Identification of Functional and Druggable Sites in Aspergillus fumigatus Essential Phosphatases by Virtual Screening


Abstract
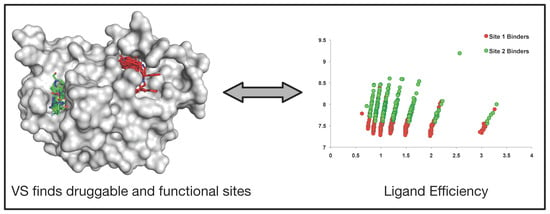
:
1. Introduction
2. Results
2.1. Identification and Classification of Fungal Phosphatases
2.2. Characterisation of the Essential Phosphatase Cohort in A. fumigatus
2.3. Building Molecular Models for A. fumigatus Phosphatases
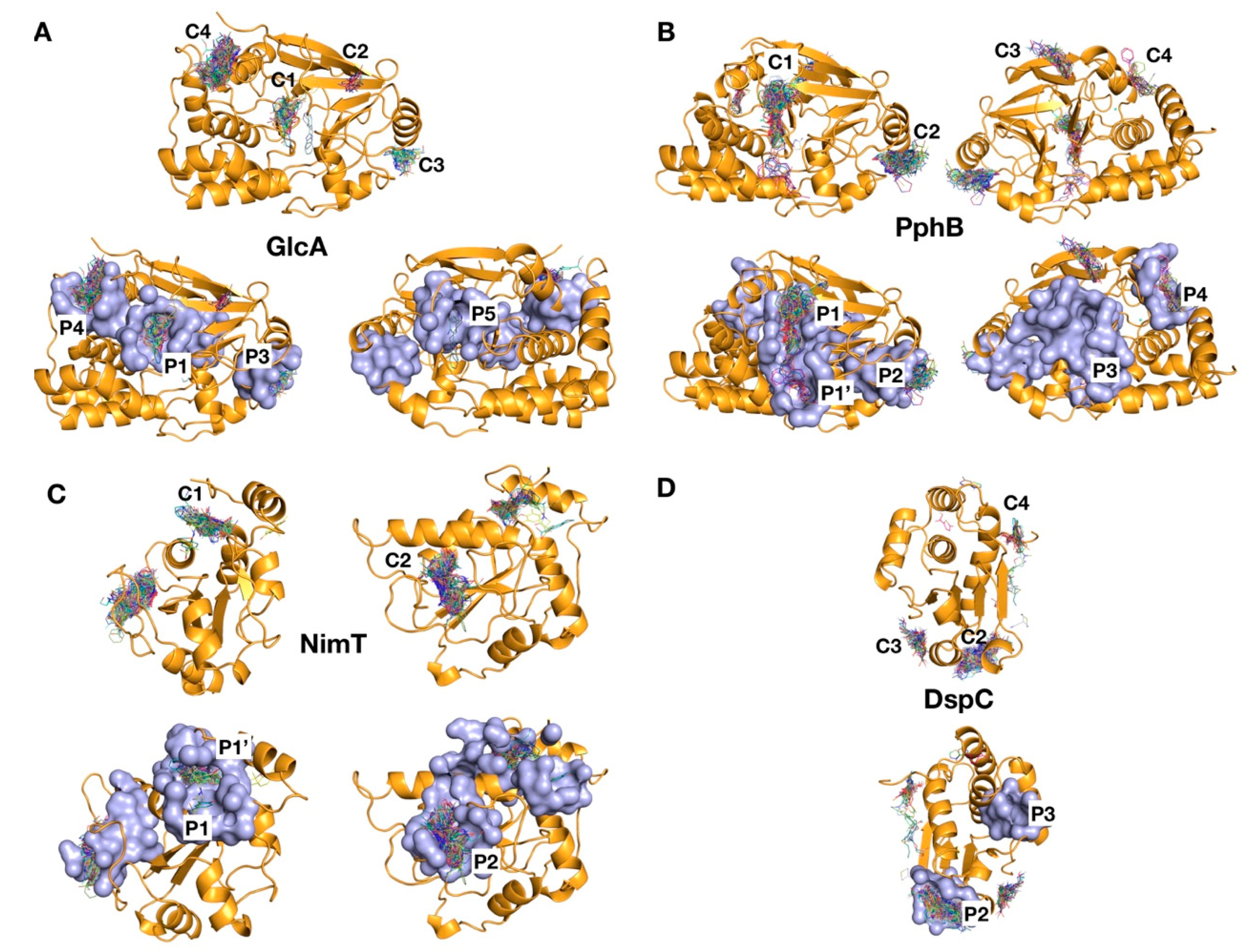
2.4. Identification of Ligand Binding Sites by Virtual Screening
2.5. Surface Pocket Prediction
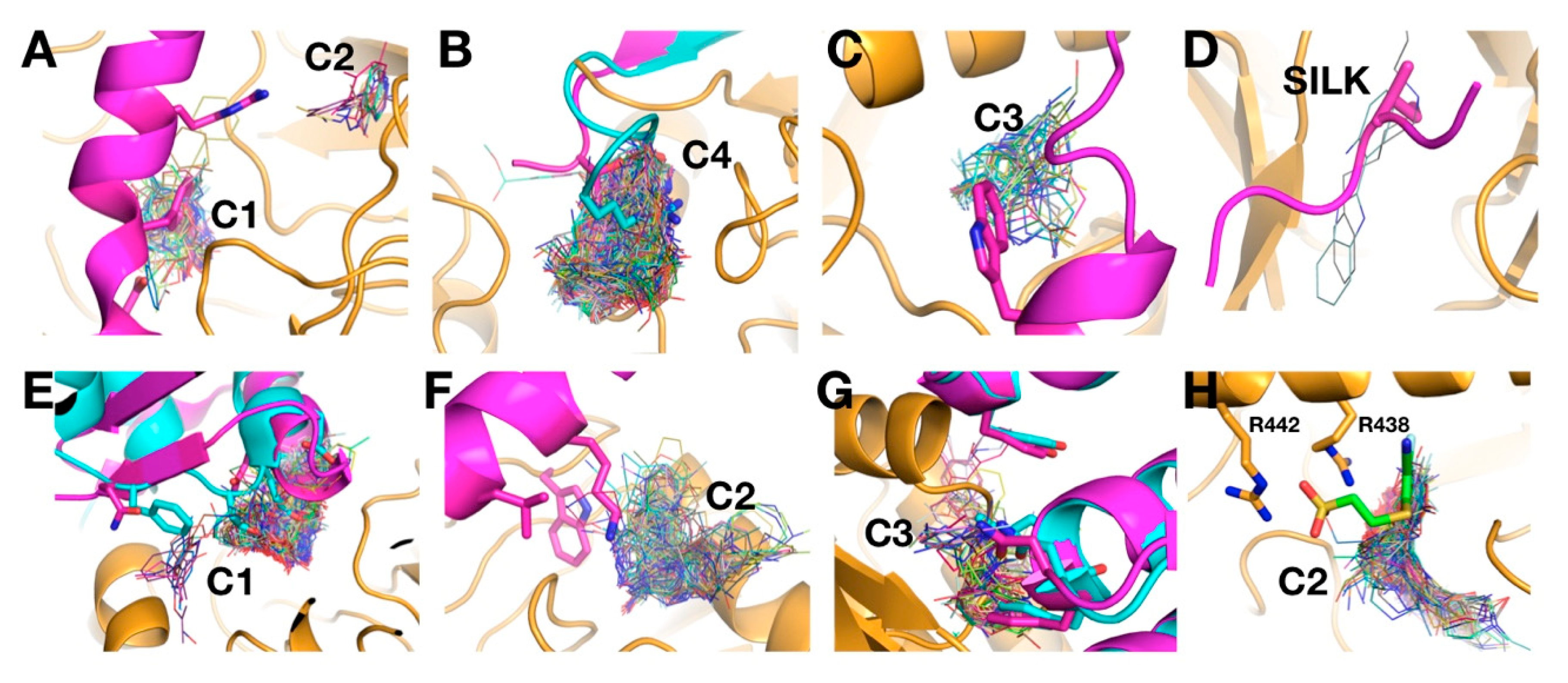
2.6. Analysis of Targeted Docking at Druggable Sites
2.7. Protein–Protein Interaction Sites and Matching with VS Clusters
3. Materials and Methods
3.1. Protein Phosphatase Classification
3.2. Generation of Protein Phosphatase Null Mutants
3.3. Molecular Homology Models
3.4. Virtual Screening with VSpipe
3.5. Pocket Predictions
3.6. Sample Availability
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Dodds Ashley, E.; Drew, R.; Johnson, M.; Danna, R.; Dabrowski, D.; Walker, V.; Prasad, M.; Alexander, B.; Papadopoulos, G.; Perfect, J. Cost of invasive fungal infections in the era of new diagnostics and expanded treatment options. Pharmacotherapy 2012, 32, 890–901. [Google Scholar] [CrossRef] [PubMed]
- Brown, G.D.; Denning, D.W.; Gow, N.A.; Levitz, S.M.; Netea, M.G.; White, T.C. Hidden killers: Human fungal infections. Sci. Transl. Med. 2012, 4, 165rv113. [Google Scholar] [CrossRef] [PubMed]
- Arino, J.; Velazquez, D.; Casamayor, A. Ser/thr protein phosphatases in fungi: Structure, regulation and function. Microb. Cell 2019, 6, 217–256. [Google Scholar] [CrossRef] [PubMed]
- Offley, S.R.; Schmidt, M.C. Protein phosphatases of saccharomyces cerevisiae. Curr. Genet. 2019, 65, 41–55. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Rubio, G.; Fernandez-Acero, T.; Martin, H.; Molina, M. Mitogen-activated protein kinase phosphatases (mkps) in fungal signaling: Conservation, function, and regulation. Int. J. Mol. Sci. 2019, 20, 1709. [Google Scholar] [CrossRef] [PubMed]
- Manfiolli, A.O.; de Castro, P.A.; Dos Reis, T.F.; Dolan, S.; Doyle, S.; Jones, G.; Riano Pachon, D.M.; Ulas, M.; Noble, L.M.; Mattern, D.J.; et al. Aspergillus fumigatus protein phosphatase ppza is involved in iron assimilation, secondary metabolite production, and virulence. Cell. Microbiol. 2017, 19, e12770. [Google Scholar] [CrossRef] [PubMed]
- Juvvadi, P.R.; Lee, S.C.; Heitman, J.; Steinbach, W.J. Calcineurin in fungal virulence and drug resistance: Prospects for harnessing targeted inhibition of calcineurin for an antifungal therapeutic approach. Virulence 2017, 8, 186–197. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.M.; Nantel, A.; Jiang, L.; Whiteway, M.; Shen, S.H. The serine/threonine protein phosphatase sit4 modulates yeast-to-hypha morphogenesis and virulence in candida albicans. Mol. Microbiol. 2004, 51, 691–709. [Google Scholar] [CrossRef] [PubMed]
- Hanaoka, N.; Umeyama, T.; Ueno, K.; Ueda, K.; Beppu, T.; Fugo, H.; Uehara, Y.; Niimi, M. A putative dual-specific protein phosphatase encoded by yvh1 controls growth, filamentation and virulence in candida albicans. Microbiology 2005, 151, 2223–2232. [Google Scholar] [CrossRef] [PubMed]
- Winkelstroter, L.K.; Bom, V.L.; de Castro, P.A.; Ramalho, L.N.; Goldman, M.H.; Brown, N.A.; Rajendran, R.; Ramage, G.; Bovier, E.; Dos Reis, T.F.; et al. High osmolarity glycerol response ptcb phosphatase is important for aspergillus fumigatus virulence. Mol. Microbiol. 2015, 96, 42–54. [Google Scholar] [CrossRef]
- Steinbach, W.J.; Schell, W.A.; Blankenship, J.R.; Onyewu, C.; Heitman, J.; Perfect, J.R. In vitro interactions between antifungals and immunosuppressants against aspergillus fumigatus. Antimicrob. Agents Chemother. 2004, 48, 1664–1669. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Lee, K.T.; Lee, S.J.; Beom, J.Y.; Hwangbo, A.; Jung, J.A.; Song, M.C.; Yoo, Y.J.; Kang, S.H.; Averette, A.F.; et al. In vitro and in vivo assessment of fk506 analogs as novel antifungal drug candidates. Antimicrob. Agents Chemother. 2018, 62, e01627-18. [Google Scholar] [CrossRef] [PubMed]
- Winkelstroter, L.K.; Dolan, S.K.; Fernanda Dos Reis, T.; Bom, V.L.; Alves de Castro, P.; Hagiwara, D.; Alowni, R.; Jones, G.W.; Doyle, S.; Brown, N.A.; et al. Systematic global analysis of genes encoding protein phosphatases in aspergillus fumigatus. G3 (Bethesda) 2015, 5, 1525–1539. [Google Scholar] [CrossRef] [PubMed]
- Wolstencroft, K.; Lord, P.; Tabernero, L.; Brass, A.; Stevens, R. Protein classification using ontology classification. Bioinformatics 2006, 22, e530–e538. [Google Scholar] [CrossRef] [Green Version]
- Alvarez-Carretero, S.; Pavlopoulou, N.; Adams, J.; Gilsenan, J.; Tabernero, L. Vspipe, an integrated resource for virtual screening and hit selection: Applications to protein tyrosine phospahatase inhibition. Molecules 2018, 23, 353. [Google Scholar] [CrossRef] [PubMed]
- Brenchley, R.; Tariq, H.; McElhinney, H.; Szoor, B.; Huxley-Jones, J.; Stevens, R.; Matthews, K.; Tabernero, L. The tritryp phosphatome: Analysis of the protein phosphatase catalytic domains. BMC Genomics 2007, 8, 434. [Google Scholar] [CrossRef]
- Hussein, H.A.; Borrel, A.; Geneix, C.; Petitjean, M.; Regad, L.; Camproux, A.C. Pockdrug-server: A new web server for predicting pocket druggability on holo and apo proteins. Nucleic Acids Res. 2015, 43, W436–W442. [Google Scholar] [CrossRef]
- Johnson, M.; Zaretskaya, I.; Raytselis, Y.; Merezhuk, Y.; McGinnis, S.; Madden, T.L. Ncbi blast: A better web interface. Nucleic Acids Res. 2008, 36, W5–W9. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Fiser, A.; Sali, A. Modeller: Generation and refinement of homology-based protein structure models. Methods Enzymol. 2003, 374, 461–491. [Google Scholar]
- Lund, G.; Dudkin, S.; Borkin, D.; Ni, W.; Grembecka, J.; Cierpicki, T. Inhibition of cdc25b phosphatase through disruption of protein-protein interaction. ACS Chem. Biol. 2015, 10, 390–394. [Google Scholar] [CrossRef] [PubMed]
- Abad-Zapatero, C.; Metz, J.T. Ligand efficiency indices as guideposts for drug discovery. Drug Discov. Today 2005, 10, 464–469. [Google Scholar] [CrossRef]
- Abad-Zapatero, C. Ligand efficiency indices for effective drug discovery. Expert Opin. Drug Discov. 2007, 2, 469–488. [Google Scholar] [CrossRef] [PubMed]
- Abad-Zapatero, C.; Blasi, D. Ligand efficiency indices (leis): More than a simple efficiency yardstick. Mol. Inform. 2011, 30, 122–132. [Google Scholar] [CrossRef]
- Abad-Zapatero, C.; Perisic, O.; Wass, J.; Bento, A.P.; Overington, J.; Al-Lazikani, B.; Johnson, M.E. Ligand efficiency indices for an effective mapping of chemico-biological space: The concept of an atlas-like representation. Drug Discov. Today 2010, 15, 804–811. [Google Scholar] [CrossRef] [PubMed]
- Chen, E.; Choy, M.S.; Petrenyi, K.; Konya, Z.; Erdodi, F.; Dombradi, V.; Peti, W.; Page, R. Molecular insights into the fungus-specific serine/threonine protein phosphatase z1 in candida albicans. MBio 2016, 7, e00872-16. [Google Scholar] [CrossRef] [PubMed]
- Sohn, J.; Parks, J.M.; Buhrman, G.; Brown, P.; Kristjansdottir, K.; Safi, A.; Edelsbrunner, H.; Yang, W.; Rudolph, J. Experimental validation of the docking orientation of cdc25 with its cdk2-cyca protein substrate. Biochemistry 2005, 44, 16563–16573. [Google Scholar] [CrossRef] [PubMed]
- Sohn, J.; Buhrman, G.; Rudolph, J. Kinetic and structural studies of specific protein-protein interactions in substrate catalysis by cdc25b phosphatase. Biochemistry 2007, 46, 807–818. [Google Scholar] [CrossRef]
- Stuart, J.S.; Frederick, D.L.; Varner, C.M.; Tatchell, K. The mutant type 1 protein phosphatase encoded by glc7-1 from saccharomyces cerevisiae fails to interact productively with the gac1-encoded regulatory subunit. Mol. Cell. Biol. 1994, 14, 896–905. [Google Scholar] [CrossRef]
- Alonso, A.; Sasin, J.; Bottini, N.; Friedberg, I.; Friedberg, I.; Osterman, A.; Godzik, A.; Hunter, T.; Dixon, J.; Mustelin, T. Protein tyrosine phosphatases in the human genome. Cell 2004, 117, 699–711. [Google Scholar] [CrossRef]
- Alonso, A.; Pulido, R. The extended human ptpome: A growing tyrosine phosphatase family. FEBS J. 2016, 283, 1404–1429. [Google Scholar] [CrossRef] [PubMed]
- Laporte, J.; Blondeau, F.; Buj-Bello, A.; Tentler, D.; Kretz, C.; Dahl, N.; Mandel, J.L. Characterization of the myotubularin dual specificity phosphatase gene family from yeast to human. Hum. Mol. Genet. 1998, 7, 1703–1712. [Google Scholar] [CrossRef] [PubMed]
- Sakumoto, N.; Mukai, Y.; Uchida, K.; Kouchi, T.; Kuwajima, J.; Nakagawa, Y.; Sugioka, S.; Yamamoto, E.; Furuyama, T.; Mizubuchi, H.; et al. A series of protein phosphatase gene disruptants in saccharomyces cerevisiae. Yeast 1999, 15, 1669–1679. [Google Scholar] [CrossRef]
- Fraczek, M.G.; Bromley, M.; Buied, A.; Moore, C.B.; Rajendran, R.; Rautemaa, R.; Ramage, G.; Denning, D.W.; Bowyer, P. The cdr1b efflux transporter is associated with non-cyp51a-mediated itraconazole resistance in aspergillus fumigatus. J. Antimicrob. Chemother. 2013, 68, 1486–1496. [Google Scholar] [CrossRef]
- Zhao, C.; Fraczek, M.G.; Dineen, L.; Lebedinec, R.; Macheleidt, J.; Heinekamp, T.; Delneri, D.; Bowyer, P.; Brakhage, A.A.; Bromley, M. High-throughput gene replacement in aspergillus fumigatus. Curr. Protoc. Microbiol. 2019, 54, e88. [Google Scholar] [CrossRef]
- Stajich, J.E.; Harris, T.; Brunk, B.P.; Brestelli, J.; Fischer, S.; Harb, O.S.; Kissinger, J.C.; Li, W.; Nayak, V.; Pinney, D.F.; et al. Fungidb: An integrated functional genomics database for fungi. Nucleic Acids Res. 2012, 40, D675–D681. [Google Scholar] [CrossRef]
- De Castro, E.; Sigrist, C.J.; Gattiker, A.; Bulliard, V.; Langendijk-Genevaux, P.S.; Gasteiger, E.; Bairoch, A.; Hulo, N. Scanprosite: Detection of prosite signature matches and prorule-associated functional and structural residues in proteins. Nucleic Acids Res. 2006, 34, W362–W365. [Google Scholar] [CrossRef]
- Drozdetskiy, A.; Cole, C.; Procter, J.; Barton, G.J. Jpred4: A protein secondary structure prediction server. Nucleic Acids Res. 2015, 43, W389–W394. [Google Scholar] [CrossRef]
- Slabinski, L.; Jaroszewski, L.; Rychlewski, L.; Wilson, I.A.; Lesley, S.A.; Godzik, A. Xtalpred: A web server for prediction of protein crystallizability. Bioinformatics 2007, 23, 3403–3405. [Google Scholar] [CrossRef]
- Yang, Z.R.; Thomson, R.; McNeil, P.; Esnouf, R.M. Ronn: The bio-basis function neural network technique applied to the detection of natively disordered regions in proteins. Bioinformatics 2005, 21, 3369–3376. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. Autodock4 and autodocktools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open babel: An open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. Autodock vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
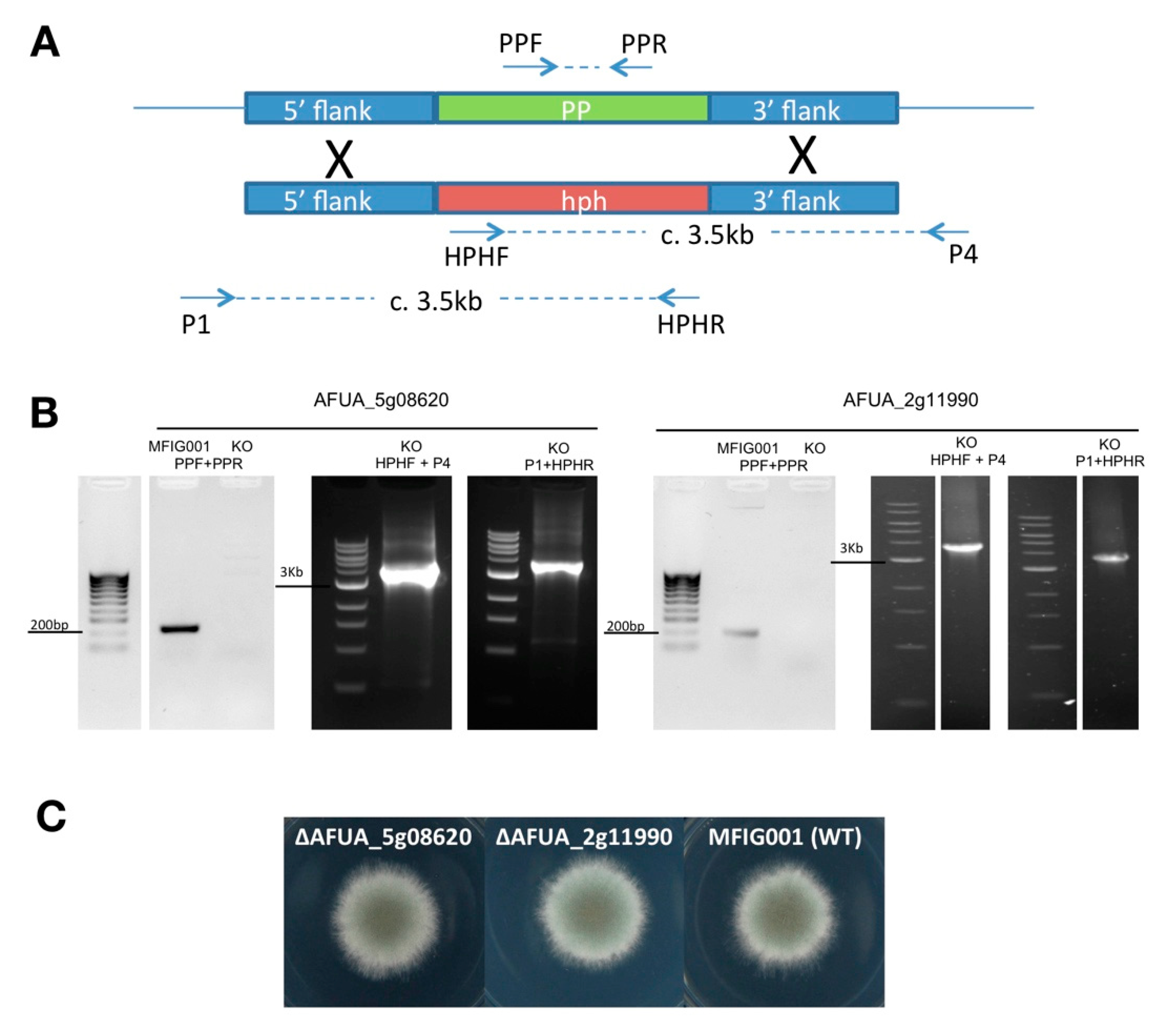{kind=link}
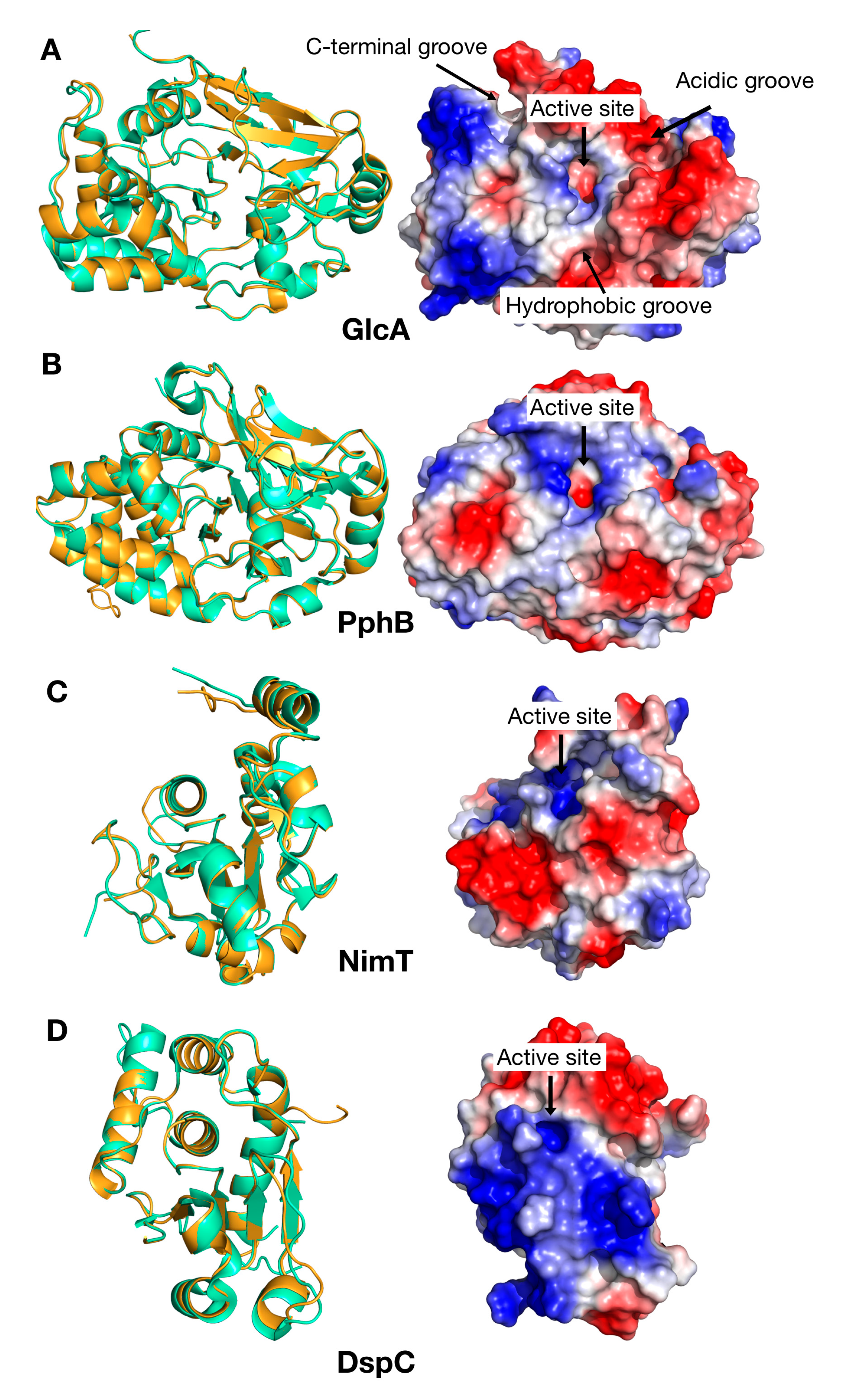{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Af PPase | Human Homologue | Full-Length Identity/Coverage (%) | Model Identity/Coverage (%) | Template (PDB ID) | Model Boundaries | Model Scores |
|---|---|---|---|---|---|---|
| PphB | PP2AC | 84/94 (H) | 86/96 | 2NYL, 3DW8, 2NYM | S19-P311 | −37,801.93 |
| GlcA | PP1CB | 84/100 (H) | 86/100 | 4G9J, 5IOH, 1S70 | M1-E299 | −39,831.18 |
| FcpA | Fcp1 | 44/75 (Sp) | 44/99 | 3EF0 | R145-P602 | −34,085.44 |
| NimT | CDC25B | 43/44 (H) | 43/87 | 1C25, 1QB0, 3OP3 | D333-K504 | −18,191.54 |
| DspC | * Yvh1/DUSP12 | 40/95 (Ct) | 39/97 | 5M43 | M1-H153 | −17,151.59 |
| SsuA | Ssu72 | 45/75 (H) | 45/87 | 3O2S | S48-L287 | −22,385.70 |
| YmrA | MTMR2 | 48/77 (H) | 34/89 | 5GNH | 121-647 | −65,336.31 |
| PPase | P1/P1′ | P 2 | P3 | P 4 | P5 |
|---|---|---|---|---|---|
| NimT | 0.55 */0.68 * | 0.60 * | |||
| DspC | 0.55 * | 0.69 * | |||
| PphB | 0.64 */0.6 * | 0.73 * | 0.63 * | 0.70 * | |
| GlcA | 0.57 * | 0.64 * | 0.63 * | 0.71 * |
| AfPPase | Conservation (%) | ||||
|---|---|---|---|---|---|
| P1/P1′ | P2 | P3 | P4 | P5 | |
| NimT | 38 | 30 | |||
| DspC | 36 | 31 | |||
| GlcA | 100 | 96 | 100 | 82 | |
| PphB | 100 | 100 | 66 | 90 | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thornton, B.P.; Johns, A.; Al-Shidhani, R.; Álvarez-Carretero, S.; Storer, I.S.R.; Bromley, M.J.; Tabernero, L. Identification of Functional and Druggable Sites in Aspergillus fumigatus Essential Phosphatases by Virtual Screening. Int. J. Mol. Sci. 2019, 20, 4636. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20184636
Thornton BP, Johns A, Al-Shidhani R, Álvarez-Carretero S, Storer ISR, Bromley MJ, Tabernero L. Identification of Functional and Druggable Sites in Aspergillus fumigatus Essential Phosphatases by Virtual Screening. International Journal of Molecular Sciences. 2019; 20(18):4636. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20184636
Chicago/Turabian StyleThornton, Benjamin P., Anna Johns, Reem Al-Shidhani, Sandra Álvarez-Carretero, Isabelle S. R. Storer, Michael J. Bromley, and Lydia Tabernero. 2019. "Identification of Functional and Druggable Sites in Aspergillus fumigatus Essential Phosphatases by Virtual Screening" International Journal of Molecular Sciences 20, no. 18: 4636. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20184636