The Role of Adipose Tissue Mitochondria: Regulation of Mitochondrial Function for the Treatment of Metabolic Diseases

Abstract
:1. Introduction
2. Mitochondrial Properties and Adipocytes
2.1. Mitochondria in White Adipocytes
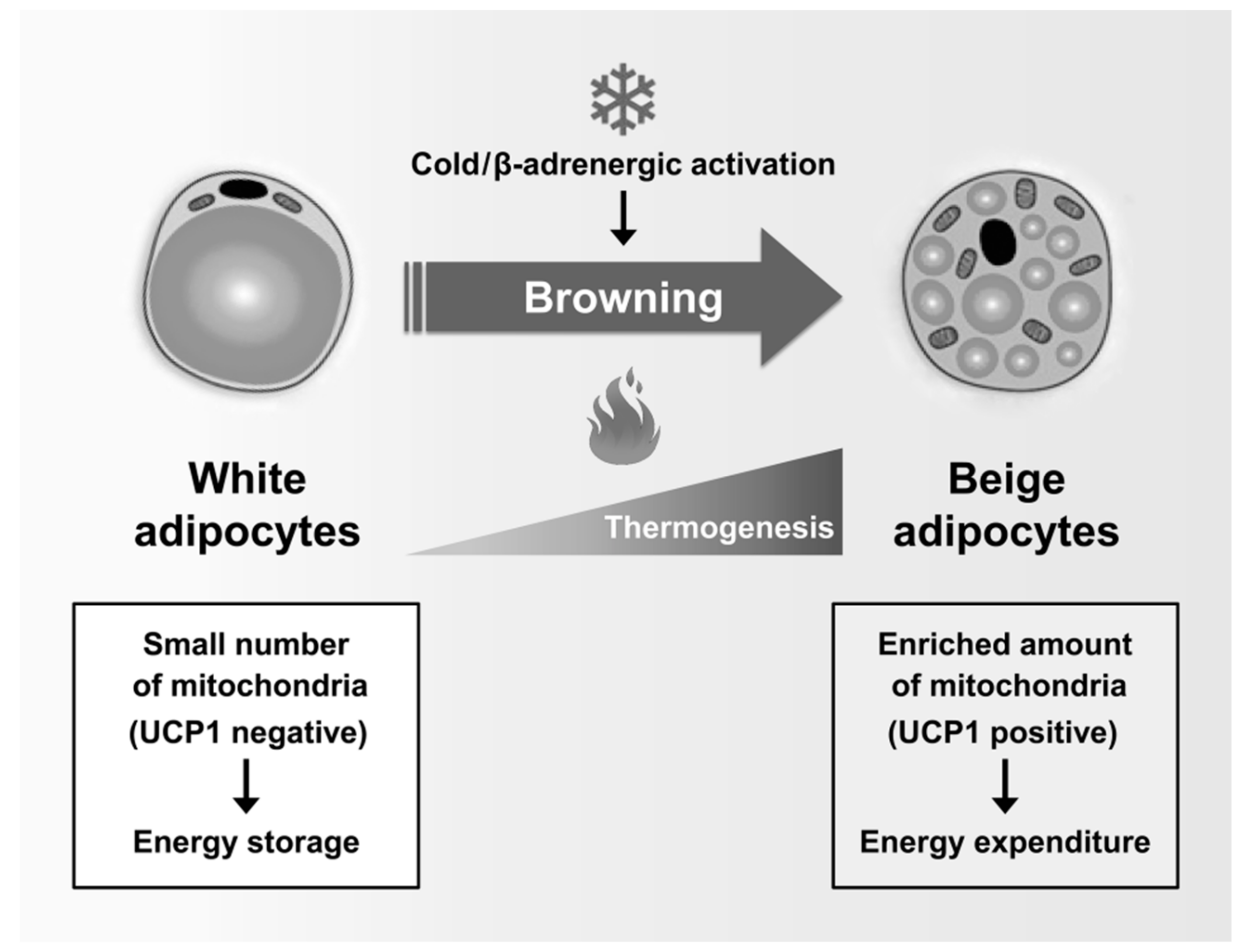
2.2. Mitochondria in Brown and Beige Adipocytes
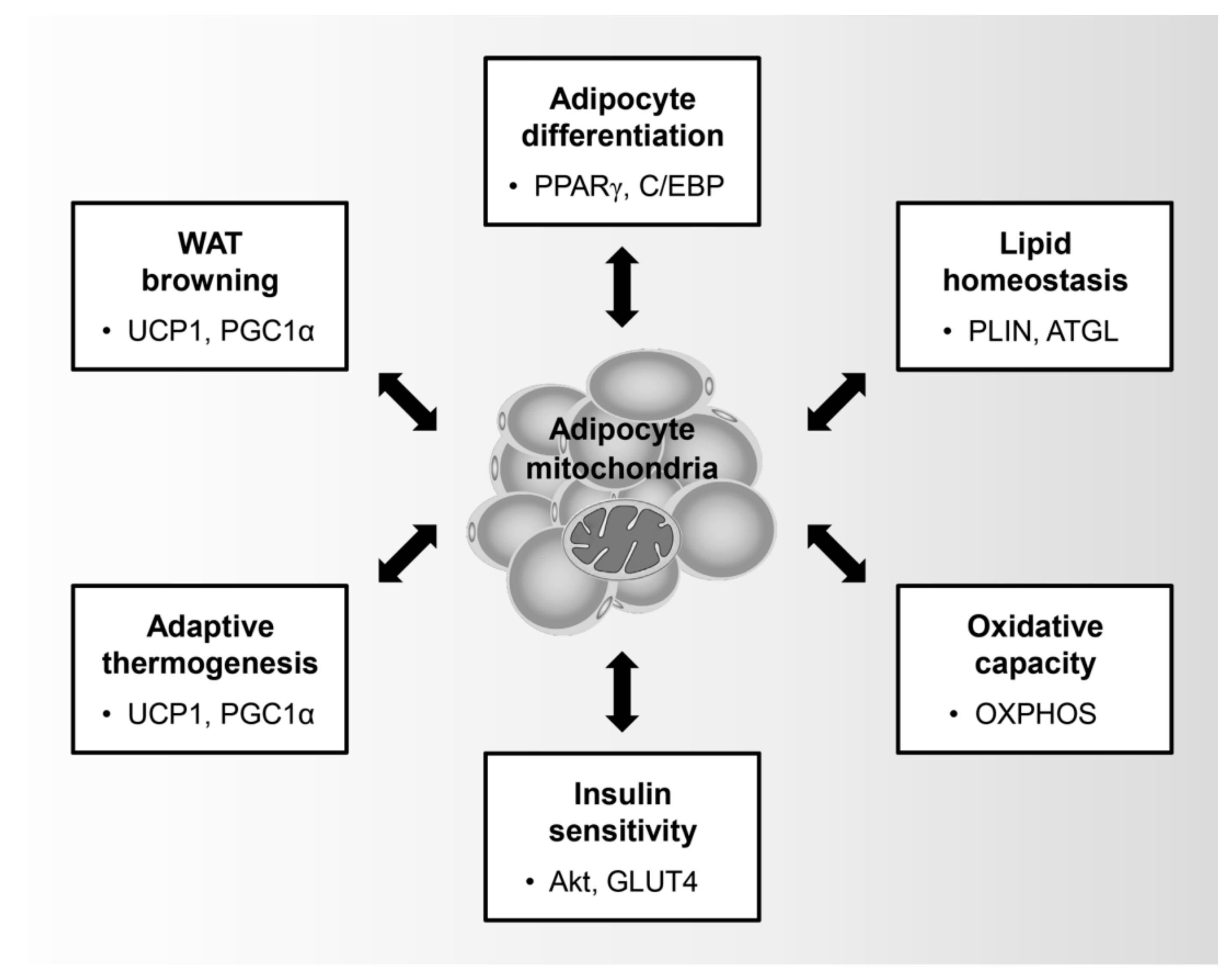
3. Role of the Mitochondria in Adipocytes
3.1. Adipocyte Differentiation
3.2. Lipid Homeostasis and Oxidative Capacity
3.3. Glucose Utilization and Insulin Sensitivity
3.4. Thermogenesis and WAT Browning
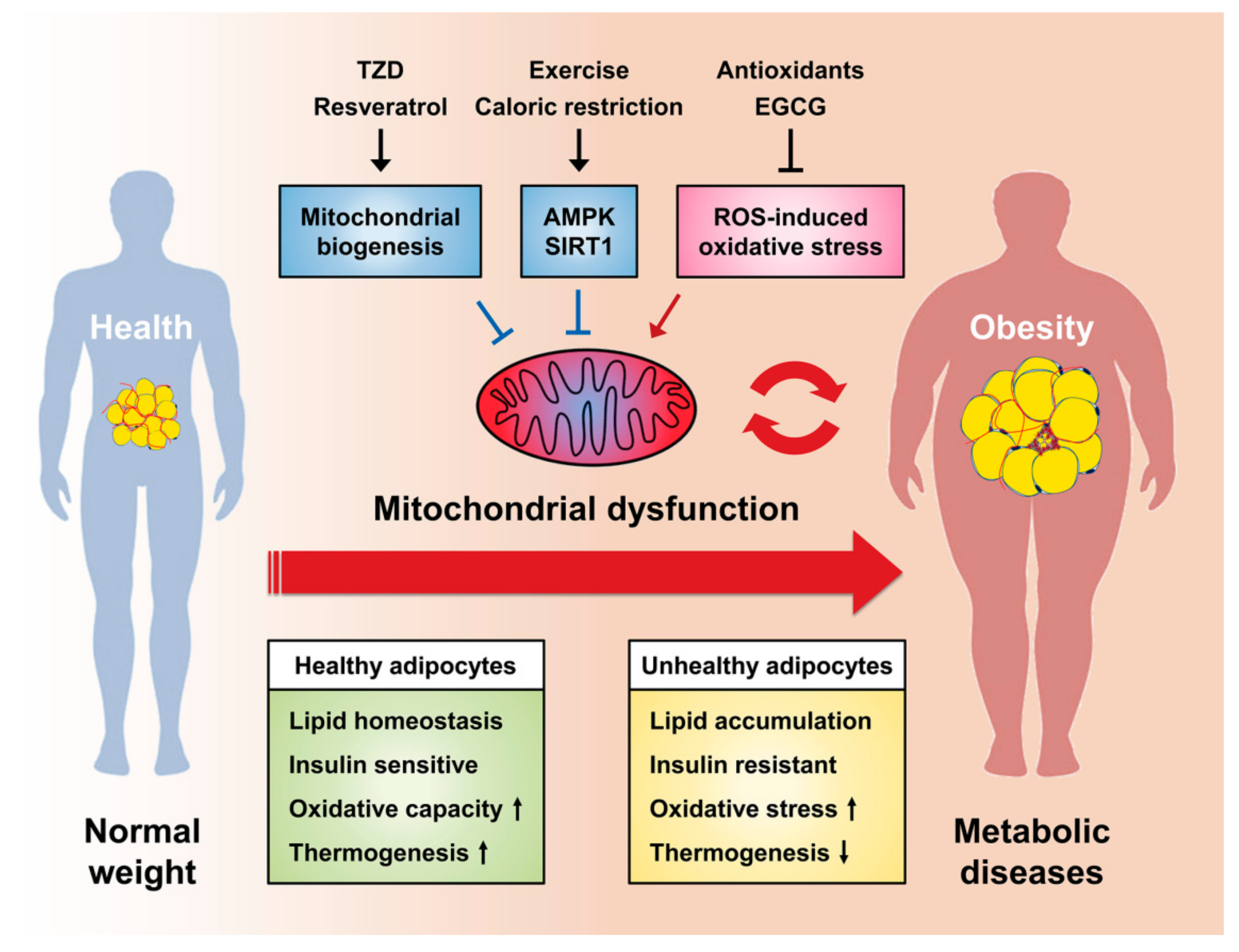
4. Mitochondrial Dysfunction in Adipocytes
4.1. Increased Production of ROS
4.2. Alteration of the Mitochondrial Genome
4.3. Dysregulated Mitochondrial Dynamics
4.4. Altered Mitophagy and Mitochondrial Turnover
5. Targeting Mitochondria as a Therapeutic Strategy for Metabolic Diseases
5.1. Activation of BAT Thermogenesis and WAT Browning
5.2. Thiazolidinediones
5.3. Mitochondria-Targeted Antioxidants
5.4. Exercise and Caloric Restriction
5.5. Dietary Natural Compounds
6. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Altmann, R. Die elementarorganismen und ihre beziehungen zu den zellen; Veit & Comp.: Leipzig, Germany, 1890; p. 145. [Google Scholar]
- Benda, C. Ueber die Spermatogenese der Vertebraten und höherer Evertebraten, II Theil: Die Histiogenese der Spermien. Arch. Anat. Physiol. 1898, 73, 393–398. [Google Scholar]
- Warburg, O. The Chemical Constitution of Respiration Ferment. Science 1928, 68, 437–443. [Google Scholar] [CrossRef] [PubMed]
- Dixon, M.; Keilin, D. An improved method for the measurement of tissue respiration. Biochem. J. 1933, 27, 86–95. [Google Scholar] [PubMed]
- Krebs, H.A. The history of the tricarboxylic acid cycle. Perspect. Biol. Med. 1970, 14, 154–170. [Google Scholar] [CrossRef]
- Claude, A.; Fullam, E.F. An Electron Microscope Study of Isolated Mitochondria: Method and Preliminary Results. J. Exp. Med. 1945, 81, 51–62. [Google Scholar] [CrossRef] [PubMed]
- Ernster, L.; Schatz, G. Mitochondria: A historical review. J. Cell Biol. 1981, 91 (3 Pt 2), 227s–255s. [Google Scholar]
- Willis, E.J. The powerhouse of the cell. Ultrastruct. Pathol. 1992, 16. [Google Scholar] [CrossRef]
- Pagliarini, D.J.; Rutter, J. Hallmarks of a new era in mitochondrial biochemistry. Genes Dev. 2013, 27, 2615–2627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McBride, H.M.; Neuspiel, M.; Wasiak, S. Mitochondria: more than just a powerhouse. Curr. Biol. 2006, 16, R551–R560. [Google Scholar] [CrossRef]
- Wang, X. The expanding role of mitochondria in apoptosis. Genes Dev. 2001, 15, 2922–2933. [Google Scholar] [PubMed]
- Green, D.R.; Galluzzi, L.; Kroemer, G. Mitochondria and the autophagy-inflammation-cell death axis in organismal aging. Science 2011, 333, 1109–1112. [Google Scholar] [CrossRef] [PubMed]
- Granneman, J.G.; Li, P.; Zhu, Z.; Lu, Y. Metabolic and cellular plasticity in white adipose tissue I: Effects of beta3-adrenergic receptor activation. Am. J. Physiol. Endocrinol. Metab. 2005, 289, E608–E616. [Google Scholar] [CrossRef] [PubMed]
- Park, A.; Kim, W.K.; Bae, K.H. Distinction of white, beige and brown adipocytes derived from mesenchymal stem cells. World J. Stem Cells 2014, 6, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Johannsen, D.L.; Ravussin, E. The role of mitochondria in health and disease. Curr. Opin. Pharmacol. 2009, 9, 780–786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gregoire, F.M.; Smas, C.M.; Sul, H.S. Understanding adipocyte differentiation. Physiol. Rev. 1998, 78, 783–809. [Google Scholar] [CrossRef] [PubMed]
- Boudina, S.; Graham, T.E. Mitochondrial function/dysfunction in white adipose tissue. Exp. Physiol. 2014, 99, 1168–1178. [Google Scholar] [CrossRef]
- Vernochet, C.; Damilano, F.; Mourier, A.; Bezy, O.; Mori, M.A.; Smyth, G.; Rosenzweig, A.; Larsson, N.G.; Kahn, C.R. Adipose tissue mitochondrial dysfunction triggers a lipodystrophic syndrome with insulin resistance, hepatosteatosis, and cardiovascular complications. FASEB J. 2014, 28, 4408–4419. [Google Scholar] [CrossRef] [Green Version]
- Keuper, M.; Jastroch, M.; Yi, C.X.; Fischer-Posovszky, P.; Wabitsch, M.; Tschop, M.H.; Hofmann, S.M. Spare mitochondrial respiratory capacity permits human adipocytes to maintain ATP homeostasis under hypoglycemic conditions. FASEB J. 2014, 28, 761–770. [Google Scholar] [CrossRef] [PubMed]
- Cinti, S. Transdifferentiation properties of adipocytes in the adipose organ. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E977–E986. [Google Scholar] [CrossRef]
- De Pauw, A.; Tejerina, S.; Raes, M.; Keijer, J.; Arnould, T. Mitochondrial (dys)function in adipocyte (de)differentiation and systemic metabolic alterations. Am. J. Pathol. 2009, 175, 927–939. [Google Scholar] [CrossRef]
- Forner, F.; Kumar, C.; Luber, C.A.; Fromme, T.; Klingenspor, M.; Mann, M. Proteome differences between brown and white fat mitochondria reveal specialized metabolic functions. Cell Metab. 2009, 10, 324–335. [Google Scholar] [CrossRef] [PubMed]
- Tormos, K.V.; Anso, E.; Hamanaka, R.B.; Eisenbart, J.; Joseph, J.; Kalyanaraman, B.; Chandel, N.S. Mitochondrial complex III ROS regulate adipocyte differentiation. Cell Metab. 2011, 14, 537–544. [Google Scholar] [CrossRef] [PubMed]
- Orava, J.; Nuutila, P.; Lidell, M.E.; Oikonen, V.; Noponen, T.; Viljanen, T.; Scheinin, M.; Taittonen, M.; Niemi, T.; Enerback, S.; et al. Different metabolic responses of human brown adipose tissue to activation by cold and insulin. Cell Metab. 2011, 14, 272–279. [Google Scholar] [CrossRef] [PubMed]
- Cannon, B.; Nedergaard, J. Brown adipose tissue: function and physiological significance. Physiol. Rev. 2004, 84, 277–359. [Google Scholar] [CrossRef] [PubMed]
- Yehuda-Shnaidman, E.; Buehrer, B.; Pi, J.; Kumar, N.; Collins, S. Acute stimulation of white adipocyte respiration by PKA-induced lipolysis. Diabetes 2010, 59, 2474–2483. [Google Scholar] [CrossRef] [PubMed]
- Rosell, M.; Kaforou, M.; Frontini, A.; Okolo, A.; Chan, Y.W.; Nikolopoulou, E.; Millership, S.; Fenech, M.E.; MacIntyre, D.; Turner, J.O.; et al. Brown and white adipose tissues: intrinsic differences in gene expression and response to cold exposure in mice. Am. J. Physiol. Endocrinol. Metab. 2014, 306, E945–E964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Enerback, S. The origins of brown adipose tissue. N. Engl. J. Med. 2009, 360, 2021–2023. [Google Scholar] [CrossRef] [PubMed]
- Bartelt, A.; Heeren, J. Adipose tissue browning and metabolic health. Nat. Rev. Endocrinol. 2014, 10, 24–36. [Google Scholar] [CrossRef]
- Nicholls, D.G.; Locke, R.M. Thermogenic mechanisms in brown fat. Physiol. Rev. 1984, 64, 1–64. [Google Scholar] [CrossRef] [PubMed]
- Kajimura, S.; Saito, M. A new era in brown adipose tissue biology: molecular control of brown fat development and energy homeostasis. Annu. Rev. Physiol. 2014, 76, 225–249. [Google Scholar] [CrossRef]
- Nicholls, D.G. Stoicheiometries of proton translocation by mitochondria. Biochem. Soc. Trans. 1977, 5, 200–203. [Google Scholar] [CrossRef] [PubMed]
- Lowell, B.B.; Spiegelman, B.M. Towards a molecular understanding of adaptive thermogenesis. Nature 2000, 404, 652–660. [Google Scholar] [CrossRef] [PubMed]
- Bouillaud, F.; Alves-Guerra, M.C.; Ricquier, D. UCPs, at the interface between bioenergetics and metabolism. Biochim. Biophys. Acta 2016, 1863, 2443–2456. [Google Scholar] [CrossRef] [PubMed]
- Rosen, E.D.; Spiegelman, B.M. Molecular regulation of adipogenesis. Annu. Rev. Cell Dev. Biol. 2000, 16, 145–171. [Google Scholar] [CrossRef] [PubMed]
- Forni, M.F.; Peloggia, J.; Trudeau, K.; Shirihai, O.; Kowaltowski, A.J. Murine Mesenchymal Stem Cell Commitment to Differentiation Is Regulated by Mitochondrial Dynamics. Stem Cells 2016, 34, 743–755. [Google Scholar] [CrossRef] [PubMed]
- Wilson-Fritch, L.; Burkart, A.; Bell, G.; Mendelson, K.; Leszyk, J.; Nicoloro, S.; Czech, M.; Corvera, S. Mitochondrial biogenesis and remodeling during adipogenesis and in response to the insulin sensitizer rosiglitazone. Mol. Cell Biol. 2003, 23, 1085–1094. [Google Scholar] [CrossRef] [PubMed]
- Si, Y.; Palani, S.; Jayaraman, A.; Lee, K. Effects of forced uncoupling protein 1 expression in 3T3-L1 cells on mitochondrial function and lipid metabolism. J. Lipid Res. 2007, 48, 826–836. [Google Scholar] [CrossRef] [Green Version]
- Kajimoto, K.; Terada, H.; Baba, Y.; Shinohara, Y. Essential role of citrate export from mitochondria at early differentiation stage of 3T3-L1 cells for their effective differentiation into fat cells, as revealed by studies using specific inhibitors of mitochondrial di- and tricarboxylate carriers. Mol. Genet. Metab. 2005, 85, 46–53. [Google Scholar] [CrossRef]
- Boneh, A. Regulation of mitochondrial oxidative phosphorylation by second messenger-mediated signal transduction mechanisms. Cell Mol. Life Sci. 2006, 63, 1236–1248. [Google Scholar] [CrossRef]
- Carriere, A.; Carmona, M.C.; Fernandez, Y.; Rigoulet, M.; Wenger, R.H.; Penicaud, L.; Casteilla, L. Mitochondrial reactive oxygen species control the transcription factor CHOP-10/GADD153 and adipocyte differentiation: A mechanism for hypoxia-dependent effect. J. Biol. Chem. 2004, 279, 40462–40469. [Google Scholar] [CrossRef]
- Zhang, Y.; Marsboom, G.; Toth, P.T.; Rehman, J. Mitochondrial respiration regulates adipogenic differentiation of human mesenchymal stem cells. PLoS ONE 2013, 8, e77077. [Google Scholar] [CrossRef] [PubMed]
- Brasaemle, D.L. Thematic review series: adipocyte biology. The perilipin family of structural lipid droplet proteins: stabilization of lipid droplets and control of lipolysis. J. Lipid Res. 2007, 48, 2547–2559. [Google Scholar] [CrossRef] [PubMed]
- Duncan, R.E.; Ahmadian, M.; Jaworski, K.; Sarkadi-Nagy, E.; Sul, H.S. Regulation of lipolysis in adipocytes. Annu. Rev. Nutr. 2007, 27, 79–101. [Google Scholar] [CrossRef] [PubMed]
- Sun, K.; Kusminski, C.M.; Scherer, P.E. Adipose tissue remodeling and obesity. J. Clin. Investig. 2011, 121, 2094–2101. [Google Scholar] [CrossRef] [Green Version]
- Vankoningsloo, S.; Piens, M.; Lecocq, C.; Gilson, A.; De Pauw, A.; Renard, P.; Demazy, C.; Houbion, A.; Raes, M.; Arnould, T. Mitochondrial dysfunction induces triglyceride accumulation in 3T3-L1 cells: role of fatty acid beta-oxidation and glucose. J. Lipid Res. 2005, 46, 1133–1149. [Google Scholar] [CrossRef]
- Vamecq, J.; Dessein, A.F.; Fontaine, M.; Briand, G.; Porchet, N.; Latruffe, N.; Andreolotti, P.; Cherkaoui-Malki, M. Mitochondrial dysfunction and lipid homeostasis. Curr. Drug Metab. 2012, 13, 1388–1400. [Google Scholar] [CrossRef] [PubMed]
- Bournat, J.C.; Brown, C.W. Mitochondrial dysfunction in obesity. Curr. Opin. Endocrinol. Diabetes Obes. 2010, 17, 446–452. [Google Scholar] [CrossRef] [Green Version]
- Wilson-Fritch, L.; Nicoloro, S.; Chouinard, M.; Lazar, M.A.; Chui, P.C.; Leszyk, J.; Straubhaar, J.; Czech, M.P.; Corvera, S. Mitochondrial remodeling in adipose tissue associated with obesity and treatment with rosiglitazone. J. Clin. Investig. 2004, 114, 1281–1289. [Google Scholar] [CrossRef]
- Rong, J.X.; Qiu, Y.; Hansen, M.K.; Zhu, L.; Zhang, V.; Xie, M.; Okamoto, Y.; Mattie, M.D.; Higashiyama, H.; Asano, S.; et al. Adipose mitochondrial biogenesis is suppressed in db/db and high-fat diet-fed mice and improved by rosiglitazone. Diabetes 2007, 56, 1751–1760. [Google Scholar] [CrossRef]
- Chen, X.H.; Zhao, Y.P.; Xue, M.; Ji, C.B.; Gao, C.L.; Zhu, J.G.; Qin, D.N.; Kou, C.Z.; Qin, X.H.; Tong, M.L.; et al. TNF-alpha induces mitochondrial dysfunction in 3T3-L1 adipocytes. Mol. Cell Endocrinol. 2010, 328, 63–69. [Google Scholar] [CrossRef]
- Dahlman, I.; Forsgren, M.; Sjogren, A.; Nordstrom, E.A.; Kaaman, M.; Naslund, E.; Attersand, A.; Arner, P. Downregulation of electron transport chain genes in visceral adipose tissue in type 2 diabetes independent of obesity and possibly involving tumor necrosis factor-alpha. Diabetes 2006, 55, 1792–1799. [Google Scholar] [CrossRef] [PubMed]
- Chattopadhyay, M.; Guhathakurta, I.; Behera, P.; Ranjan, K.R.; Khanna, M.; Mukhopadhyay, S.; Chakrabarti, S. Mitochondrial bioenergetics is not impaired in nonobese subjects with type 2 diabetes mellitus. Metabolism 2011, 60, 1702–1710. [Google Scholar] [CrossRef] [PubMed]
- Heinonen, S.; Buzkova, J.; Muniandy, M.; Kaksonen, R.; Ollikainen, M.; Ismail, K.; Hakkarainen, A.; Lundbom, J.; Lundbom, N.; Vuolteenaho, K.; et al. Impaired Mitochondrial Biogenesis in Adipose Tissue in Acquired Obesity. Diabetes 2015, 64, 3135–3145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischer, B.; Schottl, T.; Schempp, C.; Fromme, T.; Hauner, H.; Klingenspor, M.; Skurk, T. Inverse relationship between body mass index and mitochondrial oxidative phosphorylation capacity in human subcutaneous adipocytes. Am. J. Physiol. Endocrinol. Metab. 2015, 309, E380–E387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taniguchi, C.M.; Emanuelli, B.; Kahn, C.R. Critical nodes in signalling pathways: insights into insulin action. Nat. Rev. Mol. Cell Biol. 2006, 7, 85–96. [Google Scholar] [CrossRef]
- Abel, E.D.; Peroni, O.; Kim, J.K.; Kim, Y.B.; Boss, O.; Hadro, E.; Minnemann, T.; Shulman, G.I.; Kahn, B.B. Adipose-selective targeting of the GLUT4 gene impairs insulin action in muscle and liver. Nature 2001, 409, 729–733. [Google Scholar] [CrossRef]
- Wang, C.H.; Wang, C.C.; Huang, H.C.; Wei, Y.H. Mitochondrial dysfunction leads to impairment of insulin sensitivity and adiponectin secretion in adipocytes. FEBS J. 2013, 280, 1039–1050. [Google Scholar] [CrossRef]
- Sutherland, L.N.; Capozzi, L.C.; Turchinsky, N.J.; Bell, R.C.; Wright, D.C. Time course of high-fat diet-induced reductions in adipose tissue mitochondrial proteins: potential mechanisms and the relationship to glucose intolerance. Am. J. Physiol. Endocrinol. Metab. 2008, 295, E1076–E1083. [Google Scholar] [CrossRef] [Green Version]
- Gao, C.L.; Zhu, C.; Zhao, Y.P.; Chen, X.H.; Ji, C.B.; Zhang, C.M.; Zhu, J.G.; Xia, Z.K.; Tong, M.L.; Guo, X.R. Mitochondrial dysfunction is induced by high levels of glucose and free fatty acids in 3T3-L1 adipocytes. Mol. Cell Endocrinol. 2010, 320, 25–33. [Google Scholar] [CrossRef]
- Ryu, M.J.; Kim, S.J.; Kim, Y.K.; Choi, M.J.; Tadi, S.; Lee, M.H.; Lee, S.E.; Chung, H.K.; Jung, S.B.; Kim, H.J.; et al. Crif1 deficiency reduces adipose OXPHOS capacity and triggers inflammation and insulin resistance in mice. PLoS Genet. 2013, 9, e1003356. [Google Scholar] [CrossRef]
- Wang, W.; Seale, P. Control of brown and beige fat development. Nat. Rev. Mol. Cell Biol. 2016, 17, 691–702. [Google Scholar] [CrossRef] [PubMed]
- Zafrir, B. Brown adipose tissue: Research milestones of a potential player in human energy balance and obesity. Horm. Metab. Res. 2013, 45, 774–785. [Google Scholar] [CrossRef] [PubMed]
- Stanford, K.I.; Middelbeek, R.J.; Townsend, K.L.; An, D.; Nygaard, E.B.; Hitchcox, K.M.; Markan, K.R.; Nakano, K.; Hirshman, M.F.; Tseng, Y.H.; et al. Brown adipose tissue regulates glucose homeostasis and insulin sensitivity. J. Clin. Investig. 2013, 123, 215–223. [Google Scholar] [CrossRef] [PubMed]
- Cypess, A.M.; Kahn, C.R. Brown fat as a therapy for obesity and diabetes. Curr. Opin. Endocrinol. Diabetes Obes. 2010, 17, 143–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanssen, M.J.; Hoeks, J.; Brans, B.; van der Lans, A.A.; Schaart, G.; van den Driessche, J.J.; Jorgensen, J.A.; Boekschoten, M.V.; Hesselink, M.K.; Havekes, B.; et al. Short-term cold acclimation improves insulin sensitivity in patients with type 2 diabetes mellitus. Nat. Med. 2015, 21, 863–865. [Google Scholar] [CrossRef] [PubMed]
- Harms, M.; Seale, P. Brown and beige fat: development, function and therapeutic potential. Nat. Med. 2013, 19, 1252–1263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uldry, M.; Yang, W.; St-Pierre, J.; Lin, J.; Seale, P.; Spiegelman, B.M. Complementary action of the PGC-1 coactivators in mitochondrial biogenesis and brown fat differentiation. Cell Metab. 2006, 3, 333–341. [Google Scholar] [CrossRef] [Green Version]
- Puigserver, P.; Wu, Z.; Park, C.W.; Graves, R.; Wright, M.; Spiegelman, B.M. A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell 1998, 92, 829–839. [Google Scholar] [CrossRef]
- Wikstrom, J.D.; Mahdaviani, K.; Liesa, M.; Sereda, S.B.; Si, Y.; Las, G.; Twig, G.; Petrovic, N.; Zingaretti, C.; Graham, A.; et al. Hormone-induced mitochondrial fission is utilized by brown adipocytes as an amplification pathway for energy expenditure. EMBO J. 2014, 33, 418–436. [Google Scholar] [CrossRef]
- Giralt, M.; Villarroya, F. White, brown, beige/brite: different adipose cells for different functions? Endocrinology 2013, 154, 2992–3000. [Google Scholar] [CrossRef]
- Shabalina, I.G.; Petrovic, N.; de Jong, J.M.; Kalinovich, A.V.; Cannon, B.; Nedergaard, J. UCP1 in brite/beige adipose tissue mitochondria is functionally thermogenic. Cell Rep. 2013, 5, 1196–1203. [Google Scholar] [CrossRef] [PubMed]
- De Matteis, R.; Lucertini, F.; Guescini, M.; Polidori, E.; Zeppa, S.; Stocchi, V.; Cinti, S.; Cuppini, R. Exercise as a new physiological stimulus for brown adipose tissue activity. Nutr. Metab. Cardiovasc. Dis. 2013, 23, 582–590. [Google Scholar] [CrossRef] [PubMed]
- Bostrom, P.; Wu, J.; Jedrychowski, M.P.; Korde, A.; Ye, L.; Lo, J.C.; Rasbach, K.A.; Bostrom, E.A.; Choi, J.H.; Long, J.Z.; et al. A PGC1-alpha-dependent myokine that drives brown-fat-like development of white fat and thermogenesis. Nature 2012, 481, 463–468. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.H.; Wang, C.C.; Wei, Y.H. Mitochondrial dysfunction in insulin insensitivity: implication of mitochondrial role in type 2 diabetes. Ann. N.Y. Acad. Sci. 2010, 1201, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.A.; Wei, Y.; Sowers, J.R. Role of mitochondrial dysfunction in insulin resistance. Circ. Res. 2008, 102, 401–414. [Google Scholar] [CrossRef] [PubMed]
- Loh, K.; Deng, H.; Fukushima, A.; Cai, X.; Boivin, B.; Galic, S.; Bruce, C.; Shields, B.J.; Skiba, B.; Ooms, L.M.; et al. Reactive oxygen species enhance insulin sensitivity. Cell Metab. 2009, 10, 260–272. [Google Scholar] [CrossRef] [PubMed]
- Chouchani, E.T.; Kazak, L.; Spiegelman, B.M. Mitochondrial reactive oxygen species and adipose tissue thermogenesis: Bridging physiology and mechanisms. J. Biol. Chem. 2017, 292, 16810–16816. [Google Scholar] [CrossRef] [Green Version]
- Chouchani, E.T.; Kazak, L.; Jedrychowski, M.P.; Lu, G.Z.; Erickson, B.K.; Szpyt, J.; Pierce, K.A.; Laznik-Bogoslavski, D.; Vetrivelan, R.; Clish, C.B.; et al. Mitochondrial ROS regulate thermogenic energy expenditure and sulfenylation of UCP1. Nature 2016, 532, 112–116. [Google Scholar] [CrossRef] [Green Version]
- Bloch-Damti, A.; Bashan, N. Proposed mechanisms for the induction of insulin resistance by oxidative stress. Antioxid. Redox. Signal. 2005, 7, 1553–1567. [Google Scholar] [CrossRef]
- Evans, J.L.; Goldfine, I.D.; Maddux, B.A.; Grodsky, G.M. Are oxidative stress-activated signaling pathways mediators of insulin resistance and beta-cell dysfunction? Diabetes 2003, 52, 1–8. [Google Scholar] [CrossRef]
- Frank, G.D.; Eguchi, S.; Motley, E.D. The role of reactive oxygen species in insulin signaling in the vasculature. Antioxid. Redox. Signal. 2005, 7, 1053–1061. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.K. Fat uses a TOLL-road to connect inflammation and diabetes. Cell Metab. 2006, 4, 417–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choo, H.J.; Kim, J.H.; Kwon, O.B.; Lee, C.S.; Mun, J.Y.; Han, S.S.; Yoon, Y.S.; Yoon, G.; Choi, K.M.; Ko, Y.G. Mitochondria are impaired in the adipocytes of type 2 diabetic mice. Diabetologia 2006, 49, 784–791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pietilainen, K.H.; Naukkarinen, J.; Rissanen, A.; Saharinen, J.; Ellonen, P.; Keranen, H.; Suomalainen, A.; Gotz, A.; Suortti, T.; Yki-Jarvinen, H.; et al. Global transcript profiles of fat in monozygotic twins discordant for BMI: pathways behind acquired obesity. PLoS Med. 2008, 5, e51. [Google Scholar] [CrossRef] [PubMed]
- Scarpulla, R.C. Metabolic control of mitochondrial biogenesis through the PGC-1 family regulatory network. Biochim. Biophys. Acta 2011, 1813, 1269–1278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pardo, R.; Enguix, N.; Lasheras, J.; Feliu, J.E.; Kralli, A.; Villena, J.A. Rosiglitazone-induced mitochondrial biogenesis in white adipose tissue is independent of peroxisome proliferator-activated receptor gamma coactivator-1alpha. PLoS ONE 2011, 6, e26989. [Google Scholar] [CrossRef] [PubMed]
- Rong, J.X.; Klein, J.L.; Qiu, Y.; Xie, M.; Johnson, J.H.; Waters, K.M.; Zhang, V.; Kashatus, J.A.; Remlinger, K.S.; Bing, N.; et al. Rosiglitazone Induces Mitochondrial Biogenesis in Differentiated Murine 3T3-L1 and C3H/10T1/2 Adipocytes. PPAR Res. 2011, 2011, 179454. [Google Scholar] [CrossRef] [PubMed]
- Kleiner, S.; Mepani, R.J.; Laznik, D.; Ye, L.; Jurczak, M.J.; Jornayvaz, F.R.; Estall, J.L.; Chatterjee Bhowmick, D.; Shulman, G.I.; Spiegelman, B.M. Development of insulin resistance in mice lacking PGC-1alpha in adipose tissues. Proc. Natl. Acad. Sci. USA 2012, 109, 9635–9640. [Google Scholar] [CrossRef]
- Kujoth, G.C.; Bradshaw, P.C.; Haroon, S.; Prolla, T.A. The role of mitochondrial DNA mutations in mammalian aging. PLoS Genet. 2007, 3, e24. [Google Scholar] [CrossRef] [PubMed]
- Okura, T.; Koda, M.; Ando, F.; Niino, N.; Tanaka, M.; Shimokata, H. Association of the mitochondrial DNA 15497G/A polymorphism with obesity in a middle-aged and elderly Japanese population. Hum. Genet. 2003, 113, 432–436. [Google Scholar] [CrossRef]
- Liguori, R.; Mazzaccara, C.; Pasanisi, F.; Buono, P.; Oriani, G.; Finelli, C.; Contaldo, F.; Sacchetti, L. The mtDNA 15497 G/A polymorphism in cytochrome b in severe obese subjects from Southern Italy. Nutr. Metab. Cardiovasc. Dis. 2006, 16, 466–470. [Google Scholar] [CrossRef] [PubMed]
- Larsson, N.G.; Tulinius, M.H.; Holme, E.; Oldfors, A. Pathogenetic aspects of the A8344G mutation of mitochondrial DNA associated with MERRF syndrome and multiple symmetric lipomas. Muscle Nerve Suppl. 1995, 3, S102–S106. [Google Scholar] [CrossRef] [PubMed]
- Westermann, B. Mitochondrial fusion and fission in cell life and death. Nat. Rev. Mol. Cell Biol. 2010, 11, 872–884. [Google Scholar] [CrossRef] [PubMed]
- Wai, T.; Langer, T. Mitochondrial Dynamics and Metabolic Regulation. Trends Endocrinol. Metab. 2016, 27, 105–117. [Google Scholar] [CrossRef] [PubMed]
- Wada, J.; Nakatsuka, A. Mitochondrial Dynamics and Mitochondrial Dysfunction in Diabetes. Acta Med. Okayama 2016, 70, 151–158. [Google Scholar] [PubMed]
- Hales, K.G.; Fuller, M.T. Developmentally regulated mitochondrial fusion mediated by a conserved, novel, predicted GTPase. Cell 1997, 90, 121–129. [Google Scholar] [CrossRef]
- Cipolat, S.; Martins de Brito, O.; Dal Zilio, B.; Scorrano, L. OPA1 requires mitofusin 1 to promote mitochondrial fusion. Proc. Natl. Acad. Sci. USA 2004, 101, 15927–15932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koshiba, T.; Detmer, S.A.; Kaiser, J.T.; Chen, H.; McCaffery, J.M.; Chan, D.C. Structural basis of mitochondrial tethering by mitofusin complexes. Science 2004, 305, 858–862. [Google Scholar] [CrossRef] [PubMed]
- Song, Z.; Ghochani, M.; McCaffery, J.M.; Frey, T.G.; Chan, D.C. Mitofusins and OPA1 mediate sequential steps in mitochondrial membrane fusion. Mol. Biol. Cell 2009, 20, 3525–3532. [Google Scholar] [CrossRef] [PubMed]
- Elgass, K.; Pakay, J.; Ryan, M.T.; Palmer, C.S. Recent advances into the understanding of mitochondrial fission. Biochim. Biophys. Acta 2013, 1833, 150–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benard, G.; Bellance, N.; James, D.; Parrone, P.; Fernandez, H.; Letellier, T.; Rossignol, R. Mitochondrial bioenergetics and structural network organization. J. Cell Sci. 2007, 120 (Pt 5), 838–848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liesa, M.; Shirihai, O.S. Mitochondrial dynamics in the regulation of nutrient utilization and energy expenditure. Cell Metab. 2013, 17, 491–506. [Google Scholar] [CrossRef] [PubMed]
- Blake, R.; Trounce, I.A. Mitochondrial dysfunction and complications associated with diabetes. Biochim. Biophys. Acta 2014, 1840, 1404–1412. [Google Scholar] [CrossRef] [PubMed]
- Bhatti, J.S.; Bhatti, G.K.; Reddy, P.H. Mitochondrial dysfunction and oxidative stress in metabolic disorders—A step towards mitochondria based therapeutic strategies. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1066–1077. [Google Scholar] [CrossRef]
- Kita, T.; Nishida, H.; Shibata, H.; Niimi, S.; Higuti, T.; Arakaki, N. Possible role of mitochondrial remodelling on cellular triacylglycerol accumulation. J. Biochem. 2009, 146, 787–796. [Google Scholar] [CrossRef] [PubMed]
- Boutant, M.; Kulkarni, S.S.; Joffraud, M.; Ratajczak, J.; Valera-Alberni, M.; Combe, R.; Zorzano, A.; Canto, C. Mfn2 is critical for brown adipose tissue thermogenic function. EMBO J. 2017, 36, 1543–1558. [Google Scholar] [CrossRef] [Green Version]
- Mahdaviani, K.; Benador, I.Y.; Su, S.; Gharakhanian, R.A.; Stiles, L.; Trudeau, K.M.; Cardamone, M.; Enriquez-Zarralanga, V.; Ritou, E.; Aprahamian, T.; et al. Mfn2 deletion in brown adipose tissue protects from insulin resistance and impairs thermogenesis. EMBO Rep. 2017, 18, 1123–1138. [Google Scholar] [CrossRef]
- Mancini, G.; Pirruccio, K.; Yang, X.; Bluher, M.; Rodeheffer, M.; Horvath, T.L. Mitofusin 2 in Mature Adipocytes Controls Adiposity and Body Weight. Cell Rep. 2019, 26, 2849–2858. [Google Scholar] [CrossRef]
- Kim, I.; Rodriguez-Enriquez, S.; Lemasters, J.J. Selective degradation of mitochondria by mitophagy. Arch. Biochem. Biophys. 2007, 462, 245–253. [Google Scholar] [CrossRef] [Green Version]
- Ashrafi, G.; Schwarz, T.L. The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ. 2013, 20, 31–42. [Google Scholar] [CrossRef]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narendra, D.P.; Jin, S.M.; Tanaka, A.; Suen, D.F.; Gautier, C.A.; Shen, J.; Cookson, M.R.; Youle, R.J. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010, 8, e1000298. [Google Scholar] [CrossRef] [PubMed]
- Altshuler-Keylin, S.; Kajimura, S. Mitochondrial homeostasis in adipose tissue remodeling. Sci. Signal 2017, 10. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Xiang, Y.; Wang, Y.; Baikati, K.; Cuervo, A.M.; Luu, Y.K.; Tang, Y.; Pessin, J.E.; Schwartz, G.J.; Czaja, M.J. Autophagy regulates adipose mass and differentiation in mice. J. Clin. Investig. 2009, 119, 3329–3339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Goldman, S.; Baerga, R.; Zhao, Y.; Komatsu, M.; Jin, S. Adipose-specific deletion of autophagy-related gene 7 (atg7) in mice reveals a role in adipogenesis. Proc. Natl. Acad. Sci. USA 2009, 106, 19860–19865. [Google Scholar] [CrossRef] [PubMed]
- Kovsan, J.; Bluher, M.; Tarnovscki, T.; Kloting, N.; Kirshtein, B.; Madar, L.; Shai, I.; Golan, R.; Harman-Boehm, I.; Schon, M.R.; et al. Altered autophagy in human adipose tissues in obesity. J. Clin. Endocrinol. Metab. 2011, 96, E268–E277. [Google Scholar] [CrossRef] [PubMed]
- Kraunsoe, R.; Boushel, R.; Hansen, C.N.; Schjerling, P.; Qvortrup, K.; Stockel, M.; Mikines, K.J.; Dela, F. Mitochondrial respiration in subcutaneous and visceral adipose tissue from patients with morbid obesity. J. Physiol. 2010, 588 (Pt 12), 2023–2032. [Google Scholar] [CrossRef]
- Chattopadhyay, M.; Khemka, V.K.; Chatterjee, G.; Ganguly, A.; Mukhopadhyay, S.; Chakrabarti, S. Enhanced ROS production and oxidative damage in subcutaneous white adipose tissue mitochondria in obese and type 2 diabetes subjects. Mol. Cell Biochem. 2015, 399, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.H.; Jeong, Y.T.; Oh, H.; Kim, S.H.; Cho, J.M.; Kim, Y.N.; Kim, S.S.; Kim, D.H.; Hur, K.Y.; Kim, H.K.; et al. Autophagy deficiency leads to protection from obesity and insulin resistance by inducing Fgf21 as a mitokine. Nat. Med. 2013, 19, 83–92. [Google Scholar] [CrossRef]
- Altshuler-Keylin, S.; Shinoda, K.; Hasegawa, Y.; Ikeda, K.; Hong, H.; Kang, Q.; Yang, Y.; Perera, R.M.; Debnath, J.; Kajimura, S. Beige Adipocyte Maintenance Is Regulated by Autophagy-Induced Mitochondrial Clearance. Cell Metab. 2016, 24, 402–419. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.; Fujioka, H.; Joshi, D.; Li, Q.; Sangwung, P.; Hsieh, P.; Zhu, J.; Torio, J.; Sweet, D.; Wang, L.; et al. Mitophagy is required for brown adipose tissue mitochondrial homeostasis during cold challenge. Sci. Rep. 2018, 8, 8251. [Google Scholar] [CrossRef] [PubMed]
- Powell, T.M.; Khera, A. Therapeutic approaches to obesity. Curr. Treat. Options Cardiovasc. Med. 2010, 12, 381–395. [Google Scholar] [CrossRef] [PubMed]
- Bartelt, A.; Bruns, O.T.; Reimer, R.; Hohenberg, H.; Ittrich, H.; Peldschus, K.; Kaul, M.G.; Tromsdorf, U.I.; Weller, H.; Waurisch, C.; et al. Brown adipose tissue activity controls triglyceride clearance. Nat. Med. 2011, 17, 200–205. [Google Scholar] [CrossRef]
- Vegiopoulos, A.; Muller-Decker, K.; Strzoda, D.; Schmitt, I.; Chichelnitskiy, E.; Ostertag, A.; Berriel Diaz, M.; Rozman, J.; Hrabe de Angelis, M.; Nusing, R.M.; et al. Cyclooxygenase-2 controls energy homeostasis in mice by de novo recruitment of brown adipocytes. Science 2010, 328, 1158–1161. [Google Scholar] [CrossRef] [PubMed]
- Seale, P.; Conroe, H.M.; Estall, J.; Kajimura, S.; Frontini, A.; Ishibashi, J.; Cohen, P.; Cinti, S.; Spiegelman, B.M. Prdm16 determines the thermogenic program of subcutaneous white adipose tissue in mice. J. Clin. Investig. 2011, 121, 96–105. [Google Scholar] [CrossRef] [Green Version]
- Petrovic, N.; Walden, T.B.; Shabalina, I.G.; Timmons, J.A.; Cannon, B.; Nedergaard, J. Chronic peroxisome proliferator-activated receptor gamma (PPARgamma) activation of epididymally derived white adipocyte cultures reveals a population of thermogenically competent, UCP1-containing adipocytes molecularly distinct from classic brown adipocytes. J. Biol. Chem. 2010, 285, 7153–7164. [Google Scholar] [PubMed]
- Sidossis, L.; Kajimura, S. Brown and beige fat in humans: thermogenic adipocytes that control energy and glucose homeostasis. J. Clin. Investig. 2015, 125, 478–486. [Google Scholar] [CrossRef]
- Christian, M. Transcriptional fingerprinting of “browning” white fat identifies NRG4 as a novel adipokine. Adipocyte 2015, 4, 50–54. [Google Scholar] [CrossRef]
- Fisher, F.M.; Kleiner, S.; Douris, N.; Fox, E.C.; Mepani, R.J.; Verdeguer, F.; Wu, J.; Kharitonenkov, A.; Flier, J.S.; Maratos-Flier, E.; et al. FGF21 regulates PGC-1alpha and browning of white adipose tissues in adaptive thermogenesis. Genes Dev. 2012, 26, 271–281. [Google Scholar] [CrossRef]
- Wu, J.; Bostrom, P.; Sparks, L.M.; Ye, L.; Choi, J.H.; Giang, A.H.; Khandekar, M.; Virtanen, K.A.; Nuutila, P.; Schaart, G.; et al. Beige adipocytes are a distinct type of thermogenic fat cell in mouse and human. Cell 2012, 150, 366–376. [Google Scholar] [CrossRef]
- Hauner, H. The mode of action of thiazolidinediones. Diabetes Metab. Res. Rev. 2002, 18 (Suppl. 2), S10–S15. [Google Scholar] [CrossRef]
- Tonelli, J.; Li, W.; Kishore, P.; Pajvani, U.B.; Kwon, E.; Weaver, C.; Scherer, P.E.; Hawkins, M. Mechanisms of early insulin-sensitizing effects of thiazolidinediones in type 2 diabetes. Diabetes 2004, 53, 1621–1629. [Google Scholar] [CrossRef] [PubMed]
- Hock, M.B.; Kralli, A. Transcriptional control of mitochondrial biogenesis and function. Annu. Rev. Physiol. 2009, 71, 177–203. [Google Scholar] [CrossRef] [PubMed]
- Bogacka, I.; Xie, H.; Bray, G.A.; Smith, S.R. Pioglitazone induces mitochondrial biogenesis in human subcutaneous adipose tissue in vivo. Diabetes 2005, 54, 1392–1399. [Google Scholar] [CrossRef] [PubMed]
- Fridlyand, L.E.; Philipson, L.H. Reactive species, cellular repair and risk factors in the onset of type 2 diabetes mellitus: review and hypothesis. Curr. Diabetes. Rev. 2006, 2, 241–259. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.; Liu, K.; Tian, C.; Yang, L.; Li, X.; Ren, J.; Packer, L.; Cotman, C.W.; Liu, J. R-alpha-lipoic acid and acetyl-L-carnitine complementarily promote mitochondrial biogenesis in murine 3T3-L1 adipocytes. Diabetologia 2008, 51, 165–174. [Google Scholar] [CrossRef]
- Kelso, G.F.; Porteous, C.M.; Coulter, C.V.; Hughes, G.; Porteous, W.K.; Ledgerwood, E.C.; Smith, R.A.; Murphy, M.P. Selective targeting of a redox-active ubiquinone to mitochondria within cells: antioxidant and antiapoptotic properties. J. Biol. Chem. 2001, 276, 4588–4596. [Google Scholar] [CrossRef]
- Smith, R.A.; Porteous, C.M.; Coulter, C.V.; Murphy, M.P. Selective targeting of an antioxidant to mitochondria. Eur. J. Biochem. 1999, 263, 709–716. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.A.; Murphy, M.P. Mitochondria-targeted antioxidants as therapies. Discov. Med. 2011, 11, 106–114. [Google Scholar]
- Feillet-Coudray, C.; Fouret, G.; Ebabe Elle, R.; Rieusset, J.; Bonafos, B.; Chabi, B.; Crouzier, D.; Zarkovic, K.; Zarkovic, N.; Ramos, J.; et al. The mitochondrial-targeted antioxidant MitoQ ameliorates metabolic syndrome features in obesogenic diet-fed rats better than Apocynin or Allopurinol. Free Radic. Res. 2014, 48, 1232–1246. [Google Scholar] [CrossRef]
- Skulachev, V.P. A biochemical approach to the problem of aging: “megaproject” on membrane-penetrating ions. The first results and prospects. Biochemistry (Mosc.) 2007, 72, 1385–1396. [Google Scholar] [CrossRef] [PubMed]
- Jeong, E.M.; Chung, J.; Liu, H.; Go, Y.; Gladstein, S.; Farzaneh-Far, A.; Lewandowski, E.D.; Dudley, S.C., Jr. Role of Mitochondrial Oxidative Stress in Glucose Tolerance, Insulin Resistance, and Cardiac Diastolic Dysfunction. J. Am. Heart Assoc. 2016, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lonn, E.; Bosch, J.; Yusuf, S.; Sheridan, P.; Pogue, J.; Arnold, J.M.; Ross, C.; Arnold, A.; Sleight, P.; Probstfield, J.; et al. Effects of long-term vitamin E supplementation on cardiovascular events and cancer: A randomized controlled trial. JAMA 2005, 293, 1338–1347. [Google Scholar] [PubMed]
- Sakellariou, G.K.; Pearson, T.; Lightfoot, A.P.; Nye, G.A.; Wells, N.; Giakoumaki, I.I.; Griffiths, R.D.; McArdle, A.; Jackson, M.J. Long-term administration of the mitochondria-targeted antioxidant mitoquinone mesylate fails to attenuate age-related oxidative damage or rescue the loss of muscle mass and function associated with aging of skeletal muscle. FASEB J. 2016, 30, 3771–3785. [Google Scholar] [CrossRef] [PubMed]
- Toledo, F.G.; Menshikova, E.V.; Ritov, V.B.; Azuma, K.; Radikova, Z.; DeLany, J.; Kelley, D.E. Effects of physical activity and weight loss on skeletal muscle mitochondria and relationship with glucose control in type 2 diabetes. Diabetes 2007, 56, 2142–2147. [Google Scholar] [CrossRef] [PubMed]
- Jager, S.; Handschin, C.; St-Pierre, J.; Spiegelman, B.M. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proc. Natl. Acad. Sci. USA 2007, 104, 12017–12022. [Google Scholar] [CrossRef]
- Rodgers, J.T.; Lerin, C.; Haas, W.; Gygi, S.P.; Spiegelman, B.M.; Puigserver, P. Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature 2005, 434, 113–118. [Google Scholar] [CrossRef]
- Omodei, D.; Fontana, L. Calorie restriction and prevention of age-associated chronic disease. FEBS Lett. 2011, 585, 1537–1542. [Google Scholar] [CrossRef] [Green Version]
- Civitarese, A.E.; Carling, S.; Heilbronn, L.K.; Hulver, M.H.; Ukropcova, B.; Deutsch, W.A.; Smith, S.R.; Ravussin, E.; Team, C.P. Calorie restriction increases muscle mitochondrial biogenesis in healthy humans. PLoS Med. 2007, 4, e76. [Google Scholar] [CrossRef]
- Wang, H.; Arias, E.B.; Cartee, G.D. Calorie restriction leads to greater Akt2 activity and glucose uptake by insulin-stimulated skeletal muscle from old rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2016, 310, R449–R458. [Google Scholar] [CrossRef] [Green Version]
- Heilbronn, L.K.; Gan, S.K.; Turner, N.; Campbell, L.V.; Chisholm, D.J. Markers of mitochondrial biogenesis and metabolism are lower in overweight and obese insulin-resistant subjects. J. Clin. Endocrinol. Metab. 2007, 92, 1467–1473. [Google Scholar] [CrossRef] [PubMed]
- Keijer, J.; van Schothorst, E.M. Adipose tissue failure and mitochondria as a possible target for improvement by bioactive food components. Curr. Opin. Lipidol. 2008, 19, 4–10. [Google Scholar] [CrossRef] [PubMed]
- Lalia, A.Z.; Lanza, I.R. Insulin-Sensitizing Effects of Omega-3 Fatty Acids: Lost in Translation? Nutrients 2016, 8, 329. [Google Scholar] [CrossRef] [PubMed]
- Flachs, P.; Horakova, O.; Brauner, P.; Rossmeisl, M.; Pecina, P.; Franssen-van Hal, N.; Ruzickova, J.; Sponarova, J.; Drahota, Z.; Vlcek, C.; et al. Polyunsaturated fatty acids of marine origin upregulate mitochondrial biogenesis and induce beta-oxidation in white fat. Diabetologia 2005, 48, 2365–2375. [Google Scholar] [CrossRef] [PubMed]
- De la Lastra, C.A.; Villegas, I. Resveratrol as an antioxidant and pro-oxidant agent: mechanisms and clinical implications. Biochem. Soc. Trans. 2007, 35 (Pt 5), 1156–1160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lagouge, M.; Argmann, C.; Gerhart-Hines, Z.; Meziane, H.; Lerin, C.; Daussin, F.; Messadeq, N.; Milne, J.; Lambert, P.; Elliott, P.; et al. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1alpha. Cell 2006, 127, 1109–1122. [Google Scholar] [CrossRef] [PubMed]
- Wood dos Santos, T.; Pereira, Q.C.; Teixeira, L.; Gambero, A.; Villena, J.A.; Ribeiro, M.L. Effects of Polyphenols on Thermogenesis and Mitochondrial Biogenesis. Int. J. Mol. Sci. 2018, 19, 2757. [Google Scholar] [CrossRef] [PubMed]
- Andrade, J.M.; Frade, A.C.; Guimaraes, J.B.; Freitas, K.M.; Lopes, M.T.; Guimaraes, A.L.; de Paula, A.M.; Coimbra, C.C.; Santos, S.H. Resveratrol increases brown adipose tissue thermogenesis markers by increasing SIRT1 and energy expenditure and decreasing fat accumulation in adipose tissue of mice fed a standard diet. Eur. J. Nutr. 2014, 53, 1503–1510. [Google Scholar] [CrossRef] [PubMed]
- Moon, H.S.; Lee, H.G.; Choi, Y.J.; Kim, T.G.; Cho, C.S. Proposed mechanisms of (-)-epigallocatechin-3-gallate for anti-obesity. Chem. Biol. Interact. 2007, 167, 85–98. [Google Scholar] [CrossRef]
- Lee, M.S.; Lee, S.; Doo, M.; Kim, Y. Green Tea (-)-Epigallotocatechin-3-Gallate Induces PGC-1alpha Gene Expression in HepG2 Cells and 3T3-L1 Adipocytes. Prev. Nutr. Food Sci. 2016, 21, 62–67. [Google Scholar] [CrossRef]
- Yoneshiro, T.; Matsushita, M.; Hibi, M.; Tone, H.; Takeshita, M.; Yasunaga, K.; Katsuragi, Y.; Kameya, T.; Sugie, H.; Saito, M. Tea catechin and caffeine activate brown adipose tissue and increase cold-induced thermogenic capacity in humans. Am. J. Clin. Nutr. 2017, 105, 873–881. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Mitochondrial Property | White | Brown and Activated Beige | References |
|---|---|---|---|
| Morphology | [20] | ||
|
|
| |
| Content |
|
| [21] |
| Development |
|
| [21] |
| Major function |
|
| [22] |
| UCP1 expression |
|
| [21] |
| Tissue-specific mitochondrial genes |
|
| [22] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, J.H.; Park, A.; Oh, K.-J.; Lee, S.C.; Kim, W.K.; Bae, K.-H. The Role of Adipose Tissue Mitochondria: Regulation of Mitochondrial Function for the Treatment of Metabolic Diseases. Int. J. Mol. Sci. 2019, 20, 4924. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20194924
Lee JH, Park A, Oh K-J, Lee SC, Kim WK, Bae K-H. The Role of Adipose Tissue Mitochondria: Regulation of Mitochondrial Function for the Treatment of Metabolic Diseases. International Journal of Molecular Sciences. 2019; 20(19):4924. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20194924
Chicago/Turabian StyleLee, Jae Ho, Anna Park, Kyoung-Jin Oh, Sang Chul Lee, Won Kon Kim, and Kwang-Hee Bae. 2019. "The Role of Adipose Tissue Mitochondria: Regulation of Mitochondrial Function for the Treatment of Metabolic Diseases" International Journal of Molecular Sciences 20, no. 19: 4924. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20194924