The Anti-Amyloidogenic Action of Doxycycline: A Molecular Dynamics Study on the Interaction with Aβ42

 , , ,
, , ,
Abstract
:
1. Introduction
2. Results and Discussion
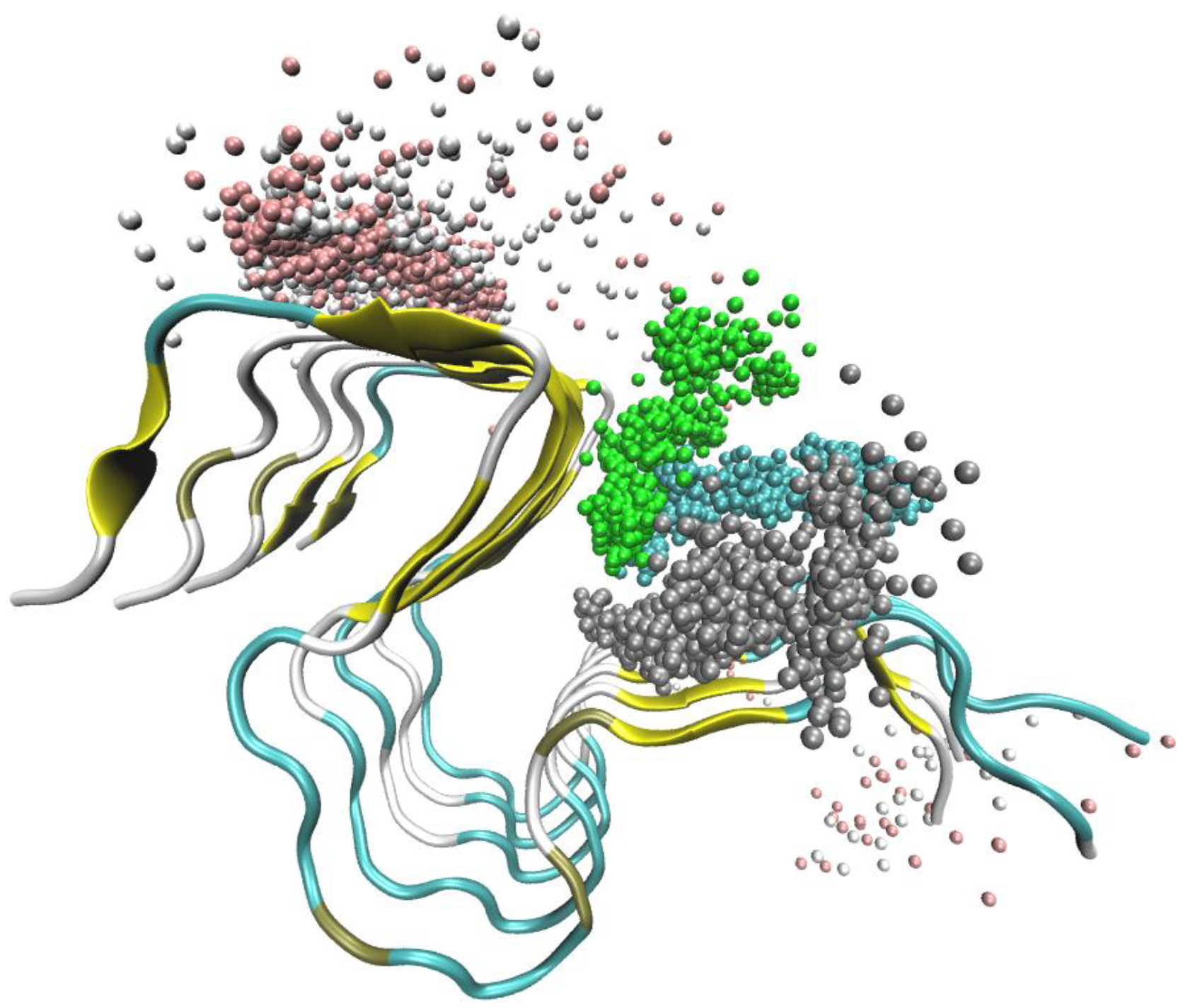
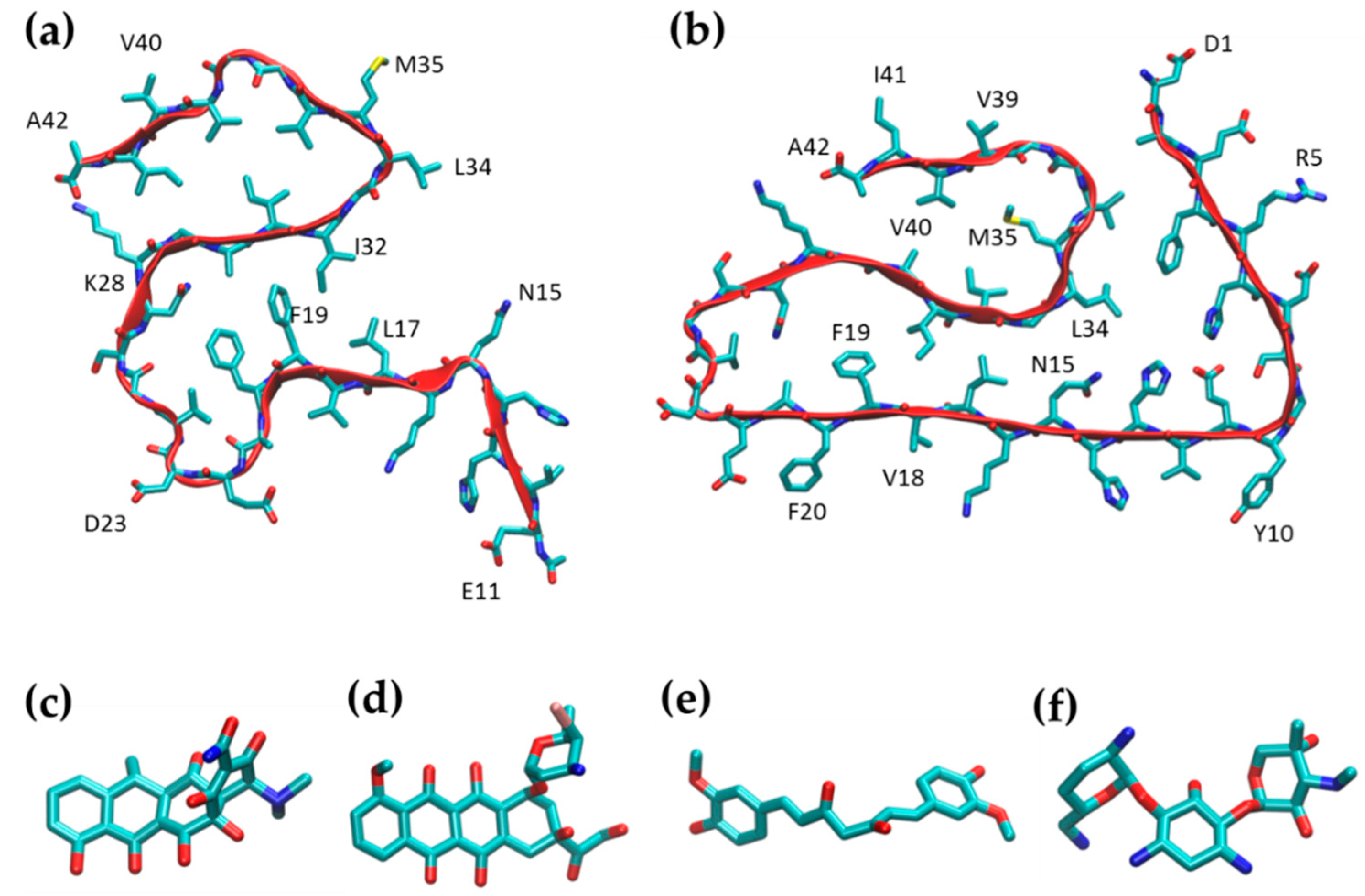
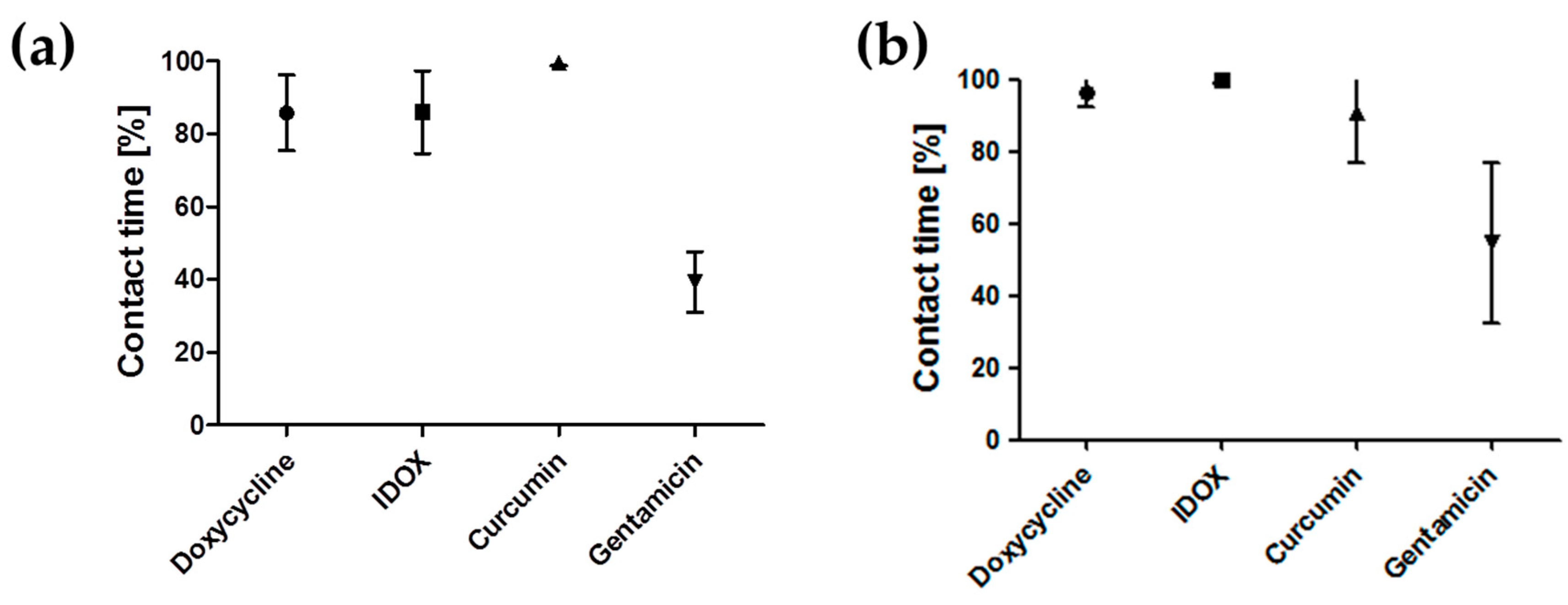
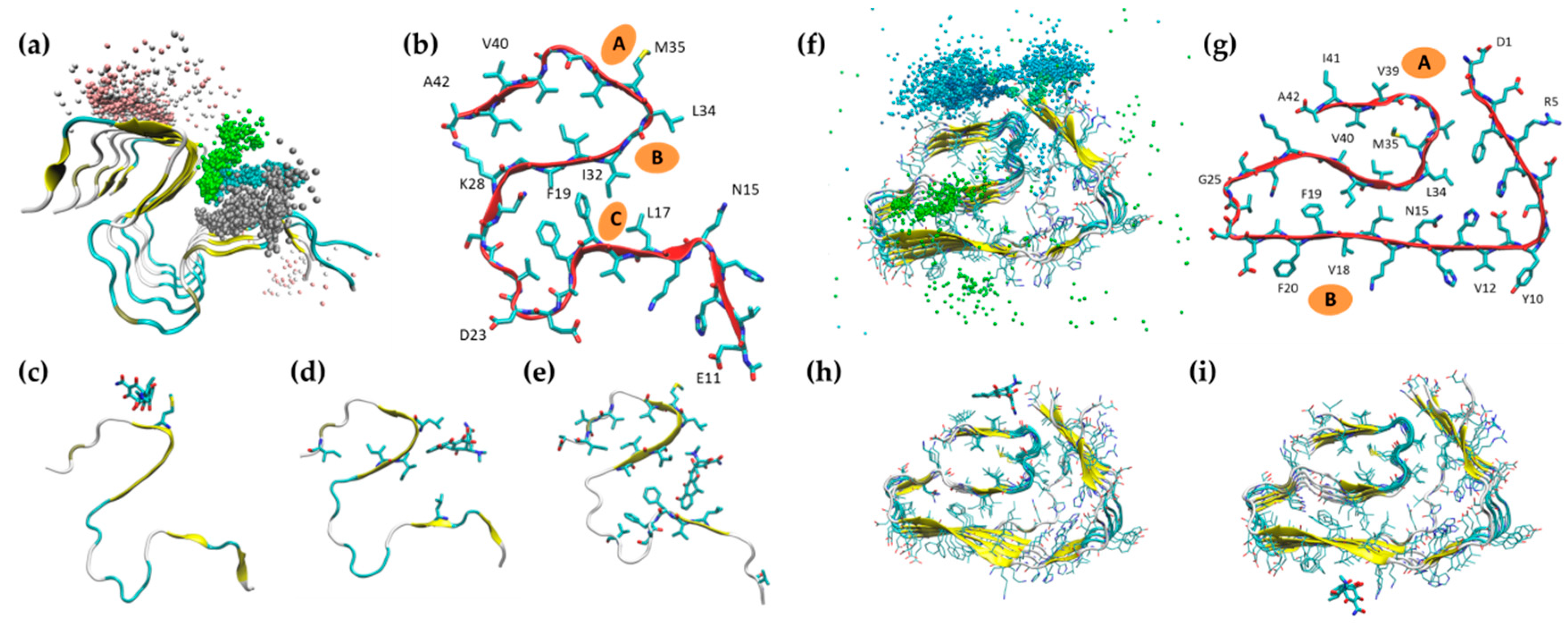
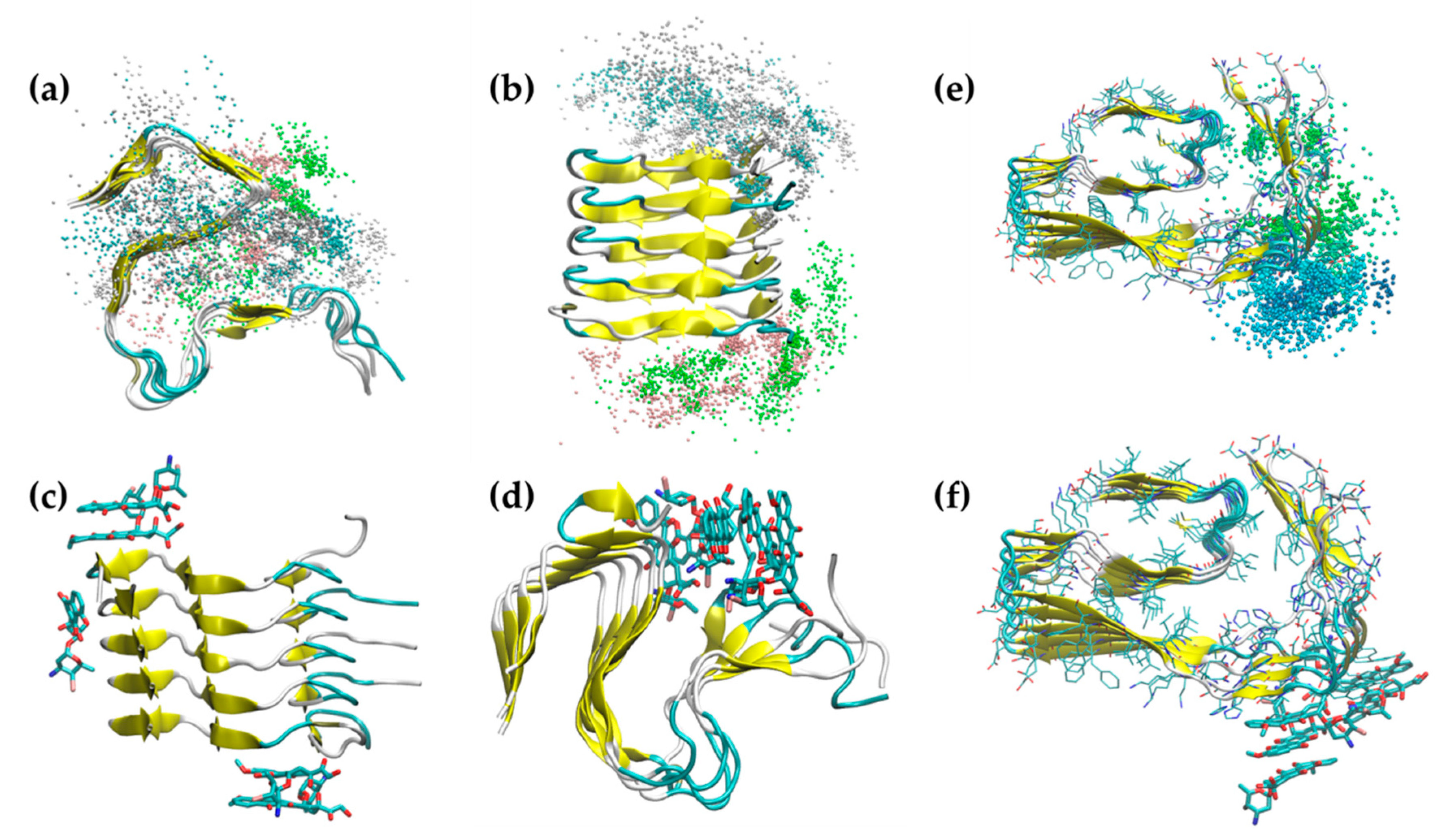
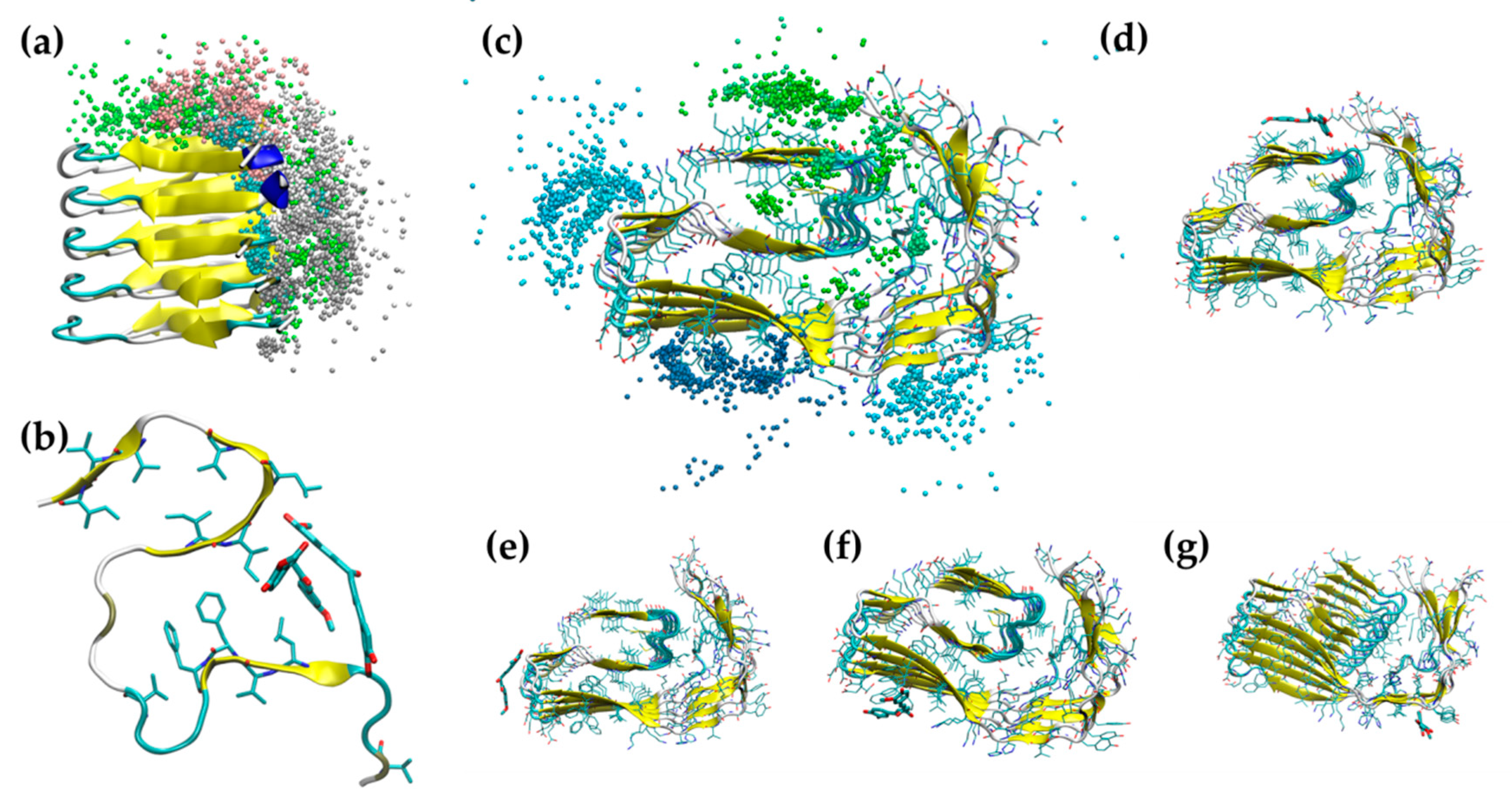
2.1. Simulation of Aβ Fibrils
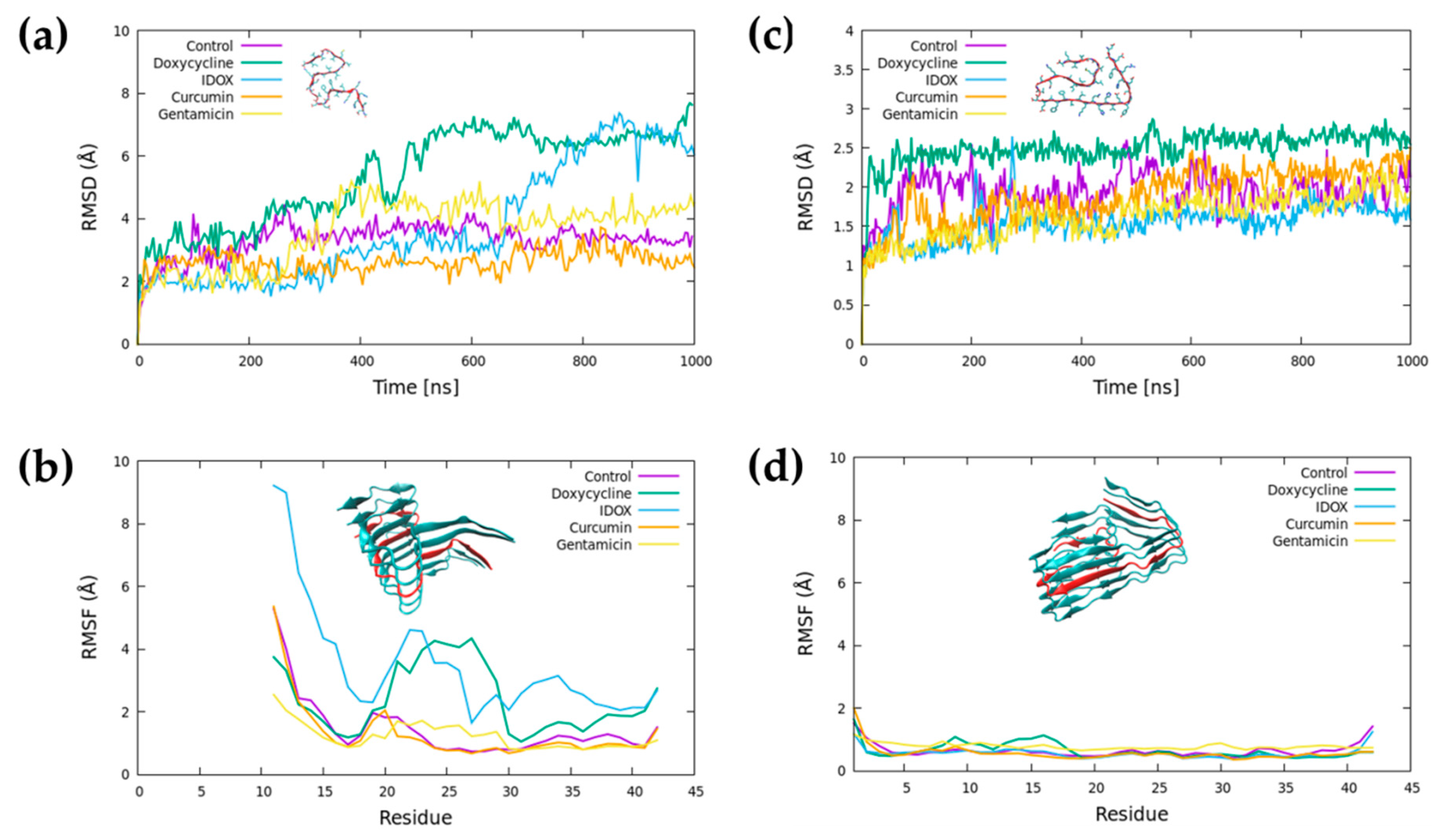
2.2. Doxycycline Binding
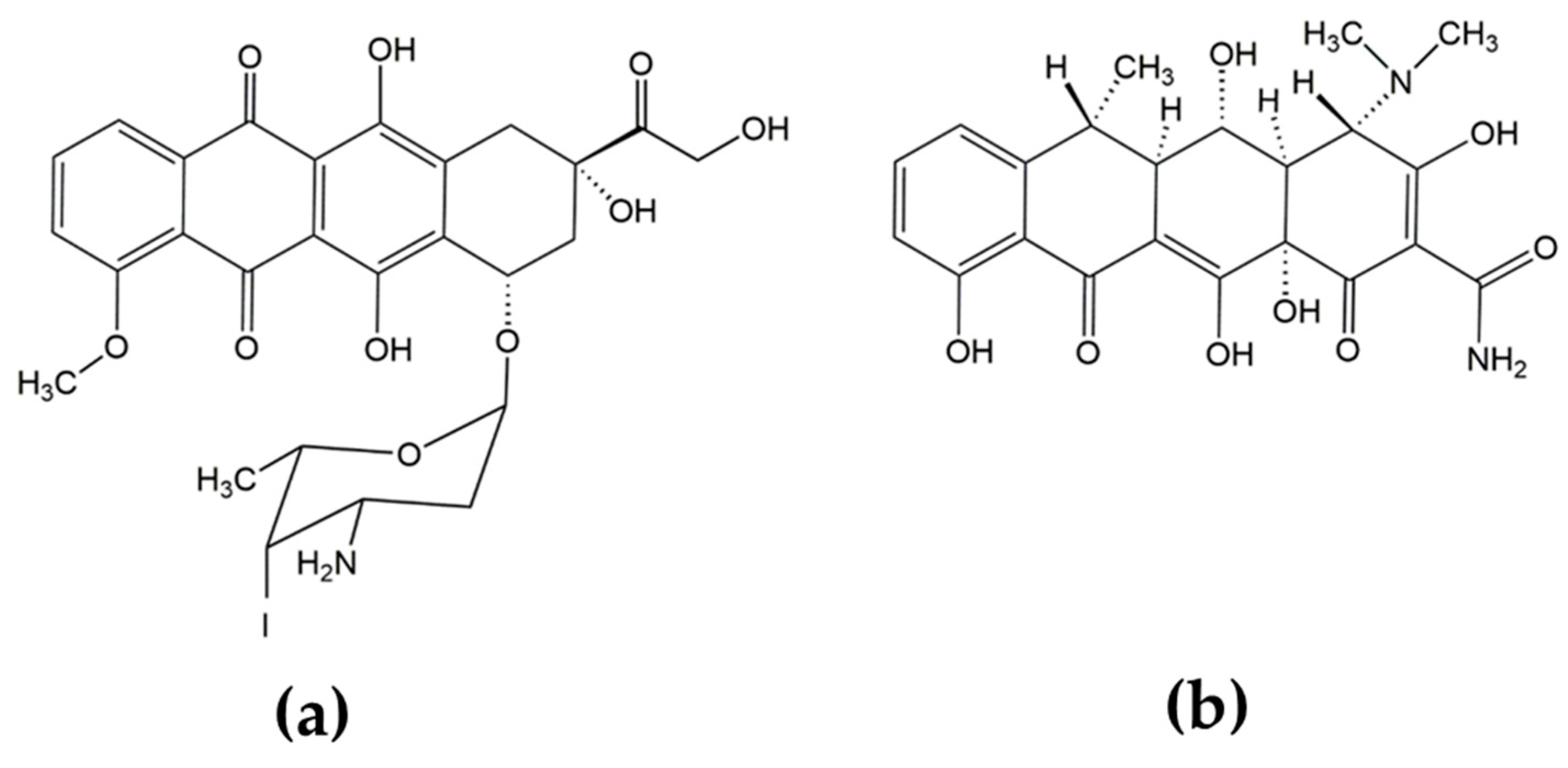
2.3. Iododoxorubicin Binding
2.4. Curcumin Binding
3. Materials and Methods
3.1. Molecular Models
3.2. Molecular Dynamics
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| IDOX | 4′-do-4′deoxydoxorubicin |
| TCs | tetracyclines |
References
- Gianni, B.L.; Bellotti, V.; Gianni, A.M.; Merlini, G. New Drug Therapy of Amyloidoses: Resorption of AL-Type Deposits with 4′-Iodo-4′-Deoxydoxorubicin. Blood 1995, 86, 855–861. [Google Scholar]
- Merlini, G.; Ascari, E.; Amboldi, N.; Bellotti, V.; Arbustini, E.; Perfetti, V.; Ferrari, M.; Zorzoli, I.; Marinone, M.G.; Garini, P. Interaction of the anthracycline 4′-iodo-4′-deoxydoxorubicin with amyloid fibrils: Inhibition of amyloidogenesis. Proc. Natl. Acad. Sci. USA 1995, 92, 2959–2963. [Google Scholar] [CrossRef]
- Palha, J.A.; Ballinari, D.; Amboldi, N.; Cardoso, I.; Fernandes, R.; Bellotti, V.; Merlini, G.; Saraiva, M.J. 4′-iodo-4′-deoxydoxorubicin disrupts the fibrillar structure of transthyretin amyloid. Am. J. Pathol. 2000, 156, 1919–1925. [Google Scholar] [CrossRef]
- Tagliavini, F.; McArthur, R.A.; Canciani, B.; Giaccone, G.; Porro, M.; Bugiani, M.; Lievens, P.M.J.; Bugiani, O.; Peri, E.; Dall’Ara, P.; et al. Effectiveness of anthracycline against experimental prion disease in Syrian hamsters. Science 1997, 276, 1119–1122. [Google Scholar] [CrossRef]
- Tagliavini, F.; Forloni, G.; Colombo, L.; Rossi, G.; Girola, L.; Canciani, B.; Angeretti, N.; Giampaolo, L.; Peressini, E.; Awan, T.; et al. Tetracycline affects abnormal properties of synthetic PrP peptides and PrP(Sc) in vitro. J. Mol. Biol. 2000, 300, 1309–1322. [Google Scholar] [CrossRef]
- Forloni, G.; Iussich, S.; Awan, T.; Colombo, L.; Angeretti, N.; Girola, L.; Bertani, I.; Poli, G.; Caramelli, M.; Grazia Bruzzone, M.; et al. Tetracyclines affect prion infectivity. Proc. Natl. Acad. Sci. USA 2002, 99, 10849–10854. [Google Scholar] [CrossRef] [Green Version]
- Cardoso, I.; Merlini, G.; Saraiva, M.J. 4′-iodo-4′-Deoxydoxorubicin and tetracyclines disrupt transthyretin amyloid fibrils in vitro producing noncytotoxic species: Screening for TTR fibril disrupters. FASEB J. 2003, 17, 803–809. [Google Scholar] [CrossRef]
- Cardoso, I.; Saraiva, M.J. Doxycycline disrupts transthyretin amyloid: Evidence from studies in a FAP transgenic mice model. FASEB J. 2006, 20, 234–239. [Google Scholar] [CrossRef]
- Obici, L.; Cortese, A.; Lozza, A.; Lucchetti, J.; Gobbi, M.; Palladini, G.; Perlini, S.; Saraiva, M.J.; Merlini, G. Doxycycline plus tauroursodeoxycholic acid for transthyretin amyloidosis: A phase II study. Amyloid 2012, 19, 34–36. [Google Scholar] [CrossRef]
- Ward, J.E.; Ren, R.; Toraldo, G.; SooHoo, P.; Guan, J.; O’Hara, C.; Jasuja, R.; Trinkaus-Randall, V.; Liao, R.; Connors, L.H.; et al. Doxycycline reduces fibril formation in a transgenic mouse model of AL amyloidosis. Blood 2011, 118, 6610–6617. [Google Scholar] [CrossRef] [Green Version]
- Giorgetti, S.; Raimondi, S.; Pagano, K.; Relini, A.; Bucciantini, M.; Corazza, A.; Fogolari, F.; Codutti, L.; Salmona, M.; Mangione, P.; et al. Effect of tetracyclines on the dynamics of formation and destructuration of β2-microglobulin amyloid fibrils. J. Biol. Chem. 2011, 286, 2121–2131. [Google Scholar] [CrossRef]
- Montagna, G.; Cazzulani, B.; Obici, L.; Uggetti, C.; Giorgetti, S.; Porcari, R.; Ruggiero, R.; Patrizia Mangione, P.; Brambilla, M.; Lucchetti, J.; et al. Benefit of doxycycline treatment on articular disability caused by dialysis related amyloidosis. Amyloid 2013, 20, 173–178. [Google Scholar] [CrossRef]
- Diomede, L.; Cassata, G.; Fiordaliso, F.; Salio, M.; Ami, D.; Natalello, A.; Doglia, S.M.; De Luigi, A.; Salmona, M. Tetracycline and its analogues protect Caenorhabditis elegans from β amyloid-induced toxicity by targeting oligomers. Neurobiol. Dis. 2010, 40, 424–431. [Google Scholar] [CrossRef]
- Balducci, C.; Santamaria, G.; La Vitola, P.; Brandi, E.; Grandi, F.; Viscomi, A.R.; Beeg, M.; Gobbi, M.; Salmona, M.; Ottonello, S.; et al. Doxycycline counteracts neuroinflammation restoring memory in Alzheimer’s disease mouse models. Neurobiol. Aging 2018, 70, 128–139. [Google Scholar] [CrossRef]
- Forloni, G.; Colombo, L.; Girola, L.; Tagliavini, F.; Salmona, M. Anti-amyloidogenic activity of tetracyclines: Studies in vitro. FEBS Lett. 2001, 487, 404–407. [Google Scholar] [CrossRef]
- Griffin, M.O.; Fricovsky, E.; Ceballos, G.; Villarreal, F. Tetracyclines: A pleitropic family of compounds with promising therapeutic properties. Review of the literature. AJP Cell Physiol. 2010, 299, C539–C548. [Google Scholar] [CrossRef]
- Stoilova, T.; Colombo, L.; Forloni, G.; Tagliavini, F.; Salmona, M. A new face for old antibiotics: Tetracyclines in treatment of amyloidoses. J. Med. Chem. 2013, 56, 5987–6006. [Google Scholar] [CrossRef]
- Lucchetti, J.; Fracasso, C.; Balducci, C.; Passoni, A.; Forloni, G.; Salmona, M.; Gobbi, M. Plasma and Brain Concentrations of Doxycycline after Single and Repeated Doses in Wild-Type and APP23 Mice. J. Pharmacol. Exp. Ther. 2019, 368, 32–40. [Google Scholar] [CrossRef]
- Doxycycline Hyclate for the Treatment of Familial Amyloid Polyneuropathy. Available online: http://www.emea.europa.eu/docs/en_GB/document_library/Orphan_designation/2012/05/WC500127736.pdf (accessed on 1 May 2012).
- Mourtas, S.; Canovi, M.; Zona, C.; Aurilia, D.; Niarakis, A.; La Ferla, B.; Salmona, M.; Nicotra, F.; Gobbi, M.; Antimisiaris, S.G. Curcumin-decorated nanoliposomes with very high affinity for amyloid-β1-42 peptide. Biomaterials 2011, 32, 1635–1645. [Google Scholar] [CrossRef]
- Luo, Y.; Smith, J.V.; Paramasivam, V.; Burdick, A.; Curry, K.J.; Buford, J.P.; Khan, I.; Netzer, W.J.; Xu, H.; Butko, P. Inhibition of amyloid-beta aggregation and caspase-3 activation by the Ginkgo biloba extract EGb761. Proc. Natl. Acad. Sci. USA. 2002, 99, 12197–12202. [Google Scholar] [CrossRef]
- Chakrabortee, S.; Liu, Y.; Zhang, L.; Matthews, H.R.; Zhang, H.; Pan, N.; Cheng, C.; Guan, S.; Guo, D.; Huang, Z.; et al. Macromolecular and small-molecule modulation of intracellular Aβ42 aggregation and associated toxicity. Biochem. J. 2012, 442, 507–515. [Google Scholar] [CrossRef]
- Ringman, J.; Frautschy, S.; Cole, G.; Masterman, D.; Cummings, J. A Potential Role of the Curry Spice Curcumin in Alzheimers Disease. Curr. Alzheimer Res. 2005, 2, 131–136. [Google Scholar] [CrossRef] [Green Version]
- De Luigi, A.; Colombo, L.; Diomede, L.; Capobianco, R.; Mangieri, M.; Miccolo, C.; Limido, L.; Forloni, G.; Tagliavini, F.; Salmona, M. The efficacy of tetracyclines in peripheral and intracerebral prion infection. PLoS ONE 2008, 3, e1888. [Google Scholar] [CrossRef]
- Lührs, T.; Ritter, C.; Adrian, M.; Riek-Loher, D.; Bohrmann, B.; Döbeli, H.; Schubert, D.; Riek, R. 3D structure of Alzheimer’s amyloid-beta(1–42) fibrils. Proc. Natl. Acad. Sci. USA 2005, 102, 17342–17347. [Google Scholar] [CrossRef]
- Colvin, M.T.; Silvers, R.; Ni, Q.Z.; Can, T.V.; Sergeyev, I.; Rosay, M.; Donovan, K.J.; Michael, B.; Wall, J.; Linse, S.; et al. Atomic Resolution Structure of Monomorphic Aβ 42 Amyloid Fibrils. J. Am. Chem. Soc. 2016, 138, 9663–9674. [Google Scholar] [CrossRef]
- Gremer, L.; Schölzel, D.; Schenk, C.; Reinartz, E.; Labahn, J.; Ravelli, R.B.G.; Tusche, M.; Lopez-iglesias, C.; Hoyer, W.; Heise, H.; et al. Fibril structure of amyloid- b (1–42) by cryo – electron microscopy. Science 2017, 358, 116–119. [Google Scholar] [CrossRef]
- Martin, T.D.; Malagodi, A.J.; Chi, E.Y.; Evans, D.G. Computational Study of the Driving Forces and Dynamics of Curcumin Binding to Amyloid-β Protofibrils. J. Phys. Chem. B 2019, 123, 551–560. [Google Scholar] [CrossRef]
- Fan, H.M.; Gu, R.X.; Wang, Y.J.; Pi, Y.L.; Zhang, Y.H.; Xu, Q.; Wei, D.Q. Destabilization of Alzheimer’s Aβ42 Protofibrils with a Novel Drug Candidate wgx-50 by Molecular Dynamics Simulations. J. Phys. Chem. B 2015, 119, 11196–11202. [Google Scholar] [CrossRef]
- Xiao, Y.; Ma, B.; McElheny, D.; Parthasarathy, S.; Long, F.; Hoshi, M.; Nussinov, R.; Ishii, Y. Aβ(1–42) fibril structure illuminates self-recognition and replication of amyloid in Alzheimer’s disease. Nat. Struct. Mol. Biol. 2015, 22, 499–505. [Google Scholar] [CrossRef]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.M.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Gautieri, A.; Ionita, M.; Silvestri, D.; Votta, E.; Vesentini, S.; Fiore, G.B.; Barbani, N.; Ciardelli, G.; Redaelli, A. Computer-Aided Molecular Modeling and experimental validation of water permeability properties biosynthetic materials. J. Comput. Theor. Nanosci. 2010, 7, 1–7. [Google Scholar] [CrossRef]
- Gautieri, A.; Vesentini, S.; Redaelli, A.; Buehler, M.J. Osteogenesis imperfecta mutations lead to local tropocollagen unfolding and disruption of H-bond network. RSC Adv. 2012, 2, 3890. [Google Scholar] [CrossRef]
- Rigoldi, F.; Donini, S.; Giacomina, F.; Sorana, F.; Redaelli, A.; Bandiera, T.; Parisini, E.; Gautieri, A. Thermal stabilization of the deglycating enzyme Amadoriase i by rational design. Sci. Rep. 2018, 8. [Google Scholar] [CrossRef]
- Rigoldi, F.; Spero, L.; Dalle Vedove, A.; Redaelli, A.; Parisini, E.; Gautieri, A. Molecular dynamics simulations provide insights into the substrate specificity of FAOX family members. Mol. BioSyst. 2016, 12, 2622–2633. [Google Scholar] [CrossRef] [Green Version]
- Rigoldi, F.; Donini, S.; Redaelli, A.; Parisini, E.; Gautieri, A.; Rigoldi, F.; Donini, S.; Redaelli, A.; Parisini, E. Engineering of thermostable enzymes for industrial applications Review: Engineering of thermostable enzymes for industrial applications. APL Bioeng. 2018, 2, 011501. [Google Scholar] [CrossRef]
- Nelson, M.T.; Humphrey, W.; Gursoy, A.; Dalke, A.; Kale, L.V.; Skeel, R.D.; Schulten, K. NAMD: A parallel, object oriented molecular dynamics program. Int. J. Supercomput. Appl. High Perform. Comput. 1996, 10, 251–268. [Google Scholar] [CrossRef]
- Harvey, M.J.; Giupponi, G.; De Fabritiis, G. ACEMD: Accelerating Biomolecular Dynamics in the Microsecond Time Scale. J. Chem. Theory Comput. 2009, 5, 1632–1639. [Google Scholar] [CrossRef] [Green Version]
- Rigoldi, F.; Gautieri, A.; Dalle Vedove, A.; Lucarelli, A.P.; Vesentini, S.; Parisini, E. Crystal structure of the deglycating enzyme amadoriase i in its free form and substrate-bound complex. Proteins 2016, 84, 744–758. [Google Scholar] [CrossRef]
- Rigoldi, F.; Metrangolo, P.; Redaelli, A.; Gautieri, A. Nanostructure and stability of calcitonin amyloids. J. Biol. Chem. 2017, 292, 744–758. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Miller, B.R.; McGee, T.D.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA.py: An efficient program for end-state free energy calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef]
- Salomon-Ferrer, R.; Case, D.A.; Walker, R.C. An overview of the Amber biomolecular simulation package. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2013, 3, 198–210. [Google Scholar] [CrossRef]
- Airoldi, C.; Colombo, L.; Manzoni, C.; Sironi, E.; Natalello, A.; Doglia, S.M.; Forloni, G.; Tagliavini, F.; Del Favero, E.; Cantù, L.; et al. Tetracycline prevents Aβ oligomer toxicity through an atypical supramolecular interaction. Org. Biomol. Chem. 2011, 9, 463–472. [Google Scholar] [CrossRef]
- Cohen, S.I.A.; Linse, S.; Luheshi, L.M.; Hellstrand, E.; White, D.A.; Rajah, L.; Otzen, D.E.; Vendruscolo, M.; Dobson, C.M.; Knowles, T.P.J. Proliferation of amyloid-β42 aggregates occurs through a secondary nucleation mechanism. Proc. Natl. Acad. Sci. USA 2013, 110, 9758–9763. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Binding Site | ΔGvdW | ΔGEL | ΔGSol-P | ΔGSol-NP | ΔGbind |
|---|---|---|---|---|---|
| M35 | −39.4 ± 3.7 | −19.7 ± 5.9 | 46.9 ± 6.7 | −3.3 ± 0.2 | −15.5 ± 3.4 |
| I32–L34 | −35.2 ± 2.5 | −17.3 ± 4.8 | 38.7 ± 4.8 | −3.2 ± 0.1 | −16.8 ± 3.3 |
| F19–L17 | −44.9 ± 9.8 | −15.7 ± 9.4 | 40.7 ± 5.5 | −3.8 ± 0.4 | −23.7 ± 5.0 |
| Binding Region | ΔGvdW | ΔGEL | ΔGSol-P | ΔGSol-NP | ΔGbind |
|---|---|---|---|---|---|
| K28–A42 | −65.6 ± 5.5 | −12.6 ± 7.8 | 45.4 ± 7.4 | −5.1 ± 0.2 | −38.0 ± 5.3 |
| L17–I32 | −60.6 ± 5.7 | −18.1 ± 11.3 | 47.9 ± 10.3 | −4.9 ± 0.4 | −35.9 ± 4.3 |
| Hydrophobic pocket | −52.4 ± 5.4 | −10.8 ± 4.6 | 36.0 ± 5.2 | −4.2 ± 0.4 | −31.6 ± 4.4 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gautieri, A.; Beeg, M.; Gobbi, M.; Rigoldi, F.; Colombo, L.; Salmona, M. The Anti-Amyloidogenic Action of Doxycycline: A Molecular Dynamics Study on the Interaction with Aβ42. Int. J. Mol. Sci. 2019, 20, 4641. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20184641
Gautieri A, Beeg M, Gobbi M, Rigoldi F, Colombo L, Salmona M. The Anti-Amyloidogenic Action of Doxycycline: A Molecular Dynamics Study on the Interaction with Aβ42. International Journal of Molecular Sciences. 2019; 20(18):4641. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20184641
Chicago/Turabian StyleGautieri, Alfonso, Marten Beeg, Marco Gobbi, Federica Rigoldi, Laura Colombo, and Mario Salmona. 2019. "The Anti-Amyloidogenic Action of Doxycycline: A Molecular Dynamics Study on the Interaction with Aβ42" International Journal of Molecular Sciences 20, no. 18: 4641. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20184641