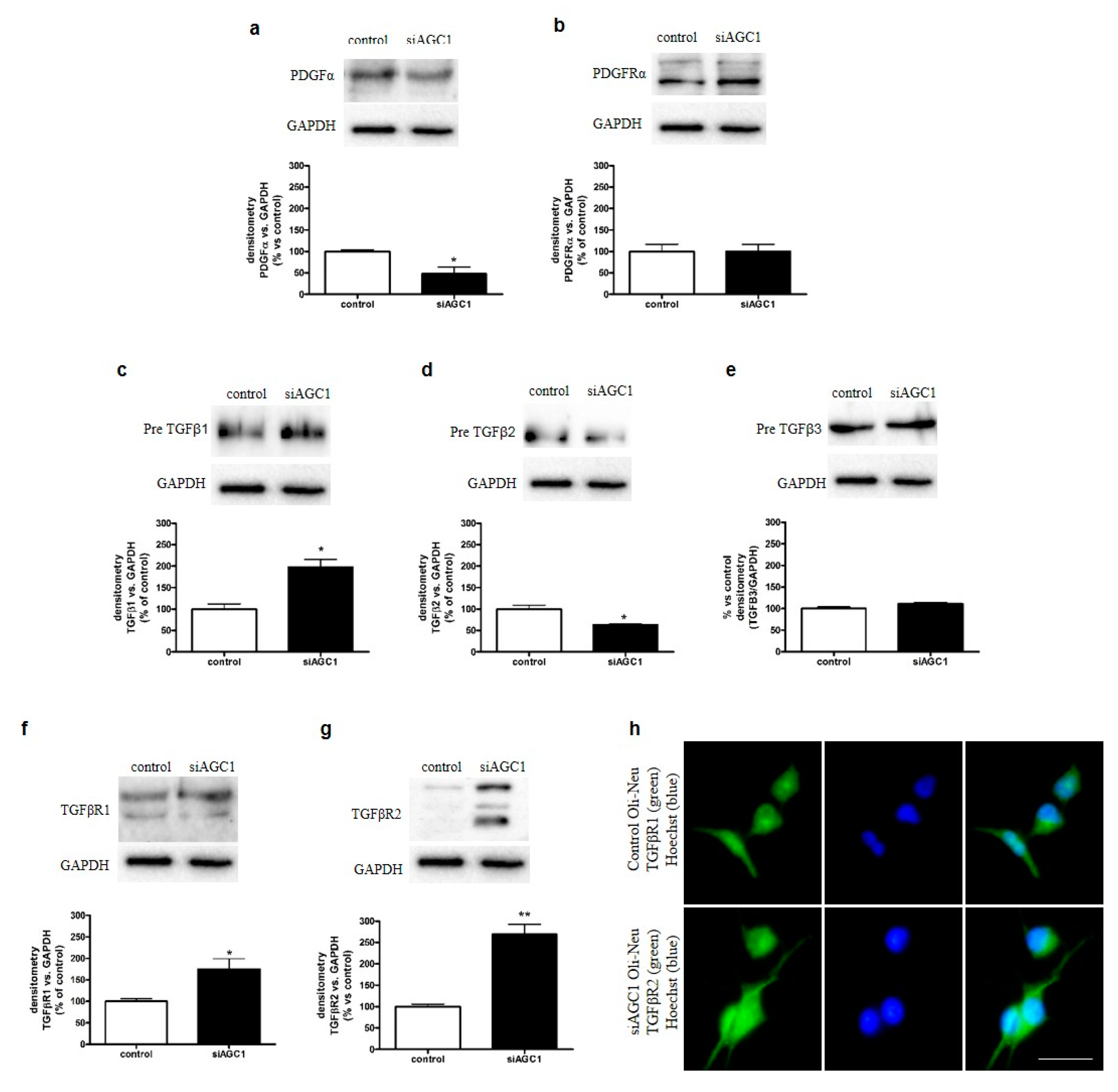
2.3. Effect of AGC1 Silencing on PDGFα and TGFβ Pathways in Oli-Neu Cells
2.4. Effects of TGFβ2 Treatment on Oli-Neu Cell Proliferation and Differentiation
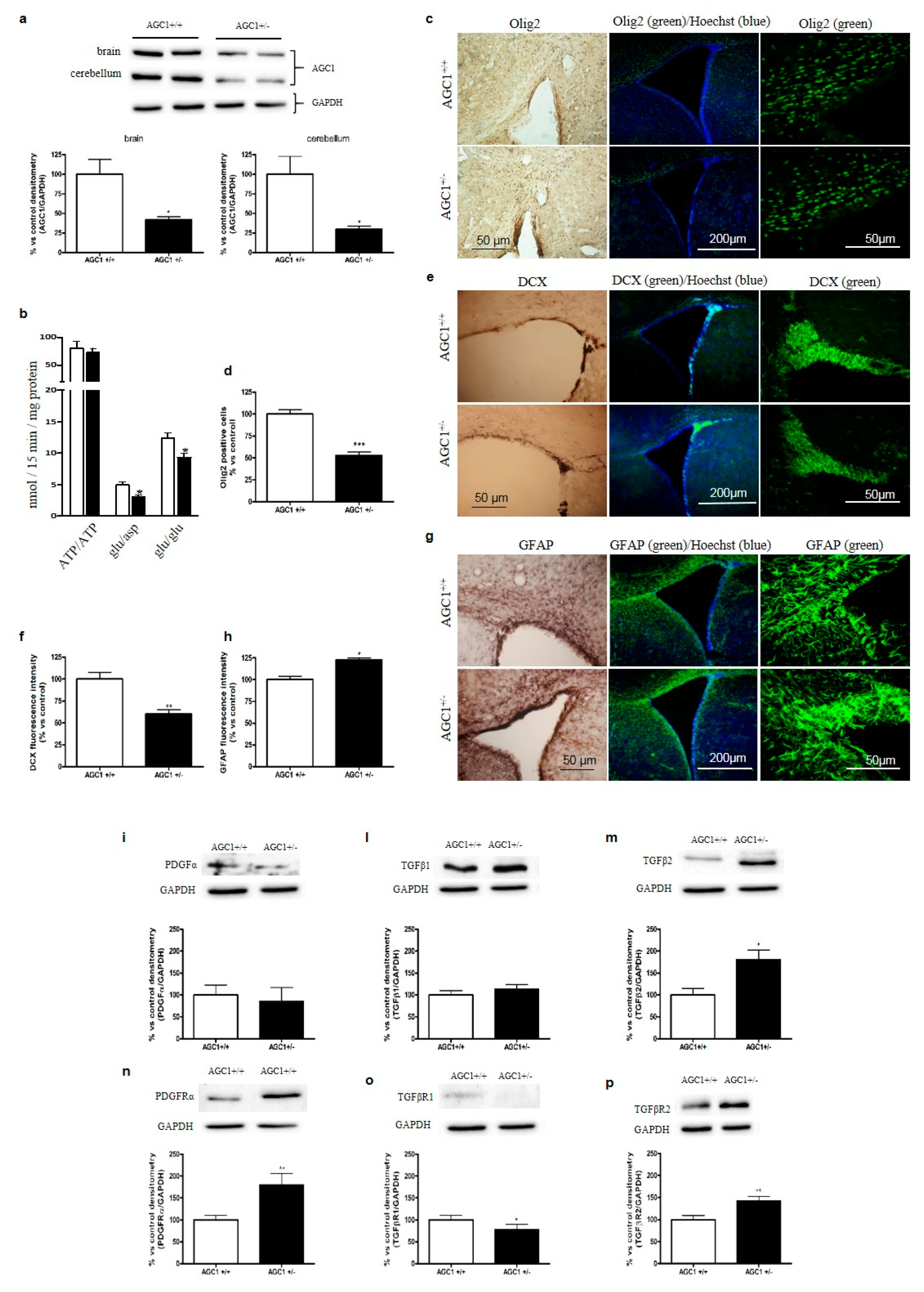
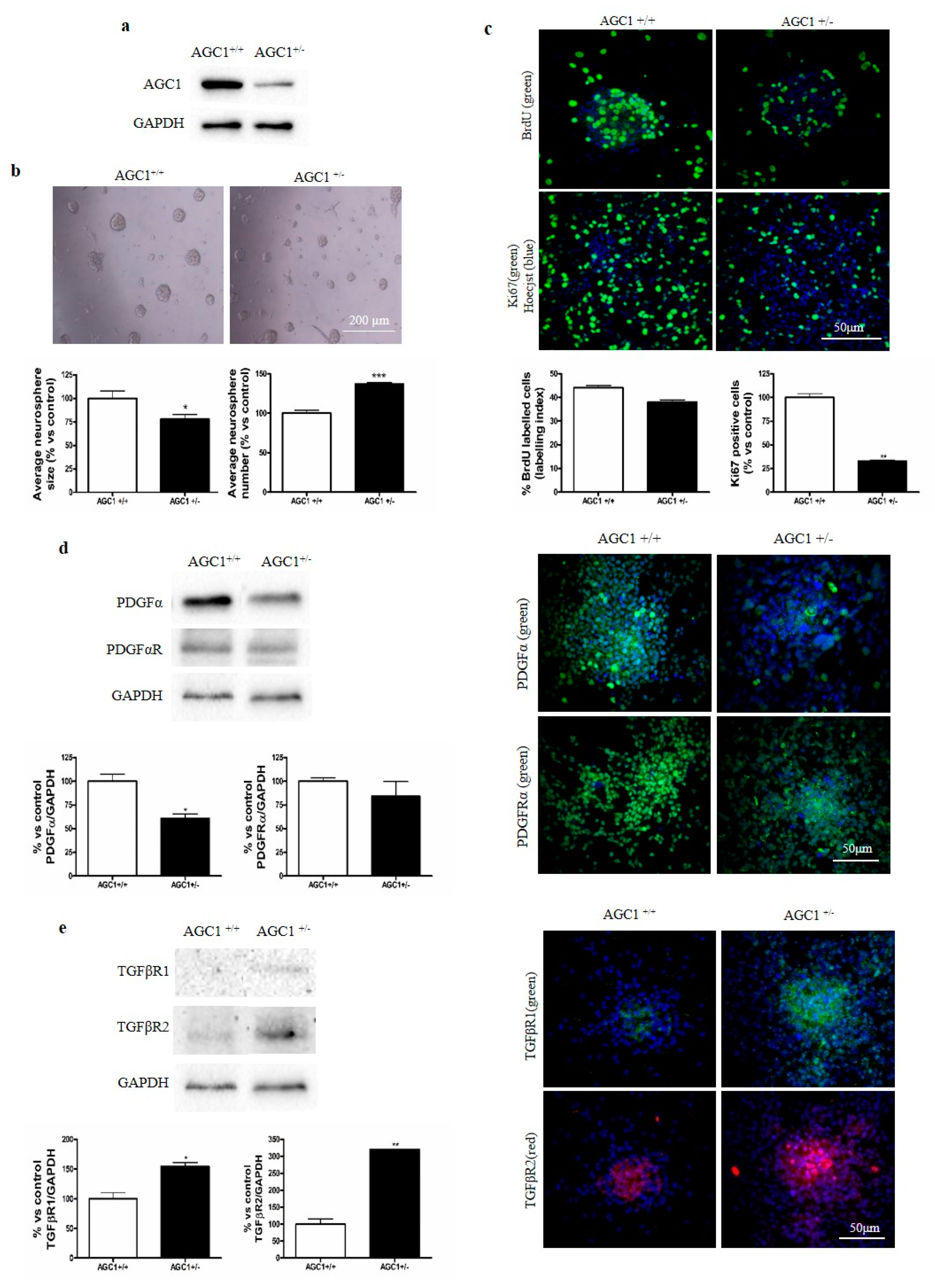
2.5. OPC Proliferation and PDGFα/TGFβs Expression in AGC1+/− Mice as an AGC1 Deficiency In Vivo Model
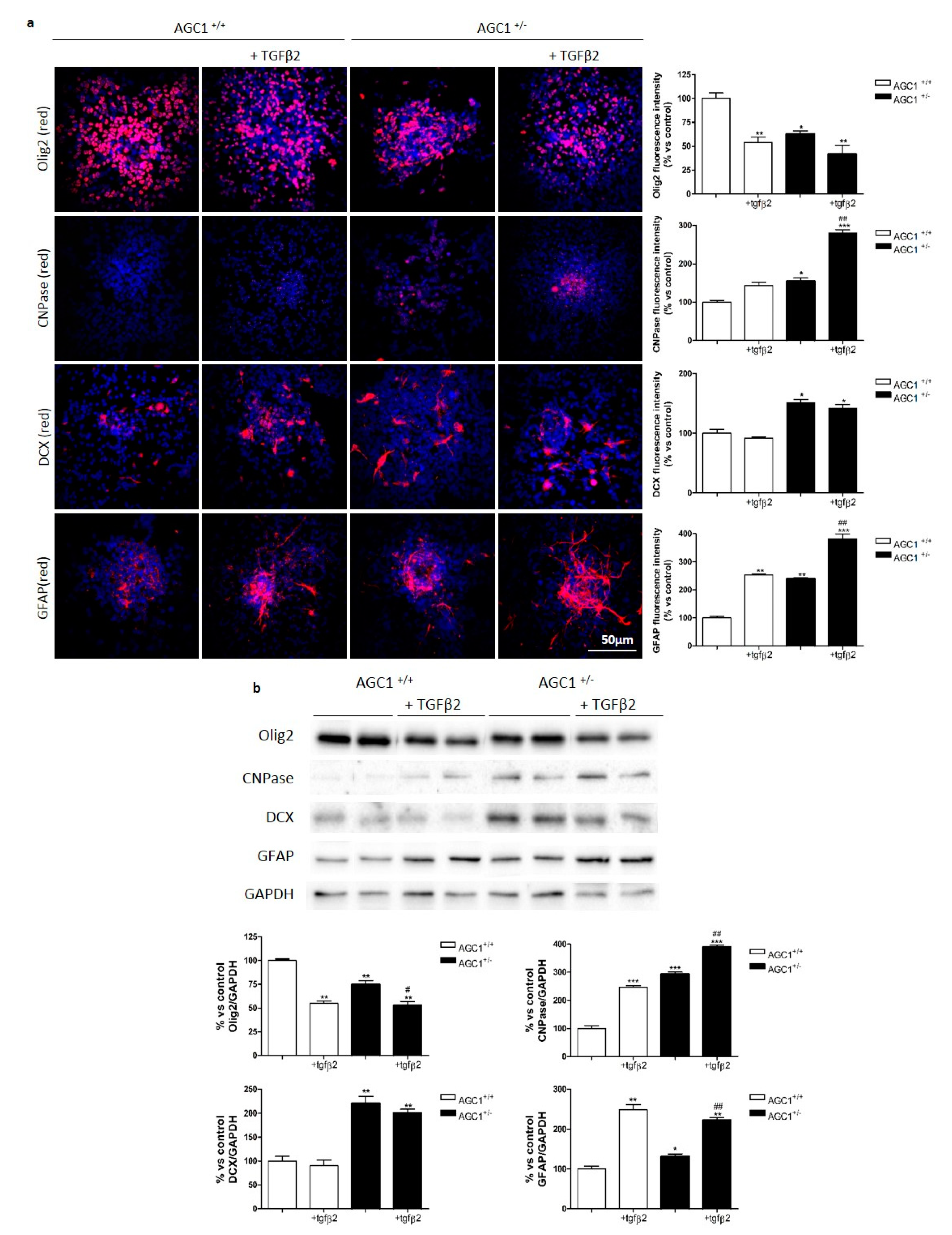
2.6. OPC Proliferation and PDGFα/TGFβs Expression in Neurospheres From the SVZ of AGC1+/+ and AGC1+/− Mice
3. Discussion
4. Materials and Methods
4.1. Cell Cultures
4.2. Stable Clone Generation
- 5′-CCGGTACAACCAACGCACGCTAATCTCGAGATTAGCGTGCGTTGGTTGTTTTTTG-3′
- 5′-CCGGTGCTTGCAGACCTATATAATGCctcgagGCATTATATAGGTCTGCAAGCTTTTT-3′)
4.3. TGFβ2 Treatment
4.4. Cell Count
4.5. Cell Filament Number and Length Measurement
4.6. Oli-Neu Bromodeoxyuridine (BrdU) Counting Following Immunofluorescence and Elisa Incorporation Assay
4.7. Lactic Acid Measurements
4.8. Aequorin and Luciferase Luminescence Measurement
4.9. Cell Fluorescence Analysis
4.10. Brain Mitochondria Isolation and Measurement of AGC1 Activity
4.11. AGC1+/− Mice Experiments
4.12. Neurosphere Generation
4.13. Neurosphere Proliferation and BrdU Assay
4.14. Neurosphere Treatment with Exogenous TGFβ
4.15. Cell and Tissue Sample Preparation and Western Blot Analysis
4.16. Cell and Tissue Immunohistochemistry and Immunofluorescence
4.17. Cell Counting after Immunohistochemistry and Immunofluorescence
4.18. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AEQ | aequorin |
| AGC1 | mitochondrial aspartate-glutamate carrier isoform 1 |
| BrdU | Bromodeoxyuridine |
| CNPase | 2′,3′-Cyclic-nucleotide 3′-phosphodiesterase |
| CTLN2 | type II citrullinemia |
| db-cAMP | N6,2′-o-dibutyryl)-adenosine-3′,5′-mono-phosphate |
| DCX | doublecortin |
| FCCP | Carbonyl cyanide-4-(trifluoromethoxy)phenylhydrazone |
| GFAP | Glial fibrillary acidic protein |
| HBSS | Hank’s Balanced Salt Solution |
| MAG | myelin associated glycoprotein |
| MBP | myelin basic protein |
| NAA | n-acetyl-aspartate |
| NICCD | neonatal intrahepatic cholestasis |
| NSCs | neural stem cells |
| OPCs | oligodendrocyte precursors cells |
| PDGFα | Platelet-Derived Growth Factor α |
| PBS | Phosphate Buffer Solution |
| PFA | paraformaldehyde |
| RT | room temperature |
| SLC25A12 | solute carrier family 25, member 12 |
| SVZ | subventricular zone |
| TGFβ | Transforming Growth Factor β |
| TMRM | tetramethyl rhodamine methyl ester |
References
- Wibom, R.; Lasorsa, F.M.; Tohonen, V.; Barbaro, M.; Sterky, F.H.; Kucinski, T.; Naess, K.; Jonsson, M.; Pierri, C.L.; Palmieri, F.; et al. AGC1 Deficiency Associated with Global Cerebral Hypomyelination. New Engl. J. Med. 2009, 361, 489–495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falk, M.J.; Li, D.; Gai, X.; McCormick, E.; Place, E.; Lasorsa, F.M.; Otieno, F.G.; Hou, C.; Kim, C.E.; Abdel-Magid, N.; et al. AGC1 Deficiency Causes Infantile Epilepsy, Abnormal Myelination and Reduced N-Acetylaspartate. JIMD Rep. 2014, 14, 77–85. [Google Scholar] [PubMed]
- Palmieri, F. Mitochondrial transporters of the SLC25 family and associated diseases: A review. J. Inherit. Metab. Dis. 2014, 37, 565–575. [Google Scholar] [CrossRef] [PubMed]
- Indiveri, C.; Krämer, R.; Palmieri, F. Reconstitution of the malate/aspartate shuttle from mitochondria. J. Boil. Chem. 1987, 262, 15979–15983. [Google Scholar]
- Palmieri, L.; Pardo, B.; Lasorsa, F.M.; del Arco, A.; Kobayashi, K.; Iijima, M.; Runswick, M.J.; Walker, J.E.; Saheki, T.; Satrústegui, J.; et al. Citrin and aralar1 are Ca(2+)-stimulated aspartate/glutamate transporters in mitochondria. EMBO J. 2001, 20, 5060–5069. [Google Scholar] [CrossRef] [PubMed]
- Del Arco, A.; Satrústegui, J. Molecular Cloning of Aralar, a New Member of the Mitochondrial Carrier Superfamily That Binds Calcium and Is Present in Human Muscle and Brain. J. Boil. Chem. 1998, 273, 23327–23334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saheki, T.; Kobayashi, K. Mitochondrial aspartate glutamate carrier (citrin) deficiency as the cause of adult-onset type II citrullinemia (CTLN2) and idiopathic neonatal hepatitis (NICCD). J. Hum. Genet. 2002, 47, 333–341. [Google Scholar] [CrossRef] [PubMed]
- Ledeen, R.W.; Wang, J.; Wu, G.; Lu, Z.H.; Chakraborty, G.; Meyenhofer, M.; Tyring, S.K.; Matalon, R. Physiological role of N-acetylaspartate: Contribution to myelinogenesis. Adv. Exp. Med. Biol. 2006, 576, 131–143. [Google Scholar]
- Jalil, M.A.; Begum, L.; Contreras, L.; Pardo, B.; Iijima, M.; Li, M.X.; Ramos, M.; Marmol, P.; Horiuchi, M.; Shimotsu, K.; et al. ReducedN-Acetylaspartate Levels in Mice Lacking Aralar, a Brain- and Muscle-type Mitochondrial Aspartate-glutamate Carrier. J. Boil. Chem. 2005, 280, 31333–31339. [Google Scholar] [CrossRef]
- Ramos, M.; Pardo, B.; Saheki, T.; Del Arco, A.; Satrústegui, J.; Llorente-Folch, I.; Llorente-Folch, I. Deficiency of the mitochondrial transporter of aspartate/glutamate aralar/AGC1 causes hypomyelination and neuronal defects unrelated to myelin deficits in mouse brain. J. Neurosci. Res. 2011, 89, 2008–2017. [Google Scholar] [CrossRef]
- Gómez-Galán, M.; Makarova, J.; Llorente-Folch, I.; Saheki, T.; Pardo, B.; Satrústegui, J.; Herreras, O. Altered postnatal development of cortico-hippocampal neuronal electric activity in mice deficient for the mitochondrial aspartate-glutamate transporter. J. Cereb. Blood Flow Metab. 2012, 32, 306–317. [Google Scholar] [CrossRef] [PubMed]
- Llorente-Folch, I.; Rueda, C.B.; Amigo, I.; Del Arco, A.; Saheki, T.; Pardo, B.; Satrustegui, J. Calcium-Regulation of Mitochondrial Respiration Maintains ATP Homeostasis and Requires ARALAR/AGC1-Malate Aspartate Shuttle in Intact Cortical Neurons. J. Neurosci. 2013, 33, 13957–13971. [Google Scholar] [CrossRef] [PubMed]
- Llorente-Folch, I.; Sahún, I.; Contreras, L.; Casarejos, M.J.; Grau, J.M.; Saheki, T.; Mena, M.A.; Satrústegui, J.; Dierssen, M.; Pardo, B.; et al. AGC1-malate aspartate shuttle activity is critical for dopamine handling in the nigrostriatal pathway. J. Neurochem. 2013, 124, 347–362. [Google Scholar] [CrossRef] [PubMed]
- Contreras, L.; Ramirez, L.; Du, J.; Hurley, J.B.; Satrústegui, J.; De La Villa, P. Deficient glucose and glutamine metabolism in Aralar/AGC1/Slc25a12 knockout mice contributes to altered visual function. Mol. Vis. 2016, 22, 1198–1212. [Google Scholar] [PubMed]
- Juaristi, I.; García-Martín, M.L.; Satrústegui, J.; Llorente-Folch, I.; Pardo, B.; Rodrigues, T.B. ARALAR/AGC1 deficiency, a neurodevelopmental disorder with severe impairment of neuronal mitochondrial respiration, does not produce a primary increase in brain lactate. J. Neurochem. 2017, 142, 132–139. [Google Scholar] [CrossRef]
- Boulanger, J.; Messier, C. From precursors to myelinating oligodendrocytes: Contribution of intrinsic and extrinsic factors to white matter plasticity in the adult brain. Neuroscience 2014, 269, 343–366. [Google Scholar] [CrossRef]
- Chamberlain, K.A.; Nanescu, S.E.; Psachoulia, K.; Huang, J.K. Oligodendrocyte regeneration: Its significance in myelin replacement and neuroprotection in multiple sclerosis. Neuropharmacol 2016, 110, 633–643. [Google Scholar] [CrossRef]
- Sakurai, T.; Ramoz, N.; Barreto, M.; Gazdoiu, M.; Takahashi, N.; Gertner, M.; Dorr, N.; Gama Sosa, M.A.; De Gasperi, R.; Perez, G.; et al. Slc25a12 disruption alters myelination and neurofilaments: A model for a hypomyelination syndrome and childhood neurodevelopmental disorders. Biol. Psychiatry 2010, 679, 887–894. [Google Scholar] [CrossRef]
- Profilo, E.; Peña-Altamira, L.E.; Corricelli, M.; Castegna, A.; Danese, A.; Agrimi, G.; Petralla, S.; Giannuzzi, G.; Porcelli, V.; Sbano, L.; et al. Down-regulation of the mitochondrial aspartate-glutamate carrier isoform 1 AGC1 inhibits proliferation and N-acetylaspartate synthesis in Neuro2A cells. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2017, 1863, 1422–1435. [Google Scholar] [CrossRef]
- Belousov, V.V.; Fradkov, A.F.; Lukyanov, K.A.; Staroverov, D.B.; Shakhbazov, K.S.; Terskikh, A.V.; Lukyanov, S. Genetically encoded fluorescent indicator for intracellular hydrogen peroxide. Nat. Methods 2006, 3, 281–286. [Google Scholar] [CrossRef]
- Hendzel, M.J.; Wei, Y.; Mancini, M.A.; Van Hooser, A.; Ranalli, T.; Brinkley, B.R.; Bazett-Jones, D.P.; Allis, C.D. Mitosis-specific phosphorylation of histone H3 initiates primarily within pericentromeric heterochromatin during G2 and spreads in an ordered fashion coincident with mitotic chromosome condensation. Chromosom 1997, 106, 348–360. [Google Scholar] [CrossRef] [PubMed]
- Takebayashi, H.; Nabeshima, Y.; Yoshida, S.; Chisaka, O.; Ikenaka, K.; Nabeshima, Y.-I. The Basic Helix-Loop-Helix Factor Olig2 Is Essential for the Development of Motoneuron and Oligodendrocyte Lineages. Curr. Boil. 2002, 12, 1157–1163. [Google Scholar] [CrossRef] [Green Version]
- Francis, F.; Koulakoff, A.; Boucher, D.; Chafey, P.; Schaar, B.; Vinet, M.-C.; Friocourt, G.; McDonnell, N.; Reiner, O.; Kahn, A.; et al. Doublecortin Is a Developmentally Regulated, Microtubule-Associated Protein Expressed in Migrating and Differentiating Neurons. Neuron 1999, 23, 247–256. [Google Scholar] [CrossRef] [Green Version]
- Brunner, C.; Lassmann, H.; Waehneldt, T.V.; Matthieu, J.; Linington, C. Differential Ultrastructural Localization of Myelin Basic Protein, Myelin/Oligodendroglial Glycoprotein and 2′,3′-Cyclic Nucleotide 3′-Phosphodiesterase in the CNS of Adult Rats. J. Neurochem. 1989, 52, 296–304. [Google Scholar] [CrossRef] [PubMed]
- Baumann, N.; Pham-Dinh, D. Biology of Oligodendrocyte and Myelin in the Mammalian Central Nervous System. Physiol. Rev. 2001, 81, 871–927. [Google Scholar] [CrossRef] [PubMed]
- Accetta, R.; Damiano, S.; Morano, A.; Mondola, P.; Paternò, R.; Avvedimento, E.V.; Santillo, M. Reactive Oxygen Species Derived from NOX3 and NOX5 Drive Differentiation of Human Oligodendrocytes. Front. Cell. Neurosci. 2016, 10, 146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diemel, L.T.; Jackson, S.J.; Cuzner, M.L. Role for TGF-β1, FGF-2 and PDGF-AA in a myelination of CNS aggregate cultures enriched with macrophages. J. Neurosci. Res. 2003, 74, 858–867. [Google Scholar] [CrossRef] [PubMed]
- Palazuelos, J.; Klingener, M.; Aguirre, A. TGFβ Signalling Regulates the Timing of CNS Myelination by Modulating Oligodendrocyte Progenitor Cell Cycle Exit through SMAD3/4/FoxO1/Sp1. J. Neurosci. 2014, 34, 7917–7930. [Google Scholar] [CrossRef]
- Koch, H.B.; Zhang, R.; Verdoodt, B.; Bailey, A.; Zhang, C.D.; Yates, J.R.; Menssen, A.; Hermeking, H. Large-scale identification of c-MYC-associated proteins using a combined TAP/MudPIT approach. Cell Cycle 2007, 6, 205–217. [Google Scholar] [CrossRef]
- Xie, D.; Shen, F.; He, S.; Chen, M.; Han, Q.; Fang, M.; Zeng, H.; Chen, C.; Deng, Y. IL-1β induces hypomyelination in the periventricular white matter through inhibition of oligodendrocyte progenitor cell maturation via FYN/MEK/ERK signaling pathway in septic neonatal rats. Glia 2016, 64, 583–602. [Google Scholar] [CrossRef]
- Remaud, S.; Ortiz, F.C.; Perret-Jeanneret, M.; Aigrot, M.S.; Gothié, J.D.; Fekete, C.; Kvárta-Papp, Z.; Gereben, B.; Langui, D.; Lubetzki, C.; et al. Transient hypothyroidism favors oligodendrocyte generation providing functional remyelination in the adult mouse brain. Elife 2017, e29996. [Google Scholar] [CrossRef] [PubMed]
- Ayanlaja, A.A.; Xiong, Y.; Gao, Y.; Ji, G.; Tang, C.; Abdikani, A.Z.; Gao, D. Distinct Features of Doublecortin as a Marker of Neuronal Migration and Its Implications in Cancer Cell Mobility. Front. Mol. Neurosci. 2017, 10, 199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gil-Perotín, S.; Duran-Moreno, M.; Ramirez, M.; García-Verdugo, J.M.; Gil-Perotin, S.; Duran-Moreno, M.; Cebrián-Silla, A.; García-Belda, P.; Garcia-Verdugo, J.M.; Gil-Perotin, S.; et al. Adult Neural Stem Cells from the Subventricular Zone: A Review of the Neurosphere Assay. Anat. Rec. Adv. Integr. Anat. Evol. Boil. 2013, 296, 1435–1452. [Google Scholar] [CrossRef] [PubMed]
- Walker, T.L.; Kempermann, G. One Mouse, Two Cultures: Isolation and Culture of Adult Neural Stem Cells from the Two Neurogenic Zones of Individual Mice. J. Vis. Exp. 2014, e51225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hart, I.K.; Richardson, W.D.; Bolsover, S.R.; Raff, M.C. PDGF and intracellular signaling in the timing of oligodendrocyte differentiation. J. Cell Boil. 1989, 109, 3411–3417. [Google Scholar] [CrossRef] [PubMed]
- Noble, M.; Murray, K.; Stroobant, P.; Waterfield, M.D.; Riddle, P. Platelet-derived growth factor promotes division and motility and inhibits premature differentiation of the oligodendrocyte/type-2 astrocyte progenitor ceil. Nature 1988, 333, 560–562. [Google Scholar] [CrossRef] [PubMed]
- Raff, M.C.; Lillien, L.E.; Richardson, W.D.; Burne, J.F.; Noble, M.D. Platelet-derived growth factor from astrocytes drives the clock that times oligodendrocyte development in culture. Nature 1988, 333, 562–565. [Google Scholar] [CrossRef]
- Dahlin, M.; Mártin, D.A.; Hedlund, Z.; Jönsson, M.; Von Döbeln, U.; Wedell, A. The ketogenic diet compensates for AGC1 deficiency and improves myelination. Epilepsia 2015, 56, 176–181. [Google Scholar] [CrossRef]
- Heo, G.; Kim, S.H.; Chang, M.J. Effect of ketogenic diet and other dietary therapies on anti-epileptic drug concentrations in patients with epilepsy. J. Clin. Pharm. Ther. 2017, 42, 758–764. [Google Scholar] [CrossRef]
- Barañano, K.W.; Hartman, A.L. The Ketogenic Diet: Uses in Epilepsy and Other Neurologic Illnesses. Curr. Treat. Options Neurol. 2008, 10, 410–419. [Google Scholar] [CrossRef]
- Vozza, A.; Parisi, G.; De Leonardis, F.; Lasorsa, F.M.; Castegna, A.; Amorese, D.; Marmo, R.; Calcagnile, V.M.; Palmieri, L.; Ricquier, D.; et al. UCP2 transports C4 metabolites out of mitochondria, regulating glucose and glutamine oxidation. Proc. Natl. Acad. Sci. USA 2014, 111, 960–965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lasorsa, F.M.; Pinton, P.; Palmieri, L.; Fiermonte, G.; Rizzuto, R.; Palmieri, F. Recombinant expression of the Ca(2+)-sensitive aspartate/glutamate carrier increases mitochondrial ATP production in agonist-stimulated Chinese hamster ovary cells. J. Biol. Chem. 2003, 278, 38686–38692. [Google Scholar] [CrossRef]
- Bonora, M.; Giorgi, C.; Bononi, A.; Marchi, S.; Patergnani, S.; Rimessi, A.; Rizzuto, R.; Pinton, P. Subcellular calcium measurements in mammalian cells using jellyfish photoprotein aequorin-based probes. Nat. Protoc. 2013, 8, 2105–2118. [Google Scholar] [CrossRef] [PubMed]
- Suski, J.M.; Lebiedzinska, M.; Bonora, M.; Pinton, P.; Duszynski, J.; Wieckowski, M.R. Relation Between Mitochondrial Membrane Potential and ROS Formation. Mitochondrial Bioenerg. 2012, 810, 183–205. [Google Scholar]
- Grove, B.D.; Bruchey, A.K. Intracellular Distribution of Gravin, a PKA and PKC Binding Protein, in Vascular Endothelial Cells. J. Vasc. Res. 2001, 38, 163–175. [Google Scholar] [CrossRef]
- Lowry, O.H.; Rosebrough, N.J.; Farr, A.L.; Randall, R.J. Protein measurement with the Folin phenol reagent. J. Boil. Chem. 1951, 193. [Google Scholar]
- Goubert, E.; Mircheva, Y.; Lasorsa, F.M.; Melon, C.; Profilo, E.; Sutera, J.; Becq, H.; Palmieri, F.; Palmieri, L.; Aniksztejn, L.; et al. Inhibition of the Mitochondrial Glutamate Carrier SLC25A22 in Astrocytes Leads to Intracellular Glutamate Accumulation. Front. Cell. Neurosci. 2017, 11, 149. [Google Scholar] [CrossRef]
- Choudhry, P. High-Throughput Method for Automated Colony and Cell Counting by Digital Image Analysis Based on Edge Detection. PLoS ONE 2016, 11. [Google Scholar] [CrossRef]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Petralla, S.; Peña-Altamira, L.E.; Poeta, E.; Massenzio, F.; Virgili, M.; Barile, S.N.; Sbano, L.; Profilo, E.; Corricelli, M.; Danese, A.; et al. Deficiency of Mitochondrial Aspartate-Glutamate Carrier 1 Leads to Oligodendrocyte Precursor Cell Proliferation Defects Both In Vitro and In Vivo. Int. J. Mol. Sci. 2019, 20, 4486. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20184486
Petralla S, Peña-Altamira LE, Poeta E, Massenzio F, Virgili M, Barile SN, Sbano L, Profilo E, Corricelli M, Danese A, et al. Deficiency of Mitochondrial Aspartate-Glutamate Carrier 1 Leads to Oligodendrocyte Precursor Cell Proliferation Defects Both In Vitro and In Vivo. International Journal of Molecular Sciences. 2019; 20(18):4486. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20184486
Chicago/Turabian StylePetralla, Sabrina, Luis Emiliano Peña-Altamira, Eleonora Poeta, Francesca Massenzio, Marco Virgili, Simona Nicole Barile, Luigi Sbano, Emanuela Profilo, Mariangela Corricelli, Alberto Danese, and et al. 2019. "Deficiency of Mitochondrial Aspartate-Glutamate Carrier 1 Leads to Oligodendrocyte Precursor Cell Proliferation Defects Both In Vitro and In Vivo" International Journal of Molecular Sciences 20, no. 18: 4486. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20184486