The Link of the Prion Protein with Ca2+ Metabolism and ROS Production, and the Possible Implication in Aβ Toxicity

 , , and
, , and
Abstract
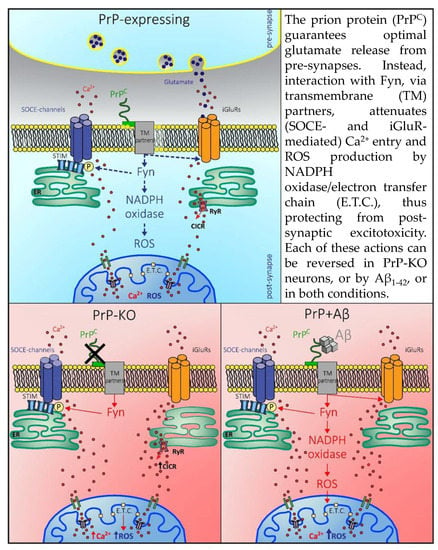
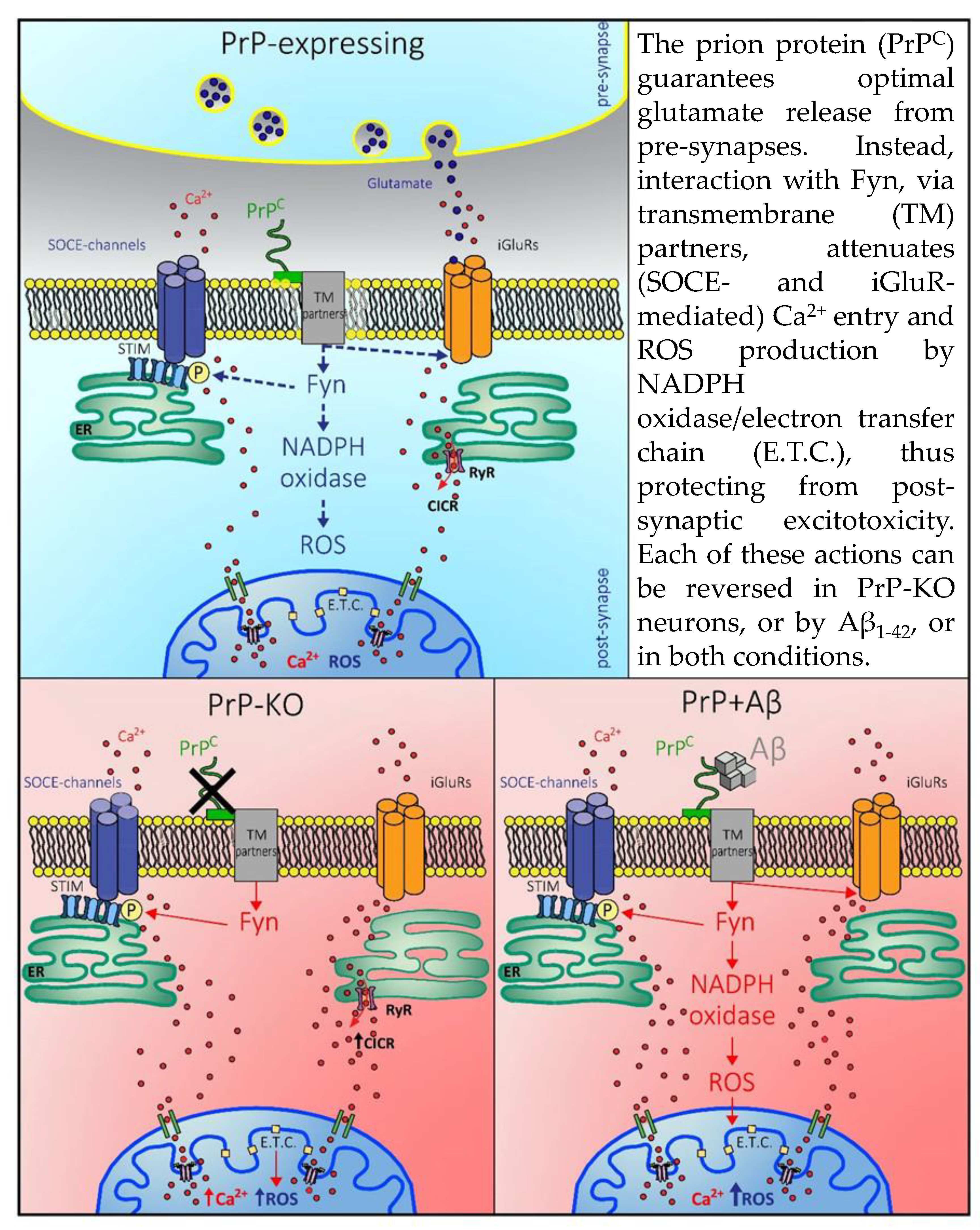
:
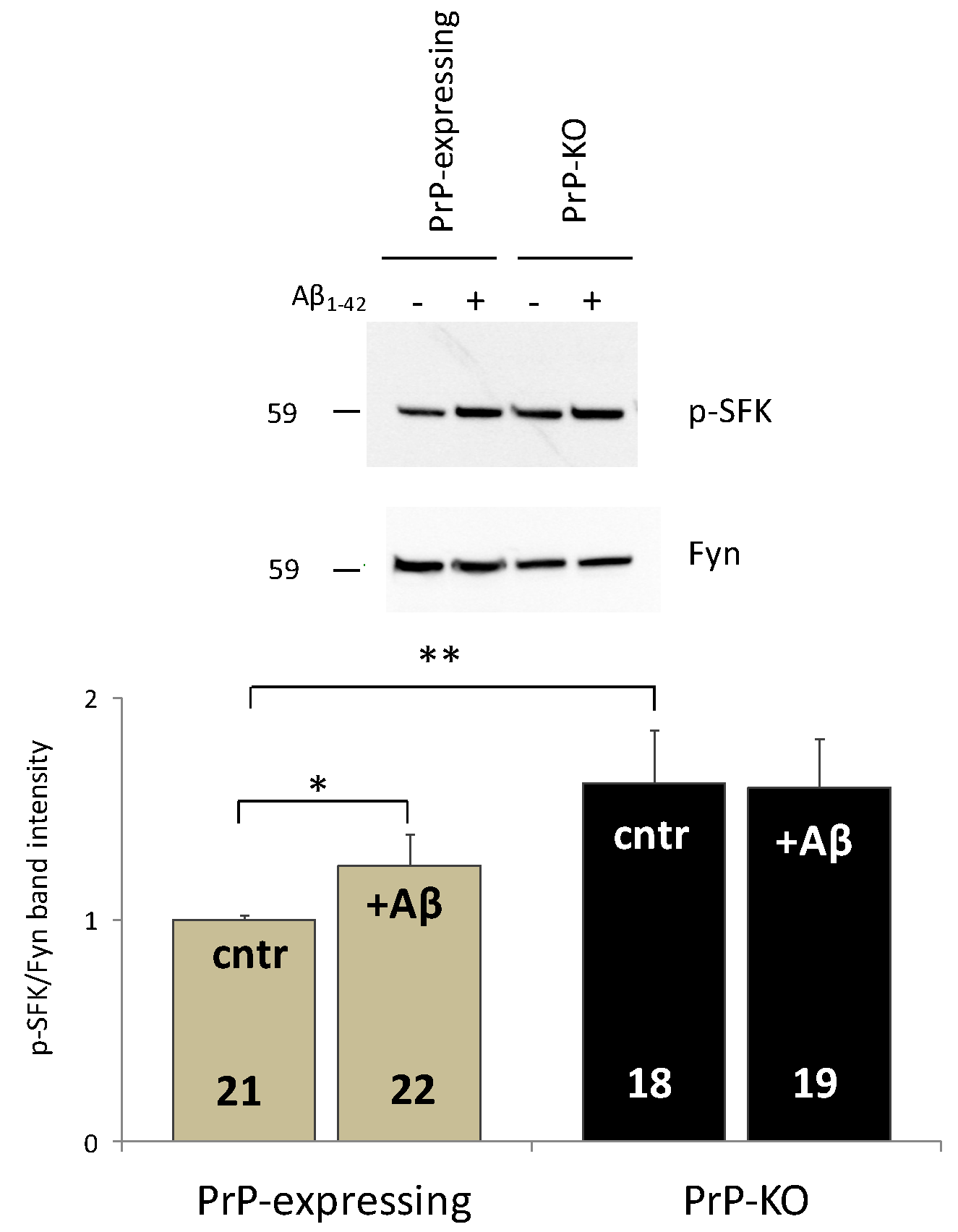
{kind=link}
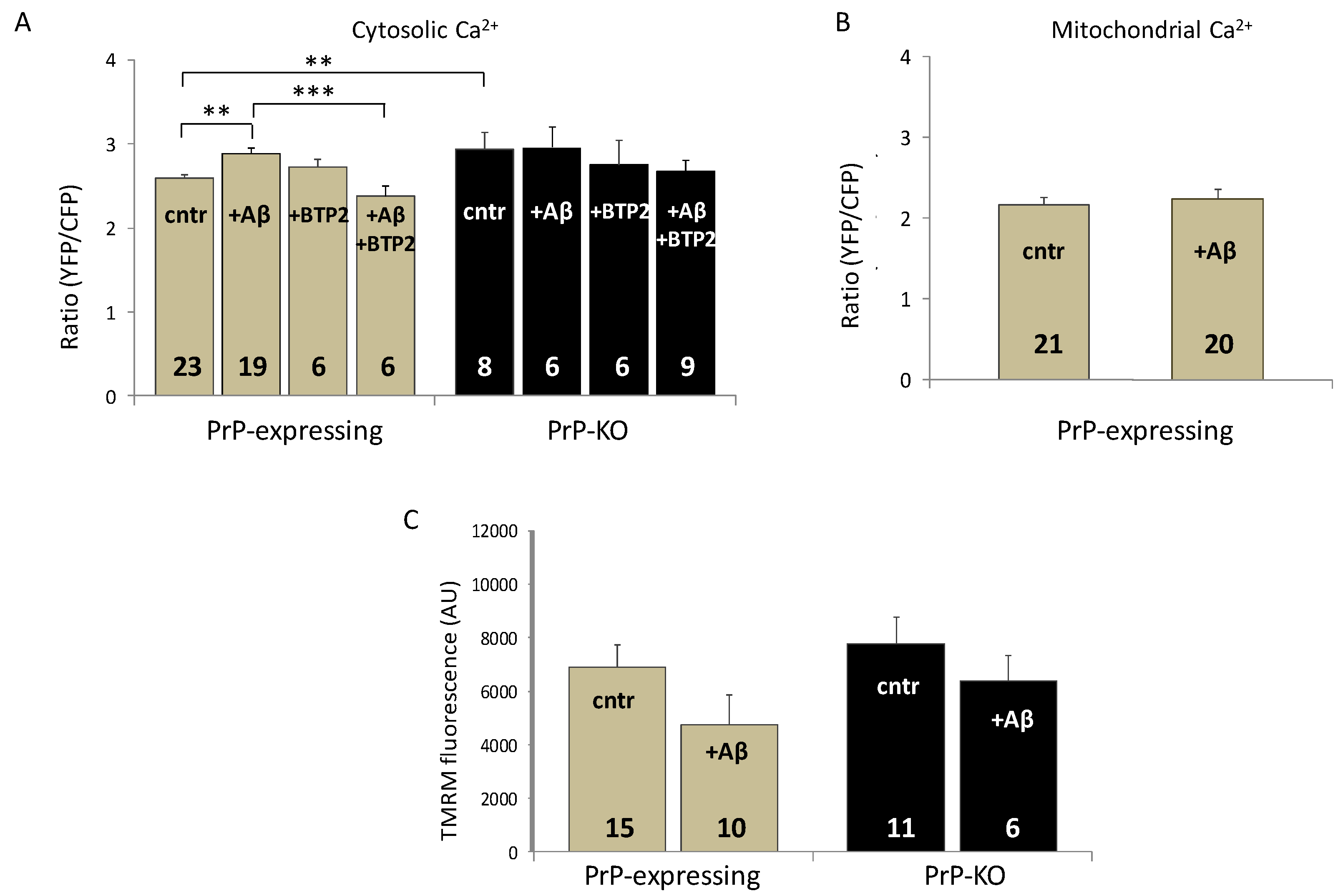
{kind=link}
{kind=link}
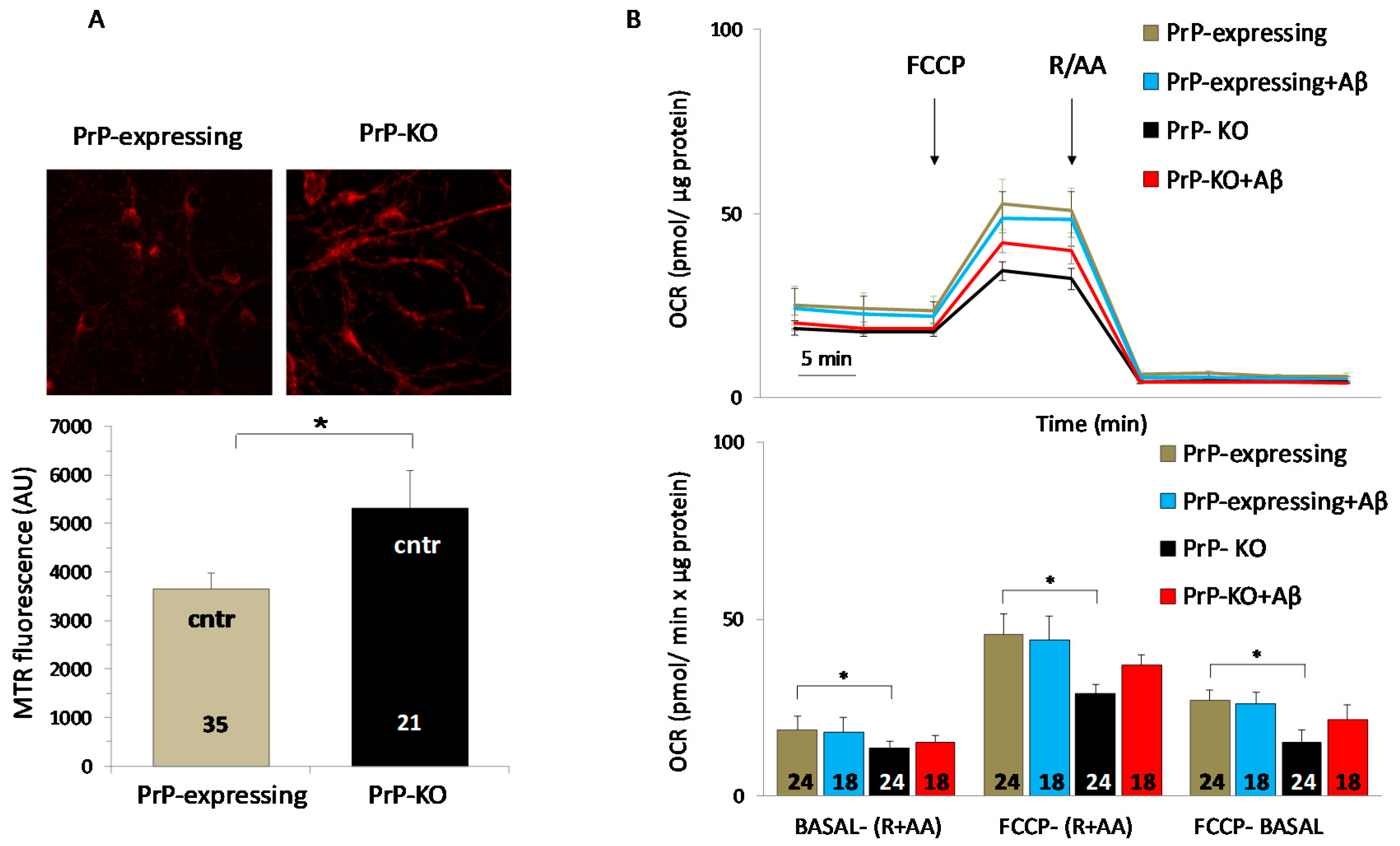{kind=link}
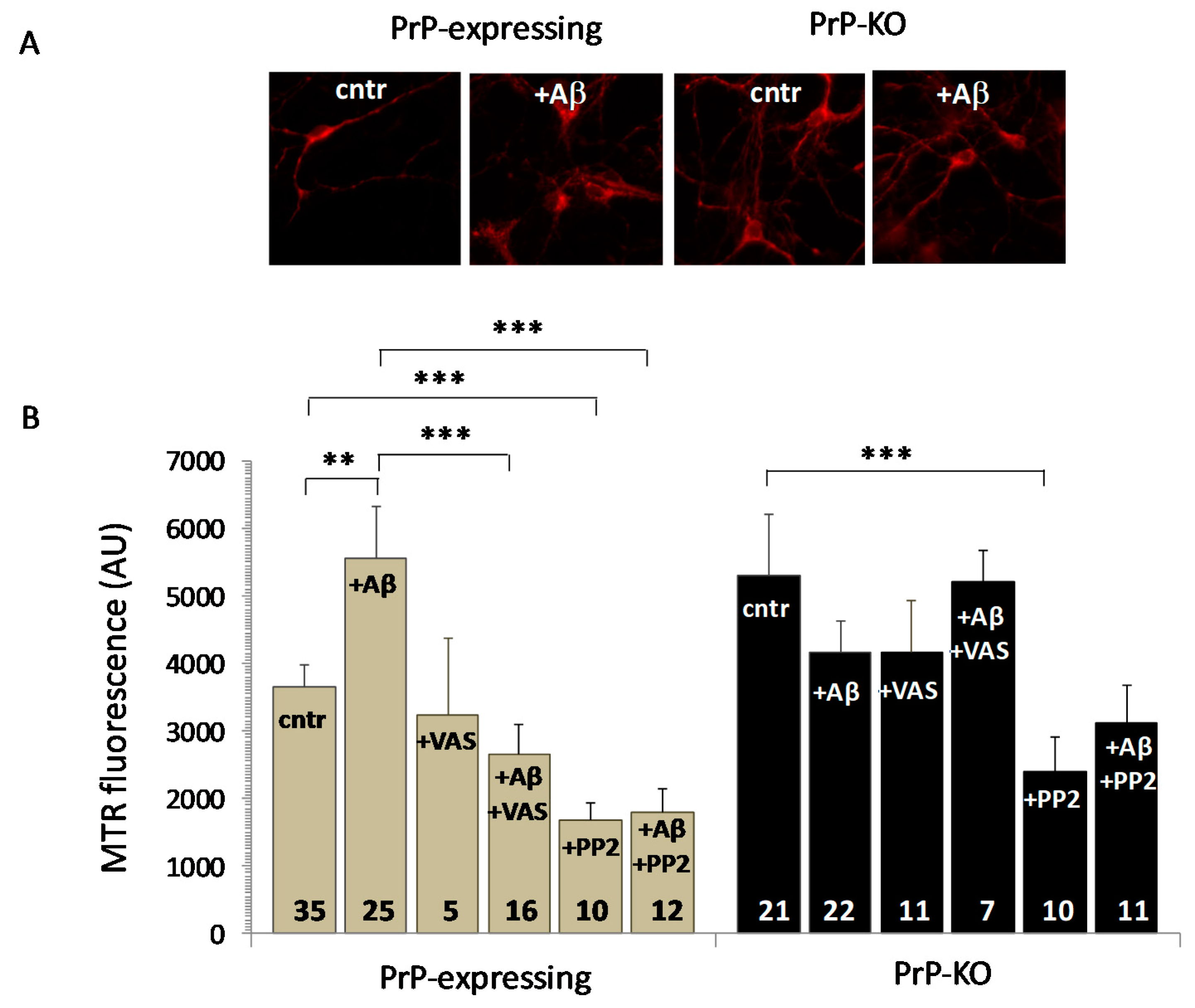{kind=link}
1. Introduction
2. PrPC Supports Correct Neuronal Activities by Regulating Gene Expression and Ca2+ Metabolism
2.1. Action of PrPC at Pre-Synapses
2.2. Action of PrPC on SOCE and iGluRs
3. Effects of Aβ Oligomers on PrPC-Linked Neuronal Controls
4. Conclusions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AA | antimycin |
| AD | Alzheimer′s disease |
| AMPA | α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid |
| BTP2 | N-{4-[3,5-bis(Trifluoromethyl)-1H-pyrazol-1-yl]phenyl}-4-methyl-1,2,3-thiadiazole-5-carboxamide, |
| CFP | cyan fluorescent protein |
| CGN | cerebellar granule neurons |
| CICR | Ca2+-induced Ca2+-release |
| CREB | cAMP-response element binding protein |
| ER | endoplasmic reticulum |
| ERK | extracellular signal-regulated kinase |
| FCCP | carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone |
| GPI | glycosylphosphatidylinositol |
| iGluR | ionotropic glutamatergic receptor |
| IP3 | inositol triphosphate |
| mGluR | metabotropic glutamatergic receptor |
| MTR | MitoTracker Red |
| NMDA | N-methyl-D-aspartate |
| OCR | oxygen consumption rate |
| PM | plasma membrane |
| PP2 | 3-(4-chlorophenyl) 1-(1,1-dimethylethyl)-1H-pyrazolo(3,4-d)pyrimidin-4-amine |
| PrPC | cellular prion protein |
| PrP-KO | PrP-knockout |
| PrPSc | abnormal PrPC conformer present in prions |
| R | Rotenone |
| ROS | reactive oxygen species |
| SEM | standard error of the mean |
| SFK | Src family kinases |
| SOCE | store-operated Ca2+ entry |
| STIM | stromal interaction molecules |
| TMRM | tetramethylrhodamine methyl ester |
| VAS 2870 | 3-benzyl-7-(2-benzoxazolyl)thio-1,2,3-triazolo(4,5-d)pyrimidine |
| YFP | yellow fluorescent protein |
| WB | western blot |
References
- Wells, G.A.; Scott, A.C.; Johnson, C.T.; Gunning, R.F.; Hancock, R.D.; Jeffrey, M.; Dawson, M.; Bradley, R. A novel progressive spongiform encephalopathy in cattle. Vet. Rec. 1987, 121, 419–420. [Google Scholar] [CrossRef] [PubMed]
- Bruce, M.E.; Will, R.G.; Ironside, J.W.; McConnell, I.; Drummond, D.; Suttie, A.; McCardle, L.; Chree, A.; Hope, J.; Birkett, C.; et al. Transmissions to mice indicate that ′new variant′ CJD is caused by the BSE agent. Nature 1997, 389, 498–501. [Google Scholar] [CrossRef] [PubMed]
- Aguzzi, A.; Lakkaraju, A.K.K. Cell Biology of Prions and Prionoids: A Status Report. Trends Cell Biol. 2016, 26, 40–51. [Google Scholar] [CrossRef] [PubMed]
- Sales, N.; Rodolfo, K.; Hassig, R.; Faucheux, B.; Di Giamberardino, L.; Moya, K.L. Cellular prion protein localization in rodent and primate brain. Eur. J. Neurosci. 1998, 10, 2464–2471. [Google Scholar] [CrossRef] [PubMed]
- Herms, J.; Tings, T.; Gall, S.; Madlung, A.; Giese, A.; Siebert, H.; Schurmann, P.; Windl, O.; Brose, N.; Kretzschmar, H. Evidence of presynaptic location and function of the prion protein. J. Neurosci. 1999, 19, 8866–8875. [Google Scholar] [CrossRef] [PubMed]
- Um, J.W.; Nygaard, H.B.; Heiss, J.K.; Kostylev, M.A.; Stagi, M.; Vortmeyer, A.; Wisniewski, T.; Gunther, E.C.; Strittmatter, S.M. Alzheimer amyloid-beta oligomer bound to postsynaptic prion protein activates Fyn to impair neurons. Nat. Neurosci. 2012, 15, 1227–1235. [Google Scholar] [CrossRef]
- Um, J.W.; Strittmatter, S.M. Amyloid-β induced signaling by cellular prion protein and Fyn kinase in Alzheimer disease. Prion 2013, 7, 37–41. [Google Scholar] [CrossRef]
- Walmsley, A.R.; Zeng, F.; Hooper, N.M. The N-terminal region of the prion protein ectodomain contains a lipid raft targeting determinant. J. Biol. Chem. 2003, 278, 37241–37248. [Google Scholar] [CrossRef]
- Baron, G.S.; Caughey, B. Effect of glycosylphosphatidylinositol anchor-dependent and -independent prion protein association with model raft membranes on conversion to the protease-resistant isoform. J. Biol. Chem. 2003, 278, 14883–14892. [Google Scholar] [CrossRef]
- Prusiner, S.B. Prions. Proc. Natl. Acad. Sci. USA 1998, 95, 13363–13383. [Google Scholar] [CrossRef] [Green Version]
- Kara, E.; Marks, J.D.; Aguzzi, A. Toxic Protein Spread in Neurodegeneration: Reality versus Fantasy. Trends Mol. Med. 2018, 24, 1007–1020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bueler, H.; Aguzzi, A.; Sailer, A.; Greiner, R.A.; Autenried, P.; Aguet, M.; Weissmann, C. Mice devoid of PrP are resistant to scrapie. Cell 1993, 73, 1339–1347. [Google Scholar] [CrossRef]
- Brandner, S.; Raeber, A.; Sailer, A.; Blattler, T.; Fischer, M.; Weissmann, C.; Aguzzi, A. Normal host prion protein (PrPC) is required for scrapie spread within the central nervous system. Proc. Natl. Acad. Sci. USA 1996, 93, 13148–13151. [Google Scholar] [CrossRef] [PubMed]
- Mallucci, G.; Dickinson, A.; Linehan, J.; Klöhn, P.; Brandner, S.; Collinge, J. Depleting neuronal PrP in Prion Infection Prevents Disease and Reverses spongiosis. Science 2003, 302, 871–874. [Google Scholar] [CrossRef] [PubMed]
- Chesebro, B.; Trifilo, M.; Race, R.; Meade-White, K.; Teng, C.; LaCasse, R.; Raymond, L.; Favara, C.; Baron, G.; Priola, S.; et al. Anchorless prion protein results in infectious amyloid disease without clinical scrapie. Science 2005, 308, 1435–1439. [Google Scholar] [CrossRef] [PubMed]
- White, M.D.; Farmer, M.; Mirabile, I.; Brandner, S.; Collinge, J.; Mallucci, G.R. Single treatment with RNAi against prion protein rescues early neuronal dysfunction and prolongs survival in mice with prion disease. Proc. Natl. Acad. Sci. USA 2008, 105, 10238–10243. [Google Scholar] [CrossRef] [Green Version]
- Barton, K.A.; Caughey, B. Is PrP the road to ruin? Embo J. 2011, 30, 1882–1884. [Google Scholar] [CrossRef]
- Hirsch, T.Z.; Hernandez-Rapp, J.; Martin-Lanneree, S.; Launay, J.-M.; Mouillet-Richard, S. PrP(C) signalling in neurons: From basics to clinical challenges. Biochimie 2014, 104, 2–11. [Google Scholar] [CrossRef]
- Wulf, M.-A.; Senatore, A.; Aguzzi, A. The biological function of the cellular prion protein: An update. BMC Biol. 2017, 15, 34. [Google Scholar] [CrossRef]
- Linden, R. The Biological Function of the Prion Protein: A Cell Surface Scaffold of Signaling Modules. Front. Mol. Neurosci. 2017, 10, 77. [Google Scholar] [CrossRef] [Green Version]
- Watts, J.C.; Bourkas, M.E.C.; Arshad, H. The function of the cellular prion protein in health and disease. Acta Neuropathol. 2018, 135, 159–178. [Google Scholar] [CrossRef] [PubMed]
- Resenberger, U.K.; Harmeier, A.; Woerner, A.C.; Goodman, J.L.; Muller, V.; Krishnan, R.; Vabulas, R.M.; Kretzschmar, H.A.; Lindquist, S.; Hartl, F.U.; et al. The cellular prion protein mediates neurotoxic signalling of beta-sheet-rich conformers independent of prion replication. Embo. J. 2011, 30, 2057–2070. [Google Scholar] [CrossRef] [PubMed]
- Scheckel, C.; Aguzzi, A. Prions, prionoids and protein misfolding disorders. Nat. Rev. Genet. 2018, 19, 405–418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peggion, C.; Stella, R.; Chemello, F.; Massimino, M.L.; Arrigoni, G.; Cagnin, S.; Biancotto, G.; Franchin, C.; Sorgato, M.C.; Bertoli, A. The Prion Protein Regulates Synaptic Transmission by Controlling the Expression of Proteins Key to Synaptic Vesicle Recycling and Exocytosis. Mol. Neurobiol. 2019, 56, 3420–3436. [Google Scholar] [CrossRef] [PubMed]
- Pradines, E.; Loubet, D.; Schneider, B.; Launay, J.M.; Kellermann, O.; Mouillet-Richard, S. CREB-dependent gene regulation by prion protein: Impact on MMP-9 and β-dystroglycan. Cell. Signal. 2008, 20, 2050–2058. [Google Scholar] [CrossRef] [PubMed]
- Pradines, E.; Hernandez-Rapp, J.; Villa-Diaz, A.; Dakowski, C.; Ardila-Osorio, H.; Haik, S.; Schneider, B.; Launay, J.-M.; Kellermann, O.; Torres, J.-M.; et al. Pathogenic prions deviate PrP(C) signaling in neuronal cells and impair A-beta clearance. Cell Death Dis. 2013, 4, e456. [Google Scholar] [CrossRef] [PubMed]
- Laurén, J.; Gimbel, D.A.; Nygaard, H.B.; Gilbert, J.W.; Strittmatter, S.M. Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature 2009, 457, 1128–1132. [Google Scholar] [CrossRef]
- Gimbel, D.A.; Nygaard, H.B.; Coffey, E.E.; Gunther, E.C.; Laurén, J.; Gimbel, Z.A.; Strittmatter, S.M. Memory Impairment in Transgenic Alzheimer Mice Requires Cellular Prion Protein. J. Neurosci. 2010, 30, 6367–6374. [Google Scholar] [CrossRef]
- Hu, N.-W.; Nicoll, A.J.; Zhang, D.; Mably, A.J.; O′Malley, T.; Purro, S.A.; Terry, C.; Collinge, J.; Walsh, D.M.; Rowan, M.J. mGlu5 receptors and cellular prion protein mediate amyloid-beta-facilitated synaptic long-term depression in vivo. Nat. Commun. 2014, 5, 3374. [Google Scholar] [CrossRef]
- Balducci, C.; Beeg, M.; Stravalaci, M.; Bastone, A.; Sclip, A.; Biasini, E.; Tapella, L.; Colombo, L.; Manzoni, C.; Borsello, T.; et al. Synthetic amyloid-beta oligomers impair long-term memory independently of cellular prion protein. Proc. Natl. Acad. Sci. USA 2010, 107, 2295–2300. [Google Scholar] [CrossRef]
- Kessels, H.W.; Nguyen, L.N.; Nabavi, S.; Malinow, R. The prion protein as a receptor for amyloid-beta. Nature 2010, 466, E3–E4. discussion E4–E5. [Google Scholar] [CrossRef] [PubMed]
- Shankar, G.M.; Li, S.; Mehta, T.H.; Garcia-Munoz, A.; Shepardson, N.E.; Smith, I.; Brett, F.M.; Farrell, M.A.; Rowan, M.J.; Lemere, C.A.; et al. Amyloid-beta protein dimers isolated directly from Alzheimer′s brains impair synaptic plasticity and memory. Nat. Med. 2008, 14, 837–842. [Google Scholar] [CrossRef] [PubMed]
- Masters, C.L.; Selkoe, D.J. Biochemistry of amyloid beta-protein and amyloid deposits in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2012, 2, a006262. [Google Scholar] [CrossRef] [PubMed]
- Cowan, C.M.; Quraishe, S.; Mudher, A. What is the pathological significance of tau oligomers? Biochem. Soc. Trans. 2012, 40, 693–697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aulic, S.; Masperone, L.; Narkiewicz, J.; Isopi, E.; Bistaffa, E.; Ambrosetti, E.; Pastore, B.; De Cecco, E.; Scaini, D.; Zago, P.; et al. alpha-Synuclein Amyloids Hijack Prion Protein to Gain Cell Entry, Facilitate Cell-to-Cell Spreading and Block Prion Replication. Sci. Rep. 2017, 7, 10050. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, D.G.; Temido-Ferreira, M.; Miranda, H.V.; Batalha, V.L.; Coelho, J.E.; Szego, E.M.; Marques-Morgado, I.; Vaz, S.H.; Rhee, J.S.; Schmitz, M.; et al. alpha-synuclein interacts with PrP(C) to induce cognitive impairment through mGluR5 and NMDAR2B. Nat. Neurosci. 2017, 20, 1569–1579. [Google Scholar] [CrossRef] [PubMed]
- Sorgato, M.C.; Bertoli, A. From cell protection to death: May Ca2+ signals explain the chameleonic attributes of the mammalian prion protein? Biochem. Biophys. Res. Commun. 2009, 379, 171–174. [Google Scholar] [CrossRef] [PubMed]
- Sorgato, M.C.; Peggion, C.; Bertoli, A. Is, indeed, the prion protein a Harlequin servant of ′many′ masters? Prion 2009, 3, 202–205. [Google Scholar] [CrossRef]
- Peggion, C.; Bertoli, A.; Sorgato, M.C. Possible role for Ca2+ in the pathophysiology of the prion protein? Biofactors 2011, 37, 241–249. [Google Scholar] [CrossRef] [PubMed]
- Filadi, R.; Pozzan, T. Generation and functions of second messengers microdomains. Cell Calcium 2015, 58, 405–414. [Google Scholar] [CrossRef]
- Green, K.N.; LaFerla, F.M. Linking calcium to Abeta and Alzheimer′s disease. Neuron 2008, 59, 190–194. [Google Scholar] [CrossRef] [PubMed]
- Demuro, A.; Parker, I.; Stutzmann, G.E. Calcium signaling and amyloid toxicity in Alzheimer disease. J. Biol. Chem. 2010, 285, 12463–12468. [Google Scholar] [CrossRef] [PubMed]
- Lim, D.; Bertoli, A.; Sorgato, M.C.; Moccia, F. Generation and usage of aequorin lentiviral vectors for Ca(2+) measurement in sub-cellular compartments of hard-to-transfect cells. Cell Calcium 2016, 59, 228–239. [Google Scholar] [CrossRef] [PubMed]
- Lazzari, C.; Peggion, C.; Stella, R.; Massimino, M.L.; Lim, D.; Bertoli, A.; Sorgato, M.C. Cellular prion protein is implicated in the regulation of local Ca2+ movements in cerebellar granule neurons. J. Neurochem. 2011, 116, 881–890. [Google Scholar] [CrossRef] [PubMed]
- De Mario, A.; Castellani, A.; Peggion, C.; Massimino, M.L.; Lim, D.; Hill, A.F.; Sorgato, M.C.; Bertoli, A. The prion protein constitutively controls neuronal store-operated Ca(2+) entry through Fyn kinase. Front. Cell. Neurosci. 2015, 9, 416. [Google Scholar] [CrossRef] [PubMed]
- De Mario, A.; Peggion, C.; Massimino, M.L.; Viviani, F.; Castellani, A.; Giacomello, M.; Lim, D.; Bertoli, A.; Sorgato, M.C. The prion protein regulates glutamate-mediated Ca(2+) entry and mitochondrial Ca(2+) accumulation in neurons. J. Cell Sci. 2017, 130, 2736–2746. [Google Scholar] [CrossRef] [PubMed]
- Soboloff, J.; Rothberg, B.S.; Madesh, M.; Gill, D.L. STIM proteins: Dynamic calcium signal transducers. Nat. Rev. Mol. Cell Biol. 2012, 13, 549–565. [Google Scholar] [CrossRef]
- Lopez, E.; Jardin, I.; Berna-Erro, A.; Bermejo, N.; Salido, G.M.; Sage, S.O.; Rosado, J.A.; Redondo, P.C. STIM1 tyrosine-phosphorylation is required for STIM1-Orai1 association in human platelets. Cell. Signal. 2012, 24, 1315–1322. [Google Scholar] [CrossRef]
- Berna-Erro, A.; Braun, A.; Kraft, R.; Kleinschnitz, C.; Schuhmann, M.K.; Stegner, D.; Wultsch, T.; Eilers, J.; Meuth, S.G.; Stoll, G.; et al. STIM2 Regulates Capacitive Ca2+ Entry in Neurons and Plays a Key Role in Hypoxic Neuronal Cell Death. Sci. Signal. 2009, 2, 1–11. [Google Scholar] [CrossRef]
- Zuo, W.-L.; Du, J.-Y.; Huang, J.-H.; Li, S.; Zhang, G.; Chen, S.-L.; Ruan, Y.-C.; Cheng, C.H.K.; Zhou, W.-L. Tyrosine phosphorylation modulates store-operated calcium entry in cultured rat epididymal basal cells. J. Cell. Physiol. 2011, 226, 1069–1073. [Google Scholar] [CrossRef]
- Lopez, E.; Frischauf, I.; Jardin, I.; Derler, I.; Muik, M.; Cantonero, C.; Salido, G.M.; Smani, T.; Rosado, J.A.; Redondo, P.C. STIM1 phosphorylation at Y(316) modulates its interaction with SARAF and the activation of SOCE and I CRAC. J. Cell Sci. 2019, 132. [Google Scholar]
- Erpel, T.; Courtneidge, S. a Src family protein tyrosine kinases and cellular signal transduction pathways. Curr. Opin. Cell Biol. 1995, 7, 176–182. [Google Scholar] [CrossRef]
- Larson, M.; Sherman, M.A.; Amar, F.; Nuvolone, M.; Schneider, J.A.; Bennett, D.A.; Aguzzi, A.; Lesné, S.E. The complex PrP(c)-Fyn couples human oligomeric Aβ with pathological tau changes in Alzheimer′s disease. J. Neurosci. 2012, 32, 16857–16871. [Google Scholar] [CrossRef]
- Goniotaki, D.; Lakkaraju, A.K.K.; Shrivastava, A.N.; Bakirci, P.; Sorce, S.; Senatore, A.; Marpakwar, R.; Hornemann, S.; Gasparini, F.; Triller, A.; et al. Inhibition of group-I metabotropic glutamate receptors protects against prion toxicity. PLoS Pathog. 2017, 13, e1006733. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Ren, C.H.; Gunawardana, C.G.; Schmitt-Ulms, G. Overcoming barriers and thresholds-signaling of oligomeric Abeta through the prion protein to Fyn. Mol. Neurodegener. 2013, 8, 24. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, P.; Rasola, A.; Forte, M.; Lippe, G. The Mitochondrial Permeability Transition Pore: Channel Formation by F-ATP Synthase, Integration in Signal Transduction, and Role in Pathophysiology. Physiol. Rev. 2015, 95, 1111–1155. [Google Scholar] [CrossRef]
- Mallucci, G.R.; Ratté, S.; Asante, E.A.; Linehan, J.; Gowland, I.; Jefferys, J.G.R.; Collinge, J. Post-natal knockout of prion protein alters hippocampal CA1 properties, but does not result in neurodegeneration. Embo. J. 2002, 21, 202–210. [Google Scholar] [CrossRef] [PubMed]
- Khosravani, H.; Zhang, Y.; Tsutsui, S.; Hameed, S.; Altier, C.; Hamid, J.; Chen, L.; Villemaire, M.; Ali, Z.; Jirik, F.R.; et al. Prion protein attenuates excitotoxicity by inhibiting NMDA receptors. J. Cell Biol. 2008, 181, 551–555. [Google Scholar] [CrossRef]
- You, H.; Tsutsui, S.; Hameed, S.; Kannanayakal, T.J.; Chen, L.; Xia, P.; Engbers, J.D.T.; Lipton, S.A.; Stys, P.K.; Zamponi, G.W. Aβ neurotoxicity depends on interactions between copper ions, prion protein, and N-methyl-D-aspartate receptors. Proc. Natl. Acad. Sci. USA 2012, 109, 1737–1742. [Google Scholar] [CrossRef]
- Song, R.S.; Massenburg, B.; Wenderski, W.; Jayaraman, V.; Thompson, L.; Neves, S.R. ERK regulation of phosphodiesterase 4 enhances dopamine-stimulated AMPA receptor membrane insertion. Proc. Natl. Acad. Sci. USA 2013, 110, 15437–15442. [Google Scholar] [CrossRef] [Green Version]
- Diering, G.H.; Heo, S.; Hussain, N.K.; Liu, B.; Huganir, R.L. Extensive phosphorylation of AMPA receptors in neurons. Proc. Natl. Acad. Sci. USA 2016, 113, E4920–E4927. [Google Scholar] [CrossRef] [PubMed]
- Rizzuto, R.; Pinton, P.; Carrington, W.; Fay, F.S.; Fogarty, K.E.; Lifshitz, L.M.; Tuft, R.A.; Pozzan, T. Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science 1998, 280, 1763–1766. [Google Scholar] [CrossRef] [PubMed]
- Sneyd, J. Calcium buffering and diffusion: On the resolution of an outstanding problem. Biophys. J. 1994, 67, 4–5. [Google Scholar] [CrossRef]
- Roderick, H.L.; Berridge, M.J.; Bootman, M.D. Calcium-induced calcium release. Curr. Biol. 2003, 13, R425. [Google Scholar] [CrossRef]
- Collins, M.O.; Husi, H.; Yu, L.; Brandon, J.M.; Anderson, C.N.G.; Blackstock, W.P.; Choudhary, J.S.; Grant, S.G.N. Molecular characterization and comparison of the components and multiprotein complexes in the postsynaptic proteome. J. Neurochem. 2006, 97 (Suppl. 1), 16–23. [Google Scholar] [CrossRef]
- Lalonde, J.; Saia, G.; Gill, G. Store-operated calcium entry promotes the degradation of the transcription factor Sp4 in resting neurons. Sci. Signal. 2014, 7, ra51. [Google Scholar] [CrossRef]
- Hartmann, J.; Karl, R.M.; Alexander, R.P.D.; Adelsberger, H.; Brill, M.S.; Ruhlmann, C.; Ansel, A.; Sakimura, K.; Baba, Y.; Kurosaki, T.; et al. STIM1 controls neuronal Ca(2+) signaling, mGluR1-dependent synaptic transmission, and cerebellar motor behavior. Neuron 2014, 82, 635–644. [Google Scholar] [CrossRef]
- Lin, Y.-P.; Bakowski, D.; Mirams, G.R.; Parekh, A.B. Selective recruitment of different Ca2+-dependent transcription factors by STIM1-Orai1 channel clusters. Nat. Commun. 2019, 10, 2516. [Google Scholar] [CrossRef]
- LaFerla, F.M.; Green, K.N.; Oddo, S. Intracellular amyloid-beta in Alzheimer’s disease. Nat. Rev. Neurosci. 2007, 8, 499–509. [Google Scholar] [CrossRef]
- Brody, A.H.; Strittmatter, S.M. Synaptotoxic Signaling by Amyloid Beta Oligomers in Alzheimer′s Disease Through Prion Protein and Mgluradv. Adv. Pharmacol. 2018, 82, 293–323. [Google Scholar]
- Palmer, A.E.; Tsien, R.Y. Measuring calcium signaling using genetically targetable fluorescent indicators. Nat. Protoc. 2006, 1, 1057–1065. [Google Scholar] [CrossRef] [PubMed]
- Demuro, A.; Parker, I. Cytotoxicity of intracellular abeta42 amyloid oligomers involves Ca2+ release from the endoplasmic reticulum by stimulated production of inositol trisphosphate. J. Neurosci. 2013, 33, 3824–3833. [Google Scholar] [CrossRef] [PubMed]
- Jensen, L.E.; Bultynck, G.; Luyten, T.; Amijee, H.; Bootman, M.D.; Roderick, H.L. Alzheimer′s disease-associated peptide Abeta42 mobilizes ER Ca(2+) via InsP3R-dependent and independent mechanisms. Front. Mol. Neurosci. 2013, 6, 36. [Google Scholar] [CrossRef] [PubMed]
- Jairaman, A.; Prakriya, M. Molecular pharmacology of store-operated CRAC channels. Channels 2013, 7, 402–414. [Google Scholar] [CrossRef] [Green Version]
- Norante, R.P.; Massimino, M.L.; Lorenzon, P.; De Mario, A.; Peggion, C.; Vicario, M.; Albiero, M.; Sorgato, M.C.; Lopreiato, R.; Bertoli, A. Generation and validation of novel adeno-associated viral vectors for the analysis of Ca2+ homeostasis in motor neurons. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef]
- Lobao-Soares, B.; Bianchin, M.M.; Linhares, M.N.; Carqueja, C.L.; Tasca, C.I.; Souza, M.; Marques, W.J.; Brentani, R.; Martins, V.R.; Sakamoto, A.C.; et al. Normal brain mitochondrial respiration in adult mice lacking cellular prion protein. Neurosci. Lett. 2005, 375, 203–206. [Google Scholar] [CrossRef]
- Rui, Y.; Zheng, J.Q. Amyloid beta oligomers elicit mitochondrial transport defects and fragmentation in a time-dependent and pathway-specific manner. Mol. Brain 2016, 9, 79. [Google Scholar] [CrossRef]
- Han, X.-J.; Hu, Y.-Y.; Yang, Z.-J.; Jiang, L.-P.; Shi, S.-L.; Li, Y.-R.; Guo, M.-Y.; Wu, H.-L.; Wan, Y.-Y. Amyloid beta-42 induces neuronal apoptosis by targeting mitochondria. Mol. Med. Rep. 2017, 16, 4521–4528. [Google Scholar] [CrossRef]
- Calvo-Rodriguez, M.; Hernando-Perez, E.; Nuñez, L.; Villalobos, C. Amyloid β Oligomers Increase ER-Mitochondria Ca(2+) Cross Talk in Young Hippocampal Neurons and Exacerbate Aging-Induced Intracellular Ca(2+) Remodeling. Front. Cell. Neurosci. 2019, 13, 22. [Google Scholar] [CrossRef]
- Eckert, A.; Hauptmann, S.; Scherping, I.; Rhein, V.; Muller-Spahn, F.; Gotz, J.; Muller, W.E. Soluble beta-amyloid leads to mitochondrial defects in amyloid precursor protein and tau transgenic mice. Neurodegener. Dis. 2008, 5, 157–159. [Google Scholar] [CrossRef]
- Lei, Y.; Yang, L.; Ye, C.Y.; Qin, M.Y.; Yang, H.Y.; Jiang, H.L.; Tang, X.C.; Zhang, H.Y. Involvement of Intracellular and Mitochondrial Aβ in the Ameliorative Effects of Huperzine A against Oligomeric Aβ42-Induced Injury in Primary Rat Neurons. PLoS ONE 2015, 10, e0128366. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.T.; Beal, M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006, 443, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Sun, L.; Chen, X.; Zhang, D. Oxidative stress, mitochondrial damage and neurodegenerative diseases. Neural Regen. Res. 2013, 8, 2003–2014. [Google Scholar] [PubMed]
- Bhat, A.H.; Dar, K.B.; Anees, S.; Zargar, M.A.; Masood, A.; Sofi, M.A.; Ganie, S.A. Oxidative stress, mitochondrial dysfunction and neurodegenerative diseases; a mechanistic insight. Biomed. Pharm. 2015, 74, 101–110. [Google Scholar] [CrossRef]
- Perry, G.; Cash, A.D.; Smith, M.A. Alzheimer Disease and Oxidative Stress. J. Biomed. Biotechnol. 2002, 2, 120–123. [Google Scholar] [CrossRef]
- Guglielmotto, M.; Giliberto, L.; Tamagno, E.; Tabaton, M. Oxidative stress mediates the pathogenic effect of different Alzheimer′s disease risk factors. Front. Aging Neurosci. 2010, 2, 3. [Google Scholar] [PubMed]
- Wang, J.; Chen, G.-J. Mitochondria as a therapeutic target in Alzheimer′s disease. Genes Dis. 2016, 3, 220–227. [Google Scholar] [CrossRef]
- Manoharan, S.; Guillemin, G.J.; Abiramasundari, R.S.; Essa, M.M.; Akbar, M.; Akbar, M.D. The Role of Reactive Oxygen Species in the Pathogenesis of Alzheimer′s Disease, Parkinson′s Disease, and Huntington′s Disease: A Mini Review. Oxid. Med. Cell. Longev. 2016, 2016, 8590578. [Google Scholar] [CrossRef]
- Wong, B.S.; Pan, T.; Liu, T.; Li, R.; Gambetti, P.; Sy, M.S. Differential contribution of superoxide dismutase activity by prion protein in vivo. Biochem. Biophys. Res. Commun. 2000, 273, 136–139. [Google Scholar] [CrossRef]
- Paterson, A.W.J.; Curtis, J.C.; Macleod, N.K. Complex I specific increase in superoxide formation and respiration rate by PrP-null mouse brain mitochondria. J. Neurochem. 2008, 105, 177–191. [Google Scholar] [CrossRef]
- Sorce, S.; Krause, K.-H.; Jaquet, V. Targeting NOX enzymes in the central nervous system: Therapeutic opportunities. Cell. Mol. Life Sci. 2012, 69, 2387–2407. [Google Scholar] [CrossRef] [PubMed]
- Schneider, B.; Mutel, V.; Pietri, M.; Ermonval, M.; Mouillet-Richard, S.; Kellermann, O. NADPH oxidase and extracellular regulated kinases 1/2 are targets of prion protein signaling in neuronal and nonneuronal cells. Proc. Natl. Acad. Sci. USA 2003, 100, 13326–13331. [Google Scholar] [CrossRef] [Green Version]
- ten Freyhaus, H.; Huntgeburth, M.; Wingler, K.; Schnitker, J.; Baumer, A.T.; Vantler, M.; Bekhite, M.M.; Wartenberg, M.; Sauer, H.; Rosenkranz, S. Novel Nox inhibitor VAS2870 attenuates PDGF-dependent smooth muscle cell chemotaxis, but not proliferation. Cardiovasc. Res. 2006, 71, 331–341. [Google Scholar] [CrossRef] [PubMed]
- Dikalov, S. Cross talk between mitochondria and NADPH oxidases. Free Radic. Biol. Med. 2011, 51, 1289–1301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, L.; Aon, M.A.; Almas, T.; Cortassa, S.; Winslow, R.L.; O’Rourke, B. A reaction-diffusion model of ROS-induced ROS release in a mitochondrial network. PLoS Comput. Biol. 2010, 6, e1000657. [Google Scholar] [CrossRef] [PubMed]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef] [PubMed]
- Bain, J.; McLauchlan, H.; Elliott, M.; Cohen, P. The specificities of protein kinase inhibitors: An update. Biochem. J. 2003, 371, 199–204. [Google Scholar] [PubMed]
- Chowdhury, A.K.; Watkins, T.; Parinandi, N.L.; Saatian, B.; Kleinberg, M.E.; Usatyuk, P.V.; Natarajan, V. Src-mediated tyrosine phosphorylation of p47phox in hyperoxia-induced activation of NADPH oxidase and generation of reactive oxygen species in lung endothelial cells. J. Biol. Chem. 2005, 280, 20700–20711. [Google Scholar] [CrossRef]
- Koo, J.H.; Lee, W.H.; Lee, C.G.; Kim, S.G. Fyn inhibition by cycloalkane-fused 1,2-dithiole-3-thiones enhances antioxidant capacity and protects mitochondria from oxidative injury. Mol. Pharm. 2012, 82, 27–36. [Google Scholar] [CrossRef]
- Malenka, R.C.; Bear, M.F. LTP and LTD: An embarrassment of riches. Neuron 2004, 44, 5–21. [Google Scholar] [CrossRef]
- Henley, J.M.; Wilkinson, K.A. AMPA receptor trafficking and the mechanisms underlying synaptic plasticity and cognitive aging. Dialogues Clin. Neurosci. 2013, 15, 11–27. [Google Scholar] [PubMed]
- Castle, A.R.; Gill, A.C. Physiological Functions of the Cellular Prion Protein. Front. Mol. Biosci. 2017, 4, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewerenz, J.; Maher, P. Chronic Glutamate Toxicity in Neurodegenerative Diseases-What is the Evidence? Front. Neurosci. 2015, 9, 469. [Google Scholar] [CrossRef] [PubMed]
- Bezprozvanny, I.; Mattson, M.P. Neuronal calcium mishandling and the pathogenesis of Alzheimer’s disease. Trends Neurosci. 2008, 31, 454–463. [Google Scholar] [CrossRef] [PubMed]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Mario, A.; Peggion, C.; Massimino, M.L.; Norante, R.P.; Zulian, A.; Bertoli, A.; Sorgato, M.C. The Link of the Prion Protein with Ca2+ Metabolism and ROS Production, and the Possible Implication in Aβ Toxicity. Int. J. Mol. Sci. 2019, 20, 4640. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20184640
De Mario A, Peggion C, Massimino ML, Norante RP, Zulian A, Bertoli A, Sorgato MC. The Link of the Prion Protein with Ca2+ Metabolism and ROS Production, and the Possible Implication in Aβ Toxicity. International Journal of Molecular Sciences. 2019; 20(18):4640. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20184640
Chicago/Turabian StyleDe Mario, Agnese, Caterina Peggion, Maria Lina Massimino, Rosa Pia Norante, Alessandra Zulian, Alessandro Bertoli, and Maria Catia Sorgato. 2019. "The Link of the Prion Protein with Ca2+ Metabolism and ROS Production, and the Possible Implication in Aβ Toxicity" International Journal of Molecular Sciences 20, no. 18: 4640. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20184640